

Abstract
Pharmacokinetic (PK) data describing a prolonged time course of antiretrovirals in plasma and peripheral blood mononuclear cells (PBMCs) are important for understanding and managing late or missed doses and to assess the appropriateness of compounds for preexposure prophylaxis (PrEP). This study aimed to evaluate the PK of coformulated tenofovir disoproxil fumarate (DF), emtricitabine, and rilpivirine in plasma and of the intracellular (IC) anabolites tenofovir diphosphate (TFV-DP) and emtricitabine triphosphate (FTC-TP) in healthy volunteers up to 9 days after drug cessation. Individuals received daily tenofovir DF-emtricitabine-rilpivirine (245/200/25 mg) for 14 days. Drug intake was stopped, and serial sampling occurred prior to the final dose and up to 216 h (9 days) after stopping drug intake. Concentrations were quantified and PK parameters calculated. Eighteen volunteers completed the study. The terminal elimination plasma half-lives for tenofovir and emtricitabine over 216 h (geometric mean [90% confidence interval]) were higher than those seen over 0 to 24 h (for tenofovir, 31 h [27 to 40 h] versus 13.3 h [12.5 to 15.1 h]; for emtricitabine, 41 h [36 to 54 h] versus 6.4 h (5.9 to 7.6 h]). Model-predicted IC half-lives (0 to 168 h) were 116 h (TFV-DP) and 37 h (FTC-TP). The plasma rilpivirine concentration at 216 h was 4.5 ng/ml (4.2 to 6.2 ng/ml), and half-lives over 0 to 216 h and 0 to 24 h were 47 h (41 to 59 h) and 35 h (28 to 46 h), respectively. These data contribute to our understanding of drug behavior following treatment interruption; however, adherence to therapy should be promoted. Validated plasma and IC target concentrations are necessary to allow interpretation with respect to sustained virus suppression or HIV prevention. (The trial was conducted in accordance with the Declaration of Helsinki [EudraCT 2012-002781-13].)
INTRODUCTION
The challenge of maintaining a high level of adherence to antiretroviral therapy has been met, in part, by the development of fixed-dose-combination tablets suitable for once-daily dosing, such as tenofovir (TFV) disoproxil fumarate (DF)-emtricitabine (FTC)-efavirenz (EFV) tablets (Atripla; Gilead Sciences Ltd., London, United Kingdom] and tenofovir DF-emtricitabine-rilpivirine (RPV) tablets (Eviplera [or Complera]; Gilead Sciences Ltd., London, United Kingdom, and Gilead Sciences Inc., Foster City, CA, USA). Eviplera, containing rilpivirine, a nonnucleoside reverse transcriptase inhibitor (NNRTI), and tenofovir DF and emtricitabine (a nucleotide reverse transcriptase inhibitor and a nucleoside reverse transcriptase inhibitor, respectively), is approved in Europe for treatment of HIV-infected adults without NNRTI-associated resistance mutations or mutations associated with tenofovir or emtricitabine resistance and with viral loads of ≤100,000 copies/ml (1). It has been approved in the United States for therapy-naive adults with a viral load of ≤100,000 copies/ml and for switching treatments of virologically suppressed patients (viral load of <50 copies/ml) under certain conditions (2). Moreover, encouraging therapeutic outcomes and improved lipid profiles have been observed in switching suppressed patients to tenofovir DF-emtricitabine-rilpivirine from tenofovir DF-emtricitabine-efavirenz or raltegravir-based regimens (3).
Despite the inroads made into improving patient adherence to therapy, delays in drug intake or missed doses can occur as a result of individual circumstances, e.g., busy lifestyle or personal problems, risking viral rebound and resistance emergence. Key pharmacokinetic (PK) characteristics, such as a prolonged elimination half-life, are likely to be more forgiving of late or missed doses; however, PK data under these conditions are lacking, particularly for coformulated regimens. Although patients are instructed to maintain a high level of adherence, information regarding drug persistence in plasma and cells following treatment interruption could potentially improve management of late or missed doses.
Data describing persistence of drugs within plasma, cells, and other physiological compartments are also essential for HIV prevention strategies such as preexposure prophylaxis (PrEP), determining which drugs may have suitable PK properties. Coformulated tenofovir DF-emtricitabine (Truvada; Gilead Sciences Ltd., London, United Kingdom) was approved by the U.S. Food and Drug Administration in 2012 for use as PrEP in high-risk individuals and in those engaging in sexual activity with HIV-infected partners (4). An intramuscularly administered, long-acting formulation of rilpivirine is also under investigation as a PrEP agent (5).
Tenofovir (administered as tenofovir DF and rapidly converted by esterases following absorption to tenofovir) and emtricitabine are prodrugs that require intracellular (IC) phosphorylation to their active anabolites. While tenofovir is a monophosphate analogue requiring two phosphorylation steps to form tenofovir diphosphate (TFV-DP), emtricitabine triphosphate (FTC-TP) is formed by three endogenous enzymatic steps (6). Concentrations of parent compounds in plasma and of TFV-DP and FTC-TP within peripheral blood mononuclear cells (PMBC) have been reported in combination with efavirenz (Atripla) over 9.5 days after stopping therapy in healthy volunteers (7); however, their PK profiles coformulated with rilpivirine after stopping medication have not been evaluated. Moreover, rilpivirine plasma PK and terminal half-life after drug cessation have not been previously investigated.
The primary aim of this study was to evaluate plasma PK of tenofovir, emtricitabine, and rilpivirine and IC PK of TFV-DP and FTC-TP in healthy, HIV-negative volunteers over 9 days following drug intake cessation.
MATERIALS AND METHODS
Study population.
