

Abstract
Tenofovir alafenamide (TAF) is an investigational oral prodrug of the HIV-1 nucleotide reverse transcriptase inhibitor tenofovir (TFV). Tenofovir disoproxil fumarate (TDF) is another TFV prodrug, widely used for the treatment of HIV-1 infection. TAF is converted mostly intracellularly to TFV and, in comparison to TDF, achieves higher tenofovir diphosphate (TFV-DP) levels in peripheral blood mononuclear cells. As a result, TAF has demonstrated potent anti-HIV-1 activity at lower doses than TDF in monotherapy studies. Here, the in vitro virology profile of TAF was evaluated and compared to that of TDF. TAF displayed potent antiviral activity against all HIV-1 groups/subtypes, as well as HIV-2. TAF exhibited minimal changes in the drug concentration needed to inhibit 50% of viral spread (EC50) upon removal of the prodrug, similar to TDF, demonstrating intracellular antiviral persistence. While TAF and TDF exhibited comparable potencies in the absence of serum pretreatment, TAF maintained activity in the presence of human serum, whereas TDF activity was significantly reduced. This result demonstrates TAF's improved plasma stability over TDF, which is driven by the different metabolic pathways of the two prodrugs and is key to TAF's improved in vivo antiviral activity. The activity of TAF is specific for HIV, as TAF lacked activity against a large panel of human viruses, with the exception of herpes simplex virus 2, where weak TAF antiviral activity was observed, as previously observed with TFV. Finally, in vitro combination studies with antiretroviral drugs from different classes showed additive to synergistic interactions with TAF, consistent with ongoing clinical studies with TAF in fixed-dose combinations with multiple other antiretroviral drugs for the treatment of HIV.
INTRODUCTION
Tenofovir [(R)-9-(2-phosphonomethoxypropyl)adenine] (TFV) is a human immunodeficiency virus (HIV) nucleotide reverse transcriptase (RT) inhibitor (NtRTI), also frequently referred to as member of the nucleoside RT inhibitor (NRTI) class (1). TFV contains a phosphonate group that is equivalent to the first phosphate group present in natural monophosphate nucleotides (2). In comparison to the natural phosphoric group, the TFV phosphonate moiety has the C—O bond switched, which is not recognized by host enzymes and thus makes it less prone to phosphatase activities. Consequently, TFV activation requires only two phosphorylation steps (3). Tenofovir is intracellularly phosphorylated to the active metabolite tenofovir diphosphate (TFV-DP), which is incorporated into viral DNA by HIV RT and acts as a DNA chain terminator (4). However, the negative charges harbored by TFV from its phosphonate moiety reduce its cellular permeability, limiting its absorption and oral bioavailability. Tenofovir disoproxil fumarate (TDF) (Fig. 1) is a prodrug of TFV widely used for the treatment of HIV-1 infection and is the preferred NtRTI for use in combination with other antiretroviral agents for the treatment of HIV-1 infection (5–7). TDF was developed to improve TFV permeability and to allow systemic delivery of TFV via oral administration. However, TDF is quickly metabolized to TFV by gut and serum esterases in vivo, and high drug exposures are necessary to allow proper loading of target cells.
FIG 1.
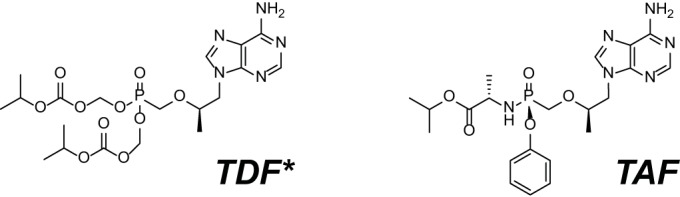
Chemical structures of TAF and TDF. *, the fumarate salt is not represented for TDF.
Tenofovir alafenamide (TAF) (formerly GS-7340) (Fig. 1) is an investigational prodrug of TFV. In contrast to TDF, TAF has been shown to be significantly more stable in blood and plasma (Fig. 2) and is quickly converted to TFV within lymphocytes, improving intracellular accumulation of TFV-DP in HIV target cells (8). The cellular enzyme mostly responsible for the intracellular conversion of TAF has previously been identified as the hydrolase cathepsin A (CatA) or carboxyesterase 1 (CES1), depending on the cell type (9, 10). In preclinical studies in dogs, TAF (administered orally) has been shown to distribute preferentially into peripheral lymphocytes and lymphatic tissues (8). In clinical studies, TAF has demonstrated favorable pharmacological properties, with higher intracellular levels of TFV-DP and lower circulating levels of TFV than of TDF. The higher TFV-DP levels in peripheral blood mononuclear cells (PBMCs) have resulted in improved antiviral activity at lower doses than TDF in monotherapy studies (11, 12). The efficacy of low doses of TAF has been confirmed in phase 2 clinical trials in combination with elvitegravir (EVG, or E), cobicistat (COBI, or C), and emtricitabine (FTC, or F). Moreover, patients receiving E/C/F/TAF experienced decreased changes in kidney function (estimated glomerular filtration rate [eGFR] and tubular proteinuria) and bone mineral density compared to the control arm of E/C/F/TDF (13). No resistance was detected in the E/C/F/TAF arm compared to 3.4% of patients in the E/C/F/TDF arm, supporting the potential improved resistance profile of TAF over TDF (13).
FIG 2.
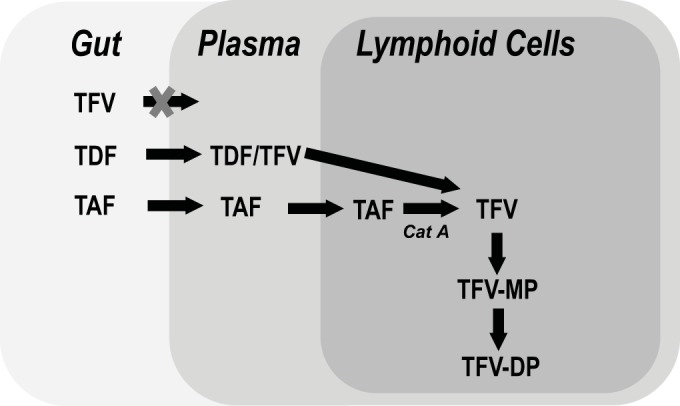
TFV conversion pathways for TAF and TDF. The schematic shows the different pathways by which TAF and TDF are converted to TFV. TAF remains stable in plasma and is mostly converted intracellularly to TFV by cathepsin A (CatA). In contrast, TDF is mostly converted to TFV by gut and plasma hydrolases. For both TAF and TDF, TFV is then converted by cellular kinases to TFV monophosphate (TFV-MP), followed by TFV-DP, the active form of the drug.
