

Abstract
Although suramin (Sur) is suggested as a potential drug candidate in the management of Chagas disease, this issue has not been objectively tested. In this study, we examined the applicability of concomitant treatment with benznidazole (Bz) and suramin in mice infected with a virulent strain of Trypanosoma cruzi. Eighty 12-week-old male C57BL/6 mice were equally randomized in eight groups: (i) noninfected mice (negative control) and mice infected with T. cruzi Y strain receiving (ii) no treatment (positive control), (iii) Bz, 100 mg/kg of body weight per day, (iv) Sur, 20 mg/kg/day, and (v to viii) Sur, 20 mg/kg/day, combined with Bz, 100, 50, 25, or 5 mg/kg/day. Bz was administered by gavage, and Sur was administered intraperitoneally. Sur dramatically increased the parasitemia, cardiac content of parasite DNA, inflammation, oxidative tissue damage, and mortality. In response to high parasitic load in cardiac tissue, Sur stimulated the immune system in a manner typical of the acute phase of Chagas disease, increasing tissue levels of gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α) and inducing a preferential IgG2a anti-T. cruzi serum pattern. When Sur and Bz were combined, the infection severity was attenuated, showing a dose-dependent Bz response. Sur therapy had a more harmful effect on the host than on the parasite and reduced the efficacy of Bz against T. cruzi infection. Considering that Sur drastically reinforced the infection evolution, potentiating the inflammatory process and the severity of cardiac lesions, the in vivo findings contradicted the in vitro anti-T. cruzi potential described for this drug.
INTRODUCTION
More than a century after its discovery, Chagas disease still represents a neglected parasitic infection responsible for the most common form of nonischemic cardiomyopathy worldwide (1, 2), with 14,000 annual deaths induced by heart failure in South America (3). It is estimated that 8 to 10 million people are infected with Trypanosoma cruzi in Mexico and Central and South America, with 28 million remaining at risk of infection (3). Population migration and the lack of immunoprophylactic agents have resulted in an increasing number of infected individuals in areas where Chagas disease is nonendemic, especially in North America and European countries (2, 3). There are estimates that 90 million people are at risk of contracting the infection worldwide (3, 4).
Current specific chemotherapy for Chagas disease, based on nitroheterocyclic compounds, is unsatisfactory. Since the 1960s, the compound N-benzyl-2-nitroimidazole acetamide (benznidazole [Bz]) has been the first-line drug against T. cruzi infection. Although chemotherapy with Bz is not always successful, no drugs with therapeutic efficiency superior to that of Bz are available (5–7). Clinical studies have also reported marked side effects of Bz associated with low specificity and systemic toxicity (1, 5). These limitations have highlighted the need for more effective and suitable strategies for Chagas disease control (1, 7).
An important mechanism associated with T. cruzi virulence involves the parasite's ability to interfere with cell signaling triggered by extracellular ATP and other nucleotides (8, 9). Extracellular ATP originating during lysis of T. cruzi-infected cells is a direct target of parasite ectonucleotidases like ecto-nucleoside triphosphate diphosphohydrolase (ecto-NTPDase) (9, 10). ATP hydrolysis by ecto-NTPDase has the potential to subvert and avoid host defense mechanisms because, in normal conditions, ATP triggers the release of proinflammatory cytokines mediated by the activation of type 2 (P2) purinergic receptors to control the infection (8, 9). ATP hydrolysis is also important for the acquisition of purine rings, which cannot be synthesized by T. cruzi but are essential to its survival and replication (11). A study conducted by our research group showed that suramin (Sur), a symmetrical polysulfonated derivative of urea used in the treatment of human African trypanosomiasis, beyond being a broad-spectrum antagonist of P2X and P2Y purinergic receptors in mammalian cells (12, 13), is also a T. cruzi ATPase inhibitor (12). In that study, we found that Sur significantly reduced the parasitism of Vero cells. Furthermore, mice infected with parasites pretreated with this drug presented increased survival (12). Although Sur is suggested as a potential drug candidate in the management of Chagas disease, this issue has not been objectively investigated. Thus, the present study was designed to investigate the applicability of concomitant treatment with Bz and Sur using different therapeutic schemes in mice infected with a virulent strain of T. cruzi.
MATERIALS AND METHODS
Animals and ethics.
Male C57BL/6 mice (age, 10 weeks; weight, 23.5 ± 3.2 g) were obtained from the Central Animal Laboratory of the Center of Biosciences and Health of Federal University of Viçosa (Brazil) and maintained under conditions of controlled temperature at 21 ± 2°C, relative humidity of 60% to 70%, and 12-h light/dark cycles. The animals received food and water ad libitum. All protocols were conducted in two independent experimental replicates and the experimental protocols were approved by the Ethics Committee of Animal Use of the Federal University of Viçosa (CEUA/UFV, protocol 77/2012) and carried out in compliance with the guidelines issued by the National Council for the Control of Animal Experimentation (CONCEA).
Infection, parasitemia, and mortality.
Groups of 10 animals were inoculated intraperitoneally (i.p.) with T. cruzi Y strain (5,000 trypomastigote forms in 0.1 ml of infected mouse blood). Inocula were obtained from mice that had been previously infected with metacyclic trypomastigote forms obtained from late-stationary-phase cultures on liver infusion tryptose (LIT) medium. The number of parasites in each inoculum was determined according to the method of Toledo et al. (14). The parasitemia was determined daily with 5-μl blood samples obtained from the tail according to Brener (15). Curves were plotted using the mean of the parasitemia, and mortality rate was expressed as a percentage of the accumulated deaths within the experimental period. Parasitemia and mortality were additionally investigated in a third independent experiment due to wide variability in these parameters comparing the two previous experimental replicates.
Benznidazole and suramin therapy.
Twenty-four hours after inoculation, tail blood was examined for the presence of parasites. After confirmation of the infection by microscopic identification of trypomastigotes in fresh blood samples from mouse tails, 70 animals were randomized into seven equal groups. The animals were submitted to a specific treatment with Bz (Pernambuco State Pharmaceutical Laboratory [LAFEPE], Recife, Pernambuco, Brazil) and Sur (Sigma Chemical Co., St. Louis, MO, USA) alone or in different combinations. Standardized therapeutic doses applied to murine models of American trypanosomiasis (Bz, 100 mg/kg of body weight per day) (16) and African trypanosomiasis (Sur, 20 mg/kg/day) (17) were used. Sur, administered in a fixed dose of 20 mg/kg/day, was associated with Bz in four selected doses: (i) 100 mg/kg/day, the dose able to induce parasitological cure in a longer treatment course, (ii) 50 mg/kg/day, (iii) 25 mg/kg/day (16), and (iv) 5 mg/kg/day. The natural evolution of T. cruzi infection was evaluated in infected animals receiving no treatment (positive control). Noninfected mice were used as negative controls.