Males or nonlactating, nonpregnant females aged 18 to 65 years with a body mass index (BMI) of 18 to 35 kg/m2 who provided written informed consent were eligible for enrollment. Exclusion criteria included the presence of any significant acute or chronic medical illness; a positive screen result for hepatitis B virus, hepatitis C virus, or HIV; evidence of organ dysfunction or abnormal physical examination results; abnormalities in vital signs or electrocardiogram (ECG) or clinical laboratory parameters; current or recent (within 3 months) gastrointestinal disease; clinically relevant alcohol or drug use (including positive urine drug screen results) or comparable activities considered by the investigator to affect compliance with trial procedures; exposure to any investigational drug or placebo within 3 months of administration of the first dose of the study drug; use of any other drugs, including over-the-counter medications and herbal preparations within 2 weeks of administration of the first dose of the study drug; known allergy to any constituents of the study drug; or (for females of childbearing potential) nonuse of effective nonhormonal birth control methods.
Study design.
This was a 23-day (excluding screening and follow-up), open-label, single-treatment-arm, PK study, carried out at the PK Unit of St Stephen's Centre, Chelsea & Westminster Foundation Trust (London, United Kingdom). The study was reviewed and approved by the National Research Ethics Service (NRES Chelsea, London, United Kingdom).
Routine laboratory tests were performed at screening, and drug safety and tolerability were assessed throughout the study period according to the NIAID Division of AIDS grading scale for adverse events (from grade 1 [mild] to grade 4 [life-threatening]) in addition to monitoring of vital signs, physical examinations, and clinical laboratory investigations.
Following a 10-h overnight fast on study day 1 (baseline visit), participants were administered tenofovir DF-emtricitabine-rilpivirine (245/200/25 mg) with a 533-kcal breakfast. All participants continued tenofovir DF-emtricitabine-rilpivirine once daily at home, and adherence was monitored by questionnaire and pill counting. On day 14, individuals were admitted to the research unit and blood collection for drug quantification commenced immediately before (within 10 min) administration of the final tenofovir DF-emtricitabine-rilpivirine dose (predose, 0 h). Samples were drawn at 2, 4, 8, and 12 h after stopping the drug intake. Subjects were discharged thereafter, returning to provide 24-, 36-, 48-, 60-, 72-, 96-, 120-, 144-, 168-, 192-, and 216-h samples. All visits to the unit included documentation of concomitant medications and adverse events. A final follow-up visit between days 30 and 36 was used to review adverse events, vital signs, and clinical laboratory assessments.
Analytical methods. (i) Plasma collection for tenofovir, emtricitabine, and rilpivirine quantification.
Blood was collected into lithium heparin Vacutainer blood collection tubes which were immediately inverted several times, placed in a light-protective container, and kept on ice or refrigerated until centrifugation. Samples were centrifuged (10 min, 1,200 × g, 4°C) within 30 min of collection, and plasma was stored in light-protective amber-colored tubes (at −20°C) prior to shipping on dry ice to the Good Clinical Laboratory Practice (GCLP)-accredited Liverpool Bioanalytical Facility (Liverpool, United Kingdom) for analysis.
(ii) PBMC isolation for TFV-DP and FTC-TP quantification.
PBMCs were obtained as previously described (7). There was a technical issue encountered in generating the cell counts which meant that IC TFV-DP and FTC-TP data could not be determined by bioanalytical methods.
(iii) Quantification of tenofovir and emtricitabine and rilpivirine in plasma.
Plasma tenofovir, emtricitabine, and rilpivirine concentrations were determined using fully validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) methods (7, 8). The lower limit of quantification (LLQ) was 0.5 ng/ml, and assay precision was <15% for all three drugs.
(iv) Modeling and prediction of TFV-DP and FTC-TP concentrations in peripheral blood mononuclear cells.
Modeling of plasma tenofovir and emtricitabine linked to their IC anabolites (TFV-DP and FTC-TP) using various approaches has been previously described (9–11). This methodology was explored to allow prediction of TFV-DP and FTC-TP concentrations, up to 168 h (7 days) following drug cessation, from plasma data.
Separate models were developed for tenofovir and emtricitabine using nonlinear mixed-effects modeling (NONMEM v. 7.2; Icon Development Solutions, Ellicott City, MD, USA) (12), and initial parameter estimates for plasma data were taken from the literature (9, 13).
Plasma tenofovir and emtricitabine and time-matched TFV-DP and FTC-TP concentrations from a previous study investigating tenofovir, emtricitabine, and efavirenz (Atripla) PK following drug cessation in healthy volunteers (EFV study) (7) were used as prior information to describe the relationship between plasma and IC anabolite concentrations. All data from both studies were modeled simultaneously. Plasma and IC concentrations between 0 and 156 h (6.5 days) for the EFV study and plasma concentrations between 0 and 168 h (7 days) for the present study were included, as this provided the majority of samples with concentrations above the assay LLQ. Samples with concentrations less than the LLQ between 0 and 156 h and between 0 and 168 h were excluded from the modeling process.
The influences of covariates, including age, weight, BMI, serum creatinine level, creatinine clearance (CrCL; calculated using the Chronic Kidney Disease Epidemiology Collaboration [CKD-EPI] formula [14]), sex, ethnicity, and food intake (drug intake under fasted versus fed conditions), on plasma tenofovir and emtricitabine PK were investigated. To accept a model with one extra parameter, a decrease in the minimal objective function value (OFV) of at least 3.84 units was required (P = 0.05; χ2 distribution; 1 degree of freedom [df]). A backward-elimination step was performed once significant covariates were included; biologically plausible covariates producing an increase in the OFV (>6.64 units, P = 0.01, χ2 distribution, 1 df) upon removal were retained.
To evaluate the models, 90% prediction intervals (P5 to P95) using final parameter estimates were generated from 1,000 simulated individuals with the same distribution of covariates as the original data set, and the observed data were superimposed. At least 90% of observed data being within the prediction interval was representative of an adequate model.