This study evaluated the in vitro virology profile of TAF, including its antiviral activity against laboratory and clinical isolates, as well as its selectivity, in vitro potency, stability, persistence, and suitability to be coformulated with other antiretroviral drugs (ARVs).
MATERIALS AND METHODS
TAF, TFV, and TDF were evaluated in parallel for their antiviral activities in several assays using multiple cell types. In assays not specifically evaluating the role of the prodrug moiety, TFV was evaluated rather than TDF due to its better stability in vitro.
Antiviral drugs.
TAF (GS-7340), TDF (GS-4331), TFV (GS-1278), EVG (GS-9137), COBI (GS-9350), and emtricitabine (FTC) (GS-9036) were synthesized by Gilead Sciences. Nevirapine (NVP), raltegravir (RAL), dolutegravir (DTG), atazanavir (ATV), and efavirenz (EFV) were isolated from therapeutic formulations. Darunavir (DRV) was purchased from Toronto Research Chemicals (North York, ON, Canada). Ribavirin (RBV) and didanosine (ddI) were purchased from Sigma (St. Louis, MO). Stavudine (d4T) was supplied by Bristol-Myers Squib (Princeton, NJ). The antiviral control compounds used in the human virus panel were provided by Southern Research Institute (SRI) (Frederick, MD, USA).
Cells and viruses.
MT-2 cells (provided by Stanford University, Palo Alto, CA) and MT-4 cells (AIDS Research and Reference Reagent Program) were maintained in RPMI 1640 medium supplemented with antibiotics and 10% fetal bovine serum (FBS). The cells were passaged twice a week and kept at a density of <0.6 × 106 cells/ml. Human PBMCs were isolated from donor buffy coats (Stanford Blood Bank, Palo Alto, CA), using centrifugation in Ficoll Paque Plus (Amersham Biosciences, Piscataway, NJ), and activated for 5 days in RPMI 1640 medium with 20% FBS, antibiotics, interleukin-2 (IL-2) (20 units/ml), and phytohemagglutinin (PHA) (1 μg/ml). Human CD4 T lymphocytes were purified from activated PBMCs. Macrophages were removed by cell adhesion. CD4+ T cells were purified from the nonadherent fraction (peripheral blood lymphocytes [PBLs]) by negative magnetic bead sorting with an AutoMACS using a CD4+ T cell Isolation Kit II (Miltenyi Biotec). Non-CD4+ T cells were indirectly labeled by using a cocktail of biotin-conjugated antibodies, in combination with anti-biotin microbeads. Purified CD4+ T cells from several donors (up to 4) were pooled to reduce donor variability, improve assay reproducibility, and increase total cell numbers. The HIV-1 strains HIV-1IIIB and BaL (Advanced Biotechnologies, Columbia, MD) were used for infection of MT-2 or MT-4 cells and of primary CD4 cells, respectively. SRI provided the panel of human viruses and cells.
HIV antiviral assay.
MT-2 cells (2.4 million) were incubated with virus for 3 h at 37°C in 1-ml screw-cap tubes. Virus was normalized to yield a signal-to-noise ratio (uninfected cells to infected cells) between 4 and 7, which was equivalent to a multiplicity of infection (MOI) of ∼0.003 for wild-type HIV-1IIIB. Fivefold drug dilutions were transferred in triplicate to the inside wells of 96-well plates (50 μl per well). The signal-to-noise ratio for each plate was calculated from the no-drug control (maximum cell killing) and the 300 μM TFV control (minimum cell killing). After incubation, infected MT-2 cells were seeded at 8,500 per well in 100 μl of medium. After incubation for 5 days at 37°C, 100 μl of CellTiterGlo reagent (Promega, Madison, WI, USA) was added to each well of the assay plates, and luminescence was measured using an Envision plate reader (PerkinElmer, Shelton, CT, USA).
Serum treatment.
Potency experiments were performed in MT-2 cells as described above in the presence of ARV drugs pretreated or mock treated for 30 min in type AB human serum (HS) (catalog number 14-491E; Lonza, Walkersville, MD). The HS was pooled from multiple donors. The final concentration of HS was adjusted to 5% for all samples prior to infection.
Cytotoxicity assays.
Cells (MT-2 and MT-4 cells) were mixed in 96-well plates with 5-fold serial dilutions of compounds at a density of 20,000 cells/well in a final volume of 200 μl. After a 5-day incubation at 37°C, the cells were mixed with CellTiterGlo as described above. Cell viability was expressed as a percentage of the signal from untreated samples (0% cytotoxicity) after subtraction of the signal from samples treated with 1 μM podophyllotoxin (Sigma) (100% cytotoxicity). The drug concentration that reduced cell viability by 50% (CC50) was determined by nonlinear regression analysis (Prism 4; GraphPad, San Diego, CA).
HIV-1 and HIV-2 primary isolate assays.
Infections with HIV-1 primary isolates were conducted at SRI using freshly isolated human PBMCs seronegative for HIV and hepatitis B virus (HBV) (14). Wild-type HIV primary isolates representing HIV-1 groups M (subtypes A to G), N, and O, as well as HIV-2 isolates, were tested for susceptibility to TAF and AZT. Assay readout was performed using an RT activity assay as described below.
CD4 single-cycle infection assay.
Primary CD4+ T lymphocytes were infected with a high-titer stock of HIV-1 BaL at a high multiplicity of infection (MOI = 1) to achieve a strong synchronous infection and virus detection during a single viral cycle (15, 16). After 2 h of virus incubation, the cells were washed twice with medium containing 50% fetal calf serum (FCS) and seeded into 96-well plates at 100,000 cells/well. Serial drug dilutions were added, and 36 h later, the supernatants were collected and virus production was measured using a HIV-1 p24 antigen enzyme-linked immunosorbent assay (ELISA) (Beckman Coulter, Miami, FL). Regression analysis was used to determine the drug concentrations needed to inhibit 50% of viral spread (EC50s).
Antiviral persistence assay in CD4 primary cells.