After confirmation of the infection (4 days after inoculation), all treatments were administered for 15 days, and the animals were euthanized 24 h after the last treatment by deep anesthesia (ketamine, 45 mg/kg, and xylazine, 5 mg/kg, i.p.) followed by cardiac puncture. Bz was suspended in 0.5% methyl cellulose (wt/vol) dissolved in distilled water and administered orally by gavage. Sur was dissolved in sterile distilled water and administered intraperitoneally.
Quantitative PCR for T. cruzi DNA.
Extraction of the total genomic DNA cardiac tissue of control mice and those infected with T. cruzi was performed using a commercial kit (Assistente Genomic DNA purification kit; Promega) according to Caldas et al. (18). The DNA was quantified by spectrophotometry (GeneQuant; Pharmacia Biotech, Piscataway, NJ, USA), and the concentrations were adjusted to 25 ng/μl.
The PCR was performed in a 10-μl volume containing 50 ng of genomic DNA, 5 μl of SYBR green PCR master mix (Applied Biosystems, Carlsbad, CA, USA), and either 0.35 μM T. cruzi 195-bp repeat DNA-specific primers or 0.50 μM murine-specific tumor necrosis factor alpha (TNF-α) primers. The primers for T. cruzi repetitive DNA (TCZ-F, 5′-GCTCTTGCCCACAMGGGTGC-3′, where M = A or C, and TCZ-R, 5′-CCAAGCAGCGGATAGTTCAGG-3′) amplify a 182-bp fragment. Primers for murine TNF-α (TNF-5241, 5′-TCCCTCTCATCAGTTCTATGGCCCA-3′, and TNF-5411, 5′-CAGCAAGCATCTATGCACTTAGACCCC-3′) amplify a 170-bp product (19).
The cycling program consisted of an initial denaturation at 95°C for 10 min, followed by 40 cycles of 94°C for 15 s and 64.3°C for 1 min with fluorescence acquisition at 64.3°C. Amplification was immediately followed by a melt program with an initial denaturation of 15 s at 95°C, cooling to 60°C for 1 min, and then a stepwise temperature increase of 0.3°C/s from 60°C to 95°C. Each 96-well reaction plate contained a standard curve and two negative controls. Negative controls consisted of a reaction mixture with T. cruzi-specific or murine-specific primers without DNA and also with tissue DNA from noninfected mice (negative control). Each DNA sample was quantified in duplicate. The mean quantification values for T. cruzi DNA were normalized by the data obtained with the murine-specific (TNF-α) primers as follows: normalized value = (mean T. cruzi DNA/mean TNF-α DNA) × 1,000, where “1,000” corresponds to the expected value for TNF-α from 30 mg of cardiac tissue. The efficiencies of amplification were determined automatically by the StepOne TM software v2.0 by calculating efficiency (E) = 10(−1/slope) (20).
Cytometric bead array for cytokines.
Fragments of cardiac tissue were homogenized in the presence of a protease inhibitor (protease inhibitor cocktails; Sigma-Aldrich, St. Louis, MO, USA) in a portable tissue homogenizer (YO-04727-09; LabGEN) and centrifuged at 3,000 × g for 10 min. The supernatant was collected for the cytokine assay. Analysis was conducted using a BD cytometric bead array (CBA) mouse Th1/Th2 cytokine kit (BD Biosciences, San Diego, CA, USA), and data were collected using FACSVerse and analyzed with FCAP 3.0 software. Standard curves were determined for each cytokine from a range of 20 to 5,000 pg/ml. According to the manufacturer, the lower limit of cytokine detection for the CBA was 2.5 to 52.7 pg/ml, depending on the analyte. The following cytokines were measured: tumor necrosis factor alpha (TNF-α), gamma interferon (IFN-γ), interleukin-2 (IL-2), interleukin-6 (IL-6), and interleukin-10 (IL-10).
Serological assay for immunoglobulin.
Blood samples from all mice were collected from orbital venous sinus (0.5 ml) and centrifuged at 3,000 × g for 10 min. T.-cruzi-specific antibodies were detected by the technique described by Voller et al. (21) using an enzyme-linked immunosorbent assay (ELISA). Polystyrene microplates (96 well) were sensitized overnight at 4°C with 100 μl of T. cruzi Y-strain antigen (4.0 μg/ml) prepared by alkaline extraction at exponential growth in LIT medium. Following washing steps and blocking with phosphate-buffered saline (PBS) and 1% gelatin, 100 μl of the diluted serum (1:40) from all groups was added to the selected wells and the plate incubated for 45 min at 37°C. After three washes with 0.05% PBS and 20% Tween 20, 100 μl of rabbit anti-mouse IgG, IgG1, and IgG2a (Sigma, St. Louis, MO, USA) was added to specific wells, and the plate was incubated for 45 min at 37°C. The plate was washed and incubated with peroxidase-conjugated goat anti-rabbit IgG (Sigma Chemical Co., St. Louis, MO, USA). The optical density (OD) was determined using a 490-nm filter (SoftMax Pro 4.0, life sciences edition).
Lipid and protein oxidation.
Oxidation of tissue lipids was measured by enzyme-linked immunosorbent assay (ELISA). Fragments of the cardiac tissue were homogenized in the presence of a protease inhibitor (protease inhibitor cocktails; Sigma Chemical Co., St. Louis, MO, USA) in a portable tissue homogenizer (YO-04727-09; LabGEN) and centrifuged at 3,000 × g for 10 min. The supernatant was collected for the 8-isoprostane assay. The concentration of isoprostane was measured using a 96-well commercial kit according to the manufacturer's instructions (Cayman Chemical, Ann Arbor, MI, USA). All samples were measured simultaneously in duplicate.
Protein carbonyl content was measured biochemically in the cardiac tissue pellets by adding 0.5 ml of 10 mM 2,4-dinitrophenylhydrazine (DNPH). The reaction involved derivatization of the carbonyl group with DNPH, leading to the formation of a stable 2,4-dinitrophenyl (DNP) hydrazone product, which indicates the content of protein carbonyl in the samples. The optical density (OD) was measured spectrophotometrically at 370 nm (22). The total protein levels in the heart tissues were determined by using the Bradford method (23).
Heart parasitism and inflammation.
Fragments of the cardiac ventricles from each animal were immersed in histological fixative for 48 h (freshly prepared 10% [wt/vol] formaldehyde in 0.1 M phosphate buffer at pH 7.2) and embedded in methacrylate resin. Blocks were cut into 3-μm-thick histological sections and stained with hematoxylin and eosin (H&E) for general histopathology. To avoid repeated analysis of the same histological area, the sections were evaluated in semiseries, using one out of every 20 sections. For each group, 40 fields from H&E slides were randomly chosen at ×400 magnification for a total of 6.09 × 105 μm2 analyzed myocardium area. Images were captured using a light microscope (Olympus BX-60; Tokyo, Japan) equipped with a digital camera (Olympus QColor-3) (24). The inflammatory process was evaluated by the correlation index among the number of cells observed in myocardium muscle from nontreated and treated animals (16).