Final model parameters were used to predict IC TFV-DP and FTC-TP concentration-time profiles between 0 and 168 h for the present study. Plasma PK parameters were fixed to individual Bayesian estimates for the present study, and population parameters obtained for the relationship between drug in plasma and IC anabolites were used as prior information. Predictions were made utilizing the $SIMULATION option of NONMEM.
(v) Statistical analysis.
This was an exploratory study, and no formal sample size calculation was performed. It was estimated that a total of 16 subjects completing the study would be sufficient to allow relevant conclusions.
Area under the concentration-time curve from 0 to 24 h postdose (AUC0–24) and from h 0 to the last measureable time point within 216 h (AUC0–last), the maximum concentration (Cmax), and the concentration 24 h postdose (C24) were calculated for plasma tenofovir, emtricitabine, and rilpivirine using noncompartmental methods (WinNonlin Phoenix v. 6.3; Pharsight Corporation, Mountain View, CA, USA). Terminal elimination half-life was determined to the last measureable time point within 216 h.
AUC0–24, AUC0–168, Cmax, and C24 were calculated as outlined above for TFV-DP and FTC-TP using model-predicted concentrations. Terminal elimination half-life was calculated using the following formula: ln(2)/k40 (k40, rate constant for loss or elimination of TFV-DP or FTC-TP; see Fig. S1 in the supplemental material).
Pharmacokinetic parameters were summarized as geometric means (90% confidence interval [CI]), and interindividual variability was expressed as percent coefficient of variation (CV%) using the following equation: CV% = (standard deviation/mean) × 100.
RESULTS
Study population.
Eighteen participants (11 [61%] female) completed the study. Median (range) age, weight, BMI, serum creatinine level, and CrCL were 31 years (19 to 47), 75 kg (60 to 105), 24 kg/m2 (21 to 31), 73 μmol/liter (57 to 104), and 103 ml/min/1.73 m2 (78 to 146), respectively. Participants described themselves as Caucasian (n = 10), black Caribbean (n = 2), black African (n = 2), Asian (n = 1), Hispanic (n = 1), or mixed ethnicity (n = 2). The study drug was well tolerated, and no grade 3 or 4 adverse events were reported.
Plasma tenofovir, emtricitabine, and rilpivirine pharmacokinetics.
Of 288 samples, 20 (7%; 120 to 216 h), 5 (2%; 168 to 216 h), and 1 (0.3%; 192 h) were below the LLQ for tenofovir, emtricitabine, and rilpivirine, respectively. Nine, 15, and 17 individuals had quantifiable tenofovir, emtricitabine, and rilpivirine concentrations at all sampling time points between 0 and 216 h after stopping drug intake.
Geometric mean plasma concentrations over time for tenofovir, emtricitabine, and rilpivirine are shown in Fig. 1, and PK parameters are summarized in Table 1. The geometric mean (in hours [90% CI]) terminal elimination half-life to the last measureable time point within 216 h was markedly longer than that corresponding to 0 to 24 h for tenofovir and emtricitabine (30.7 h [27.2 to 39.7] versus 13.3 h [12.5 to 15.1] and 40.5 h [35.8 to 53.5] versus 6.44 h [5.88 to 7.55]). The elimination half-life corresponding to 0 to 216 h was slightly longer than that corresponding to 0 to 24 h for rilpivirine (47.2 h [41.3 to 59.3] versus 34.6 h [28.4 to 45.9]). The rilpivirine geometric mean (90% CI) C216 was 4.53 ng/ml (4.22 to 6.15). A therapeutic cutoff has not been defined for rilpivirine, but 50 ng/ml has been suggested based on unpublished data (5). At 24 and 36 h (representing a 12-h-delayed dose), 2/18 (11%) and 6/18 (33%) of the participants had concentrations below 50 ng/ml; 7/18 (39%) and 11/18 (61%) of the participants had concentrations that were below this value 48 h and 72 h after stopping drug intake.
FIG 1.
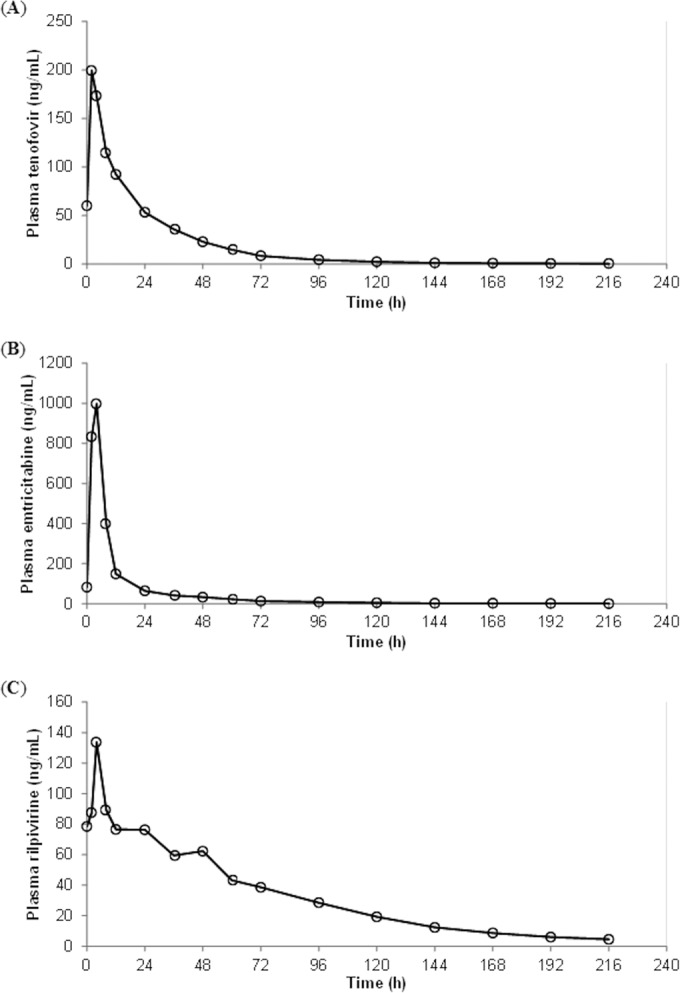
Geometric mean plasma tenofovir (A), emtricitabine (B) and rilpivirine (C) concentrations over 216 h following drug intake cessation in healthy volunteers (n = 18).
