

Abstract
The hepatitis C virus (HCV) NS4B protein is an antiviral therapeutic target for which small-molecule inhibitors have not been shown to exhibit in vivo efficacy. We describe here the in vitro and in vivo antiviral activity of GSK8853, an imidazo[1,2-a]pyrimidine inhibitor that binds NS4B protein. GSK8853 was active against multiple HCV genotypes and developed in vitro resistance mutations in both genotype 1a and genotype 1b replicons localized to the region of NS4B encoding amino acids 94 to 105. A 20-day in vitro treatment of replicons with GSK8853 resulted in a 2-log drop in replicon RNA levels, with no resistance mutation breakthrough. Chimeric replicons containing NS4B sequences matching known virus isolates showed similar responses to a compound with genotype 1a sequences but altered efficacy with genotype 1b sequences, likely corresponding to the presence of known resistance polymorphs in those isolates. In vivo efficacy was tested in a humanized-mouse model of HCV infection, and the results showed a 3-log drop in viral RNA loads over a 7-day period. Analysis of the virus remaining at the end of in vivo treatment revealed resistance mutations encoding amino acid changes that had not been identified by in vitro studies, including NS4B N56I and N99H. Our findings provide an in vivo proof of concept for HCV inhibitors targeting NS4B and demonstrate both the promise and potential pitfalls of developing NS4B inhibitors.
INTRODUCTION
Hepatitis C virus (HCV) is a significant public health threat, with up to 3% of the world population estimated to be harboring the infection. Although the virus can be naturally cleared, it more often becomes established as a chronic infection with an increased risk of poor long-term outcomes, including liver cirrhosis and cancer (1). Treatment of the disease historically used a combination of ribavirin and pegylated interferon, a cocktail with a 50% cure rate against genotype 1 but with a high likelihood of undesirable side effects (2, 3). The regulatory approval of direct-acting small-molecule antivirals represents a major advance in therapeutics by increasing the chance of efficacious treatment (4, 5), but there is still a need for novel antiviral therapies with reduced side effects and complementary resistance profiles. HCV inhibitors of the NS3 protease, of the multifunctional protein NS5A, and of the NS5B RNA-dependent RNA polymerase have been shown to be efficacious in the clinic, but inhibitors of NS4B have not been validated in vivo. We (6–8) and others (9–12) have described compounds targeting NS4B in vitro that, if developed, would offer a complementary mechanism that may improve the standard of care for hepatitis C virus infection treatment.
HCV RNA replication occurs in vesicle-induced intracellular membranes derived from the cellular endoplasmic reticulum (ER), catalyzed by a complex of proteins encoded by the virus and host factors. NS4B is an integral membrane protein (13) that induces the rearrangement of intracellular membranes into a “membranous web,” a collection of vesicles thought to comprise the scaffold for HCV replication (14). In this environment, NS4B protein likely provides a platform for interactions of proteins that comprise the HCV replication complex, as supported by evidence of genetic and physical interactions with other proteins in the complex (15–19). Several biochemical functions of NS4B have been described, including protein multimerization (20–22), ATPase/GTPase activity (23, 24), RNA binding (25), and interaction with membranes or lipid droplets (26–28). Previous reports have identified small-molecule binders of NS4B that are associated with inhibition of particular biochemical functions. The reported RNA binding of NS4B can be inhibited by clemizole, a weak inhibitor of HCV replication (25), while a pyrazolopyrimidine compound referred to as anguizole was able to disrupt dimerization of NS4B protein (21) and cause intracellular rearrangements of NS4B protein (12). Both anguizole and an unrelated amiloride compound have also been reported to affect the association of membrane vesicles with NS4B peptides in vitro (26). Compounds that show either direct binding to NS4B or genetic evidence of interaction with NS4B are referred to here as “NS4B inhibitors” regardless of the specific evidence about the biochemical or viral processes inhibited.
We recently reported the discovery of an imidazo[1,2-a]pyrimidine compound series that inhibited HCV by targeting of the viral NS4B protein as validated both by direct binding of a compound to NS4B protein and by genetic resistance data (7). We present here a detailed characterization of GSK8853, one of the most potent compounds in that chemical series. The in vivo efficacy and resistance profile of GSK8853 were studied using a humanized-mouse model, and in vitro replicon assays were used to measure genotype specificity, resistance, and the variability of efficacy in naturally occurring variants of NS4B.
MATERIALS AND METHODS
Compounds.
The purity of GSK8853 and the purity of GSK9547 (6) were determined to be ≥95% by 1H nuclear magnetic resonance (NMR) analysis and liquid chromatography-mass spectrometry (LC/MS); structural assignments were consistent with the spectroscopic data. 1H NMR spectra were obtained on a Varian Inova 400 NMR spectrometer. Mass spectrometric analyses and compound purity determinations were conducted on a Waters Acquity ultrapressure LC (UPLC) system and Waters Acquity Single Quad detector (SQD).
Cell lines.
Stable cell lines carrying a bicistronic replicon of genotype 1a (H77), genotype 1b (Con1 ET), or genotype 2a (JFH-1) were licensed from Apath LLC (Brooklyn, NY) or ReBLikon GmbH (Mainz, Germany) or created in-house, respectively (1, 29, 30). All three replicons express luciferase, neomycin phosphotransferase, and HCV NS3-5B. Cured ET cells are a derivative of replicon-containing genotype 1b Con1 ET cells generated by treatment with alpha interferon for several passages until HCV RNA levels were undetectable. Huh-7 Lunet cells were licensed from ReBLikon GmbH (Mainz, Germany).
Chimeric replicon construction.
Replicon alterations encoding individual single and double amino acid changes were synthesized in the backbone of the parental replicon by GenScript USA Inc. (Piscataway, NJ) using Con1 ET for genotype 1b and a modified H77 sequence containing an additional adaptive mutation for genotype 1a (31). HCV genotypes were modeled in chimeric replicons constructed as previously described (32). An additional chimeric replicon containing the NS4B sequence from HCV genotype 1a strain PBC002 was constructed to test the NS4B sequence from the virus used for in vivo testing.
NS4B chimeras modeling patient isolates were constructed using sequences identified from public databases. Separate phylogenetic trees were constructed for genotypes 1a and 1b from approximately 300 available sequences each, and 10 sequences were selected from each tree to represent the diversity of the sequences. These were cloned into genotype 1a H77 or genotype 1b Con1 ET backbones as appropriate. The amino-terminal sequences encoding the first 4 residues and carboxy-terminal 6 residues of NS4B were changed to match the H77 or Con1 parental sequence to increase the chance that polypeptide maturation would function properly. Nine sequences of genotype 1a, namely, EU781788, EU781798, EU781774, D10749, EF407439, EU155278, EU781746, EU781757, and EU781780, and six of genotype 1b, namely, EU781829, AF207771, AB154177, AB049098, AF165063, and EU256088, were viable and could be tested.
Compound inhibition potency determination.
Transient and stable replicons were used for 50% inhibitory concentration (EC50) analysis as described previously (7). Unless otherwise stated, replicons were tested under transient-expression conditions. The effect of human serum on compound efficacy was modeled for dose predictions by comparing the levels of potency of the compound with respect to stable genotype 1b replicons in the presence and absence of 40% (vol/vol) human serum (Lonza catalog no. 14-402E). The modified assay format required by the human serum addition led to slight differences in compound potency in the absence of serum (0% human serum) compared to that seen with the standard replicon, so the fold change values were calculated using 0% and 40% human serum values run in parallel.
In vitro resistance selection and sequencing.
Cell lines stably carrying HCV replicons of genotype 1a or 1b were treated with GSK8853 at a concentration of 5× or 20× the EC50. Medium and compound were changed every 3 days until colonies started to form, at which point total RNA from each plate was isolated with TRIzol (Invitrogen) and cDNAs were generated with a High Capacity cDNA reverse transcription kit (Applied Biosciences). The NS4B gene was amplified with the following primers for population sequencing: for genotype 1a, 5′-TCTCAGCACTTACCGTACATC-3′ and 5′-CCCTTAGCCAGGAACCGGAGCATG-3′; and for genotype 1b, 5′-GCCTCACACCTCCCTTACATC-3′ and 5′-CTCTTAGCCACGAGCCGGAGC-3′.
Twenty-day HCV RNA reduction assay.
Replicons of genotype 1b were treated with compound over a 20-day period to measure the effect of long-term dosing on replication of viral RNA, as described previously (33). Cells were seeded in flasks containing compound at a concentration of 10× the calculated EC90. Media and compound were replenished every 2 to 3 days, and cells were reseeded every fifth day in the presence of compound. During each reseeding, samples were frozen for later analysis. At experiment completion, RNA was purified with Qiashredder and RNEasy kits (Qiagen) and was copied to cDNA with reverse transcriptase. Quantitative PCR was used to determine the amounts of cDNA at different time points, using a 7900HT sequence detection system in Fast Real Time mode. Neomycin was quantitated as a marker for HCV RNA using primers 5′-TGGATTGCACGCAGGTTCT-3′ and 5′-GTGCCCAGTCATAGCCGAAT-3′ and a 5′-(FAM)-CGGCCGCTTGGGTGGAGAGG-(TAMRA)-3′ probe (FAM, 6-carboxyfluorescein; TAMRA, 6-carboxytetramethylrhodamine), while GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as a standard for cellular RNA (Human GAPDH Endogenous TaqMan Control; Applied Biosystems, Inc.). The relative concentration of HCV RNA was calculated and compared to the concentration in an untreated sample to determine the reduction in the viral RNA load over time. Error bars correspond to multiple RNA extractions of the test culture. The experiment was repeated multiple times, with a representative curve shown.