Cardiomyocyte damage.
Cardiomyocyte damage was investigated by spectrophotometric quantification of creatine kinase-isoenzyme MB (CK-MB) serum levels by using a commercial diagnostic kit (BioClin, Belo Horizonte, Minas Gerais, Brazil).
Statistical analysis.
Data were analyzed by one-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls (SNK) post hoc test for multiple comparisons. Differences were considered significant if P was ≤0.05.
RESULTS
For all groups inoculated with T. cruzi, infection was confirmed and the parasitemia peak was detected 4 and 6 days after T. cruzi inoculation, respectively. In combined therapy, animals treated with higher doses of Bz showed a higher peak of parasitemia in the presence of Sur than untreated animals or those receiving Bz alone (P < 0.05). Bz and Sur had opposite effects on mean parasitemia. At 20 mg/kg, Sur treatment caused a high parasitemia compared to that of the untreated infected animals and those treated with 100 mg/kg Bz, for which the lowest parasitemias were detected (P < 0.05). The mean parasitemias were similar in all groups receiving Bz and Sur combined (Table 1).
TABLE 1.
Parasitemia and survival rate of C57BL/6 mice infected with T. cruzi Y straina
| Groupb | PP (parasites × 103/0.1 ml blood)c | MP (parasites × 103/0.1 ml blood)d | No. of mice with BPC/total no. (%)e | No. survived/total no. | Mortality (%) |
|---|---|---|---|---|---|
| NI | NDf | ND | ND | 10/10 | 0 |
| INF | 82.00 ± 31.08A | 25.89 ± 10.38A | 0/10 (0) | 6/10 | 40 |
| B100 | 80.13 ± 28.32A | 10.16 ± 1.52B | 10/10 (100) | 10/10 | 0 |
| S20 | 114.5 ± 22.29A,B | 40.69 ± 11.05C | 0/10 (0) | 4/10 | 60 |
| B100 + S20 | 140.63 ± 36.05B | 15.84 ± 3.06D | 10/10 (100) | 10/10 | 0 |
| B50 + S20 | 112.5 ± 33.92A,B | 16.93 ± 5.18D | 7/10 (70) | 8/10 | 20 |
| B25 + S20 | 97.88 ± 30.35A,B | 18.18 ± 9.57D | 6/10 (60) | 6/10 | 40 |
| B5 + S20 | 87.03 ± 46.29A | 17.78 ± 6.94D | 5/10 (50) | 5/10 | 50 |
Data are representative of the second experimental replicate from three independent studies.
The animals were treated with benznidazole (B) at 100, 50, 25, or 5 mg/kg alone or combined with suramin (S; 20 mg/kg) for a period of 15 days after confirmation of the infection (4 days after inoculation). NI, noninfected controls; INF, infected without treatment.
PP, peak of parasitemia. Different letters (A and B) denote a statistically significant difference among the groups (P < 0.05), and groups with a common letter are not statistically significantly different (P > 0.05).
Mean parasitemia (MP) was calculated from the daily counting of blood trypomastigotes during 15 days of treatment. Different letters (A to D) denote a statistically significant difference among the groups (P < 0.05), and groups with a common letter are not statistically significantly different (P > 0.05).
BPC, blood parasitemia clearance (fresh blood examination).
ND, not detected.
Parasitemia was suppressed in 50% to 100% of animals treated at 5 to 100 mg/kg/day. There was no mortality in the groups treated with 100 mg/kg Bz alone or combined with Sur. High mortality was observed in the group treated with Sur alone compared to that of the other groups. The rate of mortality in animals exposed to low doses of Bz (25 or 5 mg/kg) combined with Sur was similar to that observed for untreated infected animals (Table 1).
Using quantitative PCR (qPCR) analysis, parasite DNA was identified in the cardiac tissue from all infected animals (Fig. 1). A high tissue level of parasite DNA was observed in animals treated with Sur alone. The level of parasite DNA was significantly reduced in animals exposed to Bz alone or combined with Sur compared to that in untreated infected animals (P < 0.05), except in the group receiving the lowest dose of Bz (5 mg/kg) combined with Sur, which had the highest DNA level (P < 0.05).
FIG 1.
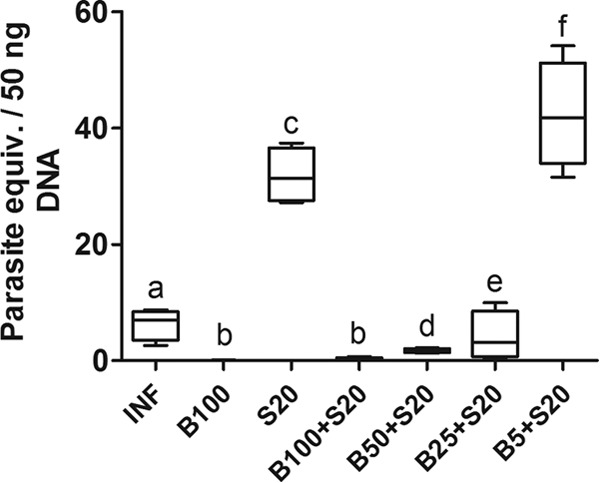
T. cruzi DNA in cardiac tissue from C57BL/6 mice infected with T. cruzi Y strain. The animals were treated with benznidazole (B) at 100, 50, 25, or 5 mg/kg alone or combined with suramin (S; 20 mg/kg) for a period of 15 days after confirmation of the infection (4 days after inoculation). INF, infected without treatment. Results for noninfected controls (NI) were suppressed in this figure since anti-T. cruzi immunoglobulins were not detected. T. cruzi DNA was determined by qPCR in cardiac samples collected 24 h after the last treatment. All animals that survived were investigated (NI, n = 10; INF, n = 6; B100, n = 10; S20, n = 4; B100 + S20, n = 10; B50 + S20, n = 8; B25 + S20, n = 6; B5 + S20, n = 5). The box represents the interquartile interval with the median indicated (horizontal line), and the whiskers represent the superior and inferior quartiles. Data are representative of the second experimental replicate from two independent studies with similar results. Different letters (a to f) denote a statistically significant difference among the groups (P < 0.05), and groups with a common letter are not statistically significantly different (P > 0.05).
