

Abstract
Skin and soft tissue infections (SSTIs) are a common occurrence in health care facilities with a heightened risk for immunocompromised patients. Klebsiella pneumoniae has been increasingly implicated as the bacterial agent responsible for SSTIs, and treatment can be challenging as more strains become multidrug resistant (MDR). Therefore, new treatments are needed to counter this bacterial pathogen. Gallium complexes exhibit antimicrobial activity and are currently being evaluated as potential treatment for bacterial infections. In this study, we tested a topical formulation containing gallium citrate (GaCi) for the treatment of wounds infected with K. pneumoniae. First, the MIC against K. pneumoniae ranged from 0.125 to 2.0 μg/ml GaCi. After this in vitro efficacy was established, two topical formulations with GaCi (0.1% [wt/vol] and 0.3% [wt/vol]) were tested in a murine wound model of MDR K. pneumoniae infection. Gross pathology and histopathology revealed K. pneumoniae-infected wounds appeared to close faster with GaCi treatment and were accompanied by reduced inflammation compared to those of untreated controls. Similarly, quantitative indications of infection remediation, such as reduced weight loss and wound area, suggested that treatment improved outcomes compared to those of untreated controls. Bacterial burdens were measured 1 and 3 days following inoculation, and a 0.5 to 1.5 log reduction of CFU was observed. Lastly, upon scanning electron microscopy analysis, GaCi treatment appeared to prevent biofilm formation on dressings compared to those of untreated controls. These results suggest that with more preclinical testing, a topical application of GaCi may be a promising alternative treatment strategy for K. pneumoniae SSTI.
INTRODUCTION
The demand for novel antibacterial treatments is growing given that many species and strains of bacteria worldwide have become increasingly resistant to the majority of clinically available antibiotic treatments. Specifically, Klebsiella pneumoniae, a member of the ESKAPE pathogens (Enterobacter spp., Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterococcus spp.) has been responsible for numerous outbreaks in a number of hospitals (1–4), including a recent, fatal outbreak at the National Institutes of Health in the United States (5). K. pneumoniae isolates were also the first bacterial species shown to harbor the NDM-1 plasmid, a plasmid that harbors the New Delhi metallo-beta-lactamase (NDM-1) responsible for extensive carbapenem resistance (6). Increases in the number of K. pneumoniae infections have been observed in U.S. military hospitals with the shift to the conflict in Afghanistan compared to the A. baumannii infections that were more prevalent during Operation Iraqi Freedom (7). Moreover, colistin-resistant K. pneumoniae strains have started to emerge around the world (8–10), suggesting that this bacterial species can evolve quickly even in the face of aggressive antibiotic treatments. The combination of nosocomial spread with multidrug-resistant (MDR) K. pneumoniae strains is a daunting prospect for clinicians, as there are few treatment options remaining on the foreseeable horizon.
Iron sequestration or competition for iron has been explored for many years as a potential therapy for bacterial infection. In fact, the human body utilizes the iron sequestration approach by limiting free iron at infection sites with molecules such as lactoferrin, transferrin, hepcidin, and other iron-binding proteins in order to limit bacterial growth (11–13). However, bacterial pathogens have evolved a number of mechanisms to counter these host defenses by capturing iron from various sources. For example, iron acquisition systems, such as the Feo, Sit, and Efe systems, transport free ferrous iron, Fe(II), into the bacterial cell in Gram-negative bacteria (13, 14) and are an essential process for many bacterial species (15–18). FeoB is well conserved across Gram-negative bacteria and was shown to localize in the inner, cytoplasmic membrane, where Fe(II) transport appears to be dependent on GTP activity (19). The Sit acquisition system appears to be specialized for intracellular, enteric bacteria such as Shigella or Salmonella species, where Fe(II) is depleted in the cytoplasm of the host (17, 20). In contrast, enterohemorrhagic Escherichia coli does not invade cells, but the presence of the Efe system helps to transport Fe(II) inside the bacteria and confer a survival advantage in the low pH environment of the gastrointestinal tract (21).
However, bacteria also utilize ferric iron, Fe(III), for survival and employ siderophores, small molecules that are secreted into the extracellular milieu in order to capture Fe(III) from the host (11–13). There are a large variety of siderophores with a high binding affinity for Fe(III), and their cognate receptors are found on the surface of the outer membrane (11–13). Siderophores are often unique to each bacterial species and are required for survival in the host environment (12, 18). Lastly, bacteria have been shown to harbor membrane-bound receptors for the human proteins transferrin, lactoferrin, and hemin to traffic the iron that is complexed by these host proteins into the infecting bacterium (11–13). All of these iron acquisition mechanisms speak to the significant importance of iron for bacterial survival in the host and were likely derived from systems used by progenitors that were fighting for iron and other nutrients in restricted, nonhost environments such as the soil.
In an attempt to counter iron-dependent bacterial processes, researchers have attempted treatment with iron chelators with high binding constants to outcompete the siderophores and other bacterial iron acquisition mechanisms (22–25). However, the use of iron chelators as an antimicrobial therapeutic approach has had mixed success and is likely reflective of the chelator choice and the indication pursued (26). Another promising antibacterial method that exploits the requirement for iron is a “Trojan horse” strategy, where siderophores are conjugated to antibiotics (sideromycins) via linkage chemistry (27, 28). This antibacterial approach has had successes in vitro and in vivo (29–33); however, resistance may also rapidly emerge because many bacteria harbor multiple siderophore receptors, and there are also “cheaters,” bacteria that usurp siderophores other than their own, which are prevalent in polymicrobial infections (34–39) A recent failure of a sideromycin in vivo also points to efflux as a mechanism of resistance (33); however, a follow-up study showed efflux can be overcome with the application of an efflux inhibitor (40).
Another approach exploiting the bacterial need for iron is the use of simple gallium(III) salts, such as gallium nitrate, gallium maltolate, or gallium citrate (Fig. 1). In this case, rather than chelating iron away from the bacterial pathogen, gallium, in excess, outcompetes the iron that is targeted by the aforementioned acquisition systems. Then, upon uptake, it replaces the iron in key chemical reactions required for survival and ultimately poisons the bacteria (41). This approach has shown efficacy against P. aeruginosa in vitro and in vivo (42–46), and treatment success may also involve antibiofilm activity (44–46). Gallium formulations have also been shown to be effective against other pathogenic bacteria (47–51), including A. baumannii (52, 53); however, it should be noted that certain strains of A. baumannii appear to circumvent gallium nitrate activity with a heme receptor in the host environment (54). Given this mixed efficacy against different bacteria, each gallium formulation needs to be thoroughly tested in vitro and in vivo against each pathogen. Moreover, there is also the challenge of toxicity. Gallium(III) has been shown to be safe in lower doses, where specifically, gallium citrate has been used as a diagnostic for cancer and gallium nitrate has been approved for human use as a cancer treatment (Ganite) (41, 55, 56). However, any formulation at a higher concentration has the potential to limit cell division (i.e., 100 μM for human keratinocytes) or cause apoptosis (i.e., 500 μM for human keratinocytes) (56).
FIG 1.