Quantitation of compound concentrations in serum and liver of PXB mice.
GSK9574, a prodrug of GSK8853, was administered twice a day (BID) to uninfected PXB mice, created by transplanting uPA+/+/SCID mice with human hepatocytes, by oral gavage at 10, 30, and 100 mg/kg of body weight in parallel with HCV-796 (100 mg/kg) and BILN-2061 (10 mg/kg). Plasma samples were taken at time points as noted and analyzed for compound concentrations. Livers were harvested after animal sacrifice and were analyzed for compound concentrations in parallel. The presence of GSK8853, HCV-796, and BILN-2061 was quantitated by LC/MS against standard curves of compound. All studies were conducted in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and with the ethical review process at PhoenixBio Co., Ltd.
Compound efficacy in the humanized-mouse model of HCV replication.
In vivo efficacy determinations were performed at PhoenixBio Co., Ltd., using PXB mice. Human serum albumin was used as a marker of successful liver colonization, and mice that expressed human serum albumin at a concentration of at least 7 mg/ml were selected for infection with an HCV genotype 1a strain (strain PBC002) developed and grown at PhoenixBio. Mice with a minimum circulating HCV level of 3.0 × 107 copies/ml were used for the experiment.
Compounds GSK9574, HCV-796, and BILN-2061 were dosed BID by oral gavage at several concentrations (10 mg/kg, 30 mg/kg, and 100 mg/kg for GSK9574, 100 mg/kg for HCV-796, and 10 mg/kg for BILN-2061) over a 7-day period. HCV RNA was sampled and quantitated by real-time PCR from a pretreatment baseline sample on day −2 and from samples taken at 2 h, 6 h, 12 h, 1 day, 2 days, 4 days, and 7 days after the first dose for efficacy measurements. Animals were sacrificed 12 h after the last dose, and the livers were used for histology and for determining compound levels. Terminal bleeds were used to extract HCV for sequencing and for determining compound levels.
Mutation sequencing and analysis.
RNA was isolated from the serum of PXB mice by using a QIAamp UltraSens virus kit (Qiagen), and cDNAs were generated with a High Capacity cDNA reverse transcription kit (Applied Biosciences). The NS4B gene was amplified by a nested PCR method in which cDNA samples were first amplified with primers F1 (5′-ACAGGCTGCGTGGTCATAGTAGG-3′) and R1 (5′-AGACCCCCCTGTACCCGCGTTGGCAGGACA-3′) and then with primers F2 (5′-CTACCGGGAGTTCGATGAGATGGA-3′) and R2 (5′-TCGCTCAGCACCTCGCATATC-3′). The second round of amplification yielded products that were purified using Ampure (Beckman Coulter), while samples containing heavy primer-dimers were gel purified.
A Roche 454 multiplexed amplicon sequencing strategy was used to characterize the amplified NS4B gene sequences. Gene-specific primers were appended to the 3′ end of 454 amplicon fusion primers with sample-specific multiplex identifier (MID) adaptor “barcode” sequences for multiplex sample analysis with subsequent bioinformatic segregation of the Pyrosequencer data output. Pooled samples at 1 ×106 molecules/μl were titrated by emulsion-based PCR (emPCR) to determine optimal enrichment percentages (8% to 10%). EmPCR was completed in a single pool per 454 titanium plate. Pyrosequencing was performed using GS FLX Titanium series reagents (454 Life Sciences), and data were analyzed with Roche software version 2.5.3 and signal processing for amplicons. 454 sequences were filtered by barcode and aligned to the reference sequence using CLC Genomics Workbench (CLC Bio, Cambridge, MA). Low-quality reads and primer sequences were removed. Variant frequencies at each codon position were generated using only codons without gaps relative to the reference sequence. A variant frequency with fewer than 100 total reads, with a 10-fold difference between the forward and reverse frequencies, or that did not pass a statistical test (34) was considered not meaningful.
RESULTS
GSK8853 inhibits multiple HCV genotypes and generates resistant mutants in NS4B.
We previously described imidazo[1,2-a]pyrimidine compounds as inhibitors of HCV replication that directly target the viral NS4B protein and identified GSK8853 (Fig. 1A) through structural optimization of the compound series (7). GSK8853 is an inhibitor of stable HCV replicons with EC50s of 0.8 nM against genotype 1a and 5.4 nM against genotype 1b (7), although it was inactive against a stable genotype 2a replicon with an EC50 of >50 μM. GSK8853 did not show any direct toxicity to cells, with a 50% toxic concentration (CC50) of >50 μM against a genotype 1b stable replicon. Modest but reproducible differences in potency between matching stable and transient replicons were noted (compare the transient EC50s in Table 1 to the stable EC50s), with the compound appearing slightly more potent against the transient replicons. All further EC50 comparisons discussed here are therefore made using transient-replicon data.
FIG 1.
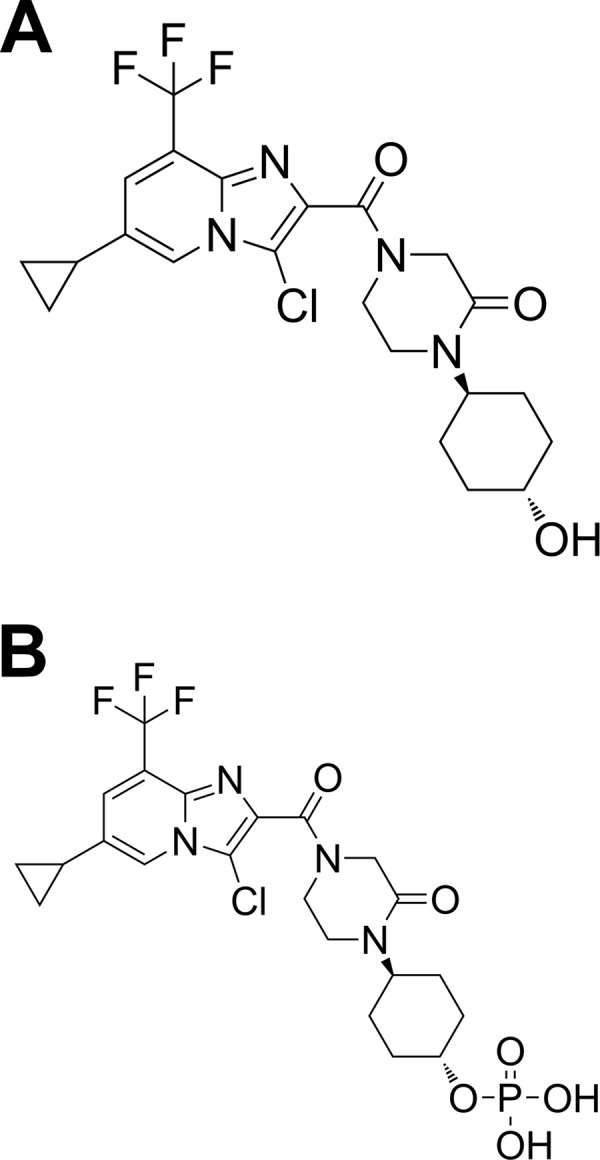
Structure of GSK8853 (A) and GSK9574 (B).
TABLE 1.
Potency of GSK8853 against transient HCV replicons
| Genotype | Residue changea | Modified protein | EC50 (nM) | Fold shiftb | 95% confidence interval (nM) |
|---|---|---|---|---|---|
| 1a | Wild type | NS4B | 0.5 | 1.0 | 0.4–0.6 |
| F98L | NS4B | 25.8 | 51.6 | 22.3–30.6 | |
| V105L | NS4B | 3.7 | 7.4 | 2.5–6.0 | |
| V105M | NS4B | 173.2 | 346.4 | 124.9–243.0 | |
| 1a PBC002 | Wild type | NS4B | 1.6 | 1.0 | 1.3–2.0 |
| N56I | NS4B | >30,000 | >18,750 | ||
| N99H | NS4B | 7,364.5 | 4,602 | 5,224–10,505 | |
| 1b | Wild type | NS4B | 1.1 | 1.0 | 1.0–1.2 |
| H94N | NS4B | 37.7 | 34.3 | 34.7–41.0 | |
| H94Rc | NS4B | 52.6 | 47.8 | 41.9–66.0 | |
| F98Lc | NS4B | 27.1 | 24.6 | 23.6–31.2 | |
| V105Lc | NS4B | 6.6 | 6.0 | 6.0–7.2 | |
| V105M | NS4B | 179.0 | 162.7 | 159.4–200.9 | |
| R155Q | NS3 | 2.6 | 2.4 | 2.3–3.4 | |
| A156Tc | NS3 | 0.9 | 0.8 | 0.60–1.5 | |
| D168A | NS3 | 1.8 | 1.6 | 1.1–7.9 | |
| L28V | NS5A | 1.6 | 1.5 | 1.5–1.8 | |
| L31Vc | NS5A | 1.4 | 1.3 | 1.0–3.3 | |
| Y93H | NS5A | 1.1 | 1.0 | 0.7–1.9 | |
| C316Y | NS5B | 1.5 | 1.4 | 1.1–3.0 | |
| M414Tc | NS5B | 1.2 | 1.1 | 0.6–8.2 | |
| P495L | NS5B | 1.1 | 1.0 | 0.9–2.0 |
Variants are numbered according to the translated peptide sequence of the gene in which the mutation is located.