Untreated infected animals presented severe cardiac lesions, evidenced by intense and diffuse infiltration of inflammatory cells, a typical pathological manifestation of the heart tissue associated with the acute phase of experimental Chagas disease. Cardiac inflammation was significantly enhanced by Sur (P < 0.05) and reduced by Bz (P < 0.05) alone compared to that in untreated infected animals. In association with Sur, Bz showed dose-dependent responses for cardiac inflammation. The drug combinations presented results equal to (Bz 100 mg/kg + Sur 20 mg/kg) or worse than (Bz 50, 25, or 5 mg/kg + Sur 20 mg/kg) those of Bz alone (Fig. 2).
FIG 2.
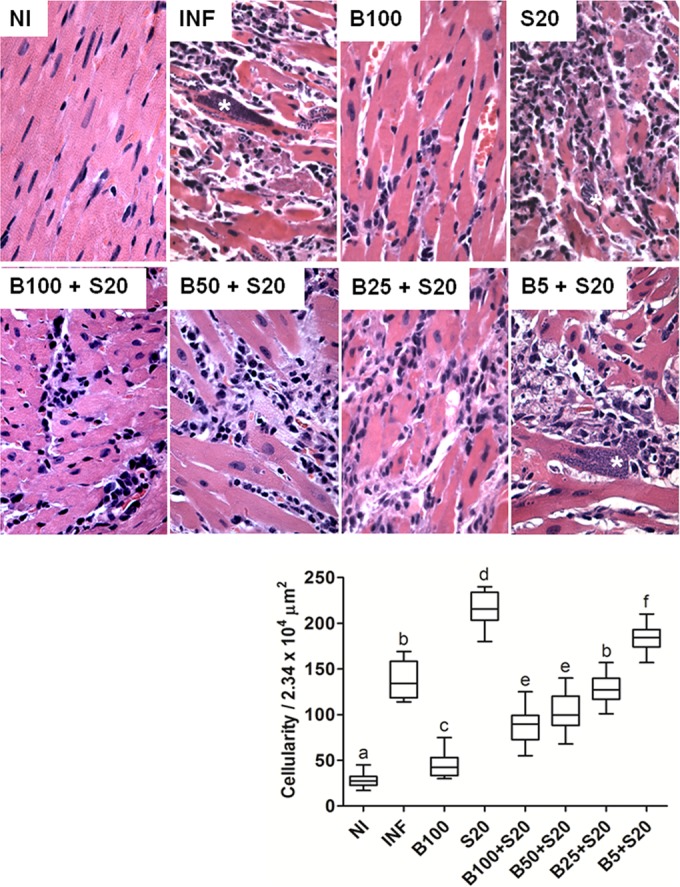
Tissue parasitism and inflammation in the ventricular myocardium of C57BL/6 mice infected with T. cruzi Y strain. The animals were treated with benznidazole (B) at 100, 50, 25, or 5 mg/kg alone or combined with suramin (S; 20 mg/kg) for a period of 15 days after confirmation of the infection (4 days after inoculation). NI, noninfected controls; INF, infected without treatment. Cardiac samples were collected 24 h after the last treatment. All animals that survived were investigated (NI, n = 10; INF, n = 6; B100, n = 10; S20, n = 4; B100 + S20, n = 10; B50 + S20, n = 8; B25 + S20, n = 6; B5 + S20, n = 5). The asterisk indicates T. cruzi nests. In the graphic, the box represents the interquartile interval with the median indicated (horizontal line), and the whiskers represent the superior and inferior quartiles. Data are representative of the second experimental replicate from two independent studies with similar results. Different letters (a to f) denote a statistically significant difference among the groups (P < 0.05), and groups with a common letter are not statistically significantly different (P > 0.05).
Untreated infected animals and those treated with Sur alone had elevated levels of proinflammatory cytokines IFN-γ, TNF-α, and IL-6 compared to those of the other groups (P < 0.05). Treatment with Bz drastically reduced serum levels of the cytokines (P < 0.05). In combined therapy, IFN-γ was enhanced in all treatments compared to that of animals treated with Bz alone. This finding was observed for IL-6 only at the lowest dose of Bz (5 mg/kg) (Fig. 3).
FIG 3.

Cytokine levels in cardiac tissue from C57BL/6 mice infected with T. cruzi Y strain. The animals were treated with benznidazole (B) at 100, 50, 25, or 5 mg/kg alone or combined with suramin (S; 20 mg/kg) for a period of 15 days after confirmation of the infection (4 days after inoculation). NI, noninfected controls; INF, infected without treatment. Cardiac samples were collected 24 h after the last treatment. All animals that survived were investigated (NI, n = 10; INF, n = 6; B100, n = 10; S20, n = 4; B100 + S20, n = 10; B50 + S20, n = 8; B25 + S20, n = 6; B5 + S20, n = 5). The box represents the interquartile interval with the median indicated (horizontal line), and the whiskers represent the superior and inferior quartiles. Data are representative of the second experimental replicate from two independent studies with similar results. Different letters (a to e) denote a statistically significant difference among the groups (P < 0.05), and groups with a common letter are not statistically significantly different (P > 0.05).
The serum levels of specific anti-T. cruzi IgG antibodies were significantly reduced in animals treated with Bz at 100 mg/kg alone or combined with Sur compared with those in untreated infected animals and those receiving Sur alone. As in the typical Th1 pattern for Chagas disease, there was a predominance of anti-T. cruzi IgG2a antibodies. In association with Sur, Bz produced dose-dependent responses for IgG2a and total IgG antibody serum levels, in which low doses of Bz were accompanied by high levels of IgG compared with those of animals treated with Bz alone. In noninfected animals, specific anti-T. cruzi IgG antibodies were not detected (Fig. 4).
FIG 4.
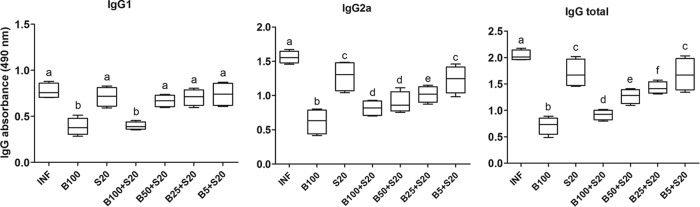
Anti-T. cruzi immunoglobulin subclasses in serum of C57BL/6 mice infected with T. cruzi Y strain. The animals were treated with benznidazole (B) at 100, 50, 25, or 5 mg/kg alone or combined with suramin (S; 20 mg/kg) for a period of 15 days after confirmation of the infection (4 days after inoculation). INF, infected without treatment. Results for noninfected controls (NI) were suppressed in this figure since anti-T. cruzi immunoglobulins were not detected. Blood samples were collected 24 h after the last treatment. All animals that survived were investigated (NI, n = 10; INF, n = 6; B100, n = 10; S20, n = 4; B100 + S20, n = 10; B50 + S20, n = 8; B25 + S20, n = 6; B5 + S20, n = 5). Data are representative of the second experimental replicate from two independent studies with similar results. Different letters (a to f) denote a statistically significant difference among the groups (P < 0.05), and groups with a common letter are not statistically significantly different (P > 0.05).