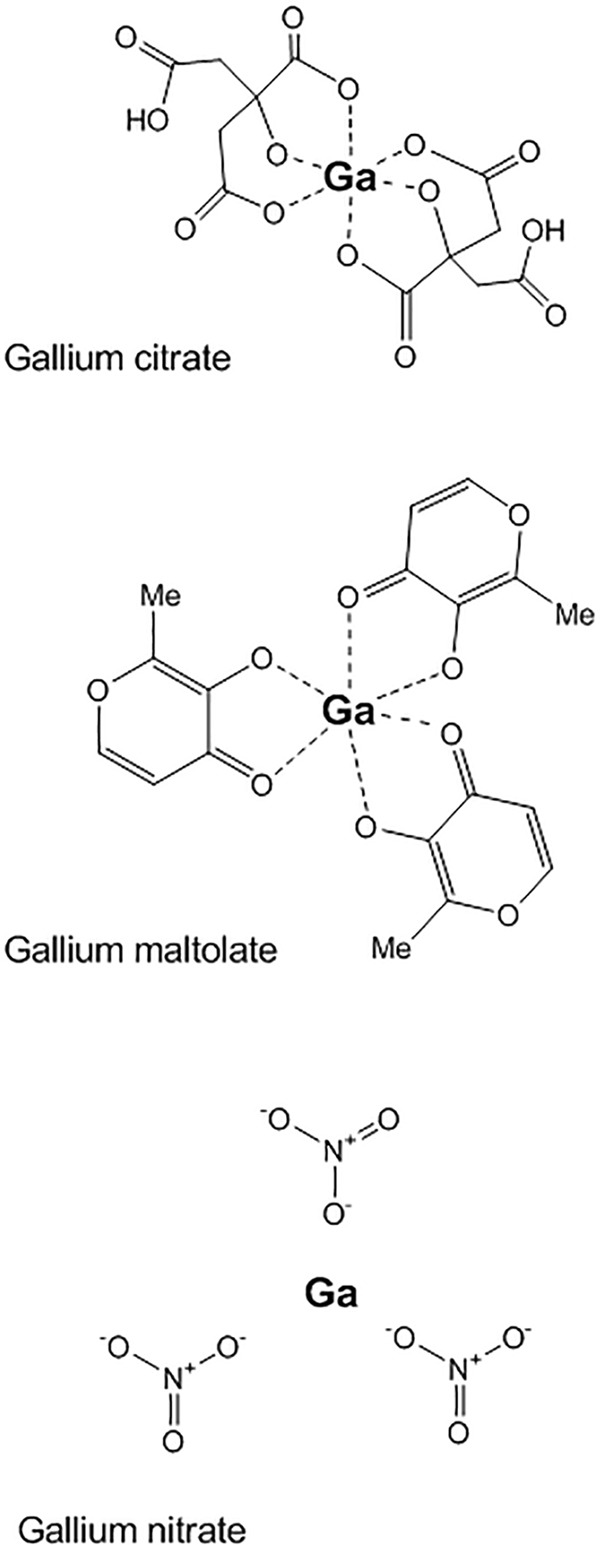
Gallium(III) salt structures. Chemical structures of commonly used gallium salts for antibacterial treatments.
In the current study, we explore the use of gallium citrate (GaCi) against K. pneumoniae. Like the other pathogenic bacterial species previously mentioned, K. pneumoniae isolates have a high demand for iron, especially in the host environment, and recently a hypervirulent strain was shown to have an increased propensity for iron acquisition (57). As K. pneumoniae has been responsible for many wound infections suffered by the U.S. military (7, 58) as well as chronic wounds suffered by diabetics (59), a topical therapy would be desirable to combat skin and soft tissue infections (SSTIs) caused by this pathogen.
MATERIALS AND METHODS
Bacterial strains and inoculum preparation.
The majority of tested K. pneumoniae isolates were wound infection clinical isolates from International Health Management Associates, Inc., with the exception of strain BAA-2146, which was obtained from the American Type Culture Collection (ATCC), and strain KP4640, which was obtained from the Multidrug-resistant Organism Repository and Surveillance Network (MRSN) at the Walter Reed Army Institute of Research (WRAIR). KP4640 was previously described (60), and this isolate was used for all of the in vivo wound infection experiments in this study. KP4640, before use in animals, was propagated on Lennox lysogeny broth (LB) (Becton, Dickinson and Co., Sparks, MD). To prepare inocula for animal infection, 100 μl of a KP4640 overnight culture was subcultured into 10 ml of fresh LB in a 250-ml Erlenmeyer flask, grown at 37°C, and shaken at 250 rpm. Cells in the midexponential growth phase were harvested at an optical density at 600 nm of 0.8. Cells were washed twice with sterile phosphate-buffered saline (PBS) and then resuspended in PBS so that 25 μl of the suspension contained approximately 5.0 × 105 cells. The cell concentration of the suspension was verified via a Petroff-Hausser counting chamber prior to inoculation of mice and confirmed by serial dilution and plating on LB agar using a spiral plating system (Autoplate; Advanced Instruments, Inc., Norwood, MA). The difference observed between each measure of cell number was never more than 2-fold.
MIC.
The MIC was determined by International Health Management Associates, Inc., or Aridis Pharmaceuticals, Inc., using an overnight culture of bacteria that was diluted to a starting inoculum of ∼2.0 × 105 CFU/ml and assessing turbidity after 24 h at 37°C and exposure to GaCi serially diluted using 2-fold dilutions with a range from 0.02 to 256 μg/ml. To mimic the in vivo, low iron concentration environs of the host, an iron-deficient culture medium (BM-2) with succinic acid as the sole carbon source [40 mM K2HPO4, 22 mM KH2PO4, 7 mM (NH4)2SO4, 34 mM succinic acid, and 1 mM MgSO4 adjusted to pH 7.0] was utilized for all of the MIC experiments instead of the standard cation-adjusted Mueller-Hinton broth, which is rich in iron and typically used.
Separately, an antibiogram for KP4640 was determined using the Phoenix automated system (Becton, Dickinson and Co., Franklin Lakes, NJ) according to the manufacturer's instructions.
Preparation of treatments.
The neutropenic agent cyclophosphamide was purchased in powdered form from Baxter (Deerfield, IL) and dissolved in sterile 0.9% sodium chloride injection solution (Hospira, Inc., Lake Forest, IL) to a final concentration of 10 mg/ml. Antibiotics, either colistin (Sigma-Aldrich), doxycycline (Sigma-Aldrich), or Primaxin (Baxter) were purchased in powdered form and dissolved in sterile 0.9% sodium chloride injection solution (Hospira, Inc.) and, if needed, sterile dimethyl sulfoxide (DMSO) was used to improve solubility and achieve final concentrations of 10 mg/ml. Placebo hydroxyethyl cellulose (HEC) formulations as well as HEC amended with either 0.1% (wt/vol) or 0.3% (wt/vol) GaCi were provided by Aridis Pharmaceuticals, Inc. Gallium citrate was prepared by mixing gallium nitrate with ammonium citrate followed by precipitation of gallium citrate and removal of nitrate-based previously known chemical reactions (56). This manufacturing process has been implemented at the kilogram scale under GMP (good manufacturing practice).
Mice and husbandry.
Female BALB/c mice were purchased from the National Cancer Institute (now Charles River) Animal Production Program (Frederick, MD). The mice used in these experiments were 6 to 10 weeks of age and weighed 14 to 20 g. All mice received sterile food and water ad libitum, and dry rodent chow was supplemented with DietGel recovery (ClearH2O, Portland, ME) during the 48 h following wounding. All mice were housed in groups of three, in sanitized cages on sterile paper bedding, and were provided with environmental enrichment, including in-cage plastic housing.
Murine dorsal wound model.