Fold shift is calculated for mutant replicons relative to the unaltered (wild-type) clone of the matching background.
Data were originally reported in reference 7.
Transient replicons were used to model genotype specificity, and in that format GSK8853 had EC50s of 0.5 nM, 1.6 nM, and 83 μM against genotypes 1a, 1b, and 2a, respectively (Table 2). To model inhibition against HCV genotypes more broadly, chimeric replicons containing NS4B cassettes of different genotypes were used (32) since viable complete replicons were not available for genotypes other than 1a, 1b, and 2a. In this system, GSK8853 showed efficacy against several HCV genotypes, including 3a (31.4 nM), 4a (2.4 nM), and 5a (2.3 nM). Low potency was seen against genotypes 2b (2.4 μM) and 6a (6.3 μM), while no activity was detectable against genotype 6o (>50 μM) (Table 2).
TABLE 2.
Genotype specificity of GSK8853
| Genotype | EC50 (nM) | Fold shift | 95% confidence interval (nM) |
|---|---|---|---|
| 1a | 0.5 | 1 | 0.4–0.6 |
| 1b | 1.6 | 3 | 1.3–2.0 |
| 2a | 83,000 | 166,600 | |
| 2b | 2,400 | 4,800 | 1,404–4,431 |
| 3a | 31.4 | 63 | 19.4–52.1 |
| 4a | 2.4 | 5 | 1.3–4.6 |
| 5a | 2.3 | 5 | 1.8–2.9 |
| 6a (75–130)a | 6,300 | 12,600 | 5,483–7,503 |
| 6o (75–130)a | >50,000 | >100,000 |
Chimeras containing full NS4B genotype 6a and 6o replacements were not viable, so analysis was performed with replicons containing the genotype 6 NS4B sequences from residues 75 to 130.
In vitro resistance selection was performed by serial passage of genotype 1b (7) and 1a stable replicons in the presence of GSK8853 at concentrations 5-fold and 20-fold above the EC50, which were intended to model both moderate and high levels of inhibition. Analysis of the genotype 1b replicon sequence after selection revealed changes from the wild type that resulted in amino acid changes H94R, F98L, and V105M, similarly to reports of studies performed with other NS4B inhibitors (7, 11, 12). These sequence changes were inserted into parental replicons and tested for GSK8853 sensitivity. The H94R and F98L changes decreased the potency of the effect of GSK8853 on genotype 1b replicons by 47.8-fold and 24.6-fold, respectively, while V105M led to a larger decrease of 162.7-fold (Table 1). GSK8853 was also profiled against genotype 1b resistance mutations encoding H94N and V105L that had been identified as implicated in resistance to compounds of similar structure and showed potency decreases of 34.3-fold and 6.0-fold, respectively. The specificity of this inhibitor series with respect to NS4B was further confirmed by the lack of a potency change when GSK8853 was tested against replicons carrying mutations known to encode amino acid changes resulting in resistance to compounds targeting NS3 (R155Q, A156T, and D168A), NS5A (L28V, L31V, and Y93H), and NS5B (C316Y, M414T, and P495L) (Table 1).
Resistance selection in genotype 1a replicons has not previously been reported for NS4B inhibitors. When analyses were performed for GSK8853, mutations were identified that encoded F98L and V105M changes, similarly to the ones identified with genotype 1b. These were inserted individually into the wild-type genotype 1a replicon, where they caused potency shifts of 51.6-fold for F98L and 346.4-fold for V105M. GSK8853 was also profiled against a resistant genotype 1a V105L replicon that had been identified using earlier compounds, and a potency decrease of 7.4-fold was observed. Overall, the genotype 1a resistance pattern was similar to that found for genotype 1b replicons with respect to both the specific amino acid changes and the magnitude of the resulting resistance, further validating the association of these residues with resistance to NS4B binding inhibitors.
The structural effect of these variants on NS4B itself is difficult to ascertain, since it is an integral membrane protein that has not been crystallized. The NS4B structure has instead been modeled with computational predictions (17, 35), and the termini of the membrane-spanning regions have been tested in a variety of experiments (13, 36–39). The expected structure remains incompletely defined but is thought to have at least 4 transmembrane (TM) domains and two amphipathic helices, one of which might form a fifth TM domain under certain conditions. In this model, the amino acid residues resulting in drug resistance to GSK8853 cluster in the ER lumen across the protein loop between the TM1 and TM2 domains. Alternative analyses suggest placement of those residues within the TM1 region itself (39). Additional experiments are needed to ascertain whether these residues form an inhibitor binding site or whether they indirectly cause a reduction in compound inhibition. Regardless, regions of NS4B reported to be involved in protein multimerization (20), RNA binding (25), ATPase/GTPase activity (23), and palmitoylation (40) do not appear to overlap the region involved in GSK8853 resistance.
Twenty-day HCV RNA reduction assay of GSK8853 treatment of genotype 1b replicons.
The effect of compound treatment on replicon RNA levels was tested in vitro by treatment of genotype 1b replicons with GSK8853. Replicon cultures were grown for 20 days in the presence of GSK8853 at a concentration of 10× EC90, with samples taken every 5 days for quantitative RNA analysis. Declines in RNA levels directly measure the effect of compound on replicon growth, while a plateau or rebound may represent outgrowth of resistant strains. A rapid decline in the viral RNA load was seen over the first 5 days of the experiment, culminating in a roughly 2.5-log drop in replicon titer (Fig. 2). This reduction in viral RNA was sustained over the 20 days of treatment with no apparent rebound.
FIG 2.

Replicon RNA is reduced by 20-day dosing with GSK8853 and BILN-2061. RNA was extracted and analyzed by quantitative PCR every 5 days, and the relative amounts after normalization are shown. DMSO, dimethyl sulfoxide.
GSK8853 is less efficacious against replicons containing NS4B sequences matching human virus isolates.
The diversity of resistant variants observed in NS4B and the variability of the NS4B protein sequence in known virus isolates suggested that naturally occurring changes in sequence might alter compound efficacy, as has been seen for inhibitors of NS3 and NS5B (41–43). This possibility was investigated by constructing a series of replicons encoding single-amino-acid polymorphs identified from public sequence databases. The efficacy of GSK8853 was tested in these constructs, showing that replicons containing sequence changes at known sites of resistance (positions 94, 98, and 105) resulted in losses of potency of between 2.7-fold and 162.7-fold (Table 3). The highest magnitude of potency loss was seen with the V105M (162.7-fold) and the H94T (125.9-fold) polymorphs. Polymorphs with mutations at E161D, M162A, and M162V were also resistant to GSK8853, with reductions of potency of between 7.3- and 37-fold. Conversely, several common polymorphs (A48S/T, S2T, and V157I) had no effect on inhibition by GSK8853.
TABLE 3.
GSK8853 efficacy against genotype 1b replicons containing NS4B sequence polymorphs
| Polymorph | EC50 (nM)a | Fold shift | 95% confidence interval (nM) | Variability at position |
|---|---|---|---|---|
| Wild type | 1.1 | 1.0 | 1.0–1.2 | |
| S2T | 1.0 | 0.9 | 0.60–1.48 | 87.7% S, 11.4% T |
| R44Q | 1.6 | 1.5 | 0.87–2.73 | 86.6% R, 11.7% Q |
| T45A | 1.1 | 1.0 | 0.65–1.88 | 10.9% T, 86.9% A |
| A48S | 0.7 | 0.6 | 0.44–1.18 | 40.9% A, 12.3% S |
| A48T | 1.7 | 1.5 | 0.83–2.34 | 40.9% A, 40.9% T |
| H94A | 37.2 | 33.8 | 20.1–39.5 | 46.2% H, 0.8% A |
| H94N | 37.7 | 34.2 | 34.7–41.0 | 46.2% H, 14.5% N |
| H94Q | 3.0 | 2.7 | 1.7–5.0 | 46.2% A, 1.9% Q |
| H94Rb | 52.6 | 47.8 | 41.9–66.0 | 46.2% A, 0% R |
| H94S | 29.7 | 27.0 | 22.7–38.8 | 46.2% A, 24.2% S |
| H94T | 138.5 | 125.9 | 70.6–270.1 | 46.2% A, 5.6% T |
| H94Y | 31.5 | 28.6 | 21.2–46.5 | 46.2% A, 6.4% Y |
| F98Lb | 27.1 | 24.6 | 23.6–31.2 | 94.7% F, 5.3% L |
| V105Lb | 6.6 | 6.0 | 6.0–7.2 | 97.8% V, 1.4% L |
| V105M | 179.0 | 162.7 | 159.4–200.9 | 97.8% V, 0.3% M |
| V157I | 1.0 | 0.9 | 0.54–1.72 | 81.6% V, 18.1% I |
| E161D | 19.4 | 17.6 | 14.3–26.2 | 89.7% E, 10.0% D |
| M162A | 8.0 | 7.3 | 5.2–12.1 | 69.9% M, 7.2% A |
| M162V | 40.7 | 37.0 | 20.4–68.0 | 69.9% M, 17.5% V |
| H94N_T45A | 52.0 | 47.3 | 35.6–74.8 | |
| H94N_I86V | 50.4 | 45.8 | 30.6–79.8 | |
| H94N_M162A | 149.3 | 135.7 | 81.5–261.4 | |
| H94N_V169I | 26.4 | 24.0 | 16.1–41.8 | |
| H94N_T45A_I86V | 34.4 | 31.3 | 25.5–45.3 | |
| H94T_Q93H | 433.2 | 393.8 | 325–569 | |
| H94T_F98L | 5,200.0 | 4,727.0 | 2,090–12,091 | |
| H94Y_R44Q | 25.4 | 23.1 | 19.2–33.2 | |
| H94Y_I86V | 155.6 | 141.5 | 128–187 | |
| H94Y_L109I | 955.0 | 868.2 | 397–1285 |
All replicons showed no change in potency in testing against NS5B inhibitor HCV-796 and/or NS3 inhibitor BILN-2061 as a control.