Bz and Sur presented opposing effects on cardiac oxidative damage. While Bz alone reduced (P < 0.05), Sur alone or combined with Bz in all doses increased (P < 0.05) lipid and protein oxidation in cardiac tissue compared to that in untreated infected animals (Fig. 5).
FIG 5.
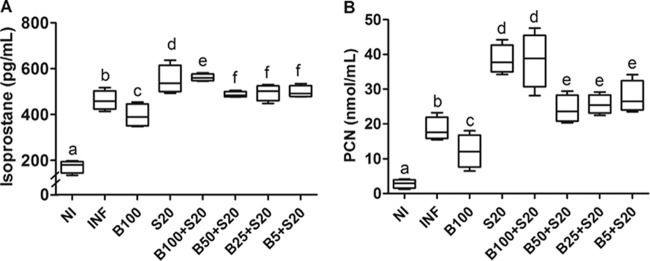
Oxidative damage in cardiac tissue from C57BL/6 mice infected with T. cruzi Y strain. Tissue contents of isoprostane (A) and protein carbonyl (PCN) (B) were used as markers of lipid and protein oxidation, respectively. The animals were treated with benznidazole (B) at 100, 50, 25, or 5 mg/kg alone or combined with suramin (S; 20 mg/kg) for a period of 15 days after confirmation of the infection (4 days after inoculation). NI, noninfected controls; INF, infected without treatment. Cardiac samples were collected 24 h after the last treatment. All animals that survived were investigated (NI, n = 10; INF, n = 6; B100, n = 10; S20, n = 4; B100 + S20, n = 10; B50 + S20, n = 8; B25 + S20, n = 6; B5 + S20, n = 5). The box represents the interquartile interval with the median indicated (horizontal line), and the whiskers represent the superior and inferior quartiles. Data are representative of the second experimental replicate from two independent studies with similar results. Different letters (a to f) denote a statistically significant difference among the groups (P < 0.05), and groups with a common letter are not statistically significantly different (P > 0.05).
The serum levels of CK-MB were significantly reduced (P < 0.05) in animals treated with Bz at 100 mg/kg alone and increased (P < 0.05) in animals receiving Sur compared to those in untreated infected animals and all groups of combined drug treatment, except for the group treated with the lowest dose of Bz (5 mg/kg) combined with Sur (Fig. 6).
FIG 6.
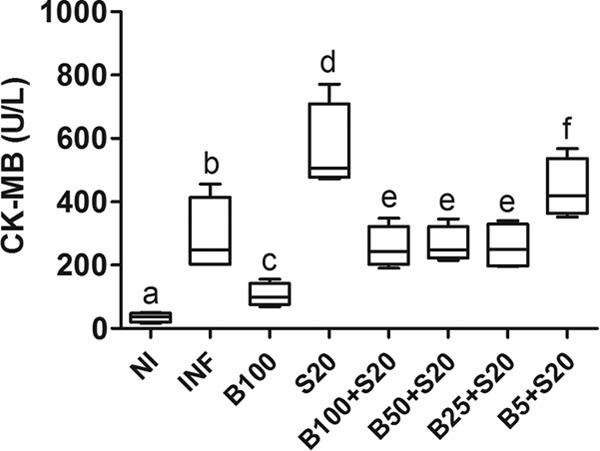
Serum levels of creatine kinase-MB (CK-MB) in C57BL/6 mice infected with T. cruzi Y strain. The animals were treated with benznidazole (B) at 100, 50, 25, or 5 mg/kg alone or combined with suramin (S; 20 mg/kg) for a period of 15 days after confirmation of the infection (4 days after inoculation). NI, noninfected controls; INF, infected without treatment. Cardiac samples were collected 24 h after the last treatment. All animals that survived were investigated (NI, n = 10; INF, n = 6; B100, n = 10; S20, n = 4; B100 + S20, n = 10; B50 + S20, n = 8; B25 + S20, n = 6; B5 + S20, n = 5). The box represents the interquartile interval with the median indicated (horizontal line), and the whiskers represent the superior and inferior quartiles. Data are representative of the second experimental replicate from two independent studies with similar results. Different letters (a to f) denote a statistically significant difference among the groups (P < 0.05), and groups with a common letter are not statistically significantly different (P > 0.05).
DISCUSSION
The present study investigated the effectiveness of a combination of Bz and Sur in treating the acute phase of experimental Chagas disease. Due to its partial resistance to Bz, the Y strain of T. cruzi was used because it induces high parasitemia and mortality, which is generally observed at days 10 to 19 postinfection (16). Our results showed that, individually, Bz and Sur presented opposing effects on the infection evolution. Bz attenuated parasitemia, cardiac content of parasite DNA, inflammation, oxidative tissue damage, and mortality. Conversely, inducing a proinflammatory status, Sur drastically enhanced all these parameters, causing a high mortality rate. In general, the infection severity was attenuated in combined therapy, showing a dose-dependent response.
Acting as an antiparasitic drug, Bz also has anti-inflammatory properties, which have been supported by previous studies (25–27). Similarly to nonsteroidal anti-inflammatory drugs (NSAIDs), BZ exerts its immunomodulatory effects by inhibiting NF-κB (a key regulator of the inflammatory response), more specifically, downregulating the proinflammatory activity of macrophages (25, 26). Immunomodulatory effects of BZ were also evidenced by its ability to increase survival and decrease serum levels of IL-6 and TNF-α in C57BL/6 mice treated with lipopolysaccharide (LPS) (25, 27). Apparently, this BZ effect is not restricted to LPS-mediated macrophage activation, and it is also observed when other stimuli are employed, such as proinflammatory cytokines (IL-1, TNF-α) and H2O2, a nonradical molecule involved in reactive tissue damage (25).
While Sur has been recently indicated as a potential anti-T. cruzi drug (12, 28), the applicability of Bz in the treatment of experimental and human Chagas disease is widely described (1, 29, 30). Since research and development of new specific medicines have been neglected for too many years, Bz remains the reference drug against T. cruzi infection. Strong evidence indicates that during Bz enzymatic processing by the NADPH-cytochrome P-450 system (5, 31–33), the production of highly reactive electrophilic metabolites overcomes the redox balance of T. cruzi, based on the enzyme dihydrotrypanothione T(SH)2, disrupting parasite metabolism and viability (29, 33). The role of Sur in T. cruzi metabolism is poorly understood. However, studies in vitro reported that its antiparasitic effects are mediated by inhibition of thymidylate kinase, an enzyme related to mitochondrial DNA replication, and ecto-NTPDase, a surface enzyme involved in ATP cleavage, adhesion, and invasion of host cells (12, 34).