The antiviral activities of TAF and TDF were determined in an HIV-1 CD4 single-cycle infection assay (see above) using either constant drug exposure (EC50) or pulsed drug exposure (50% persistence concentration [PC50] assay), as previously described (R. M. Ledford, J. E. Vela, A. S. Ray, C. Callebaut, M. D. Miller, and D. J. McColl, presented at the 19th International Conference on Antiviral Research, San Juan, Puerto Rico, 2006). Briefly, in the persistence assay, cells were pulsed with drug for 4 h, with HIV infections limited to the last 2 h; external drug was removed after 4 h by washing with medium containing 50% FCS (see Fig. S1 in the supplemental material). In the constant drug exposure assay, cells were incubated with virus for 2 h, washed twice with medium containing 50% FCS, and incubated in medium plus drugs. As the end of reverse transcription has been established in this experimental system to be ∼6 h postinfection by time-of-addition experiments (not shown), the time for the drugs to act in each setup was ∼4 h. Virus production was determined by p24 antigen ELISA as described above for the CD4 infection assay. The EC50, the PC50, and the relative antiviral persistence ratio (Prel) (PC50/EC50) were determined.
Drug combination studies.
The TAF activity in combination with various ARVs was determined in MT-2 cells infected with HIV-1IIIB. Briefly, compound serial dilutions were performed in 100% dimethyl sulfoxide (DMSO) in 384-well polypropylene plates using a Biomek FX workstation (Beckman Coulter, Inc., Brea, CA). The first compound was serially diluted in nine steps of 1:2 dilutions toward the horizontal direction in plate A, with the second compound serially diluted in seven steps of 1:2 dilutions toward the vertical direction in plate B. Equal volumes of serially diluted compounds were combined in plate C, achieving a defined set of drug concentrations and ratios. For each drug, the starting concentration was selected so that the EC50 was the midpoint concentration tested in plate C. All dilutions were performed in triplicate within the same 384-well plate. Controls containing untreated infected cells and infected cells treated with 300 μM tenofovir were included in each assay plate, representing 0% and 100% inhibition, respectively. The HIV infection was carried out as described above (see “HIV Antiviral Assay”) in a 384-well format with 2,000 cells per well. The DMSO concentration in the final assay wells was 0.4%. After a 5-day incubation at 37°C, the virus-induced cytopathic effect was determined using CellTiter Glo reagent (Promega, Madison, WI), with the signal quantified on a Victor V3 reader (PerkinElmer, Wellesley, MA) following a 15-min incubation. The combination effect (synergy, additivity, and antagonism) of each combination was determined with MacSynergy II software (17) using a previously described algorithm (18). Combination volume values were calculated at the 95% confidence level, and combination effects were ranked as previously defined (19): strong synergy (>100 μM2% [the unit represents the combination volumes resulting from each drug concentration, expressed as the percentage of the control]), moderate synergy (>50 and ≤100 μM2%), minor synergy (>25 and ≤50 μM2%), additivity (≤25 and >−25 μM2%), minor antagonism (≤−25 and >−50 μM2%), moderate antagonism (≤−50 and >−100 μM2%), and strong antagonism (≤−100 μM2%). For each combination study, three independent experiments were performed.
Human virus panel.
TAF antiviral activity against human viruses was evaluated at SRI. Viruses and cell lines were obtained from the American Type Culture Collection (ATCC) or the BEI Research Resource Repository (BEIR) or from specific sources (see Tables S1 and S2 in the supplemental material). All the methodologies utilized at SRI are summarized in the supplemental material.
RESULTS
Antiviral activity of TAF in primary lymphoid cells and T-cell lines.
Previous studies demonstrated the potent in vitro anti-HIV-1 activity of TAF under limited conditions (8). In this study, the antiviral activity of TAF was evaluated in two lymphoblastoid T-cell lines (MT-2 and MT-4) infected with HIV-1IIIB, as well as in PBMCs from multiple donors infected with HIV-1 BaL (Table 1). The drug concentrations needed to inhibit 50% of the viral spread (EC50), as well as to induce 50% cell death (CC50), were determined in 5-day assays, using TFV as a comparator. TAF antiviral activities were similar across all cell types, ranging from 5 to 7 nM, while the CC50 varied from 4.7 to 42 μM for MT-4 and MT-2 cells, respectively. The resulting selectivity index (SI) (the ratio of CC50 to EC50) for TAF was greater than 900 for all 3 cell types (903, 1,385, and 8,853, for MT-4 cells, PBMCs, and MT-2 cells, respectively). The in vitro anti-HIV-1 potency of TAF was greatly improved over that of TFV, likely due to the higher cellular permeability of TAF. TFV antiviral activity and cytotoxicity were in the micromolar (1.4 to 4.2 μM) and millimolar (>1 mM) ranges, respectively. The SI for TFV was greater than 200 for all 3 cell types (>238, >286, and >690 for MT-4 cells, PBMCs, and MT-2 cells, respectively). These results show that TAF has similar antiviral effects with high selectivity across all 3 cell types tested compared to TFV. These results are similar to those previously published showing the anti-HIV activity of TAF across a diverse donor panel of primary CD4 T cells and monocyte-derived macrophages (20). However, TAF and TFV displayed different ranges of activity, mostly driven by differences in their permeabilities, resulting in a 500-fold improvement in anti-HIV-1 activity with TAF compared to TFV in PBMCs (Table 1).
TABLE 1.
Antiviral activity and selectivity of TAF versus TFV
| Cell typea | TAF (μM) |
TFV (μM) |
||||
|---|---|---|---|---|---|---|
| EC50 | CC50 | SI | EC50 | CC50 | SI | |
| MT-2b | 0.005 ± 0.002 | 42 | 8,853 | 1.4 ± 0.5 | >1,000 | >690 |
| MT-4c | 0.005 ± 0.002 | 4.7 | 903 | 4.2 ± 0.8 | >1,000 | >238 |
| PBMCsd | 0.007 ± 0.004 | 9 | 1,385 | 3.5 ± 0.7 | >1,000e | >286 |
Cells were infected with HIV-1IIIB.
EC50 assay, n = 3 experiments; CC50 assay, n = 2 experiments.
EC50 assay, n = 2 experiments; CC50 assay, n = 1 experiment.
EC50 assay, n = 2 experiments; CC50 assay, n = 1 experiment.