Data were originally reported in reference 7.
Wild-type virus most often encodes two or more residue differences compared with a replicon, so a single polymorphic change does not adequately model a circulating virus. Chimeric replicons were therefore constructed containing NS4B sequences selected from diverse branches of the phylogenetic tree of HCV genotypes 1a and 1b. Ten genotype 1a sequences were used to replace the NS4B sequence of the genotype 1a (H77) transient-replicon backbone, and 10 genotype 1b sequences were used to replace the NS4B sequence of the genotype 1b (Con1 ET) transient-replicon backbone. Nine of the genotype 1a chimeras and six of the genotype 1b chimeras were viable and could be used to test compound efficacy (Table 4 and Table 5, respectively). The genotype 1a chimeras showed only small differences in the potency of GSK8853, with EC50s ranging from 0.1 to 1.8 nM. The response was different with the genotype 1b chimeras; 4 of the 6 chimeras gave dramatic reductions in potency of between 52.3-fold and 963-fold (Table 6). Control tests with NS5B inhibitor HCV-796 showed no significant change in efficacy.
TABLE 4.
Polymorphisms present in the NS4B gene of sequences used to make viable chimeric genotype 1a replicons
| Accession no. of sequence | Amino acid at H77 position: |
|||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M12 | R30 | I35 | T38 | A38 | V39 | Q40 | K45 | V48 | F49 | I58 | G93 | L97 | T116 | A117 | A121 | A126 | A127 | I128 | S130 | K135 | I140 | A142 | I157 | V162 | L168 | P225 | A231 | A235 | I253 | |
| EU781788 | A | R | S | S | T | V | ||||||||||||||||||||||||
| EU781798 | A | A | R | A | L | S | S | V | N | |||||||||||||||||||||
| EU781774 | A | A | T | A | S | T | ||||||||||||||||||||||||
| D10749 | A | T | A | S | S | V | R | L | L | |||||||||||||||||||||
| EF407439 | V | A | V | R | A | S | T | M | V | |||||||||||||||||||||
| EU155279 | A | V | F | S | T | Q | L | |||||||||||||||||||||||
| EU781746 | A | T | A | S | G | T | ||||||||||||||||||||||||
| EU781757 | H | A | V | T | S | V | T | V | ||||||||||||||||||||||
| EU781780 | T | S | T | T | ||||||||||||||||||||||||||
TABLE 5.
Polymorphisms present in the NS4B gene of sequences used to make viable chimeric genotype 1b replicons
| Accession no. of sequence | Amino acid at Con1 ET position: |
||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M11 | Q12 | I22 | T27 | A36 | R44 | T45 | A48 | I61 | I86 | Q93 | M94 | F98 | L109 | A115 | A124 | I131 | L133 | I140 | V157 | M162 | L168 | V169 | V185 | T234 | S239 | K247 | R248 | Q251 | |
| AB154177 | I | H | L | A | S | V | S | ||||||||||||||||||||||
| AF165063 | L | Q | A | V | Y | I | S | V | N | ||||||||||||||||||||
| AB049098 | L | A | V | N | V | V | V | A | M | I | |||||||||||||||||||
| EU781829 | L | I | I | S | G | R | |||||||||||||||||||||||
| AF207771 | L | I | V | K | |||||||||||||||||||||||||
| EU256088 | H | L | V | Q | A | N | H | T | L | F | H | ||||||||||||||||||
TABLE 6.
Chimeric replicons containing NS4B sequence from patient isolates
| Database entry | EC50 (nM)a | Fold shift | 95% confidence interval (nM) |
|---|---|---|---|
| Gt1a_WT (H77) | 0.5 | 1.0 | 0.4–0.6 |
| Gt1a_EU781788 | 0.4 | 0.8 | 0.33–0.57 |
| Gt1a_EU781798 | 0.3 | 0.6 | 0.19–0.52 |
| Gt1a_EU781774 | 0.1 | 0.2 | 0.09–0.22 |
| Gt1a_D10749 | 1.0 | 2.0 | 0.75–1.39 |
| Gt1a_EF407439 | 0.9 | 1.8 | 0.63–1.30 |
| Gt1a_EU155278 | 0.5 | 1.0 | 0.35–0.63 |
| Gt1a_EU781746 | 0.6 | 1.2 | 0.49–0.70 |
| Gt1a_EU781757 | 1.8 | 3.6 | 1.16–2.82 |
| Gt1a_EU781780 | 0.4 | 0.8 | 0.29–0.66 |
| Gt1b_WT (Con1 ET) | 1.1 | 1.0 | 1.0–1.2 |
| Gt1b_AB154177 | 57.5 | 52.3 | 50–66 |
| Gt1b_AF165063 | 1,059.3 | 963.0 | 551–2,032 |
| Gt1b_AB049098 | 615.9 | 559.9 | 425–891 |
| Gt1b_EU781829 | 0.9 | 0.8 | 0.67–1.13 |
| Gt1b_AF207771 | 0.9 | 0.8 | 0.74–1.13 |
| Gt1b_EU256088 | 701.8 | 638.0 | 353–1,381 |
All replicons showed no change in potency in testing against NS5B inhibitor HCV-796 and/or NS3 inhibitor BILN-2061 as a control.
To investigate the genetic basis of the efficacy changes, the nucleic acid sequences of the chimeras were compared and sequence differences encoding amino acid changes that might be responsible for the reduction in potency were identified in the resistant replicons (Table 4 and Table 5). None of the single polymorphs yielded potency changes of the magnitude seen in the full patient chimeras, so a series of double- and triple-polymorph genotype 1b replicons were constructed to investigate the relationship of different sequence combinations to drug efficacy. All of the double- and triple-polymorph chimeras were constructed to contain a sequence alteration at amino acid position 94 to reflect the diversity at this site. The effects of the double and triple polymorphs could be classified into three categories. The first category consisted of a series (H94N/T45A, H94N/I86V, H94N/V169I, H94Y/R44Q, and H94N/T45A/I86V) in which compound efficacy was not reduced beyond that seen with the single H94X polymorph. The second category consisted of combinations, including H94N/M162A, H94T/Q93H, and H94Y/I86V, that resulted in a decrease in efficacy that was modest (3-fold to 5-fold) but was greater than that seen with the H94X single polymorphs. The third category gave dramatic decreases in potency and included H94Y/L109I (868.2-fold decrease compared to the wild type) and H94T/F98L (4,727-fold decrease compared to the wild type).
In vivo treatment with GSK8853 leads to reductions in viral titer.
Subgenomic replicon systems are useful for optimizing and characterizing HCV drug candidates but do not recapitulate the complex environment of interactions present in an in vivo system. We therefore used a humanized-mouse model to determine the efficacy of GSK8853 as monotherapy to treat HCV infection with a genotype 1a PBC002 virus strain. Abbreviated results from this study along with data on the synthesis of prodrug GSK9574 have been previously reported (6), and this current work significantly expands on the previously reported data and analysis. Prior to compound testing performed with the in vivo infection model, the NS4B sequence from PBC002 was used to construct a chimeric replicon to ensure that the potency of GSK8853 against this strain was similar to that of our standard in vitro replicons. GSK8853 potency was shifted a relatively modest 3-fold for the PBC002 replicon compared to the standard genotype 1a replicon (Table 1), suggesting that this virus could be used to test in vivo efficacy. A target of 14.1 ng/ml for an in vivo efficacious concentration was estimated by adjusting the EC90 of the PBC002 replicon by 5.8-fold to account for binding to human serum proteins, as calculated by the change in EC50 against genotype 1b stable replicons in the presence of 40% human serum (44, 45).
PXB mice were created by transplanting human hepatocytes into uPA+/+/SCID mice. Mice expressing human serum albumin at a concentration of at least 7 mg/ml (mean value, 10.1 mg/ml) were used for the experiment and infected with the genotype 1a PBC002 virus. Mice with a minimum circulating HCV level of 3.0 × 107 copies/ml (mean value, 3.0 × 108 copies/ml) were used to test compound efficacy. The previously described preclinical drug metabolism and pharmacokinetics (DMPK) data for GSK8853 (7) suggested that an efficacious dose would not be easily attainable, so a phosphate prodrug form of this compound with improved DMPK properties, GSK9574 (Fig. 1B), was selected for use (6). A pilot PK study of GSK9574 in PXB mice determined that oral BID dosing at 10, 30, or 100 mg/kg would deliver levels of GSK8853 expected to be efficacious (see Fig. S1 and Table S2 in the supplemental material).