Based on in vitro studies conducted by our research group, we expected considerable effectiveness of Sur against T. cruzi infection in vivo. Surprisingly, Sur therapy alone presented poor outcomes, which were not compatible with the inhibitory activity of this drug on the infection of Vero cells by T. cruzi and on parasite replication (12). In the same investigation, mice infected with parasites previously treated with Sur presented reduced parasitemia and mortality. The mechanisms that explain this negative effect of Sur on T. cruzi infection in vivo are not clear. However, Bisaggio et al. (28) indicated that Sur is capable of inducing drastic morphological changes in T. cruzi trypomastigotes in vitro, leading to partial or complete flagellum detachment from the cell body. Apparently, this effect may reduce parasite motility and cell invasion, limiting parasite virulence and infection evolution (28). Thus, it is possible that the positive effect of Sur pretreatment observed in our preliminary study (12) is secondary to the reduction of parasite viability, which was not investigated. As in the present study, no pretreatment was administered and parasite viability was not compromised, as evidenced by the ability of T. cruzi to induce high parasitemia and mortality. In addition, an in vitro study conducted by Bisaggio et al. (35) showed that T. cruzi epimastigotes treated with higher doses of Sur increase ecto-ATPase activity in an effort to overcome enzyme inhibition. These parasites were then much more capable of adhering to mouse macrophages than untreated epimastigotes. Although this finding describes an adaptive response of T. cruzi to ensure its infective potential and survival, the contribution of this mechanism to the outcomes observed here requires further investigation.
The best results were observed in the group receiving Bz alone, in which inflammatory and oxidative cardiac lesions were remarkably attenuated and all animals survived the infection. Beyond aggravation of all these parameters, Sur also caused high mortality. The results for cytokines, IgG, and cardiac inflammation reinforce the proinflammatory profile stimulated by Sur. In response to high parasitemia and cardiac content of parasite DNA (i.e., high parasitic load), Sur enhanced the polarization of the immune response for a Th1-type phenotype, in which high levels of IFN-γ and TNF-α and a preferential IgG2a anti-T. cruzi pattern are a typical response to the acute phase of Chagas disease. Besides these immunological adaptations in the attempt to maximize parasitemia clearance and control the infection (36, 37), the intensity of this response was not sufficient to control parasite replication and cardiac infection. On the contrary, the satisfactory effect of Bz alone against infection evolution indicated that the antiparasitic effect of Bz occurred independently of modifications in Th1 phenotype, reinforcing its direct cytotoxic action on T. cruzi (5, 29, 38). Our results also showed that Sur markedly increased tissue levels of IL-6, a cytokine directly implicated in acute chagasic myocarditis since it induces the expression of adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), prominent mediators for the migration of inflammatory cells (39, 40).
Together with its proinflammatory effect, Sur also stimulates oxidative damage of the cardiac tissue. The tissue levels of isoprostane and carbonyl proteins were potentially related to a Bz and Sur effect on parasitemia and cardiac content of parasite DNA. There is evidence that reactive damage is directly associated with myocarditis severity in the acute phase of T. cruzi infection (41, 42). Thus, inflammatory cells recruited in the cardiac tissue activated molecular mechanisms against T. cruzi (i.e., respiratory burst) that culminate in intensive formation of reactive species (i.e., O2−, OH, NO, and H2O2) and oxidative cardiac damage (43, 44). Furthermore, due to mitochondrial alterations, the direct production of these reactive species by infected cardiomyocytes represents an additional susceptibility factor for myocardial damage, cell death, and cardiac dysfunction in Chagas disease (43–46). Apparently, this is not a random process, since it is regulated by cytokines such as IFN-γ, TNF-α, and IL-6 (43, 44). In the present study, tissue levels of these molecules were drastically reduced by Bz and enhanced by Sur. Beyond participating in parasite control, these cytokines also directly stimulate the synthesis of reactive species by leukocytes and infected cardiomyocytes, regulating cardiac redox balance and the progression of reactive myocardial damage (43, 44), a phenomenon consistent with the high CK-MB levels in our investigation. This evidence is reinforced by the findings of Gupta et al. (43, 44), showing that cardiomyocytes infected with T. cruzi and exposed to TNF-α and IFN-γ increase the production of reactive species, which does not occur when noninfected cardiomyocytes are treated with these cytokines.
Considering the opposing effects of the drugs tested, it was expected that the therapeutic combination of Bz and Sur showed no benefit compared to Bz alone. In combination with Bz, Sur reduced the therapeutic effect of the reference dose of Bz (100 mg/kg) on T. cruzi infection. On the contrary, Bz was capable of reversing the negative effects of Sur and preventing the prolonged installation of an exacerbated proinflammatory status during the infection, a condition potentially dangerous to the host since it is associated with severe cardiac lesions and death (47). Since the Sur dose was fixed, the beneficial effects observed in drug combination were not determined by Sur, and the dose-dependent outcomes were closely related to the dose of Bz.
Taken together, the results indicate that Sur therapy had a more harmful effect on the host than on the parasite and reduced the efficacy of the reference drug against T. cruzi infection. Considering that Sur drastically reinforced the infection evolution, potentiating the inflammatory process and cardiac damage, the in vivo findings contradicted the in vitro anti-T. cruzi potential described for this drug when used only on the parasite as a T. cruzi ecto-ATPDase inhibitor. Classically, beyond being an inhibitor of surface enzymes in trypanosomatids, Sur has been used as a P2 purinergic receptor antagonist in mammalian cells. Thus, further studies evaluating the participation of this pathway in Chagas disease evolution can contribute to the elucidation of new mechanisms of the host associated with tolerance and susceptibility to T. cruzi infection.
ACKNOWLEDGMENTS
This work was supported by the Brazilian funding agencies Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (552459/2011-19) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) (APQ-00176-13). A.T. and M.T.B. have fellowships from Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil.