Twenty-five percent inhibition was observed at 1,000 μM.
Antiviral activity of TAF against HIV-1 and HIV-2.
The antiviral activity of TAF was evaluated against a panel of HIV-1 and HIV-2 isolates, including HIV-1 group M subtypes A to G, as well as group N and O isolates. Overall, for the 29 primary HIV-1 isolates tested in PBMCs, TAF EC50s ranged from 0.10 to 12.0 nM, with a mean EC50 of 3.5 nM compared to a mean EC50 of 11.8 nM for AZT, which was used as an internal control. For the HIV-2 isolates, the mean EC50s were 1.8 nM for TAF and 6.4 nM for AZT. Overall, there were no significant differences in the mean TAF EC50s for any of the HIV-1 subtypes/groups evaluated, as well as HIV-2, with mean values within ∼3 times that of subtype B HIV-1 (Table 2; see Fig. 5).
TABLE 2.
TAF antiviral activities against HIV primary isolates
| HIV type | Isolate group | Isolate subtype | Isolate | EC50 (nM)a |
|
|---|---|---|---|---|---|
| TAF | AZT | ||||
| HIV-1 | M | A | 92UG029 | 8.75 | 26.4 |
| 92UG037 | 0.69 | 2.12 | |||
| 92RW016 | 0.71 | 1.53 | |||
| B | 93BR021 | 9.69 | 48.0 | ||
| JR-CSF | 1.05 | 1.95 | |||
| 90US873 | 3.87 | 6.03 | |||
| C | 92BR025 | 4.16 | 9.00 | ||
| 98BR004 | 1.75 | 7.56 | |||
| 93IN101 | 1.50 | 5.49 | |||
| D | 92UG001 | 3.79 | 11.3 | ||
| 92UG046 | 0.99 | 3.33 | |||
| 92UG024 | 6.27 | 7.21 | |||
| E | 93TH073 | 1.18 | 3.54 | ||
| CMU06 | 1.36 | 4.09 | |||
| CMU08 | 5.88 | 13.1 | |||
| F | 93BR019 | 0.73 | 4.87 | ||
| 92BR024 | 5.97 | 17.1 | |||
| 93BR029 | 0.14 | 1.28 | |||
| 93BR020 | 2.22 | 7.41 | |||
| G | G3 | 5.34 | 14.0 | ||
| RU570 | 12.0 | 66.4 | |||
| JV1083 | 9.85 | 27.4 | |||
| N | NAb | YBF30 | 1.98 | 5.23 | |
| O | NA | BCF02 | 1.30 | 1.23 | |
| BCF03 | 3.01 | 7.20 | |||
| BCF07 | 0.10 | 3.81 | |||
| HIV-2 | NA | NA | CDC310319 | 2.63 | 15.6 |
| CDC310342 | 1.96 | 1.39 | |||
| CBL-20 | 0.91 | 2.18 | |||
The mean TAF EC50 values for each subtype/group were as follows: A, 3.4 nM; B, 4.9 nM; C, 2.5 nM; D, 3.7 nM; E, 2.8 nM; F, 2.3 nM; G, 9.1 nM; N, not applicable; O, 1.5 nM; HIV-2, 1.8 nM.
NA, not applicable.
FIG 5.
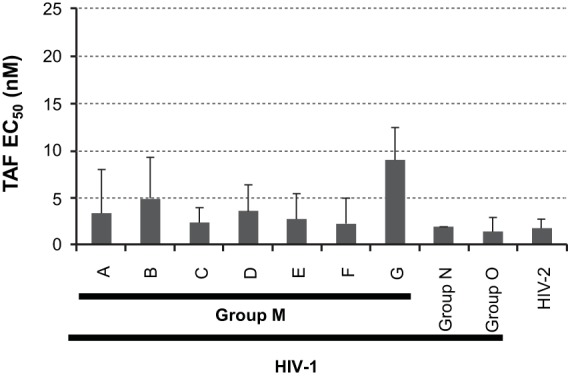
TAF activities against primary HIV isolates. The mean EC50s and standard deviations are shown for HIV-1 group M subtype A to G, group N, and group O isolates, as well as HIV-2 isolates. The mean EC50s and standard deviations were calculated from the data summarized in Table 2.
Persistence of TAF antiviral activity.
TDF has been shown to have a long intracellular half-life (as TFV-DP) in clinical studies (21). An assay was developed to assess the persistence of TAF compared to that of TDF as a prototype compound with a prolonged effect. Two experimental conditions were evaluated, in which primary CD4 T cells were subjected to either constant drug incubation or pulsed drug incubation (see Fig. S1 in the supplemental material). Under both conditions (constant drug or pulsed drug incubation), the effective drug exposure was 4 h, as drug incubations started 2 h after initial infection and reverse transcription is usually completed about 6 h postinfection. Antiviral activity was measured 48 h postinfection. The antiviral activity of TAF was measured at 7.1 nM (EC50) under constant drug incubation (“standard” conditions) (Fig. 3). Antiviral activity was maintained within 2 times the standard conditions under pulsed drug incubation (“persistence” conditions), where it was measured at 11.2 nM (PC50). Similar to TAF, the EC50 and PC50 for TDF were within 2-fold of each other (EC50, 5.5 nM; PC50, 5.4 nM). The calculated PREL (PC50/EC50) values for TAF and TDF were 1.6 and 1, respectively, indicating less than a 2-fold change in antiviral activity over a 24-h period in primary CD4 T cells. In contrast, when a protease inhibitor (PI) (22) was evaluated under the same conditions, the calculated PREL value was >1,000, indicating at least a 200-fold decrease in antiretroviral activity under the same experimental conditions (data not shown).
FIG 3.
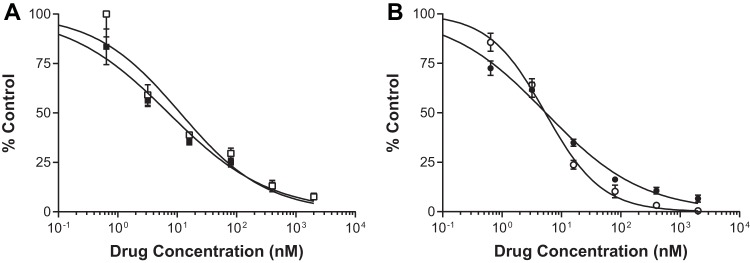
Antiviral activities for TAF (A) and TDF (B) in the persistence assay. The solid symbols indicate constant drug conditions, and the open symbols indicate pulsed drug conditions (see Fig. S1 in the supplemental material for experimental details). Samples were evaluated for antiviral efficacy with triplicate measurements; standard deviations are indicated by the error bars.