The efficacy of GSK9574 was tested in dose groups of 4 mice by orally dosing compound BID for a period of 7 days at 10, 30, and 100 mg/kg of body weight. Two control inhibitors with different inhibitory mechanisms, the NS3 inhibitor BILN-2061 (46) at 10 mg/kg and the NS5B inhibitor HCV-796 (47) at 100 mg/kg, were dosed in parallel. Serum samples were taken for quantitation of HCV RNA concentrations 2 days before dosing and at different time points after the first dose, from 2 h to 7 days. GSK9574 was well tolerated in all dose groups during the treatment period. There were no unscheduled deaths, and body weights and the ratios of liver weight to body weight were similar in all dose groups.
All three GSK9574 dose groups showed reductions in HCV RNA as early as 2 h after the first dose. The decline continued with repeated doses until a nadir was reached between day 2 and day 4 of compound dosing (Fig. 3A). The average levels of viral load reduction at the nadir were similar for the three dose groups, with 3.7 log in the 10 mg/kg GSK9574 dose group, 3.7 log in the 30 mg/kg dose group, and 3.4 log in the 100 mg/kg dose group (Fig. 3B to D and Table 7). The reductions were notably greater than the 2.3 log decrease from the HCV-796 dose group and the 2.4 log decrease from the BILN-2061 dose group (Fig. 3E and F). While the viral load in the 100 mg/kg GSK9574 dose group decreased to a stable plateau, the 10 mg/kg dose group appeared to show a postnadir viral load rebound of 1.2 logs over the final 3 days of dosing. This apparent rebound was consistent across the individual mice in the 10 mg/kg GSK9574 dose group (Fig. 3D). The 30 mg/kg dose group showed slight evidence of viral rebound, with a viral load increase of 0.2 logs that was not consistent across individual animals.
FIG 3.

In vivo reduction in viral titer. Humanized PXB mice were dosed BID with three concentrations of GSK9574 and one concentration each of comparators HCV-796 and BILN-2061. Blood samples were taken at the stated times and analyzed for the presence of HCV RNA by real-time PCR. The reduction in viral load drop was calculated by comparison to samples taken 2 days prior to compound dosing. (A) Viral RNA levels of each dose group presented as averages ± standard errors of the means (SEM). Averages of data from dose groups administered GSK9574 at doses of 10 and 30 mg/kg and HCV-796 at a dose of 100 mg/kg are from reference 6. (B to F) Quantitation of viral RNA from the individual mice within each dose group. (B) GSK9574 administered at 100 mg/kg BID. (C) GSK9574 administered at 30 mg/kg BID. (D) GSK9574 administered at 10 mg/kg BID. (E) HCV-796 administered at 100 mg/kg BID. (F) BILN-2061 administered at 10 mg/kg BID.
TABLE 7.
DMPK parameters and in vivo efficacy of GSK9574
| Dose group | Mean log VL reductiona |
Mean C12 (ng/ml)b |
Compound liver/plasma ratio | PA EC90 (ng/ml)c | Mean liver concn/PA EC90 ratio | ||
|---|---|---|---|---|---|---|---|
| At nadir | At day 7 | Plasma | Liver | ||||
| 10 mg/kg GSK9574 BID | 3.7 | 2.5 | 123 | 908 | 7.4 | 16.9 | 53.8 |
| 30 mg/kg GSK9574 BID | 3.6 | 3.4 | 380 | 2,995 | 7.9 | 16.9 | 177.2 |
| 100 mg/kg GSK9574 BID | 3.3 | 3.3 | 2,853 | 11,451 | 4.0 | 16.9 | 677.6 |
| 100 mg/kg HCV-796 BID | 2.2 | 1.5 | 127 | 3,493 | 27.4 | 14.5 | 240.9 |
| 10 mg/kg BILN-2061 BID | 2.1 | 1.8 | 3 | 86 | 25.0 | 85.5 | 1.0 |
Viral load (VL) reduction data were calculated from the serum samples relative to those taken on day −2.
Plasma and liver compound concentrations were quantitated from samples taken 12 h after the final dose (C12).
Protein-adjusted (PA) EC90 data were calculated from replicon EC50 determinations as modified by the 5.8-fold serum shift determined with the 40% human serum replicon assay.
The concentrations of inhibitor in plasma and liver were measured from samples collected 12 h after the last dose to determine the amounts required for in vivo efficacy (Table 7). Comparison of compound concentrations in liver among the different GSK9574 dose groups showed that in all cases, the amount of active compound available for viral inhibition was greater than 50-fold above the predicted efficacious concentration as defined by replicon EC90 adjusted for serum binding. For comparison, control inhibitor BILN-2061 was present at a concentration equivalent only to the predicted efficacious concentration, an observation that might explain the apparent superior antiviral activity of GSK8853. HCV-796 was present at a 240-fold ratio above the predicted efficacious concentration, similarly to the result seen with GSK8853. We speculate that the greater antiviral effect of GSK8853 than HCV-796 may have been due either to the generation of fewer viable resistance mutations or to differences in the inhibitory mechanisms of these compounds at their respective sites of action.
An additional check of GSK9574 efficacy was made by microscopically evaluating liver tissue from treated mice (i.e., routine histopathology and immunohistochemistry) compared to liver from an untreated HCV-infected PXB mouse. The livers contained in general greater than 70% human hepatocytes and had relatively normal architecture, including sinusoids and central veins. Human hepatocytes had abundant glycogen stores and lacked evidence of degeneration, necrosis, or significant steatosis (see Fig. S3 in the supplemental material). Metabolic functionality was suggested by membrane immunoreactivity for antihuman multidrug resistance-associated protein-2 (MRP-2), a hepatocyte transporter (see inset in Fig. S3). A high viral load in untreated HCV-infected PXB mice was indicated by the prominent immunoreactivity of HCV core protein in the human hepatocytes (see Fig. S4A). HCV-infected PXB mice that were treated with GSK8853 at 100 mg/kg showed decreases in HCV core protein immunoreactivity compared to untreated animals (see Fig. S4B), directly confirming the antiviral effect of the inhibitor. These microscopic observations correlated well with the viral load data at all doses.
Novel resistant variants were found in virus after in vivo treatment with GSK8853 prodrug.
Serum collected after day 7 of treatment was used for sequence analysis of the circulating virus. RNA from each individual mouse was extracted, amplified, and analyzed with ultradeep sequencing via 454 technology, with sequences compared to those of the PBC002 strain. Interestingly, only small amounts of resistant NS4B variants identified in vitro were found in the virus collected from the GSK9574-treated animals, with NS4B F98L and V105M/W variants seen in <4% of the virus population in only a few mice. The virus sequences instead presented a series of novel amino acid changes that have not been identified with in vitro replicon analysis (Table 8). Within the 10 mg/kg GSK9574 dose group, virus populations from every mouse contained high (39% to 90%) concentrations of an NS4B N99H variant, and three of the mice had small (1% to 2%) amounts of an N56I change. Virus populations from the intermediate 30 mg/kg GSK9574 dose group contained smaller proportions of NS4B N99H (4% to 13%) but larger amounts of N56I (3% to 30%) as well as an additional G60V change (6% to 11%). Sequences from the 100 mg/kg GSK9574 dose group were difficult to analyze due to the low quantity of virus remaining after dosing, but sequence changes detected in the virus populations from three of the mice included N56I (1% to 10% in three mice) and G60V (17% to 24% in two mice). Analysis of the amplicons indicated that these sequence changes occurred independently, with no linkage detected. None of these residue changes were present when pretreatment virus samples were sequenced or when the NS4B region was sequenced from virus populations of the HCV-796- and BILN-2061-treated mice.
TABLE 8.
Sequence changes identified in virus after 7-day in vivo dosing with GSK9574
| Dose group (mg/kg BID) | Mousea | % sequence changeb |
||||
|---|---|---|---|---|---|---|
| N56I | G60V | F98L | N99H | V105X | ||
| GSK9574 | ||||||
| 10 | G3-01 | 1.7 | 38.7 | |||
| G3-02 | 1.7 | 89.8 | ||||
| G3-03 | 1.9 | 1.7 | 65.7 | |||
| G3-04 | 81.0 | |||||
| 30 | G2-01 | 16.0 | 11.0 | 2.8 | 10.3 | (M) 2.6 |
| G2-02 | 30.6 | 6.5 | 6.8 | (W) 2.1 | ||
| G2-03 | 8.7 | 6.4 | 12.5 | |||
| G2-04 | 3.1 | 7.8 | 2.1 | 4.3 | (M) 1.7 | |
| 100 | G1-01 | |||||
| G1-02 | 9.6 | 17 | ||||
| G1-03 | 1.1 | |||||
| G1-04 | 10.1 | 23.9 | 3.7 | |||
| HCV-796 | ||||||
| 100 | G4-01 | |||||
| G4-02 | ||||||
| G4-03 | ||||||
| G4-04 | ||||||
| BILN-2061 | ||||||
| 10 | G5-01 | |||||
| G5-02 | ||||||
| G5-03 | ||||||
| G5-04 | ||||||
Results are separated by dose groups, designated G1 to G5, where each row refers to an individual mouse from the specific dose group.