TABLE 1.
Summary of plasma tenofovir, emtricitabine, and rilpivirine pharmacokinetic parameters obtained following drug intake cessationa
| Parameter | Values (90% CI [CV%]) (n = 18) |
||
|---|---|---|---|
| Tenofovir | Emtricitabine | Rilpivirine | |
| AUC0–24 | 2,573 ng · h/ml (2,342–3,208 [40]) | 8,537 ng · h/ml (7,860–11,955 [53]) | 2,116 ng · h/ml (1,929–2,527 [34]) |
| AUC0–last | 4,249 ng · h/ml (3,860–5,325 [41]) | 11,126 ng · h/ml (10,169–15,075 [50]) | 7,271 ng · h/ml (6,635–8,761 [36]) |
| Cmax | 227 ng/ml (208–280 [38]) | 1,260 ng/ml (1,148–1,925 [65]) | 139 ng/ml (128–168 [35]) |
| C24 | 53.3 ng/ml (48.8–71.1 [48]) | 64.7 ng/ml (58.2–97.3 [65]) | 76.3 ng/ml (68.7–94.8 [41]) |
| TE half-life | 30.7 h (27.2–39.7 [48]) | 40.5 h (35.8–53.5 [51]) | 47.2 h (41.3–59.3 [46]) |
Data are presented as geometric means (90% CI). AUC0–24, area under the curve over 24 h postdose; AUC0–last, area under the curve to the last measureable concentration within 216 h (0 to 216 for plasma rilpivirine); Cmax, maximum concentration; C24, concentration 24 h postdose; TE half-life, terminal elimination half-life to the last measureable concentration within 216 h (0 to 216 h for plasma rilpivirine).
Prediction of intracellular tenofovir diphosphate and emtricitabine triphosphate concentrations.
Sixteen healthy volunteers (5 female; n = 11 Caucasian, n = 2 black African, n = 2 mixed ethnicity, n = 1 Asian) from the EFV study were included. Median (range) age, weight, BMI, serum creatinine level, and CrCL were 32 years (21 to 57), 81 kg (54 to 109), 27 kg/m2 (21 to 35), 81 μmol/liter (60 to 112), and 102 ml/min/1.73 m2 (72 to 126), respectively. Of 206 and 207 evaluable plasma and intracellular samples, the EFV study (7) contributed 203 and 206 plasma tenofovir and emtricitabine concentrations (<LLQ, n = 3, n = 0, respectively) and 183 and 207 TFV-DP and FTC-TP concentrations (<LLQ, n = 24, n = 0, respectively). The present study contributed 245 (n = 6 <LLQ) and 250 (n = 1 <LLQ) plasma tenofovir and emtricitabine concentrations to the models.
A diagrammatic summary of the model structure used for both drugs is shown (see Fig. S1 in the supplemental material). Plasma tenofovir and emtricitabine were best described by a two-compartment oral model, parameterized by apparent oral clearance (CL/F), apparent volume of the central compartment (Vc/F) and apparent volume of the peripheral compartment (Vp/F), intercompartmental clearance (Q/F), and absorption rate constant (ka). Due to lack of information in the absorption phase, ka values for both drugs were fixed to literature values (1.05 h−1 and 0.53 h−1 for tenofovir and emtricitabine, respectively) (9, 13). Residual variability was described by a proportional-error model, and inclusion of interindividual variability (IIV) on CL/F and Vp/F was supported for tenofovir and on CL/F, Vc/F, and Vp/F for emtricitabine. TFV-DP and FTC-TP from the EFV study were described by first-order rate constants, namely, k24 (uptake and conversion to the phosphorylated form) and k40 (loss of anabolite). IIV was included in k24 determinations, and a proportional-error model described residual variability.
Inclusion of a food (or partner drug) effect on relative bioavailability (F1) significantly improved the fit for tenofovir but not for emtricitabine. F1 was fixed to a value of 1 (i.e., 100%) for the fasted state (or with efavirenz) and increased by 33% with food (or with rilpivirine; see Table S2 in the supplemental material). Inclusion of weight on the clearance and volume parameters using allometric scaling significantly improved the tenofovir model, as did addition of CrCL (linear function) on tenofovir CL/F determinations. CrCL was significantly associated with emtricitabine CL/F (linear function).
Population parameters from the final models are presented in Table S2 in the supplemental material. Diagnostic plots suggested that both models adequately described the data and were confirmed by the visual predictive checks with 90% and 92% of observed plasma tenofovir and TFV-DP concentrations, respectively, within the 90% prediction interval and with 92% and 94% of plasma emtricitabine and FTC-TP concentrations, respectively, within the prediction interval (see Fig. S3).
Intracellular tenofovir diphosphate and emtricitabine triphosphate pharmacokinetics.
Geometric mean predicted TFV-DP and FTC-TP concentrations over 168 h are illustrated (Fig. 2), and PK parameters are shown (Table 2). Model-derived terminal elimination half-lives (0 to 168 h) for TFV-DP and FTC-TP were 116 (4.8 days) and 37 h (1.5 days), respectively.
FIG 2.