Antiviral activity and stability of TAF in human serum.
In vitro, TAF has been shown to be significantly more stable in blood and plasma than TDF (8) (Fig. 2). To determine if the greater stability resulted in higher potency, TAF in vitro activity was evaluated in MT-2 cells using a 5-day infectivity assay, with drug pretreated for 30 min in medium (standard conditions) or in 100% HS to mimic plasma exposure. As shown in Fig. 4, the potencies of the two TFV prodrugs (TAF and TDF) were similar in the absence of HS pretreatment. In contrast, TAF maintained its potency after HS pretreatment, reflecting its known plasma stability, while TDF activity was more than 90% reduced after HS pretreatment.
FIG 4.
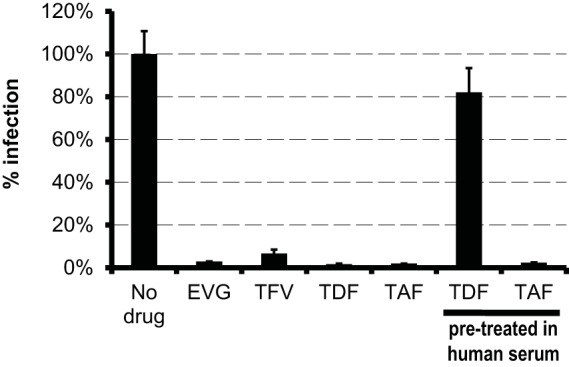
Comparative potencies and stabilities of TAF and TDF. The potency was assessed for each drug tested at 10× the EC50 against HIV-1IIIB in MT-2 cells. TAF and TDF were also pretreated in human serum for 30 min. Human serum was adjusted for all drugs to a final concentration of 5%. The error bars indicate standard deviations.
TAF antiviral activity against human viruses.
Antiviral activity assays against a panel of 18 human viruses were performed with TAF and TFV (Table 3). The result for each virus was validated with a control drug that exhibited the expected levels of antiviral activity (data not shown). With the exception of HIV-1 and simian immunodeficiency virus (SIV) isolates, which were potently inhibited by TAF and TFV, no antiviral activity was observed against 12 of the remaining 14 human viruses evaluated (Table 3). TFV weakly inhibited the herpes simplex virus 2 (HSV-2) strain KW (a clinical isolate) with an EC50 of 146 μM, which is consistent with data previously published (23). TAF had an EC50 of 424 nM for that viral isolate. Overall, TAF and TFV activities against HSV-2 KW were 150- to 200-fold weaker than those against HIV. Similarly weak activity was observed for HSV-2 MS (a laboratory isolate). The only other human virus that TAF inhibited was human parainfluenza virus, with an EC50 of 843 nM. While the EC50s against human parainfluenza virus indicate minimal TAF antiviral activity, cell growth inhibition was observed at the 1,000 nM concentration; thus, the observed effect may be due to cell inhibition. Neither TAF nor TFV exhibited cytotoxicity up to the highest concentrations used for these evaluations (1,000 nM for TAF and 1,000 μM for TFV). Overall, the results indicate TAF is a potent inhibitor of immunodeficiency viruses, such as HIV and SIV, and a weak inhibitor of HSV-2.
TABLE 3.
TAF antiviral activities against human viruses
| Virus | TAF |
Tenofovir |
Positive control | ||
|---|---|---|---|---|---|
| EC50 (nM) | CC50 (nM) | EC50 (μM) | CC50 (μM) | ||
| HIV-1 NL4-3 | 5.8 | >1,000 | 2.3 | >100 | AZT |
| HIV-1 BaL | 2.0 | >1,000 | 1.0 | >100 | AZT |
| SIV mac239 | 1.21 | >1,000 | 0.73 | >1,000 | AZT |
| SIV mac251 | 0.51 | >1,000 | 0.35 | >1,000 | AZT |
| Adenovirus | >1,000 | >1,000 | >1,000 | >1,000 | Ribavirin |
| Dengue virus | >1,000 | >1,000 | >1,000 | >1,000 | Ribavirin |
| Hepatitis C virus | >1,000 | >1,000 | >1,000 | >1,000 | rIFN-αe |
| Coxsackie B virus | >1,000 | >1,000 | >1,000 | >1,000 | Enviroxime |
| Rhinovirus | >1,000 | >1,000 | >1,000 | >1,000 | Enviroxime |
| Vaccinia virus | >1,000 | >1,000 | >1,000 | >1,000 | Cidofovir |
| Influenza A virus | >1,000 | >1,000 | >1,000 | >1,000 | Zanamivir |
| Human parainfluenza virus | 843 | >1,000c | >1,000 | >1,000 | Enviroxime |
| Respiratory syncytial virus | >1,000 | >1,000 | >1,000 | >1,000 | Ribavirin |
| VZVa | >1,000 | >1,000 | >1,000 | >1,000 | Acyclovir |
| HCMVb | >1,000 | >1,000 | >1,000 | >1,000 | Ganciclovir |
| HSV-1 | >1,000 | >1,000 | >1,000d | >1,000 | Acyclovir |
| HSV-2 (KW) | 424 | >1,000 | 146 | >1,000 | Acyclovir |
| HSV-2 (MS) | 697 | >1,000 | 278 | >1,000 | Acyclovir |
VZV, varicella-zoster virus.
HCMV, human cytomegalovirus.
Cell growth inhibition was observed at the highest concentration.
Viral inhibition was observed at the highest concentration.
rIFN-α, recombinant alpha interferon.
Combination studies with TAF and other ARVs.