All procedures were performed in accordance with protocol IB02-10 or 14-BRD-01S, which were approved by the Walter Reed Army Institute of Research (WRAIR)/Navy Medical Research Center (NMRC) Institutional Animal Care and Use Committee (Silver Spring, MD) and were previously described as a model for A. baumannii wound infection (61). Briefly, mice received 150 mg/kg of body weight and 100 mg/kg cyclophosphamide intraperitoneal (i.p.) injections before wounding and infection on days −4 and −1, respectively. On day 0, the day of wounding and inoculation, mice were anesthetized with ketamine (130 mg/kg) (Ketaset; Fort Dodge Animal Health, Fort Dodge, IA), xylazine (10 mg/kg) (AnaSed; Lloyd, Inc., Shenandoah, IA), and buprenorphine (0.05 mg/kg) (Hospira, Inc., Lake Forest, IL) injections given for pain management. Hair was clipped from the cervical to midlumbar dorsum, and the skin was scrubbed with iodine solution followed by an ethanol rinse. A 6.0-mm disposable skin biopsy punch (VisiPunch; Huot Instruments LLC, Menomonee Falls, WI) was used to create a full-thickness skin defect overlying the thoracic spinal column and the adjacent musculature. Twenty-five microliters containing 5.0 × 105 KP4640 cells in a PBS suspension was pipetted onto the wound and allowed to absorb for at least 3 min. A circular cutout (30 mm diameter) of transparent dressing (Tegaderm roll; 3M Health Care, St. Paul, MN) was placed over the wound and secured with tissue adhesive (Vetbond; 3M Animal Care, St. Paul, MN).
For experiments assessing weight loss and wound closure beginning at 4 h postinoculation, mice (3 biological replicates with 12 mice per group [a total of 144 mice]) were treated with either 25 μl of HEC placebo, 0.1% (wt/vol) GaCi, or 0.3% (wt/vol) GaCi. Subsequently and for the next 3 days, infected wounds were treated once daily (OD) with 0.3% (wt/vol) GaCi, twice daily (BID) with 0.1% (wt/vol) GaCi, or with 25 μl of HEC placebo. On day 3, after the last treatment, the transparent dressing was removed, treatment was discontinued, and the wound was monitored for closure through day 15 and sometimes to day 20.
In experiments investigating wound CFU burden, mice (3 biological replicates with 6 mice per group) were treated at 4 h postinoculation and, subsequently, treated with either placebo HEC (BID), 0.1% GaCi (wt/vol) BID, or 0.3% GaCi (wt/vol) OD in HEC. CFU enumeration data were pooled from all three experiments.
Separately, another set of mice (2 biological replicates of 5 mice per group [10 mice total]) beginning at 4 h postinoculation were treated topically with either 25 μl of sterile saline, 25 μl of colistin at a final concentration of 2.5 mg/kg applied topically, Primaxin at 2.5 mg/kg via intraperitoneal injection, or doxycycline at 25 mg/kg via i.p. injection. Subsequently, mice were dosed BID with these preparations every day for 3 days thereafter as described above.
Quantitative and qualitative wound closure assessments.
Wound area measurements were taken on the day of wounding and at subsequent time points using a Silhouette wound measurement device (Aranz Medical Limited, Christchurch, New Zealand). Time course wound photographs assessing gross pathology were taken using a five megapixel iSight camera (Apple Inc., Cupertino, CA).
CFU enumeration.
To examine CFU burden within the wound bed, mice were euthanized with ketamine (250 mg/kg) and xylazine (25 mg/kg) overdose according to protocol on day 1 or day 3. A 4-mm disposable skin biopsy punch (Acuderm Inc., Fort Lauderdale, FL) was then used to sample a disc from the wound bed. The sample was manually disrupted in PBS, and serial 10-fold dilutions were plated via a spiral plater (Autoplate; Advanced Instruments, Inc., Norwood, MA) on eosin methylene blue agar (Becton, Dickinson and Co., Sparks, MD). Plates were incubated overnight at 37°C and then enumerated.
Separately, the CFU was determined at the 4-h postinoculation time point. The dorsal wounds were removed en bloc by severing the cervical and lumbar spinal column and trimming the tissue at >2 cm beyond the wound edge. The tissue was processed using a sterile homogenizer, and serial 10-fold dilutions were plated via a spiral plater and enumerated as above.
Scanning electron microscope evaluation of wound bed and dressing biofilm.
Dressings and wound bed tissue were evaluated by scanning electron microscopy (SEM). A representative mouse from each of the three separate experiments in the placebo, 0.1% GaCi BID, and 0.3% GaCi OD treatment groups was sacrificed at 4 h postwounding and on day 7. The transparent dressings and a 4-mm tissue disc for each animal were fixed in 4% formaldehyde, 1% glutaraldehyde, and 0.1 M PBS. The samples were washed three times using 0.1 M PBS and then postfixed in 1% osmium tetroxide in 0.1 M PBS for 1 h. The samples were dehydrated in a graded series of ethanol solutions and then dried (critical point dryer, model 28000; Ladd Research Industries, Burlington, VT). The samples were mounted by double-sided carbon tape to specimen stubs and ion coated with gold/palladium (30:70) (Hummer X sputter coater; Anatech Ltd., Alexandria, VA). The samples were visualized using an Amray 3600 field emission (FE) scanning electron microscope (Bedford, MA) operated at a voltage of 3 kV. Samples were analyzed by scanning 10 or more fields at 1,000× magnification within the wounded tissue and on the portion of the dressing overlying the wounded area. Photomicrographs representative of the observed biofilm density were taken at 2,500× magnification.
Histological examination of the wound bed.
Mice from each treatment group (HEC OD, HEC BID, 0.1% GaCi BID, and 0.3% GaCi OD) were sacrificed on day 15 in order to characterize wound histopathology. The dorsal wounds were removed en bloc by severing the cervical and lumbar spinal column and trimming the tissue at >2 cm beyond the wound edge. The tissue was immediately fixed in phosphate-buffered formalin (10%) for >72 h. The wound tissue, consisting of spinal column and surrounding soft tissues, was then demineralized for 24 h using Decal Stat (Decal Chemical Corp., Tallman, NY), rinsed with water for 3 to 5 min, and trimmed in a dorsal-ventral plane bisecting the spinal column and placed back into 10% phosphate-buffered formalin. The wound tissue specimens were embedded in paraffin, cut in a dorsal-ventral plane bisecting the spinal column, mounted on positively charged glass slides (Colormark Plus; Thermo Scientific, Portsmouth, NH), and stained with hematoxylin (Astral Diagnostics, Inc., West Deptford, NJ) and eosin (Astral Diagnostics, Inc.) for light microscopic examination.
Wounds were histologically assessed for the presence of and dissemination of bacteria, host immune response, indications of wound healing, i.e., extent of epithelial migration, coverage, maturation, and amount of granulation tissue present within the wound, and if the wound and associated inflammation extended into the underlying vertebrae, spinal canal, and/or spinal cord.
Statistical analyses.
All statistical analyses were carried out using GraphPad Prism software. Wound sizes, CFU burdens, and weight changes were compared via the Mann-Whitney U test with Bonferroni corrections applied when necessary. All results were considered significant if P was <0.05.
All research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals, and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 2011 edition.
RESULTS
MICs.
The MIC of GaCi was determined for numerous bacterial species (see Table S1 in the supplemental material). Upon analysis of this data, K. pneumoniae was recognized as being more susceptible than most other bacterial species to GaCi. Therefore, the MICs for 15 K. pneumoniae clinical isolates from wound infections producing extended-spectrum beta-lactamases (ESBL) were determined (Table 1). Also, the MICs of GaCi were determined for two K. pneumoniae isolates of clinical importance: the original NDM-1 isolate (ATCC BAA-2146) (6) and KP4640, which is a Klebsiella pneumoniae carbapenemase (KPC) isolate from a wounded warrior that was previously described (60) and was also used for the subsequent animal experiments in this study (Table 1). The antimicrobial potency of GaCi was also compared to two antibiotics, tobramycin and aztreonam, which are still often used for treatment. Gallium treatment exhibited significantly lower MIC values than either the tobramycin (approximately 8-fold lower) or aztreonam (approximately 16-fold lower) (Table 1). To ensure that the different topical formulations that were to be used for application in the murine model did not impact the MICs, the activity of GaCi formulated in HEC was also tested and shown to be identical to the unformulated results found in Table 1 (data not shown).