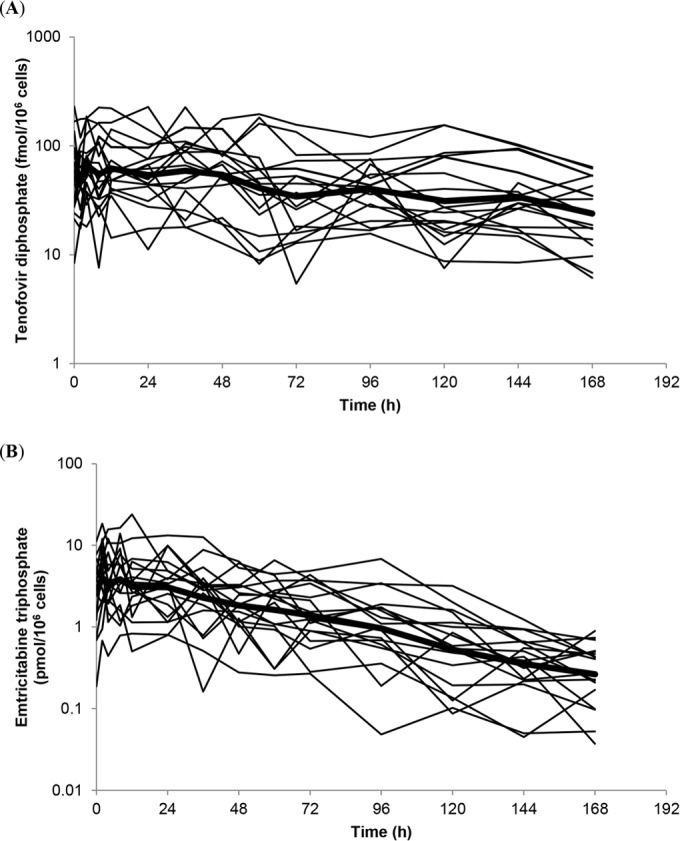
Individual predicted intracellular tenofovir diphosphate (A) and emtricitabine triphosphate (B) concentrations over 168 h following drug intake cessation in healthy volunteers on a log-linear scale (n = 18; individual concentration-time profiles generated by modeling and simulation). The bold line represents the geometric mean concentration-time profile.
TABLE 2.
Summary of intracellular tenofovir diphosphate and emtricitabine triphosphate pharmacokinetic parameters obtained following drug intake cessationa
| Parameterb | Values (90% CI [CV%]) (n = 18) |
|
|---|---|---|
| Tenofovir diphosphate | Emtricitabine triphosphate | |
| AUC0–24 | 1,456 fmol · h/106 cells (1,302–2,193 [66]) | 87.8 pmol · h/106 cells (79.2–150 [80]) |
| AUC0–168 | 7,495 fmol · h/106 cells (6,792–11,486 [66]) | 273 pmol · h/106 cells (252–440 [70]) |
| Cmax | 92.2 fmol/106 cells (83.8–135 [60]) | 6.15 pmol/106 cells (5.73–10.5 [75]) |
| C24 | 54.0 fmol/106 cells (48.2–87.9 [75]) | 3.07 pmol/106 cells (2.88–5.63 [83]) |
Data are presented as geometric means (90% CI). AUC0–24, area under the curve over 24 h postdose; AUC0–168, area under the curve over 168 h postdose; Cmax, maximum concentration; C24, concentration 24 h postdose.
Parameters were determined by noncompartmental analysis using concentration-time profiles generated by means of modeling and simulation.
IC TFV-DP and FTC-TP target concentrations for HIV suppression are not known; however, HIV prevention targets have been determined using PK data from the Pre-Exposure Prophylaxis Initiative (iPrEx) trial. Ninety percent risk reduction was associated with concentrations of 16 fmol/106 and 3.7 pmol/106 viable cells for TFV-DP and FTC-TP, respectively (15). At 24, 36, 48, and 72 h after stopping drug intake, predicted TFV-DP concentrations were <16 fmol/106 cells in 6%, 0%, 1%, and 22% of individuals, respectively, while predicted FTC-TP levels were below 3.7 pmol/106 cells in 56%, 78%, 83%, and 83% of individuals, respectively.
DISCUSSION
Concentrations in plasma of tenofovir, emtricitabine, and, for the first time, rilpivirine have been demonstrated over 9 days (216 h) after stopping tenofovir DF-emtricitabine-rilpivirine intake in healthy, HIV-negative adults. Predictions of IC TFV-DP and FTC-TP concentrations from plasma data were also achieved utilizing modeling and simulation and prior information from a previous, similar study (7).
A therapeutic cutoff for sustained viral suppression has not been defined for rilpivirine, but 50 ng/ml has been suggested based on an unpublished analysis of phase III trials in which 50 ng/ml was the upper limit of the lowest quartile of the trough concentrations in which the virological response was lowest (5). Eleven percent, 33%, and 39% of individuals had concentrations below this threshold value 24, 36, and 48 h after stopping drug intake, respectively. However, these data should be interpreted with caution given that 50 ng/ml is not a validated target concentration. The long elimination half-lives of 35 h (0 to 24) and 47 h (0 to 216) determined as part of this study are consistent with that previously reported for rilpivirine (45 h [16, 17]). The data presented indicate that rilpivirine exhibits PK properties that may allow forgiveness for delayed dosing in some patients; however, individuals should be instructed to adhere to licensed-dosing guidelines.
The tenofovir plasma exposure in the present study was higher than that obtained by Jackson et al. in healthy volunteers stopping therapy (AUC0–last, 4,249 versus 2,895 ng · h/ml [7]) and was highlighted during the modeling process. The two studies were conducted at the same research unit, and the bioanalyses occurred at the same laboratory. However, the NNRTIs used in the fixed-dose combinations were different in the two studies (efavirenz versus rilpivirine), as were the food intake conditions. The EFV study was conducted under fasting conditions (2 h prior and after drug intake); however, rilpivirine must be administered with food in order to achieve optimal absorption. Tenofovir exposure, as a component of a tenofovir DF-emtricitabine-rilpivirine fixed-dose combination, has been shown to increase by 38% following a standard meal (540 kcal) (1). A non-clinically relevant increase in plasma tenofovir of 24% has also been reported upon coadministration with rilpivirine, potentially through mild inhibition of the renal transmembrane transporters responsible for tenofovir renal elimination (18). However, an interaction has not been observed during coadministration with efavirenz (19). Inclusion of a food effect on F1 (relative bioavailability) improved the tenofovir model, resulting in an F1 value that was 33% higher for the present study than for the EFV study. This could also be attributed to the interaction with rilpivirine or to a combination of a food effect with a partner drug effect. Emtricitabine PK parameters were within the ranges previously reported and are known to be unaffected by food intake or coadministration with rilpivirine (1, 7). Terminal elimination half-lives to the last measureable time point within 216 h for both nucleosides were considerably longer than those seen over 0 to 24 h (for tenofovir, 31 versus 13 h; for emtricitabine, 41 versus 6 h) and were also in agreement with values from the earlier study (7).