The in vitro antiretroviral activity of TAF was tested in various combinations with drugs representing the major ARV classes, including NRTIs (TFV and FTC), nonnucleoside reverse transcriptase inhibitors (NNRTIs) (EFV and NVP), integrase strand transfer inhibitors (INSTIs) (EVG, RAL, and DTG), and PIs (ATV and DRV). Combinations of ddI plus RBV, d4T plus RBV, and TAF with itself were used as controls for synergy, antagonism, and additivity, respectively. The combination of TAF with TFV resulted in an additive effect, as expected, as both deliver TFV-DP to cells. When combined with any of the NRTIs or NNRTIs, TAF exhibited moderate to high synergistic effects, with synergy scores ranging from 41 to 131 (Table 4). The combination of TAF with INSTIs resulted in the highest level of synergy (271, 205, and 179 for EVG, RAL, and DTG, respectively). Moderate synergy resulted when TAF was combined with PIs, with synergy scores of 96 and 56 for ATV and DRV, respectively (Table 4). The synergy values observed for TAF with all these drugs were comparable to that of TFV, evaluated in parallel (not shown), and to the values previously reported for TFV (24). Importantly, none of the drug combinations containing TAF exhibited antagonistic antiviral effects.
TABLE 4.
TAF antiviral activities in combination with other antiretroviral drugs
| Drug combination | Class | Net effect | Synergy scorea | Antagonism scorea |
|---|---|---|---|---|
| TAF + TFV | NRTI | Additive | 24 | −14 |
| TAF + FTC | NRTI | Strong synergy | 131 | −9 |
| TAF + EFV | NNRTI | Moderate synergy | 100 | −7 |
| TAF + NVP | NNRTI | Slight synergy | 41 | −14 |
| TAF + EVG | INSTI | Strong synergy | 271 | −9 |
| TAF + RAL | INSTI | Strong synergy | 205 | −10 |
| TAF + DTG | INSTI | Strong synergy | 179 | −10 |
| TAF + ATV | PI | Moderate synergy | 96 | −10 |
| TAF + DRV | PI | Moderate synergy | 56 | −12 |
| TAF + COBI | PK enhancer | Additive | 17 | −22 |
| TAF + TAF | Control | Additive | 20 | −17 |
| ddI + RBV | Control | Strong synergy | 302 | −20 |
| d4T+ RBV | Control | Strong antagonism | 20 | −340 |
The data shown represent the means of the results of >3 independent experiments performed in triplicate.
DISCUSSION
The experiments described in this report were conducted to characterize the in vitro virology activity of TAF, an investigational prodrug of TFV, which is currently being evaluated in clinical trials in combination with other anti-HIV-1 drugs. Compared to TDF, TAF demonstrated either similar or improved characteristics in all in vitro assays. These results are aligned with phase 1/2 clinical trial results showing the superior antiviral activity of TAF compared to TDF, even with the dose of TAF being 1/10 of the TDF dose (11, 12).
The antiviral activity assays performed in multiple cell types with a panel of laboratory-derived and clinical isolates demonstrated that TAF has nanomolar potency against HIV-1 and HIV-2 (Fig. 5). These results are comparable to what has been observed with TDF, which has been shown to be effective globally in HIV-infected individuals (2). In addition, drug combination studies with TAF demonstrated synergistic effects with all drug classes evaluated and no observed antagonism. These results support combining TAF with elvitegravir, cobicistat, and emtricitabine (E/C/F/TAF), as currently evaluated in clinical studies. Furthermore, the results support the use of TAF or FTC/TAF as part of an alternative treatment regimen with the potential to be combined with ARVs from other classes, including NNRTIs, PIs, and INSTIs.
The antiviral specificity of TAF is also identical to those of TFV/TDF, with potent activity observed only with HIV and SIV isolates. Another study has recently demonstrated potent TAF activity against HBV (28). Modest antiviral activity was observed with TAF against both HSV-2 isolates. These results were not unexpected, given the results of the CAPRISA-004 study, which investigated 1% TFV vaginal gel for preexposure prophylaxis (PrEP) (25). In addition to observing a 39% reduction in HIV infection, a 51% reduction in HSV-2 infection was also observed in this PrEP study, which has been attributed to the high local concentration of TFV present in the cervicovaginal fluid and relevant tissues following treatment with 1% tenofovir gel (23). However, as these concentrations of TFV are not likely to be achieved through oral delivery, no antiherpetic activity is anticipated from the TAF oral formulation currently in clinical development.
Tenofovir has been shown to have both a long plasma half-life (17 h) and a long intracellular half-life (>60 h) in PBMCs from patients on TDF-containing regimens (21, 26). In addition, the pharmacokinetics (PK) of intracellular TFV-DP is likely the primary contributor to establishing and maintaining antiviral suppression, even after missed doses (21, 26). Similar to what has been observed in vivo, the in vitro persistence assay results from this study demonstrate that TDF maintains its antiviral activity over a full viral cycle. TAF demonstrated comparable maintenance of antiviral activity, suggesting that in the clinical setting, TAF should have an intracellular pharmacokinetic profile similar to that of TDF. A similar study found that TFV had a PREL value of 2.4, while abacavir (ABC) had a PREL value of >243 (R. M. Ledford, J. E. Vela, A. S. Ray, C. Callebaut, M. D. Miller, and D. J. McColl, presented at the 19th International Conference on Antiviral Research, San Juan, Puerto Rico, 2006), highlighting the persistence of TFV and its prodrugs compared to another NRTI. Such high intracellular persistence of TFV prodrugs may be due to the chemical linkage of the phosphonate group, which imparts a stable negative charge to TFV, thus reducing cell membrane permeability relative to those of unphosphorylated parent nucleoside analogs.
While TAF and TDF are both prodrugs of TFV, their conversion to TFV occurs by different pathways. TAF has been shown to be stable in plasma, resulting in the delivery of TAF to its target cells, followed by intracellular conversion to TFV (8), with the first step being driven by CatA or CES1, depending on the cell type (9). In contrast, most TDF is converted to TFV by gut and serum hydrolases extracellularly, resulting in delivery of the less permeable TFV to the target cells (Fig. 2). The human serum stability data in this study reflect the differential sensitivities of the two prodrugs to serum hydrolases, with TAF maintaining its potency after serum pretreatment and TDF losing most of its potency after serum pretreatment.