TABLE 1.
MICs of gallium and antibiotics against K. pneumoniae strains
| K. pneumoniae isolate | MIC (μg/ml) of: |
||
|---|---|---|---|
| GaCi | Tobramycin | Aztreonam | |
| 445761 | <0.25 | >8 | 4 |
| 480087 | 1.0 | >8 | <0.12 |
| 482375 | 1.0 | >8 | >16 |
| 517994 | 0.5 | >8 | 8 |
| 548209 | 0.5 | >8 | 2 |
| 549308 | <0.25 | >8 | >16 |
| 551800 | 0.5 | >8 | 2 |
| 551811 | 0.5 | >8 | 16 |
| 563171 | 1.0 | 2 | >16 |
| 572787 | 2.0 | >8 | 16 |
| 580621 | 0.5 | >8 | 8 |
| 589568 | 0.5 | >8 | >16 |
| 625965 | 0.5 | >8 | 2 |
| 639235 | <0.25 | 1 | >16 |
| 639240 | 0.5 | 4 | >16 |
| BAA-2146 | 0.125 | >256 | 64 |
| KP4640 | 1.0 | NDa | ND |
ND, not determined.
Time course gross pathology and wound size of treated and untreated wound infections.
The first assessment of the murine wound model included a gross pathology comparison of wounds that were infected with K. pneumoniae and received GaCi treatment versus those with no treatment. Initially, mice were treated on day 0 at 4 h postinoculation because, at this time point, we saw almost a 2 log10 increase in bacterial burden (see Fig. S1 in the supplemental material), suggesting the bacteria were not just colonizing the wound but establishing infection. Subsequently, and each day after for 3 days, wounds received either 0.1% GaCi twice daily (BID), 0.3% GaCi once daily (OD), or the HEC vehicle control. Photographs of the dorsal, full-thickness wounds were taken on days 7 and 15 (Fig. 2). On day 7, serocellular crust formation was seen in all treatment groups. In BID (Fig. 2A) and OD (Fig. 2C) placebo-treated groups, swelling and redness are more pronounced around the wounds compared to the areas around wounds of mice treated with 0.1% GaCi BID (Fig. 2B) or with 0.3% GaCi OD (Fig. 2D). By day 15 (Fig. 2E to H), wounds in all treatment groups displayed wound edge rounding, indicative of contracture. Separately, photographs were also taken on day 20 with two sets of mice. The healing trends previously observed continued, and wounds appeared to be closed with GaCi treatment in contrast to HEC vehicle-treated wounds that remained infected and open (see Fig. S2 in the supplemental material).
FIG 2.
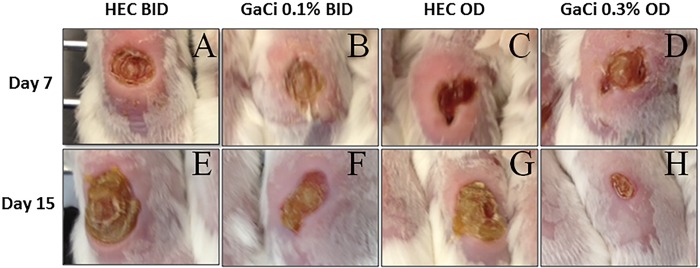
Gross pathology. Photographs of gross pathology of full-thickness wounds over time in representative BALB/c mice. (A to D) Full dorsal wounds on day 7. (E to H) Wounds on day 15 postinoculation. These images are representative of treated animals with HEC BID or OD (negative controls) or HEC with 0.1% GaCi BID or 0.3% GaCi OD from three separate experiments with 12 mice per group.
The wound area was measured on days 0, 6, 9, and 15 postinfection according to the Materials and Methods section. When wounds were measured on day 6 postinfection, the HEC twice daily group had median wound sizes of 61 mm2, while the 0.1% GaCi-treated group had median wound sizes of 38 mm2 (P < 0.05) (Fig. 3). The same trends continued, and by day 9, the HEC placebo-treated mice had a median wound size of 85 mm2, while the mice treated with 0.1% GaCi had a median wound size of 54 mm2 (P < 0.05) (Fig. 3). By day 15 postinfection, groups treated with HEC BID had median wound sizes of 105 mm2, while the 0.1% GaCi-treated mice had a median wound size of 61 mm2 (P < 0.05).
FIG 3.
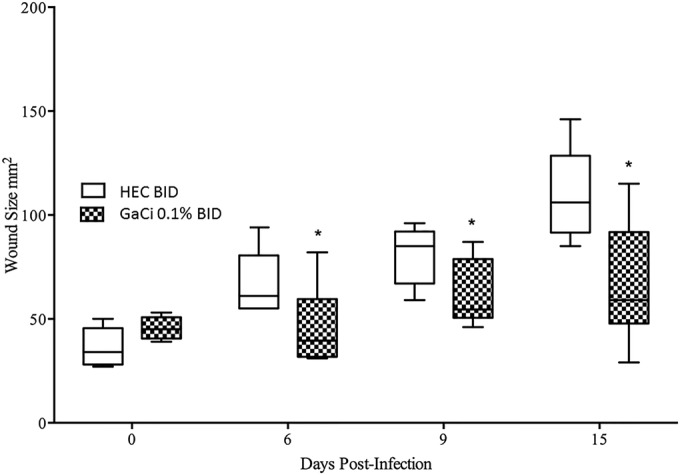
Wound area over time (0.1% GaCi versus HEC alone control). Box and whisker plots show wound sizes on days 0, 6, 9, and 15 postinfection where each wound was treated with HEC alone BID (negative control) or HEC with 0.1% GaCi BID for 3 days. Boxes show median and interquartile ranges, while whiskers represent 95% confidence intervals (CI). Treatment groups were compared each day via Mann-Whitney U test. *, P value of <0.05. These data are pooled from three separate experiments with 12 mice per group and 48 mice per test condition.
Separately, we tested 0.3% GaCi treatments, and wounds were again measured via Aranz on days 0, 6, 9, and 15 postinfection. On day 6 postinfection, the once-daily 0.3% gallium-treated group had a median wound size of 29 mm2, while the HEC once-daily group had a median wound size of 24.5. mm2, which was not statistically significant (P = 0.856). However, on day 9, the mice treated with the 0.3% GaCi formulation had a median wound size of 33 mm2, which was significantly smaller than mice treated once daily with the HEC control formulation that had a median wound size of 59 mm2 (P < 0.0001) (Fig. 4). By day 15 postinfection, groups treated with HEC once daily had a median wound size of 74 mm2, but mice treated with 0.3% GaCi had a median wound size of 34.5 mm2 (P < 0.001). Wound sizes between the two experimental groups were not statistically different on day 0 compared via Mann-Whitney tests (Fig. 3 and 4).
FIG 4.