The percentages of each variant refer to the proportion in the NS4B sequence of extracted viral RNA quantitated by deep sequencing.
To ascertain whether the sequence changes represented resistant variants, replicons were constructed that contained the changes of interest in the background of the NS4B sequence of the virus used in this experiment. Viability measurements showed that the N56I and G60V variants had very low fitness (2.9% and 1.8%, respectively) compared to the parental replicon, while the fitness of N99H was reduced only modestly (52.8%) (see Table S5 in the supplemental material). Although the viability of G60V was too low to yield efficacy information, testing of GSK8853 against the N56I and N99H replicons showed decreases in potency of >18,750-fold and 4,602-fold, respectively (Table 1).
DISCUSSION
While several groups have reported inhibitors of NS4B (6–11, 32, 48–50), the suitability of those compounds for drug development remains unclear. GSK8853 was selected for in vitro and in vivo virological evaluation to investigate the potential for compound progression. Clones with resistance to GSK8853 raised using in vitro replicons contained sequence changes encoding similar amino acid residues in genotypes 1a (F98L and V105M) and 1b (H94R, F98L, and V105M), resulting in compound efficacy changes of between 24.6-fold and 346-fold (Table 1). Prior work with other NS4B inhibitors identified analogous changes, suggesting that the specific NS4B amino acid sequence from position 94 to position 105 is critical for sensitivity to inhibitors. A focus on this region might explain the genotype specificity of these inhibitors. GSK8853 could inhibit multiple HCV genotypes, including 1a, 1b, 3a, 4a, and 5a, but showed only weak potency against genotypes 2b and 6a and essentially no activity against genotypes 2a and 6o (Table 2). Comparison of virus sequences revealed the presence of NS4B residues associated with compound resistance in genotypes 2a (T94, L98, and L105), 2b (T94 and L105), 6a (L98), and 6o (T94 and L98).
To model the breadth of GSK8853 efficacy against natural polymorphs in genotype 1a and 1b virus, replicons were constructed to encode a broad selection of single-amino-acid polymorphs identified from virus sequences deposited in public databases (Table 3). Inhibitor potency against these replicons was reduced by sequence changes, mostly at the identified sites of inhibitor resistance, including those encoding changes at positions 94, 98, and 105. Highly polymorphic residue 94 yielded potency shifts of 27.0-fold to 125.9-fold magnitude with multiple sequence changes (H94A/N/R/S/T/Y). In addition, compound efficacy was decreased by residue changes at positions 161 and 162, an effect from outside the 94-to-105 region that had not been previously seen.
Replicons constructed to encode single amino acid changes provide valuable information about drug sensitivity but do not accurately model virus isolates, as the latter often contain many residue differences from replicons rather than just one. Chimeric replicons containing NS4B sequences reflecting the diversity present in HCV subtypes were created to better predict how the drug might perform in the clinic. While nine genotype 1a replicon chimeras responded similarly to GSK8853, four of six genotype 1b chimeras showed decreased potency, implying that genotype 1b patients would not all respond similarly to inhibitor treatment (Table 6). Similar responses were seen with other compounds in this series (data not shown). The magnitude of the potency shift in the genotype 1b chimeras was greater than predicted from the individual polymorph replicons, so additional chimeras with double and triple polymorphs were made to see if the difference could be explained. Some of these replicons showed drug susceptibility that was further reduced compared to the levels seen with the single polymorphs, supporting the concept that combinations of resistant polymorphs can lead to the large reductions in GSK8853 potency seen with some of the patient cassette replicons. A recent report described NS4B chimeric replicons using sequence cloned directly from genotype 1b patient isolates (9). Those chimeras resulted in NS4B inhibitor potency reductions of lesser magnitude than those seen in the current study, a difference likely explained by the specific polymorphs present in the chimeras. The sequences used in the current work were intentionally selected to represent a greater diversity of sequence and contained polymorphs in residues 93 to 105 that were not present in the sequences studied by Rajyaguru et al. (9). Additional research on the variability of NS4B in patient populations is needed to properly model the effect of polymorphs in vivo, but their natural occurrence reveals a major development risk for NS4B inhibitors that should be addressed before the use of inhibitors can move into the clinic.
The effect of longer-term drug treatment on viral dynamics was modeled in vitro by growing genotype 1b replicon cells for 20 days in the presence of GSK8853 (Fig. 2). The resulting rapid reduction of 2.5 log in the viral RNA load, an effect slightly less than that seen with control inhibitor BILN-2061, was sustained for the length of the experiment with no obvious rebound. While this profile supports the idea of compound efficacy, similar experiments with NS5B inhibitor GSK2485852A (33) consistently showed a greater than 5-log decrease in viral load. It is possible that outgrowth of resistant NS4B polymorphs limited the reduction in virus load during this experiment, though not to the point that a viral rebound occurred. Sequencing of terminal samples from an experiment performed with a related NS4B inhibitor revealed the presence of H94R, F98L/C, and V105M, suggesting that this is likely to have been the case (data not shown).
The in vivo efficacy of NS4B inhibitors was assessed by an oral dosing of HCV-infected humanized mice with 10, 30, and 100 mg/kg BID of GSK9574, a prodrug of GSK8853. The treated mice underwent a rapid drop in HCV titer (Table 7 and Fig. 3A), while analysis of the levels of GSK8853 in the liver revealed concentrations high enough to exceed the predicted efficacious concentration. This result provided the first in vivo proof of concept for NS4B inhibitors, validating the mechanism of inhibition and strongly suggesting that an appropriately optimized NS4B inhibitor could be developed into an efficacious drug. However, although the three GSK9574 dose groups had similar early kinetics of viral load reduction, mice from the 10 mg/kg and 30 mg/kg dose groups showed apparent viral rebounds, with viral RNA levels increasing during continued treatment (Fig. 3C and D). This is suggestive of a resistant virus outgrowth that could potentially lead to the generation of viable drug-resistant virus. That risk may be of modest magnitude, as it appears the rebound could be overcome (Fig. 3B to D) by increases of the dose from 30 mg/kg to 100 mg/kg.
Sequencing of viral isolates from individual mice revealed mutations encoding NS4B N56I, G60V, and N99H across several test animals in multiple dosing groups (Table 8). The presence of mutations encoding N56I and N99H resulted in drug-resistant replicons, although the G60V replicon was not viable and could not be analyzed. Mutations resistant to small-molecule inhibitors can appear in HCV at rates related to both the alteration of inhibitor potency and the viability of viruses containing the mutation, as shown for NS3 inhibitors in vivo (51). Since viability in the PBC002 chimeric replicon is in the order WT(PBC002)>N99H>N56I>G60V and potency is in the order WT(PBC002)>N99H>N56I, the pattern of the mutant viruses identified in the humanized-mouse studies suggested that more-resistant viruses were less viable, again similarly to NS3 inhibitors (51). Further study is needed for a firm conclusion, however, given the observations that (i) while the N56I and G60V variants had very low viability in replicons, they were often found in dosed animals, suggesting that they might be more viable in a complete virus than in a replicon, and (ii) the proportion of N99H was decreased by a 3-fold-higher liver concentration of compound between the 10 mg/kg and the 30 mg/kg dose groups even though the strongly (4,600-fold) more resistant N99H should not have been affected without a much greater increase in liver drug concentration.
It is of interest that the resistance-related amino acid changes identified in vivo did not closely match those that had been generated using in vitro replicons. The different variants were not related to the 1a genotype of the virus used in the experiment, since in vitro resistance selection with a genotype 1a replicon revealed mutations encoding residue changes only at positions 98 and 105. It is possible that the NS4B sequence changes identified after in vivo treatment reflect the high micromolar concentration of GSK8853 in the livers of the dosed mice in comparison to the lower nanomolar concentration that has been used for in vitro resistance generation. It is also possible that they reflect the difference in the roles of NS4B in the replicon, which models viral RNA replication only, and in live virus, which requires a complete viral life cycle. Similar differences in resistance selection between replicon and live virus have been seen for NS3 inhibitors (52, 53), supporting this concept. A firm conclusion cannot be reached without further experimentation, but until then, the extrapolation of in vitro resistance selection to in vivo systems for NS4B inhibitors should be done with caution.
GSK8853 is an NS4B inhibitor that exhibits activity across multiple viral genotypes. In vivo dosing of HCV-infected humanized mice yielded large decreases in viral RNA levels, providing a proof of concept for the development of NS4B inhibitors. However, hurdles to drug development remain. An apparent rebound in some in vivo dose series suggested that resistant mutants could develop during treatment, while novel resistant amino acid changes identified after 7 days of dosing suggest that in vitro resistance experiments may not fully predict the development of resistance in vivo. Exploration of sequence diversity using replicons suggests that GSK8853 could have reduced potency against naturally occurring viruses, especially in genotype 1b. Overall, this body of work provides validation of NS4B inhibitors as HCV antivirals and points to risks that will need to be addressed for the further development of compounds with this mechanism.
Supplementary Material
ACKNOWLEDGMENTS
Financial support for this work was provided by GlaxoSmithKline.