The improved stability of TAF (8) leads to the improvement of two PK parameters compared to TDF in vivo. First, the increased TAF stability results in a decrease in TFV systemic exposure, reducing the potential for bone and renal adverse events, which may be related to overall TFV exposure. This is supported by the week 48 data from study GS-US-292-0102, where patients receiving E/C/F/TAF experienced reduced changes in kidney function (eGFR and tubular proteinuria) and bone mineral density compared to patients receiving E/C/F/TDF (13). Second, the improved stability of TAF results in an increase in intracellular TFV-DP, which allows a lower dose of TAF to be used in clinical settings. In a phase 1 study, 8 mg TAF produced viral load (VL) declines comparable to those with 300 mg TDF over a 10-day period (11). In the same study, the 25-mg TAF dose resulted in better reduction of the VL, as well as 5 times more TFV-DP in PBMCs, than a 300-mg TDF dose. Finally, the increase in TFV-DP levels may also lead to an improved resistance profile for TAF over TDF. In vitro, the resistance profile of TAF is similar to that of TDF/TFV (see our accompanying paper [27]). However, as previously demonstrated, the intracellular TFV-DP levels achieved with TAF were 5 times higher than those with TDF. Therefore, the TAF inhibitory quotient (IQ) should be 5 times higher than that of TDF, suggesting that TAF could have the potential to inhibit previously defined TDF-resistant viruses and may have a higher resistance barrier. In support of this observation, a 48-week phase 2 clinical study comparing E/C/F/TAF versus E/C/F/TDF has shown that no patient receiving E/C/F/TAF developed resistance, whereas 3.4% of patients receiving E/C/F/TDF developed resistance (13). Additionally, very low levels of resistance (<1%) were observed in ongoing phase 3 studies after 48 weeks of treatment (29).
In summary, this study has demonstrated that TAF either equals or improves upon the in vitro virologic profile of TDF. Ongoing clinical studies will determine the clinical benefits of TAF.
Supplementary Material
ACKNOWLEDGMENTS
We thank the personnel at SRI for drug evaluation against viral isolates, Tomas Cihlar for input into the development of the persistence assay design, and Kathryn Kitrinos for helpful discussions and critical reading of the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01152-15.
REFERENCES
- 1.Balzarini J, Holý A, Jindrich J, Naesens L, Snoeck R, Schols D, De Clercq E. 1993. Differential antiherpesvirus and antiretrovirus effects of the (S) and (R) enantiomers of acyclic nucleoside phosphonates: potent and selective in vitro and in vivo antiretrovirus activities of (R)-9-(2-phosphonomethoxypropyl)-2,6-diaminopurine. Antimicrob Agents Chemother 37:332–338. doi: 10.1128/AAC.37.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee WA, Martin JC. 2006. Perspectives on the development of acyclic nucleotide analogs as antiviral drugs. Antivir Res 71:254–259. doi: 10.1016/j.antiviral.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 3.Robbins BL, Greenhaw JJ, Connelly MC, Fridland A. 1995. Metabolic pathways for activation of the antiviral agent 9-(2-phosphonylmethoxyethyl)adenine in human lymphoid cells. Antimicrob Agents Chemother 39:2304–2308. doi: 10.1128/AAC.39.10.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suo Z, Johnson KA. 1998. Selective inhibition of HIV-1 reverse transcriptase by an antiviral inhibitor, (R)-9-(2-phosphonylmethoxypropyl)adenine. J Biol Chem 273:27250–27258. doi: 10.1074/jbc.273.42.27250. [DOI] [PubMed] [Google Scholar]
- 5.Department of Health and Human Services. 1 May 2014. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. [Google Scholar]
- 6.Williams I, Churchill D, Anderson J, Boffito M, Bower M, Cairns G, Cwynarski K, Edwards S, Fidler S, Fisher M, Freedman A, Geretti AM, Gilleece Y, Horne R, Johnson M, Khoo S, Leen C, Marshall N, Nelson M, Orkin C, Paton N, Phillips A, Post F, Pozniak A, Sabin C, Trevelion R, Ustianowski A, Walsh J, Waters L, Wilkins E, Winston A, Youle M. 2014. British HIV Association guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy 2012. HIV Med 15(Suppl 1):S1–S85. doi: 10.1111/hiv.12119. [DOI] [PubMed] [Google Scholar]
- 7.European AIDS Clinical Society. 2014. European treatment guidelines version 7.02. European AIDS Clinical Society, Brussels, Belgium. [Google Scholar]
- 8.Lee WA, He G-X, Eisenberg E, Cihlar T, Swaminathan S, Mulato A, Cundy KC. 2005. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob Agents Chemother 49:1898–1906. doi: 10.1128/AAC.49.5.1898-1906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Birkus G, Kutty N, He GX, Mulato A, Lee W, McDermott M, Cihlar T. 2008. Activation of 9-[(R)-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]phenoxyphosphinyl]-methoxy]propyl]adenine (GS-7340) and other tenofovir phosphonoamidate prodrugs by human proteases. Mol Pharmacol 74:92–100. doi: 10.1124/mol.108.045526. [DOI] [PubMed] [Google Scholar]
- 10.Birkus G, Wang R, Liu XH, Kutty N, MacArthur H, Cihlar T, Gibbs C, Swaminathan S, Lee W, McDermott M. 2007. Cathepsin A is the major hydrolase catalyzing the intracellular hydrolysis of the antiretroviral nucleotide phosphonoamidate prodrugs GS-7340 and GS-9131. Antimicrob Agents Chemother 51:543–550. doi: 10.1128/AAC.00968-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruane PJ, Dejesus E, Berger D, Markowitz M, Bredeek UF, Callebaut C, Zhong L, Ramanathan S, Rhee MS, Fordyce MW, Yale K. 2013. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of tenofovir alafenamide as 10-day monotherapy in HIV-1-positive adults. J Acquir Immune Defic Syndr 63:449–455. doi: 10.1097/QAI.0b013e3182965d45. [DOI] [PubMed] [Google Scholar]