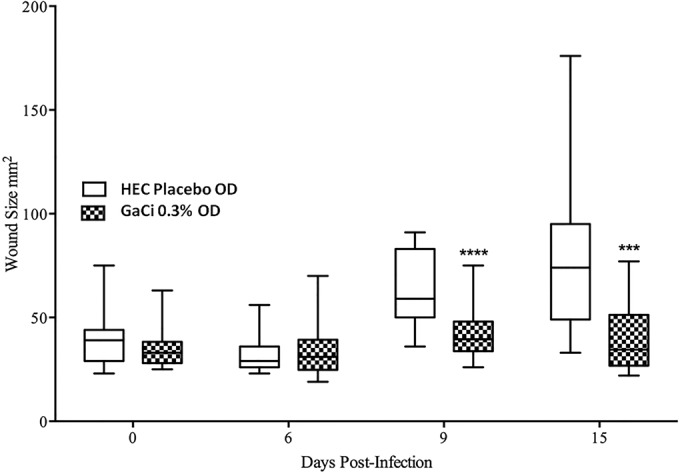
Wound area over time (0.3% GaCi versus HEC alone control). Box and whisker plots show wound sizes on days 0, 6, 9, and 15 postinfection where each wound was treated with HEC alone OD or HEC with 0.3% GaCi OD for 3 days. Boxes show median and interquartile ranges, while whiskers represent 95% CI. Treatment groups were compared each day via Mann-Whitney U test; ***, P values of <0.001; ****, P values of <0.0001. These data are pooled from three separate experiments with 12 mice per group and 48 mice per test condition.
Weight loss.
Mice treated twice daily with HEC lost a median of 10.3% of their infection day body weight 1 day postinfection, 18.52% on day 2, and median of 19.85% 3 days postinfection, while mice treated with 0.1% (wt/vol) GaCi BID lost 13.11%, 18.52%, and 19.04% on those days, respectively (see Fig. S3 in the supplemental material). There was no significant difference between twice daily treated groups.
In contrast, mice treated with 0.3% (wt/vol) GaCi OD lost significantly less weight (P < 0.01) than did mice in the HEC OD control group on day 2 postinfection. Specifically, mice treated with 0.3% (wt/vol) GaCi OD lost a median of 11.53%, 17.54%, and 16.48% of their infection day body weight on days 1, 2, and 3 postinfection, respectively. In the control group, mice treated with HEC OD lost a median of 12.57%, 19.75%, and 18.45% of their infection day body weight on the same respective days (see Fig. S3 in the supplemental material).
Histopathology of wounds.
Mice were sacrificed 15 days postinfection and wound beds preserved to evaluate histopathology (Fig. 5). BID (Fig. 5A) and OD (Fig. 5C) HEC-treated mice displayed large wound beds with necrotic debris evident, with little indication of reepithelialization. In contrast, mice in the 0.1% (wt/vol) GaCi BID-treated group (Fig. 5B) and the 0.3% (wt/vol) GaCi OD-treated group (Fig. 5D) displayed degrees of reepithelialization coupled with noticeable granulation tissue with a small amount of overlying serocellular crust. The results provide preliminary evidence of wound healing and a reduction of inflammation, which is a property of gallium that has been previously investigated by others (56, 62).
FIG 5.

Photomicrographs of hematoxylin and eosin stained, dorsal wound longitudinal sections. (A to D) Wounds on day 15 postinoculum at 12.5× magnification view. Top row images were treated with HEC BID (A) or 0.1% GaCi BID (B). Bottom row images were treated with HEC OD (C) or 0.3% GaCi OD (D). Arrows delineate the perimeter of the wound. These micrographs are representative of at least 10 images taken from samples of three mice per group per test condition from three biological replicates.
Wound bacterial burden.
The bacterial burden of placebo-treated and GaCi-treated wounds was assessed at 24 and 72 h postinfection. At 24 h, mice treated with HEC placebo OD had a median log10 CFU burden (Fig. 6) of 7.97, while mice treated with 0.1% (wt/vol) GaCi BID or with 0.3% (wt/vol) GaCi OD had a median CFU burden of 7.53 (P = 0.0044) and 6.86 (P = 0.0044), respectively. After 72 h, mice treated OD in the placebo group had a median CFU burden of 7.93, while mice treated with 0.1% (wt/vol) GaCi BID had a median CFU burden of 6.81(P = 0.0016), and mice treated with 0.3% (wt/vol) GaCi OD had a median CFU burden of 6.99 (P = 0.0044).
FIG 6.

CFU burden within the wound. Box and whisker plots of log10 CFU/4 mm2 punch on days 1 and 3 postinoculum. Mice were treated with HEC alone OD or HEC + GaCi at 0.3% OD for 3 days, or mice were treated with HEC alone BID or HEC + GaCi at 0.1% BID for 3 days. Boxes show median and interquartile ranges, while whiskers represent 95% CI. Groups were compared each day via Mann-Whitney U test followed by Bonferroni correction. **, P values of <0.01. These data are pooled from three biological replicates with 12 mice per group and 48 mice total per test condition.
Separately, we evaluated the bacterial burden after exposure to antibiotics that strain KP4640 was shown to have susceptibility and resistance to (see Table S2 in the supplemental material). After exposure to colistin, which was applied topically, statistically significant and large reductions in the CFU burden were observed (see Fig. S3 in the supplemental material). On day 1, reduction was approximately 2.5 log10. In contrast, Primaxin- (imipenem + cilastatin) or doxycycline-treated animals that were dosed via i.p. injection appeared to have no reduction of bacterial burden.
SEM analysis of biofilm formation on occlusive dressings and wound tissue.
SEM analysis at 2,500× magnification of the overlying occlusive dressings after 7 days of infection revealed a complete covering of the substratum by K. pneumoniae biofilm displaying a higher architecture with cell-to-cell linkage in mice treated twice daily with HEC placebo (Fig. 7A). In contrast, the occlusive dressings of mice treated twice daily with 0.1% (wt/vol) GaCi showed no continuous substratum K. pneumoniae biofilm presence and little evidence of higher biofilm architecture despite some single cells being present (Fig. 7B).
FIG 7.
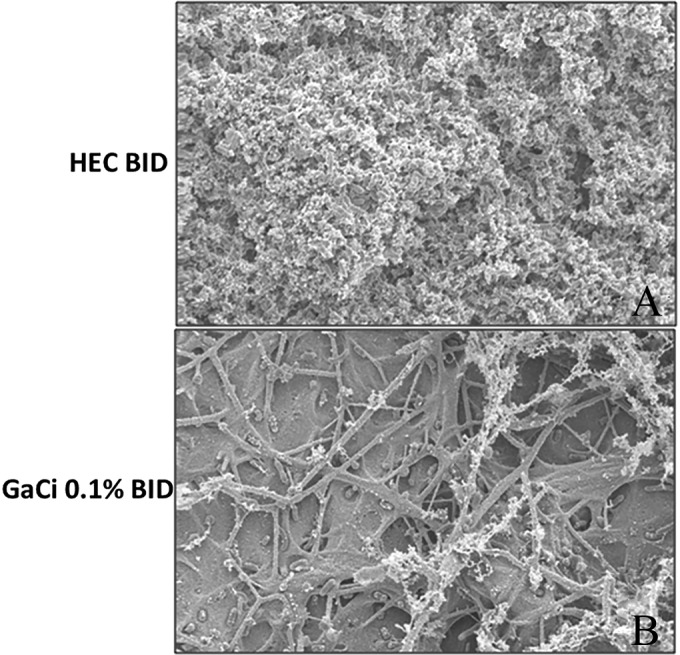
SEM analysis of occlusive dressing. Scanning electron microscopy images of Tegederm occlusive dressings 7 days postinfection. (A) Mouse treated with HEC BID; (B) mouse treated with 0.1% GaCi BID. All images are at 2,500× magnification. These micrographs are representative of three biological replicates where at least 10 images were taken from each sample with three mice total per group and per test condition.