REFERENCES
- 1.Guedes PM, Silva GK, Gutierrez FR, Silva JS. 2011. Current status of Chagas disease chemotherapy. Expert Rev Anti Infect Ther 9:609–620. doi: 10.1586/eri.11.31. [DOI] [PubMed] [Google Scholar]
- 2.Hotez PJ, Dumonteil E, Woc-Colburn L, Serpa JA, Bezek S, Edwards MS, Hallmark CJ, Musselwhite LW, Flink BJ, Bottazzi ME. 2012. Chagas disease: “the new HIV/AIDS of the Americas.” PLoS Negl Trop Dis 6(5):e1498. doi: 10.1371/journal.pntd.0001498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization. 2010. Working to overcome the global impact of neglected tropical diseases: first WHO report on neglected tropical diseases. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 4.Schofield CJ, Jannin J, Salvatella R. 2006. The future of Chagas disease control. Trends Parasitol 22:583–588. doi: 10.1016/j.pt.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 5.Maya JD, Orellana M, Ferreira J, Kemmerling U, López-Muñoz R, Morello A. 2010. Chagas disease: present status of pathogenic mechanisms and chemotherapy. Biol Res 43:323–331. [PubMed] [Google Scholar]
- 6.Rassi A, Rassi A Jr, Marin-Neto JA. 2010. Chagas disease. Lancet 375:1388–1402. doi: 10.1016/S0140-6736(10)60061-X. [DOI] [PubMed] [Google Scholar]
- 7.Bahia MT, Nascimento AFS, Mazzeti AL, Marques LF, Gonçalves KR, Motta LWR, Diniz LF, Caldas IS, Talvane A, Shackleford DM, Koltun M, Saunders J, White KL, Scandale I, Charman SA, Chatelain E. 2014. Antitrypanosomal activity of fexinidazole metabolites, potential new drug candidates for Chagas disease. Antimicrob Agents Chemother 58:4362–4370. doi: 10.1128/AAC.02754-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silverman JA, Qi H, Riehl A, Beckers C, Nakaar V, Joiner KA. 1998. Induced activation of the Toxoplasma gondii nucleoside triphosphate hydrolase leads to depletion of host cell ATP levels and rapid exit of intracellular parasites from infected cells. J Biol Chem 273:12352–12359. doi: 10.1074/jbc.273.20.12352. [DOI] [PubMed] [Google Scholar]
- 9.Sansom FM, Newton HJ, Crikis S, Cianciotto NP, Cowan PJ, d'Apice AJ, Hartland EL. 2007. A bacterial ecto-triphosphate diphosphohydrolase similar to human CD39 is essential for intracellular multiplication of Legionella pneumophila. Cell Microbiol 9:1922–1935. doi: 10.1111/j.1462-5822.2007.00924.x. [DOI] [PubMed] [Google Scholar]
- 10.Schnurr M, Then F, Galambos P, Scholz C, Siegmund B, Endres S, Eigler A. 2000. Extracellular ATP and TNF-alpha synergize in the activation and maturation of human dendritic cells. J Immunol 165:4704–4709. doi: 10.4049/jimmunol.165.8.4704. [DOI] [PubMed] [Google Scholar]
- 11.Zimmermann H. 2000. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol 362:299–309. doi: 10.1007/s002100000309. [DOI] [PubMed] [Google Scholar]
- 12.Santos RF, Pôssa MA, Bastos MS, Guedes PM, Almeida MR, Demarco R, Verjovski-Almeida S, Bahia MT, Fietto JL. 2009. Influence of ecto-nucleoside triphosphate diphosphohydrolase activity on Trypanosoma cruzi infectivity and virulence. PLoS Negl Trop Dis 3(3):e387. doi: 10.1371/journal.pntd.0000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voogd TE, Vansterkenburg EL, Wilting J, Janssen LH. 1993. Recent research on the biological activity of suramin. Pharmacol Rev 45:177–203. [PubMed] [Google Scholar]
- 14.Toledo MJ, Bahia MT, Carneiro CM, Martins-Filho OA, Tibayrenc M, Barnabé C, Tafuri WL, de Lana M. 2003. Chemotherapy with benznidazole and itraconazole for mice infected with different Trypanosoma cruzi clonal genotypes. Antimicrob Agents Chemother 47:223–230. doi: 10.1128/AAC.47.1.223-230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brener Z. 1962. Therapeutic activity and criterion of cure on mice experimentally infected with Trypanosoma cruzi. Rev Inst Med Trop Sao Paulo 4:389–396. [PubMed] [Google Scholar]
- 16.Bahia MT, de Andrade IM, Martins TA, do Nascimento Á, Diniz LDF, Caldas IS, Talvani A, Trunz BB, Torreele E, Ribeiro I. 2012. Fexinidazole: a potential new drug candidate for Chagas disease. PLoS Negl Trop Dis 6(11):e1870. doi: 10.1371/journal.pntd.0001870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amin DN, Masocha W, Ngan'dwe K, Rottenberg M, Kristensson K. 2008. Suramin and minocycline treatment of experimental African trypanososmiasis at an early stage of parasite brain invasion. Acta Trop 106:72–74. doi: 10.1016/j.actatropica.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 18.Caldas S, Caldas IS, Diniz LDF, Lima WG, Oliveira RDP, Cecílio AB, Ribeiro I, Talvani A, Bahia MT. 2012. Real-time PCR strategy for parasite quantification in blood and tissue samples of experimental Trypanosoma cruzi infection. Acta Trop 123:170–177. doi: 10.1016/j.actatropica.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Cummings KL, Tarleton RL. 2003. Rapid quantitation of Trypanosoma cruzi in host tissue by real-time PCR. Mol Biochem Parasitol 129:53–59. doi: 10.1016/S0166-6851(03)00093-8. [DOI] [PubMed] [Google Scholar]
- 20.Stordeur P, Poulin LF, Craciun L, Zhou L, Schandené L, de Lavareille A, Gorielys S, Goldman M. 2002. Cytokine mRNA quantification by real-time PCR. J Immunol Methods 259:55–64. doi: 10.1016/S0022-1759(01)00489-6. [DOI] [PubMed] [Google Scholar]
- 21.Voller A, Bidwell DE, Bartlett A. 1976. Enzyme immunoassays in diagnostic medicine. Theory and practice. Bull World Health Org 53:55–65. [PMC free article] [PubMed] [Google Scholar]
- 22.Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, Ahn BW, Shaltiel S, Stadtman ER. 1990. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol 186:464–478. doi: 10.1016/0076-6879(90)86141-H. [DOI] [PubMed] [Google Scholar]
- 23.Bradford MM. 1976. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye-binding. Anal Biochem 7:248–254. [DOI] [PubMed] [Google Scholar]