- 12.Markowitz M, Zolopa A, Squires K, Ruane P, Coakley D, Kearney B, Zhong L, Wulfsohn M, Miller MD, Lee WA. 2014. Phase I/II study of the pharmacokinetics, safety and antiretroviral activity of tenofovir alafenamide, a new prodrug of the HIV reverse transcriptase inhibitor tenofovir, in HIV-infected adults. J Antimicrob Chemother 69:1362–1369. doi: 10.1093/jac/dkt532. [DOI] [PubMed] [Google Scholar]
- 13.Sax PE, Zolopa A, Brar I, Elion R, Ortiz R, Post F, Wang H, Callebaut C, Martin H, Fordyce MW, McCallister S. 2014. Tenofovir alafenamide vs. tenofovir disoproxil fumarate in single tablet regimens for initial HIV-1 therapy: a randomized phase 2 study. J Acquir Immune Defic Syndr 67:52–58. doi: 10.1097/QAI.0000000000000225. [DOI] [PubMed] [Google Scholar]
- 14.Ptak RG, Gallay PA, Jochmans D, Halestrap AP, Ruegg UT, Pallansch LA, Bobardt MD, de Bethune MP, Neyts J, De Clercq E, Dumont JM, Scalfaro P, Besseghir K, Wenger RM, Rosenwirth B. 2008. Inhibition of human immunodeficiency virus type 1 replication in human cells by Debio-025, a novel cyclophilin binding agent. Antimicrob Agents Chemother 52:1302–1317. doi: 10.1128/AAC.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ray AS, Vela JE, Boojamra CG, Zhang L, Hui H, Callebaut C, Stray K, Lin KY, Gao Y, Mackman RL, Cihlar T. 2008. Intracellular metabolism of the nucleotide prodrug GS-9131, a potent anti-human immunodeficiency virus agent. Antimicrob Agents Chemother 52:648–654. doi: 10.1128/AAC.01209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cihlar T, Ray AS, Boojamra CG, Zhang L, Hui H, Laflamme G, Vela JE, Grant D, Chen J, Myrick F, White KL, Gao Y, Lin KY, Douglas JL, Parkin NT, Carey A, Pakdaman R, Mackman RL. 2008. Design and profiling of GS-9148, a novel nucleotide analog active against nucleoside-resistant variants of human immunodeficiency virus type 1, and its orally bioavailable phosphonoamidate prodrug, GS-9131. Antimicrob Agents Chemother 52:655–665. doi: 10.1128/AAC.01215-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prichard MN, Aseltine KR, Shipman C Jr. 1993. MacSynergy II, version 1.0. University of Michigan, Ann Arbor, MI. [Google Scholar]
- 18.Prichard MN, Prichard LE, Shipman C Jr. 1993. Strategic design and three-dimensional analysis of antiviral drug combinations. Antimicrob Agents Chemother 37:540–545. doi: 10.1128/AAC.37.3.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prichard MN, Shipman C Jr. 1990. A three-dimensional model to analyze drug-drug interactions. Antivir Res 14:181–205. doi: 10.1016/0166-3542(90)90001-N. [DOI] [PubMed] [Google Scholar]
- 20.Bam RA, Birkus G, Babusis D, Cihlar T, Yant SR. 2014. Metabolism and antiretroviral activity of tenofovir alafenamide in CD4 T-cells and macrophages from demographically diverse donors. Antivir Ther 19:669–677. doi: 10.3851/IMP2767. [DOI] [PubMed] [Google Scholar]
- 21.Back DJ, Burger DM, Flexner CW, Gerber JG. 2005. The pharmacology of antiretroviral nucleoside and nucleotide reverse transcriptase inhibitors: implications for once-daily dosing. J Acquir Immune Defic Syndr 39(Suppl 1):S1–S23. doi: 10.1097/01.qai.0000168882.67942.3f. [DOI] [PubMed] [Google Scholar]
- 22.Callebaut C, Stray K, Tsai L, Williams M, Yang ZY, Cannizzaro C, Leavitt SA, Liu X, Wang K, Murray BP, Mulato A, Hatada M, Priskich T, Parkin N, Swaminathan S, Lee W, He GX, Xu L, Cihlar T. 2011. In vitro characterization of GS-8374, a novel phosphonate-containing inhibitor of HIV-1 protease with a favorable resistance profile. Antimicrob Agents Chemother 55:1366–1376. doi: 10.1128/AAC.01183-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrei G, Lisco A, Vanpouille C, Introini A, Balestra E, van den Oord J, Cihlar T, Perno CF, Snoeck R, Margolis L, Balzarini J. 2011. Topical tenofovir, a microbicide effective against HIV, inhibits herpes simplex virus-2 replication. Cell Host Microbe 10:379–389. doi: 10.1016/j.chom.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mulato AS, Cherrington JM. 1997. Anti-HIV activity of adefovir (PMEA) and PMPA in combination with antiretroviral compounds: in vitro analyses. Antivir Res 36:91–97. doi: 10.1016/S0166-3542(97)00043-0. [DOI] [PubMed] [Google Scholar]
- 25.Abdool Karim Q, Abdool Karim SS, Frohlich JA, Grobler AC, Baxter C, Mansoor LE, Kharsany AB, Sibeko S, Mlisana KP, Omar Z, Gengiah TN, Maarschalk S, Arulappan N, Mlotshwa M, Morris L, Taylor D. 2010. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 329:1168–1174. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hawkins T, Veikley W, St Claire RLI, Guyer B, Clark N, Kearney BP. 2005. Intracellular pharmacokinetics of tenofovir diphosphate, carbovir triphosphate, and lamivudine triphosphate in patients receiving triple-nucleoside regimens. J Acquir Immune Defic Syndr 39:406–411. doi: 10.1097/01.qai.0000167155.44980.e8. [DOI] [PubMed] [Google Scholar]
- 27.Margot NA, Johnson A, Miller MD, Callebaut C. 2015. Characterization of HIV-1 resistance to tenofovir alafenamide in vitro. Antimicrob Agents Chemother 59:5917–5924. doi: 10.1128/AAC.01151-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agarwal K, Fung SK, Nguyen TT, Cheng W, Sicard E, Ryder SD, Flaherty JF, Lawson E, Zhao S, Subramanian GM, McHutchison JG, Gane EJ, Foster GR. 2015. Twenty-eight day safety, antiviral activity, and pharmacokinetics of tenofovir alafenamide for treatment of chronic hepatitis B infection. J Hepatol 62:533–540. doi: 10.1016/j.jhep.2014.10.035. [DOI] [PubMed] [Google Scholar]
- 29.Sax PE, Wohl D, Yin MT, Post F, DeJesus E, Saag M, Pozniak A, Thompson M, Podzamczer D, Molina JM, Oka S, Koenig E, Trottier B, Andrade-Villanueva J, Crofoot G, Custodio JM, Plummer A, Zhong L, Cao H, Martin H, Callebaut C, Cheng AK, Fordyce MW, McCallister S, GS-US-292-0104/0111 Study Team . 2015. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet 385:2606–2615. doi: 10.1016/S0140-6736(15)60616-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.