DISCUSSION
From the work presented in this study, it is clear that GaCi in this formulation has significant antibacterial activity in vitro (MIC50, 0.5 μg/ml) against a number of K. pneumoniae clinical isolates from wounds that include the highly resistant NDM-1 strain and other carbapenem-resistant strains in vitro (Table 1). Because of this robust in vitro activity (some <1.0 μg/ml), the efficacy of GaCi was assessed against K. pneumoniae in vivo. To this end, we employed our recently developed wound model of infection (61) and essentially substituted the A. baumannii inoculum with a strain of K. pneumoniae KP4640, which we knew was virulent in other animal models of infection (our unpublished results) and isolated from a sterile tissue site that was likely a wound infection (60) to better mimic the K. pneumoniae wound infections seen in military and civilian patient populations. We believe that this speaks to the utility of our wound infection model, as this study shows it can be used with another ESKAPE pathogen (our unpublished results).
Gallium salt toxicity has been well documented in animals and humans at high dosages (41, 56); however, at doses well below toxic levels, gallium salts have been shown to be efficacious against bacteria in vitro (49). Further, nonintravenous applications of gallium have resulted in reduced toxicity (49, 63), and topical use of gallium salts as an anti-infective is attractive, as it maximizes local concentration while minimizing systemic exposure. Therefore, we hypothesized that gallium salts used in a topical application for K. pneumoniae wound infections would also have efficacy with little or no toxicity because the compound was being applied directly to the site of infection, with limited exposure of noninfected organs. Preliminary work for this study revealed that more concentrated solutions of GaCi (from 1.0% to 3.0% [wt/vol]) resulted in a noticeable neurotoxicity in mice 3 days after treatment (data not shown); however, doses in the range of 0.1% to 0.3% (wt/vol) that displayed efficacy against K. pneumoniae had no visible evidence of toxic effects, such as the neurological deficit previously observed. We have shown previously that weight loss can be an indicator of efficacy and animal health with respect to A. baumannii infections and treatments with antibiotics (61). In testing other novel antibacterial approaches, we also found this to be a useful indicator of efficacy and overall animal health (our unpublished data). In this work, exposure to 0.3% GaCi reduced weight loss with statistical significance (see Fig. S2 in the supplemental material), which suggests this dosage is nontoxic. That said, we did not do a thorough analysis of the toxicity of GaCi at the 0.1% to 0.3% dosages utilized in this study.
While the two GaCi concentrations tested displayed a relatively small but statistically significant impact (0.5 to 1.5 log CFU reduction) on bacterial load (Fig. 6), we did observe evidence of improved wound healing by measuring wound area over time (Fig. 3 and 4). The wound area and bacterial burden experimental outcomes also appeared to be dose dependent (Fig. 3, 4, and 6). Further, wounds treated with GaCi displayed less purulent discharge and inflammation at the wound edge compared to that of untreated wounds (Fig. 2 and 5). While these are not quantitative measures, it is consistent with other reports where the application of gallium salts reduced inflammation (62, 64, 65). Further, histopathology results coincided with the gross pathology results where reepithelization of the wound bed was positively impacted by GaCi therapy compared to that of untreated controls (Fig. 5). Therefore, to our knowledge, this is the first study that has shown efficacy of a gallium(III) salt against K. pneumoniae infection, and therefore, further investigation of other gallium formulations and treatments against this bacterial species are warranted, especially given its multidrug resistance and prevalence in the hospital environment.
It should be noted that while the bacterial burden results were modest with only a 0.5 to 1.5 log reduction in CFU of K. pneumoniae (Fig. 5), the impact of GaCi on biofilm was clearly evident on the Tegaderm dressing (Fig. 7), which may contribute to the positive effect on wound healing. GaCi also exhibited some efficacy in the mouse model of A. baumannii wound infections with similar effects on biofilm and CFU reduction (our unpublished data), and Aridis scientists evaluated the efficacy of GaCi against A. baumannii infection in a porcine full-thickness wound model with encouraging results (our unpublished data). Reports have been published using gallium nitrate against a variety of bacteria to eradicate or disperse biofilms as well as significantly reduce bacterial numbers (45, 48, 52). For example, gallium maltolate was shown to have efficacy in vitro against P. aeruginosa, Staphylococcus species, and Rhodococcus equi (42, 66, 67). GaCi also exhibits single-digit MICs against isolates of many other Gram-negative bacterial species such as P. aeruginosa, Escherichia coli, Stenotrophomonas maltophilia, A. baumannii, and Burkholderia cepacia (see Table S1 in the supplemental material). Therefore, it is not surprising that gallium(III) salts would also display efficacy against K. pneumoniae or have an effect on biofilm formation, as these previous studies and our unpublished data support the results reported herein.
In conclusion, the results provide significant evidence of efficacy against K. pneumoniae in vivo and an improvement of wound healing, which warrants the further development of gallium citrate formulations as a possible wound infection treatment. Further preclinical testing of these formulations and other gallium(III) formulations in other animal models of wound infection would be the next logical step. In addition to its antimicrobial potency and antibiofilm activity, the apparent anti-inflammatory and wound healing effects of a gallium topical treatment suggest that clinically relevant applications for acute wound infections as well as chronic skin diseases such as diabetic foot ulcers and venous stasis ulcers may be possible with respect to K. pneumoniae infection. The utility of GaCi as a broad-spectrum prophylactic to prevent SSTIs rather than antibiotic prophylaxis treatment should also be considered if more compelling data can be generated.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anna Jacobs for critically reading the manuscript and assisting with some mouse experiments. We also recognize Robert Williams for the nearly 40 years of service in the Department of Pathology and for his assistance with the electron microscopy imaging. We express our gratitude to the members of the JPC-2 and the staff from the Military Infectious Diseases Research Program (MIDRP) for their continued support.
Specifically, we acknowledge the Defense Medical Research and Development Program award no. DM090034, which provided the funding for this work.
The findings and opinions expressed herein belong to the authors and do not necessarily reflect the official views of the WRAIR, the U.S. Army, or the Department of Defense.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00882-15.