We thank Katrina Creech, Luz Carballo, John Seal, III, and Lisa Stroup for their performance of stable replicon assays. We gratefully acknowledge Christopher Roberts, Andrew Spaltenstein, and Zhi Hong for their guidance and suggestions.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00813-15.
REFERENCES
- 1.Thomas DL, Seeff LB. 2005. Natural history of hepatitis C. Clin Liver Dis 9:383–398. doi: 10.1016/j.cld.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Haussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 3.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958–965. doi: 10.1016/S0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 4.Pearlman BL. 2012. Protease inhibitors for the treatment of chronic hepatitis C genotype-1 infection: the new standard of care. Lancet Infect Dis 12:717–728. doi: 10.1016/S1473-3099(12)70060-9. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization. 2014. Guidelines for the screening, care, and treatment of persons with hepatitis C infection. World Health Organization, Geneva, Switzerland. [PubMed] [Google Scholar]
- 6.Miller J, Chong P, Shotwell JB, Catalano J, Tai VW-F, Fang J, Banka A, Roberts C, Zhang H, Xiong ZZ, Mathis A, Pouliot JJ, Hamatake R, Price DJ, Seal JW III, Stroup L, Creech KL, Todd D, Spaltenstein A, Furst S, Hong Z, Peat AJ. 2014. Hepatitis C replication inhibitors that target the viral NS4B protein. J Med Chem 57:2107–2120. doi: 10.1021/jm400125h. [DOI] [PubMed] [Google Scholar]
- 7.Shotwell JB, Baskaran S, Chong P, Creech KL, Crosby RM, Dickson H, Fang J, Garrido D, Mathis A, Maung J, Parks DJ, Pouliot JJ, Price DJ, Rai R, Seal JW III, Schmitz U, Tai VW, Thomson M, Xie M, Xiong ZZ, Peat AJ. 2012. Imidazo[1, 2-a]pyridines that directly interact with hepatitis C NS4B: initial preclinical characterization. ACS Med Chem Lett 3:565–569. doi: 10.1021/ml300090x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tai VW-F, Garrido D, Price DJ, Maynard A, Pouliot JJ, Xiong ZZ, Seal JW III, Creech KL, Kryn LH, Baughman TM, Peat AJ. 2014. Design and synthesis of spirocyclic compounds as HCV replication inhibitors by targeting viral NS4B protein. Bioorg Med Chem Lett 24:2288–2294. doi: 10.1016/j.bmcl.2014.03.080. [DOI] [PubMed] [Google Scholar]
- 9.Rajyaguru S, Yang H, Martin R, Miller MD, Mo H. 2013. Development and characterization of a replicon-based phenotypic assay for assessing HCV NS4B from clinical isolates. Antiviral Res 100:328–336. doi: 10.1016/j.antiviral.2013.08.022. [DOI] [PubMed] [Google Scholar]
- 10.Dufner-Beattie J, O'Guin A, O'Guin S, Briley A, Wang B, Balsarotti J, Roth R, Starkey G, Slomczynska U, Noueiry A, Olivio PD, Rice CM. 2014. Identification of AP80978, a novel small-molecule inhibitor of hepatitis C virus replication that targets NS4B. Antimicrob Agents Chemother 58:3399–3410. doi: 10.1128/AAC.00113-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu Z, Graci JD, Lahser FC, Bresline JJ, Jung SP, Crona JH, McMonagle P, Xia E, Liu S, Karp G, Zhu J, Huang S, Nomeir A, Weetall M, Almstead NG, Peltz SW, Tong X, Ralston R, Colacino JM. 2013. Identification of PTC725, an orally bioavailable small molecule that selectively targets the hepatitis C virus NS4B protein. Antimicrob Agents Chemother 57:3250–3261. doi: 10.1128/AAC.00527-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bryson PD, Chom N-J, Einav S, Lee C, Tai V, Bechtel J, Sivaraja M, Roberts C, Schmitz U, Glenn JS. 2010. A small molecule inhibits HCV replication and alters NS4B's subcellular distribution. Antiviral Res 87:1–8. doi: 10.1016/j.antiviral.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hügle T, Fehrmann F, Bieck E, Kohara M, Krausslich H-G, Rice CM, Blum HE, Moradpour D. 2001. The hepatitis C virus nonstructural protein 4B is an integral endoplasmic reticulum membrane protein. Virology 284:70–81. doi: 10.1006/viro.2001.0873. [DOI] [PubMed] [Google Scholar]
- 14.Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aligo J, Jia S, Manna D, Konan KV. 2009. Formation and function of hepatitis C virus replication require residues in the carboxy-terminal domain of NS4B protein. Virology 393:68–83. doi: 10.1016/j.virol.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimitrova M, Imbert I, Kieny MP, Schuster C. 2003. Protein-protein interactions between hepatitis C virus nonstructural proteins. J Virol 77:5401–5414. doi: 10.1128/JVI.77.9.5401-5414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gouttenoire J, Penin F, Moradpour D. 2010. Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev Med Virol 20:117–129. doi: 10.1002/rmv.640. [DOI] [PubMed] [Google Scholar]
- 18.Paredes AM, Blight KJ. 2008. A genetic interaction between hepatitis C virus NS4B and NS3 is important for RNA replication. J Virol 82:10671–10683. doi: 10.1128/JVI.00875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piccininni S, Varaklioti A, Nardelli M, Dave B, Raney KD, McCarthy JE. 2002. Modulation of the hepatitis C virus RNA-dependent RNA polymerase activity by the non-structural (NS) 3 helicase and the NS4B membrane protein. J Biol Chem 277:45670–45679. doi: 10.1074/jbc.M204124200. [DOI] [PubMed] [Google Scholar]
- 20.Gouttenoire J, Roingeard P, Penin F, Moradpour D. 2010. Amphipathic α-helix AH2 is a major determinant for the oligomerization of hepatitis C virus nonstructural protein 4B. J Virol 84:12529–12537. doi: 10.1128/JVI.01798-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi M, Lee S, Choi T, Lee C. 2013. A hepatitis C virus NS4B inhibitor suppresses viral genome replication by disrupting NS4B's dimerization/multimerization as well as its interaction with NS5A. Virus Genes 47:395–407. doi: 10.1007/s11262-013-0956-5. [DOI] [PubMed] [Google Scholar]
- 22.Paul D, Romero-Brey I, Gouttenoire J, Stoitsova S, Krijnse-Locker J, Moradpour D, Bartenschlager R. 2011. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J Virol 85:6963–6976. doi: 10.1128/JVI.00502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson AA, Zou A, Yan J, Duggal R, Hao W, Molina D, Cronin CN, Wells PA. 2009. Biochemical characterization of recombinant hepatitis C virus nonstructural protein 4B: evidence for ATP/GTP hydrolysis and adenylate kinase activity. Biochemistry 48:906–916. doi: 10.1021/bi801747p. [DOI] [PubMed] [Google Scholar]
- 24.Einav S, Elazar M, Daneli T, Glenn JS. 2004. A nucleotide binding motif in hepatitis C virus (HCV) NS4B mediates HCV RNA replication. J Virol 78:11288–11295. doi: 10.1128/JVI.78.20.11288-11295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Einav S, Gerber D, Bryson PD, Sklan EH, Elazar M, Maerkl SJ, Glenn JS, Quake SS. 2008. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nat Biotechnol 26:1019–1027. doi: 10.1038/nbt.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho N-J, Dvory-Sobol H, Lee C, Cho S-J, Bryson P, Masek M, Elazar M, Frank CW, Glenn JS. 2010. Identification of a class of HCV inhibitors directed against the nonstructural protein NS4B. Sci Transl Med 2:15ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guillén J, González-Alvarez A, Villalaín J. 2010. A membranotropic region in the C-terminal domain of hepatitis C virus protein NS4B. Biochim Biophys Acta 1798:327–337. doi: 10.1016/j.bbamem.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka T, Kuroda K, Ikeda M, Wakita T, Kato N, Makishima M. 2013. Hepatitis C virus NS4B targets lipid droplets through hydrophobic residues in the amphipathic helices. J Lipid Res 54:881–892. doi: 10.1194/jlr.M026443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giuliano C, Fiore F, Di Marco A, Padron Velazquez J, Bishop A, Bonelle F, Gonzalez-Paz O, Marcucci I, Harper S, Narjes F, Pacini B, Monteagudo E, Migliaccio G, Rowley M, Laufer R. 2005. Preclinical pharmacokinetics and metabolism of a potent non-nucleoside inhibitor of the hepatitis C virus NS5B polymerase. Xenobiotica 35:1035–1054. doi: 10.1080/00498250500356548. [DOI] [PubMed] [Google Scholar]