Due to issues with PBMC cell counts, TFV-DP and FTC-TP could not be directly quantified; however, a modeling approach was explored using the observed plasma tenofovir and emtricitabine concentrations and data from another study as prior information. The model was relatively simplistic, using an effect compartment for TFV-DP or FTC-TP linked to the plasma compartment by a rate constant (k24) describing a number of processes, including the uptake and metabolism of tenofovir and emtricitabine. Given that tenofovir monophosphate and emtricitabine diphosphate concentrations were not measured, this helped limit problems with identifiability of model parameters. A similar model structure has recently been used to describe tenofovir and TFV-DP concentrations in healthy female volunteers (20). An indirect-response model has previously been used to describe plasma tenofovir and IC TFV-DP concentrations in HIV patients (9); however, this model was not supported by our data. A simulation study reported by Madrasi et al. investigated a mechanistic model for tenofovir, focusing more specifically on describing saturable uptake and metabolism in PBMCs using literature values (11). Despite the simplistic nature of the model used for the current analysis, it performed well for both drugs, and the parameters generally agreed with the literature but could be updated if further data became available (9, 13, 21).
The parameters describing the IC anabolites (k24, k40, variability in k24) were estimated using data generated from a previous study (7) but also incorporated the individual predicted plasma PK parameters determined for the present study. The plasma PK parameters drive the prediction of TFV-DP and FTC-TP in the model. Therefore, the predicted TFV-DP PK parameters were slightly higher than those reported by Jackson et al. (7) because the plasma tenofovir concentrations were also higher. Unsurprisingly, given the agreement in plasma PK parameters, the FTC-TP parameters were similar between studies. A limitation of the modeling is that external data sets are required to further evaluate the models; however, it is noteworthy that the TFV-DP and FTC-TP predictions are within the ranges previously reported, including the PrEP population for TFV-DP (15, 20, 22).
Evaluation of antiretroviral PK forgiveness and persistence within physiological compartments is also important for methods of HIV prevention, such as PrEP. Favorable PK characteristics, including prolonged elimination half-lives, are beneficial for PrEP agents, allowing once-daily or less-frequent dosing in order to aid adherence. Based on outcomes reported from the iPrEx and Partners PrEP trials, the use of tenofovir-emtricitabine (Truvada) has been approved as a PrEP regimen in the United States (23, 24). A long-acting, parenteral formulation of rilpivirine is under development, and investigations to determine its suitability as a PrEP compound have begun. Single-dose rilpivirine PK in plasma and in genital tracts of males (600 mg) and females (300, 600, and 1,200 mg) was assessed, and the drug was shown to persist for up to 84 days. The effect of rilpivirine concentrations in female genital tract fluid on HIV replication was also explored ex vivo (5). Studies to further evaluate long-acting rilpivirine as PrEP are planned (ClinicalTrials.gov identifier NCT02165202 [25]) or ongoing (ClinicalTrials.gov identifier NCT01656018 [26, 27]). Furthermore, a rilpivirine oral formulation (with or without tenofovir and emtricitabine) may be used in the context of PrEP for short periods of time (e.g., as an oral lead in dose for safety reasons or as an alternative to long-acting PrEP); therefore, knowledge of drug exposures after stopping drug intake and PK forgiveness may help in planning for this eventuality.
Interpretations of these data are limited by the lack of knowledge of the fully validated target concentrations at which virological suppression (or prevention) occurs for rilpivirine and IC TFV-DP and FTC-TP. Therefore, information on the time at which virological control could be lost (or transmission occurs) or how long a dose could be delayed was not attainable. Using PK data from the iPrEx study, an IC TFV-DP concentration of 16 fmol/106 viable cells was associated with 90% HIV risk reduction (15). This target was also applied to data obtained from the Cell-PrEP study which investigated the achievement and maintenance of protective concentrations of tenofovir-emtricitabine in uninfected men who have sex with men. After stopping drug intake at day 30, the samples from 80% and 48% of individuals were above this concentration at 2 and 7 days post-drug cessation, respectively (28). In comparison, predicted TFV-DP concentrations from the present study were ≥16 fmol/106 cells in 94% and 72% of volunteers at 2 and 7 days after stopping drug intake.
As this study evaluated drug PK after stopping treatment, it could not be conducted in HIV-infected patients and assessment of viral load after treatment interruption could not be performed. Translation from the present findings requires further study in patient populations where pharmacodynamic endpoints can be investigated given that PK parameters in HIV-infected and healthy individuals may differ (17). Still, this report contributes significantly to our understanding of the drug behavior that occurs when therapy is stopped, and the long elimination half-lives determined for all three drugs are encouraging.
Adherence to antiretroviral therapy should be promoted in order to maintain optimal virological control; however, the persisting plasma PK of tenofovir, emtricitabine, and rilpivirine and IC TFV-DP and FTC-TP demonstrated by this study may provide a potentially forgiving, coformulated regimen for individuals that may miss or delay an occasional dose, as well as a prospective PrEP candidate.
Supplementary Material
ACKNOWLEDGMENTS
We thank the staff of St. Stephen's Centre and the volunteers for taking part in the study.