REFERENCES
- 1.Cagnacci S, Gualco L, Roveta S, Mannelli S, Borgianni L, Docquier JD, Dodi F, Centanaro M, Debbia E, Marchese A, Rossolini GM. 2008. Bloodstream infections caused by multidrug-resistant Klebsiella pneumoniae producing the carbapenem-hydrolysing VIM-1 metallo-beta-lactamase: first Italian outbreak. J Antimicrob Chemother 61:296–300. [DOI] [PubMed] [Google Scholar]
- 2.Ho J, Tambyah PA, Paterson DL. 2010. Multiresistant Gram-negative infections: a global perspective. Curr Opin Infect Dis 23:546–553. doi: 10.1097/QCO.0b013e32833f0d3e. [DOI] [PubMed] [Google Scholar]
- 3.Webster DP, Young BC, Morton R, Collyer D, Batchelor B, Turton JF, Maharjan S, Livermore DM, Bejon P, Cookson BD, Bowler IC. 2011. Impact of a clonal outbreak of extended-spectrum beta-lactamase-producing Klebsiella pneumoniae in the development and evolution of bloodstream infections by K. pneumoniae and Escherichia coli: an 11 year experience in Oxfordshire, UK. J Antimicrob Chemother 66:2126–2135. doi: 10.1093/jac/dkr246. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. 2013. Notes from the field: hospital outbreak of carbapenem-resistant Klebsiella pneumoniae producing New Delhi metallo-beta-lactamase–Denver, Colorado, 2012. MMWR Morb Mortal Wkly Rep 62:108. [PMC free article] [PubMed] [Google Scholar]
- 5.Snitkin ES, Zelazny AM, Thomas PJ, Stock F, Henderson DK, Palmore TN, Segre JA. 2012. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med 4:148ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, Lee K, Walsh TR. 2009. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother 53:5046–5054. doi: 10.1128/AAC.00774-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weintrob AC, Murray CK, Lloyd B, Li P, Lu D, Miao Z, Aggarwal D, Carson ML, Gaskins LJ, Tribble DR. 2013. Active surveillance for asymptomatic colonization with multidrug-resistant Gram negative bacilli among injured service members–a three year evaluation. MSMR 20:17–22. [PMC free article] [PubMed] [Google Scholar]
- 8.Mammina C, Bonura C, Di Bernardo F, Aleo A, Fasciana T, Sodano C, Saporito MA, Verde MS, Tetamo R, Palma DM. 2012. Ongoing spread of colistin-resistant Klebsiella pneumoniae in different wards of an acute general hospital, Italy, June to December 2011. Euro Surveill 17(33):pii=20248 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20248. [PubMed] [Google Scholar]
- 9.van Duin D, Perez F, Rudin SD, Cober E, Hanrahan J, Ziegler J, Webber R, Fox J, Mason P, Richter SS, Cline M, Hall GS, Kaye KS, Jacobs MR, Kalayjian RC, Salata RA, Segre JA, Conlan S, Evans S, Fowler VG Jr, Bonomo RA. 2014. Surveillance of carbapenem-resistant Klebsiella pneumoniae: tracking molecular epidemiology and outcomes through a regional network. Antimicrob Agents Chemother 58:4035–4041. doi: 10.1128/AAC.02636-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halaby T, Al Naiemi N, Kluytmans J, van der Palen J, Vandenbroucke-Grauls CM. 2013. Emergence of colistin resistance in Enterobacteriaceae after the introduction of selective digestive tract decontamination in an intensive care unit. Antimicrob Agents Chemother 57:3224–3229. doi: 10.1128/AAC.02634-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassat JE, Skaar EP. 2013. Iron in infection and immunity. Cell Host Microbe 13:509–519. doi: 10.1016/j.chom.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becker KW, Skaar EP. 2014. Metal limitation and toxicity at the interface between host and pathogen. FEMS Microbiol Rev 38:1235–1249. doi: 10.1111/1574-6976.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parrow NL, Fleming RE, Minnick MF. 2013. Sequestration and scavenging of iron in infection. Infect Immun 81:3503–3514. doi: 10.1128/IAI.00602-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weaver EA, Wyckoff EE, Mey AR, Morrison R, Payne SM. 2013. FeoA and FeoC are essential components of the Vibrio cholerae ferrous iron uptake system, and FeoC interacts with FeoB. J Bacteriol 195:4826–4835. doi: 10.1128/JB.00738-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kammler M, Schon C, Hantke K. 1993. Characterization of the ferrous iron uptake system of Escherichia coli. J Bacteriol 175:6212–6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grosse C, Scherer J, Koch D, Otto M, Taudte N, Grass G. 2006. A new ferrous iron-uptake transporter, EfeU (YcdN), from Escherichia coli. Mol Microbiol 62:120–131. doi: 10.1111/j.1365-2958.2006.05326.x. [DOI] [PubMed] [Google Scholar]
- 17.Wyckoff EE, Boulette ML, Payne SM. 2009. Genetics and environmental regulation of Shigella iron transport systems. Biometals 22:43–51. doi: 10.1007/s10534-008-9188-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wyckoff EE, Mey AR, Payne SM. 2007. Iron acquisition in Vibrio cholerae. Biometals 20:405–416. doi: 10.1007/s10534-006-9073-4. [DOI] [PubMed] [Google Scholar]
- 19.Marlovits TC, Haase W, Herrmann C, Aller SG, Unger VM. 2002. The membrane protein FeoB contains an intramolecular G protein essential for Fe(II) uptake in bacteria. Proc Natl Acad Sci U S A 99:16243–16248. doi: 10.1073/pnas.242338299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher CR, Davies NM, Wyckoff EE, Feng Z, Oaks EV, Payne SM. 2009. Genetics and virulence association of the Shigella flexneri Sit iron transport system. Infect Immun 77:1992–1999. doi: 10.1128/IAI.00064-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao J, Woodhall MR, Alvarez J, Cartron ML, Andrews SC. 2007. EfeUOB (YcdNOB) is a tripartite, acid-induced and CpxAR-regulated, low-pH Fe2+ transporter that is cryptic in Escherichia coli K-12 but functional in E. coli O157:H7. Mol Microbiol 65:857–875. doi: 10.1111/j.1365-2958.2007.05802.x. [DOI] [PubMed] [Google Scholar]
- 22.Reid DW, O'May C, Kirov SM, Roddam L, Lamont IL, Sanderson K. 2009. Iron chelation directed against biofilms as an adjunct to conventional antibiotics. Am J Physiol Lung Cell Mol Physiol 296:L857–L858. doi: 10.1152/ajplung.00058.2009. [DOI] [PubMed] [Google Scholar]
- 23.Chan GC, Chan S, Ho PL, Ha SY. 2009. Effects of chelators (deferoxamine, deferiprone and deferasirox) on the growth of Klebsiella pneumoniae and Aeromonas hydrophila isolated from transfusion-dependent thalassemia patients. Hemoglobin 33:352–360. doi: 10.3109/03630260903211888. [DOI] [PubMed] [Google Scholar]
- 24.Fernandes SS, Nunes A, Gomes AR, de Castro B, Hider RC, Rangel M, Appelberg R, Gomes MS. 2010. Identification of a new hexadentate iron chelator capable of restricting the intramacrophagic growth of Mycobacterium avium. Microbes Infect 12:287–294. doi: 10.1016/j.micinf.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Thompson MG, Corey BW, Si Y, Craft DW, Zurawski DV. 2012. Antibacterial activities of iron chelators against common nosocomial pathogens. Antimicrob Agents Chemother 56:5419–5421. doi: 10.1128/AAC.01197-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zurawski DV, Jacobs AC, Thompson MG, Palys TJ. 2013. Reply to “The dual personality of iron chelators: growth inhibitors or promoters?”. Antimicrob Agents Chemother 57:2434. doi: 10.1128/AAC.00134-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Page MG. 2013. Siderophore conjugates. Ann N Y Acad Sci 1277:115–126. doi: 10.1111/nyas.12024. [DOI] [PubMed] [Google Scholar]