- 30.Howe AYM, Cheng H, Johann S, Mullen S, Chunduru SK, Young DC, Bard J, Krishnamurthy G, Mansour T, O'Connell J. 2008. Molecular mechanism of hepatitis C virus replicon variants with reduced susceptibility to a benzofuran inhibitor, HCV-796. Antimicrob Agents Chemother 52:3327–3338. doi: 10.1128/AAC.00238-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voitenleitner C, Bechtel J, Arfsten A, Hamatake R. 2012. Hepatitis C genotype 1a replicon improved through introduction of fitness mutations. Biotechniques 52:273–275. doi: 10.2144/000113841. [DOI] [PubMed] [Google Scholar]
- 32.Arico-Muendel C, Zhu Z, Dickson H, Parks DJ, Keicher J, Deng J, Aquilani L, Coppo F, Graybill T, Lind K, Peat AJ, Thomson M. 2015. Encoded library technology screening of hepatitis C virus NS4B yields a small-molecule compound series with in vitro replicon activity. Antimicrob Agents Chemother 59:3450–3459. doi: 10.1128/AAC.00070-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voitenleitner C, Crosby RM, Walker J, Remlinger K, Vamathevan J, Wang A, You S, Johnson JI, Waldu E, Van Horn S, Horton J, Creech KL, Shotwell JB, Hong Z, Hamatake R. 2013. In vitro characterization of GSK2485852, a novel hepatitis C virus polymerase inhibitor. Antimicrob Agents Chemother 57:5216–5224. doi: 10.1128/AAC.00874-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. 2007. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HCV-1 drug resistance. Genome Res 17:1195–2101. doi: 10.1101/gr.6468307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welsch C, Albrecht M, Maydt J, Herrmann E, Welker MW, Sarrazin C, Scheidig A, Lengauer T, Zeuzem S. 2007. Structural and functional comparison of the non-structural protein 4B in flaviviridae. J Mol Graph Model 26:546–557. doi: 10.1016/j.jmgm.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 36.Gouttenoire J, Montserret R, Kennel A, Penin F, Moradpour D. 2009. An amphipathic a-helix at the C terminus of hepatitis C virus nonstructural protein 4B mediates membrane association. J Virol 83:11378–11384. doi: 10.1128/JVI.01122-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elazar M, Liu P, Rice CM, Glenn JS. 2004. An N-terminal amphipathic helix in hepatitis C virus (HCV) NS4B mediates membrane association, correct localization of replication complex proteins, and HCV RNA replication. J Virol 78:11393–11400. doi: 10.1128/JVI.78.20.11393-11400.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lundin M, Lindstrom H, Gronwall C, Persson MAA. 2006. Dual topology of the processed hepatitis C virus protein NS4B is influenced by the NS5A protein. J Gen Virol 87:3263–3272. doi: 10.1099/vir.0.82211-0. [DOI] [PubMed] [Google Scholar]
- 39.Han Q, Aligo J, Manna D, Belton K, Chintapalli SV, Hong Y, Patterson RL, van Rossum DB, Konan K. 2011. Conserved GXXXG- and S/T-like motifs in the transmembrane domains of NS4B protein are required for hepatitis C virus replication. J Virol 85:6464–6479. doi: 10.1128/JVI.02298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu G-Y, Lee K-J, Gao L, Lai MMC. 2006. Palmitoylation and polymerization of hepatitis C virus NS4B protein. J Virol 80:6013–6023. doi: 10.1128/JVI.00053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Pogam S, Seshaadri A, Kosaka A, Chiu S, Kang H, Hu S, Rajyaguru S, Symons J, Cammack N, Najera I. 2008. Existence of hepatitis C NS5B variants naturally resistant to non-nucleoside, but not to nucleoside, polymerase inhibitors among untreated patients. J Antimicrob Chemother 61:1205–1216. doi: 10.1093/jac/dkn085. [DOI] [PubMed] [Google Scholar]
- 42.Aissa Larousse J, Trimoulet P, Recordon-Pinson P, Papuchon J, Azzouz MM, Ben Mami N, Cheikh I, Triki H, Fleury H. 2014. Natural prevalence of hepatitis C virus (HCV) variants resistant to protease and polymerase inhibitors in patients infected with HCV genotype 1 in Tunisia. J Med Virol 86:1350–1359. doi: 10.1002/jmv.23958. [DOI] [PubMed] [Google Scholar]
- 43.Kuntzen T, Timm J, Berical A, Lennon N, Berlin AM, Young SK, Lee B, Heckerman D, Carlson J, Reyor LL, Kleyman M, McMahon CM, Birch C, Schulze Zur Wiesch J, Ledlie T, Koehrsen M, Kodira C, Roberts AD, Lauer GM, Rosen HR, Bihl F, Cerny A, Spengler U, Liu Z, Kim AY, Xing Y, Schneidewind A, Madey MA, Fleckenstein JF, Park VM, Galagan JE, Nusbaum C, Walker BD, Lake-Bakaar GV, Daar ES, Jacobson IM, Gomperts ED, Edlin BR, Donfield SM, Chung RT, Talal AH, Marion T, Birren BW, Henn MR, Allen TM. 2008. Naturally occurring dominant resistance mutations to HCV protease and polymerase inhibitors in treatment-naïve patients. Hepatology 48:1769–1778. doi: 10.1002/hep.22549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walker J, Crosby RM, Wang A, Woldu EA, Vamathevan J, Voitenleitner C, You S, Remlinger K, Duan M, Kasmierski W, Hamatake R. 2014. Preclinical characterization of GSK2336805, a novel inhibitor of hepatitis C virus replication that selects for resistance in NS5A. Antimicrob Agents Chemother 58:38–47. doi: 10.1128/AAC.01363-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ando I, Adachi T, Ogura N, Toyonaga Y, Sugimoto K, Abe H, Kamada M, Noguchi T. 2012. Preclinical characterization of JTK-853, a novel nonnucleoside inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob Agents Chemother 56:4250–4256. doi: 10.1128/AAC.00312-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bos M, Cameron DR, Cartier M, Cordingley MG, Faucher A-M, Goudreau N, Kawai SH, Kukolj G, Lagace L, LaPlante SR, Narjes H, Poupart M-A, Rancourt J, Sentjens RE, St George R, Simoneau B, Steinmann G, Thibeault D, Tsantrizos YS, Weldon SM, Yong C-L, Llinas-Brunet M. 2003. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426:186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- 47.Kneteman NM, Howe AY, Gao T, Lewis J, Pevear D, Lund G, Douglas D, Mercer DF, Tyrrell DL, Immermann F, Chaudhary I, Speth J, Villano SA, O'Connell J, Collett M. 2009. HCV796: a selective nonstructural protein 5B polymerase inhibitor with potent anti-hepatitis C virus activity in vitro, in mice with chimeric human livers, and in humans infected with hepatitis C virus. Hepatology 49:745–752. doi: 10.1002/hep.22717. [DOI] [PubMed] [Google Scholar]
- 48.Kakarla R, Liu J, Naduthambi D, Chang W, Mosley RT, Bao D, Micolochick Steuer HM, Keilman M, Bansal S, Lam AM, Seibel W, Neilson S, Furman PA, Sofia MJ. 2014. Discovery of a novel class of potent HCV NS4B inhibitors: SAR studies on piperazinone derivatives. J Med Chem 57:2136–2160. doi: 10.1021/jm4012643. [DOI] [PubMed] [Google Scholar]
- 49.Phillips B, Cai R, Delaney W, Du Z, Ji M, Jin H, Lee J, Li J, Niedziela-Majka A, Mish M, Pyun H-J, Saugier J, Tirunagari N, Wang J, Yang H, Wu Q, Sheng C, Zonte C. 2014. Highly potent HCV NS4B inhibitors with activity against multiple genotypes. J Med Chem 57:2161–2166. doi: 10.1021/jm401646w. [DOI] [PubMed] [Google Scholar]
- 50.Zhang N, Zhang X, Zhu J, Turpoff A, Chen G, Morrill C, Huang S, Lennox W, Kakarla R, Liu R, Li C, Ren H, Almstead NG, Venkatraman S, Njorge FG, Gu Z, Clausen V, Graci J, Jung SP, Zheng Y, Colacino JM, Lasher F, Sheedy J, Molllin A, Weetall M, Nomeir A, Karp GM. 2014. Structure-activity relationship (SAR) optimization of 6-(indol-2-yl)pyridine-3-sulfonamides: identification of potent, selective, and orally bioavailable small molecules targeting hepatitis C (HCV) NS4B. J Med Chem 57:2121–2135. doi: 10.1021/jm401621g. [DOI] [PubMed] [Google Scholar]
- 51.Sarrazin C, Kieffer TL, Bartels D, Hanzelka B, Muh U, Welker M, Wincheringer D, Zhou Y, Chu H-M, Lin C, Weegink C, Reesink H, Zeuzem S, Kwong AD. 2007. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767–1777. doi: 10.1053/j.gastro.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 52.Vermehren J, Susser S, Lange C, Forestier N, Karey U, Hughes E, Ralston R, Tong X, Zeuzem S, Sarrazin C. 2012. Mutations selected in the hepatitis C virus NS3 protease domain during sequential treatment with boceprevir with and without pegylated interferon alfa-2b. J Viral Hepat 19:120–127. doi: 10.1111/j.1365-2893.2011.01449.x. [DOI] [PubMed] [Google Scholar]
- 53.Hiraga N, Imamura M, Abe H, Hayes CN, Kono T, Onishi M, Tsuge M, Takahashi S, Ochi H, Iwao E, Kamiya N, Yamada I, Tateno C, Yosizato K, Matsui H, Kanai A, Inaba T, Tanaka S, Chayama K. 2011. Rapid emergence of telaprevir resistant hepatitis C virus strain from wildtype clone in vivo. Hepatology 54:781–788. doi: 10.1002/hep.24460. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.