This study was performed with financial support from Gilead Sciences Ltd.
L.D. is supported by PreDiCT-TB and has received a travel bursary from Gilead Sciences Ltd. H.M.Y. has received travel bursaries from Gilead. Since the completion of the study, A.J. has become an employee of Gilead Sciences. G.M. has been on the Speaker Bureau for Janssen, Bristol Myers Squibb, Gilead, and Merck and has been an advisor for Tobira, Merck, and Teva. L.E., S.K., and D.B. have received research grants and/or travel bursaries from Merck, Bristol Myers and Squibb, GlaxoSmithKline, Pfizer, Abbott, ViiV, Boehringer Ingelheim, and Janssen Pharmaceuticals. M.B. has received travel and research grants from and has been an adviser for Janssen, Roche, Pfizer, ViiV, Bristol-Myers Squibb, Merck Sharp & Dohme, and Gilead.
A.A., Z.K., and C.H. declare that they have no conflicts of interest.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01441-15.
REFERENCES
- 1.Electronic Medicines Compendium. 2014. Eviplera (200 mg/25 mg/245 mg film coated tablets). http://www.medicines.org.uk/EMC/medicine/25518/SPC/Eviplera+200+mg+25+mg+245+mg+film+coated+tablets/ Accessed 23 January 2015. [Google Scholar]
- 2.Gilead Sciences. 2014Complera (emtricitabine, rilpivirine, tenofovir disoproxil fumarate) tablets, for oral use—highlights of prescribing information. Gilead Sciences, Foster City, CA. [Google Scholar]
- 3.Pinnetti C, Di Giambenedetto S, Maggiolo F, Lorenzini P, Fabbiani M, Tommasi C, Latini A, Ammassari A, Loiacono L, Sterrantino G, Bellagamba R, Boumis E, Antinori A, Zaccarelli M. 2014. Simplification to co-formulated rilpivirine/emtricitabine/tenofovir in virologically suppressed patients: data from a multicenter cohort. J Int AIDS Soc 17:19812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.US Food and Drug Administration. 2012. FDA approves first drug for reducing the risk of sexually acquired HIV infection—FDA news release. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm312210.htm Accessed 23 January 2015. [Google Scholar]
- 5.Jackson AG, Else LJ, Mesquita PM, Egan D, Back DJ, Karolia Z, Ringner-Nackter L, Higgs CJ, Herold BC, Gazzard BG, Boffito M. 2014. A compartmental pharmacokinetic evaluation of long-acting rilpivirine in HIV-negative volunteers for pre-exposure prophylaxis. Clin Pharmacol Ther 96:314–323. doi: 10.1038/clpt.2014.118. [DOI] [PubMed] [Google Scholar]
- 6.Piliero PJ. 2004. Pharmacokinetic properties of nucleoside/nucleotide reverse transcriptase inhibitors. J Acquir Immune Defic Syndr 37(Suppl 1):S2–S12. doi: 10.1097/01.qai.0000137001.40505.56. [DOI] [PubMed] [Google Scholar]
- 7.Jackson A, Moyle G, Watson V, Tjia J, Ammara A, Back D, Mohabeer M, Gazzard B, Boffito M. 2013. Tenofovir, emtricitabine intracellular and plasma, and efavirenz plasma concentration decay following drug intake cessation: implications for HIV treatment and prevention. J Acquir Immune Defic Syndr 62:275–281. doi: 10.1097/QAI.0b013e3182829bd0. [DOI] [PubMed] [Google Scholar]
- 8.Else LJ, Tjia J, Jackson A, Penchala SD, Egan D, Boffito M, Khoo SH, Back DJ. 2014. Quantification of rilpivirine in human plasma, cervicovaginal fluid, rectal fluid and genital/rectal mucosal tissues using liquid chromatography-tandem mass spectrometry. Bioanalysis 6:1907–1921. doi: 10.4155/bio.14.59. [DOI] [PubMed] [Google Scholar]
- 9.Baheti G, Kiser JJ, Havens PL, Fletcher CV. 2011. Plasma and intracellular population pharmacokinetic analysis of tenofovir in HIV-1-infected patients. Antimicrob Agents Chemother 55:5294–5299. doi: 10.1128/AAC.05317-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirt D, Pruvost A, Ekouevi DK, Urien S, Arrive E, Kone M, Nerrienet E, Nyati M, Gray G, Kruy LS, Blanche S, Dabis F, Treluyer JM. 2011. Very high concentrations of active intracellular phosphorylated emtricitabine in neonates (ANRS 12109 trial, step 2). Antimicrob Agents Chemother 55:2953–2960. doi: 10.1128/AAC.01376-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madrasi K, Burns RN, Hendrix CW, Fossler MJ, Chaturvedula A. 2014. Linking the population pharmacokinetics of tenofovir and its metabolites with its cellular uptake and metabolism. CPT Pharmacometrics Syst Pharmacol 3:e147. doi: 10.1038/psp.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beal S, Sheiner LB. 1989. –1998. NONMEM users guide. ICON Development Solutions, Ellicott City, MD, USA. [Google Scholar]
- 13.Valade E, Treluyer JM, Bouazza N, Ghosn J, Foissac F, Benaboud S, Fauchet F, Viard JP, Urien S, Hirt D. 2014. Population pharmacokinetics of emtricitabine in HIV-1-infected adult patients. Antimicrob Agents Chemother 58:2256–2261. doi: 10.1128/AAC.02058-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J, CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) . 2009. A new equation to estimate glomerular filtration rate. Ann Intern Med 150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson PL, Glidden DV, Liu A, Buchbinder S, Lama JR, Guanira JV, McMahan V, Bushman LR, Casapía M, Montoya-Herrera O, Veloso VG, Mayer KH, Chariyalertsak S, Schechter M, Bekker LG, Kallás EG, Grant RM, iPrEx Study Team. 2012. Emtricitabine-tenofovir concentrations and pre-exposure prophylaxis efficacy in men who have sex with men. Sci Transl Med 4:151ra125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garvey L, Winston A. 2009. Rilpivirine: a novel non-nucleoside reverse transcriptase inhibitor. Expert Opin Investig Drugs 18:1035–1041. doi: 10.1517/13543780903055056. [DOI] [PubMed] [Google Scholar]