- 24.Novaes RD, Penitente AR, Gonçalves RV, Talvani A, Peluzio MC, Neves CA, Natali AJ, Maldonado IR. 2013. Trypanosoma cruzi infection induces morphological reorganization of the myocardium parenchyma and stroma, and modifies the mechanical properties of atrial and ventricular cardiomyocytes in rats. Cardiovasc Pathol 22:270–279. doi: 10.1016/j.carpath.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 25.Manarin R, Pascutti MF, Ruffino JP, De Las Heras B, Boscá L, Bottasso O, Revelli S, Serra E. 2010. Benznidazole blocks NF-kappaB activation but not AP-1 through inhibition of IKK. Mol Immunol 47:2485–2491. doi: 10.1016/j.molimm.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Pascutti MF, Campodonico G, García F, Manarin R, Bottasso O, Revelli S, Serra E. 2009. Novel cytostatic activity of the trypanocidal drug benznidazole. Int Immunopharmacol 9:739–745. doi: 10.1016/j.intimp.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Pascutti MF, Pitashny M, Nocito AL, Guermonprez P, Amigorena S, Wietzerbin J, Serra E, Bottasso O, Revelli S. 2004. Benznidazole, a drug used in Chagas' disease, ameliorates LPS-induced inflammatory response in mice. Life Sci 76:685–697. doi: 10.1016/j.lfs.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 28.Bisaggio DF, Adade CM, Souto-Padron T. 2008. In vitro effects of suramin on Trypanosoma cruzi. Int J Antimicrob Agents 31:282–286. doi: 10.1016/j.ijantimicag.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 29.Urbina JA. 2010. Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Trop 115:55–68. doi: 10.1016/j.actatropica.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 30.Hall BS, Wilkinson SR. 2012. Activation of benznidazole by trypanosomal type I nitroreductases results in glyoxal formation. Antimicrob Agents Chemother 56:115–123. doi: 10.1128/AAC.05135-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreno SN, Docampo R, Mason RP, Leon W, Stoppani AO. 1982. Different behaviors of benznidazole as free radical generator with mammalian and Trypanosoma cruzi microsomal preparations. Arch Biochem Biophys 218:585–591. doi: 10.1016/0003-9861(82)90383-6. [DOI] [PubMed] [Google Scholar]
- 32.Castro JA, De Mecca MM, Bartel LC. 2006. Toxic side effects of drugs used to treat Chagas disease (American trypanosomiasis). Hum Exp Toxicol 25:471–479. doi: 10.1191/0960327106het653oa. [DOI] [PubMed] [Google Scholar]
- 33.Maya JD, Cassels BK, Iturriaga-Vásquez P, Ferreira J, Faúndez M, Galanti N, Ferreira A, Morello A. 2007. Mode of action of natural and synthetic drugs against Trypanosoma cruzi and their interaction with the mammalian host. Comp Biochem Physiol A Mol Integr Physiol 146:601–620. doi: 10.1016/j.cbpa.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 34.Chalabi KA, Gutteridge WE. 1977. Presence and properties of thimidylate synthase in trypanosomatids. Biochim Biophys Acta 481:71–79. doi: 10.1016/0005-2744(77)90138-3. [DOI] [PubMed] [Google Scholar]
- 35.Bisaggio DF, Peres-Sampaio CE, Meyer-Fernandes JR, Souto-Padron T. 2003. Ecto-ATPase activity on the surface of Trypanosoma cruzi and its possible role in the parasite-host cell interaction. Parasitol Res 91:273–282. doi: 10.1007/s00436-003-0965-8. [DOI] [PubMed] [Google Scholar]
- 36.Brener Z, Gazzinelli RT. 1997. Immunological control of Trypanosoma cruzi infection and pathogenesis of Chagas' disease. Int Arch Allergy Immunol 114:103–110. doi: 10.1159/000237653. [DOI] [PubMed] [Google Scholar]
- 37.Padilla AM, Simpson LJ, Tarleton RL. 2009. Insufficient TLR activation contributes to the slow development of CD8+ T cell responses in Trypanosoma cruzi infection. J Immunol 183:1245–1252. doi: 10.4049/jimmunol.0901178. [DOI] [PubMed] [Google Scholar]
- 38.Irigoín F, Cibils L, Comini MA, Wilkinson SR, Flohé L, Radi R. 2008. Insights into the redox biology of Trypanosoma cruzi: trypanothione metabolism and oxidant detoxification. Free Radic Biol Med 45:733–742. doi: 10.1016/j.freeradbiomed.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 39.Gao W, Pereira MA. 2002. Interleukin-6 is required for parasite specific response and host resistance to Trypanosoma cruzi. Int J Parasitol 32:167–170. doi: 10.1016/S0020-7519(01)00322-8. [DOI] [PubMed] [Google Scholar]
- 40.López L, Arai K, Giménez E, Jiménez M, Pascuzo C, Rodríguez-Bonfante C, Bonfante-Cabarcas R. 2006. C-reactive protein and interleukin-6 serum levels increase as Chagas disease progresses towards cardiac failure. Rev Esp Cardiol 59:50–56. (In Spanish.) doi: 10.1157/13083649. [DOI] [PubMed] [Google Scholar]
- 41.Ribeiro LC, Barbosa AA Jr, Andrade ZA. 2002. Pathology of intracardiac nerves in experimental Chagas disease. Mem Inst Oswaldo Cruz 97:1019–1025. doi: 10.1590/S0074-02762002000700016. [DOI] [PubMed] [Google Scholar]
- 42.Bilate AM, Teixeira PC, Ribeiro SP, Brito TD, Silva AM, Russo M, Kalil J, Cunha-Neto E. 2008. Distinct outcomes of Trypanosoma cruzi infection in hamsters are related to myocardial parasitism, cytokine/chemokine gene expression, and protein expression profile. J Infect Dis 198:614–623. doi: 10.1086/590347. [DOI] [PubMed] [Google Scholar]
- 43.Gupta S, Bhatia V, Wen JJ, Wu Y, Huang MH, Garg NJ. 2009. Trypanosoma cruzi infection disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic Biol Med 47:1414–1421. doi: 10.1016/j.freeradbiomed.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gupta S, Wen JJ, Garg NJ. 2009. Oxidative stress in Chagas disease. Interdiscip Perspect Infect Dis 2009:190354. doi: 10.1155/2009/190354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen JJ, Vyatkina G, Garg NJ. 2004. Oxidative damage during chagasic cardiomyopathy development: role of mitochondrial oxidant release and inefficient antioxidant defense. Free Radic Biol Med 37:1821–1833. doi: 10.1016/j.freeradbiomed.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 46.Wen JJ, Yachelini PC, Sembaj A, Manzur RE, Garg NJ. 2006. Increased oxidative stress is correlated with mitochondrial dysfunction in chagasic patients. Free Radic Biol Med 41:270–276. doi: 10.1016/j.freeradbiomed.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 47.Nogueira LG, Santos RH, Fiorelli AI, Mairena EC, Benvenuti LA, Bocchi EA, Stolf NA, Kalil J, Cunha-Neto E. 2014. Myocardial gene expression of T-bet, GATA-3, Ror-t, FoxP3, and hallmark cytokines in chronic Chagas disease cardiomyopathy: an essentially unopposed TH1-type response. Mediators Inflamm 2014:914326. doi: 10.1155/2014/914326. [DOI] [PMC free article] [PubMed] [Google Scholar]