- 17.Electronic Medicines Compendium. 2014. Edurant 25 mg tablets. http://www.medicines.org.uk/EMC/medicine/25490/SPC/Edurant+25+mg/ Accessed 23 January 2015. [Google Scholar]
- 18.Hoetelmans R, Van Heeswijk R, Kestens D, Stevens M, Peeters M, Williams P, Bastiaanse L, Buffels R, Woodfall B. 2005. Pharmacokinetic interaction between the novel non-nucleoside reverse transcriptase inhibitor TMC278 and tenofovir disoproxil fumarate in healthy volunteers, abstr WePe3.3C15. Abstr 3rd IAS Conf HIV Pathogenesis Treat, 24 to 27 July 2005, Rio de Janeiro, Brazil. [Google Scholar]
- 19.Droste JA, Kearney BP, Hekster YA, Burger DM. 2006. Assessment of drug-drug interactions between tenofovir disoproxil fumarate and the nonnucleoside reverse transcriptase inhibitors nevirapine and efavirenz in HIV-infected patients. J Acquir Immune Defic Syndr 41:37–43. doi: 10.1097/01.qai.0000191997.70034.80. [DOI] [PubMed] [Google Scholar]
- 20.Burns RN, Hendrix CW, Chaturvedula A. 2015. Population pharmacokinetics of tenofovir and tenofovir-diphosphate in healthy women. J Clin Pharmacol 55:629–638. doi: 10.1002/jcph.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jullien V, Treluyer JM, Rey E, Jaffray P, Krivine A, Moachon L, Lillo-Le Louet A, Lescoat A, Dupin N, Salmon D, Pons G, Urien S. 2005. Population pharmacokinetics of tenofovir in human immunodeficiency virus-infected patients taking highly active antiretroviral therapy. Antimicrob Agents Chemother 49:3361–3366. doi: 10.1128/AAC.49.8.3361-3366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang LH, Begley J, St Claire RL III, Harris J, Wakeford C, Rousseau FS. 2004. Pharmacokinetic and pharmacodynamic characteristics of emtricitabine support its once daily dosing for the treatment of HIV infection. AIDS Res Hum Retroviruses 20:1173–1182. doi: 10.1089/aid.2004.20.1173. [DOI] [PubMed] [Google Scholar]
- 23.Baeten JM, Donnell D, Ndase P, Mugo NR, Campbell JD, Wangisi J, Tappero JW, Bukusi EA, Cohen CR, Katabira E, Ronald A, Tumwesigye E, Were E, Fife KH, Kiarie J, Farquhar C, John-Stewart G, Kakia A, Odoyo J, Mucunguzi A, Nakku-Joloba E, Twesigye R, Ngure K, Apaka C, Tamooh H, Gabona F, Mujugira A, Panteleeff D, Thomas KK, Kidoguchi L, Krows M, Revall J, Morrison S, Haugen H, Emmanuel-Ogier M, Ondrejcek L, Coombs RW, Frenkel L, Hendrix C, Bumpus NN, Bangsberg D, Haberer JE, Stevens WS, Lingappa JR, Celum C, Partners PrEP Study Team. 2012. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med 367:399–410. doi: 10.1056/NEJMoa1108524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, Goicochea P, Casapía M, Guanira-Carranza JV, Ramirez-Cardich ME, Montoya-Herrera O, Fernández T, Veloso VG, Buchbinder SP, Chariyalertsak S, Schechter M, Bekker LG, Mayer KH, Kallás EG, Amico KR, Mulligan K, Bushman LR, Hance RJ, Ganoza C, Defechereux P, Postle B, Wang F, McConnell JJ, Zheng JH, Lee J, Rooney JF, Jaffe HS, Martinez AI, Burns DN, Glidden DV, iPrEx Study Team. 2010. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med 363:2587–2599. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.U.S. National Institutes of Health. 2014. Phase II safety and acceptability of an investigational injectable product, TMC278LA, for pre-exposure prophylaxis. https://clinicaltrials.gov/ct2/show/NCT02165202?term=rilpivirine&rank=6 Accessed 26 January 2015. [Google Scholar]
- 26.U.S. National Institutes of Health. 2015. A study to evaluate safety, acceptability, pharmacokinetics, and ex vivo pharmacodynamics of TMC278 long acting formulation in HIV-1 seronegative participants. https://clinicaltrials.gov/ct2/show/NCT01656018?term=rilpivirine&rank=3 Accessed 26 January 2015. [Google Scholar]
- 27.McGowan I, Siegel S, Duffill K, Shetler C, Dezzutti C, Richardson-Harman N, Abebe K, Back D, Else L, Herrick A, Williams P, Rehman KK, Cranston RD. 2014. A phase 1 open label safety, acceptability, pharmacokinetic, and pharmacodynamic study of intramuscular TMC278 LA (the MWRI-01 Study), abstr OA27.06 LB. Abstr HIV Res Prev (HIV R4P), 28 to 31 October 2014, Cape Town, South Africa. [Google Scholar]
- 28.Seifert SM, Glidden DV, Meditz AL, Castillo-Mancilla JR, Gardner EM, Predhomme JA, Rower C, Klein B, Kerr BJ, Guida LA, Zheng JH, Bushman LR, Anderson PL. 2015. Dose response for starting and stopping HIV preexposure prophylaxis for men who have sex with men. Clin Infect Dis 60:804–810. doi: 10.1093/cid/ciu916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.