- 28.Ji C, Juarez-Hernandez RE, Miller MJ. 2012. Exploiting bacterial iron acquisition: siderophore conjugates. Future Med Chem 4:297–313. doi: 10.4155/fmc.11.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tiwari R, Moraski GC, Krchnak V, Miller PA, Colon-Martinez M, Herrero E, Oliver AG, Miller MJ. 2013. Thiolates chemically induce redox activation of BTZ043 and related potent nitroaromatic anti-tuberculosis agents. J Am Chem Soc 135:3539–3549. doi: 10.1021/ja311058q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wencewicz TA, Miller MJ. 2013. Biscatecholate-monohydroxamate mixed ligand siderophore-carbacephalosporin conjugates are selective sideromycin antibiotics that target Acinetobacter baumannii. J Med Chem 56:4044–4052. doi: 10.1021/jm400265k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russo TA, Page MG, Beanan JM, Olson R, Hujer AM, Hujer KM, Jacobs M, Bajaksouzian S, Endimiani A, Bonomo RA. 2011. In vivo and in vitro activity of the siderophore monosulfactam BAL30072 against Acinetobacter baumannii. J Antimicrob Chemother 66:867–873. doi: 10.1093/jac/dkr013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hofer B, Dantier C, Gebhardt K, Desarbre E, Schmitt-Hoffmann A, Page MG. 2013. Combined effects of the siderophore monosulfactam BAL30072 and carbapenems on multidrug-resistant Gram-negative bacilli. J Antimicrob Chemother 68:1120–1129. doi: 10.1093/jac/dks527. [DOI] [PubMed] [Google Scholar]
- 33.Tomaras AP, Crandon JL, McPherson CJ, Banevicius MA, Finegan SM, Irvine RL, Brown MF, O'Donnell JP, Nicolau DP. 2013. Adaptation-based resistance to siderophore-conjugated antibacterial agents by Pseudomonas aeruginosa. Antimicrob Agents Chemother 57:4197–4207. doi: 10.1128/AAC.00629-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lv H, Hung CS, Henderson JP. 2014. Metabolomic analysis of siderophore cheater mutants reveals metabolic costs of expression in uropathogenic Escherichia coli. J Proteome Res 13:1397–1404. doi: 10.1021/pr4009749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Penn AS, Conibear TC, Watson RA, Kraaijeveld AR, Webb JS. 2012. Can Simpson's paradox explain co-operation in Pseudomonas aeruginosa biofilms? FEMS Immunol Med Microbiol 65:226–235. doi: 10.1111/j.1574-695X.2012.00970.x. [DOI] [PubMed] [Google Scholar]
- 36.Cirl C, Miethke T. 2010. Microbial Toll/interleukin 1 receptor proteins: a new class of virulence factors. Int J Med Microbiol 300:396–401. doi: 10.1016/j.ijmm.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Armstrong SK, Brickman TJ, Suhadolc RJ. 2012. Involvement of multiple distinct Bordetella receptor proteins in the utilization of iron liberated from transferrin by host catecholamine stress hormones. Mol Microbiol 84:446–462. doi: 10.1111/j.1365-2958.2012.08032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanabe T, Funahashi T, Shiuchi K, Okajima N, Nakao H, Miyamoto K, Tsujibo H, Yamamoto S. 2012. Characterization of Vibrio parahaemolyticus genes encoding the systems for utilization of enterobactin as a xenosiderophore. Microbiology 158:2039–2049. doi: 10.1099/mic.0.059568-0. [DOI] [PubMed] [Google Scholar]
- 39.Henderson JP, Crowley JR, Pinkner JS, Walker JN, Tsukayama P, Stamm WE, Hooton TM, Hultgren SJ. 2009. Quantitative metabolomics reveals an epigenetic blueprint for iron acquisition in uropathogenic Escherichia coli. PLoS Pathog 5:e1000305. doi: 10.1371/journal.ppat.1000305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomaras AP, Crandon JL, McPherson CJ, Nicolau DP. 2015. Potentiation of antibacterial activity of the MB-1 siderophore-monobactam conjugate using an efflux pump inhibitor. Antimicrob Agents Chemother 59:2439–2442. doi: 10.1128/AAC.04172-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minandri F, Bonchi C, Frangipani E, Imperi F, Visca P. 2014. Promises and failures of gallium as an antibacterial agent. Future Microbiol 9:379–397. doi: 10.2217/fmb.14.3. [DOI] [PubMed] [Google Scholar]
- 42.DeLeon K, Balldin F, Watters C, Hamood A, Griswold J, Sreedharan S, Rumbaugh KP. 2009. Gallium maltolate treatment eradicates Pseudomonas aeruginosa infection in thermally injured mice. Antimicrob Agents Chemother 53:1331–1337. doi: 10.1128/AAC.01330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frangipani E, Bonchi C, Minandri F, Imperi F, Visca P. 2014. Pyochelin potentiates the inhibitory activity of gallium on Pseudomonas aeruginosa. Antimicrob Agents Chemother 58:5572–5575. doi: 10.1128/AAC.03154-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rzhepishevska O, Hakobyan S, Ekstrand-Hammarstrom B, Nygren Y, Karlsson T, Bucht A, Elofsson M, Boily JF, Ramstedt M. 2014. The gallium(III)-salicylidene acylhydrazide complex shows synergistic anti-biofilm effect and inhibits toxin production by Pseudomonas aeruginosa. J Inorg Biochem 138:1–8. doi: 10.1016/j.jinorgbio.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 45.Kaneko Y, Thoendel M, Olakanmi O, Britigan BE, Singh PK. 2007. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J Clin Invest 117:877–888. doi: 10.1172/JCI30783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Banin E, Lozinski A, Brady KM, Berenshtein E, Butterfield PW, Moshe M, Chevion M, Greenberg EP, Banin E. 2008. The potential of desferrioxamine-gallium as an anti-Pseudomonas therapeutic agent. Proc Natl Acad Sci U S A 105:16761–16766. doi: 10.1073/pnas.0808608105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fecteau ME, Aceto HW, Bernstein LR, Sweeney RW. 2014. Comparison of the antimicrobial activities of gallium nitrate and gallium maltolate against Mycobacterium avium subsp. paratuberculosis in vitro. Vet J 202:195–197. doi: 10.1016/j.tvjl.2014.06.023. [DOI] [PubMed] [Google Scholar]
- 48.Fecteau ME, Whitlock RH, Fyock TL, McAdams SC, Boston RC, Sweeney RW. 2011. Antimicrobial activity of gallium nitrate against Mycobacterium avium subsp. paratuberculosis in neonatal calves. J Vet Intern Med 25:1152–1155. doi: 10.1111/j.1939-1676.2011.0768.x. [DOI] [PubMed] [Google Scholar]
- 49.Kelson AB, Carnevali M, Truong-Le V. 2013. Gallium-based anti-infectives: targeting microbial iron-uptake mechanisms. Curr Opin Pharmacol 13:707–716. doi: 10.1016/j.coph.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 50.Valappil SP, Owens GJ, Miles EJ, Farmer NL, Cooper L, Miller G, Clowes R, Lynch RJ, Higham SM. 2014. Effect of gallium on growth of Streptococcus mutans NCTC 10449 and dental tissues. Caries Res 48:137–146. [DOI] [PubMed] [Google Scholar]
- 51.Olakanmi O, Kesavalu B, Pasula R, Abdalla MY, Schlesinger LS, Britigan BE. 2013. Gallium nitrate is efficacious in murine models of tuberculosis and inhibits key bacterial Fe-dependent enzymes. Antimicrob Agents Chemother 57:6074–6080. doi: 10.1128/AAC.01543-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Antunes LC, Imperi F, Minandri F, Visca P. 2012. In vitro and in vivo antimicrobial activities of gallium nitrate against multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 56:5961–5970. doi: 10.1128/AAC.01519-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Leseleuc L, Harris G, KuoLee R, Chen W. 2012. In vitro and in vivo biological activities of iron chelators and gallium nitrate against Acinetobacter baumannii. Antimicrob Agents Chemother 56:5397–5400. doi: 10.1128/AAC.00778-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Leseleuc L, Harris G, KuoLee R, Xu HH, Chen W. 2014. Serum resistance, gallium nitrate tolerance and extrapulmonary dissemination are linked to heme consumption in a bacteremic strain of Acinetobacter baumannii. Int J Med Microbiol 304:360–369. doi: 10.1016/j.ijmm.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 55.Rangel-Vega A, Bernstein LR, Mandujano-Tinoco EA, Garcia-Contreras SJ, Garcia-Contreras R. 2015. Drug repurposing as an alternative for the treatment of recalcitrant bacterial infections. Front Microbiol 6:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bernstein LR. 1998. Mechanisms of therapeutic activity for gallium. Pharmacol Rev 50:665–682. [PubMed] [Google Scholar]
- 57.Russo TA, Olson R, Macdonald U, Metzger D, Maltese LM, Drake EJ, Gulick AM. 2014. Aerobactin mediates virulence and accounts for increased siderophore production under iron-limiting conditions by hypervirulent (hypermucoviscous) Klebsiella pneumoniae. Infect Immun 82:2356–2367. doi: 10.1128/IAI.01667-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tribble DR, Conger NG, Fraser S, Gleeson TD, Wilkins K, Antonille T, Weintrob A, Ganesan A, Gaskins LJ, Li P, Grandits G, Landrum ML, Hospenthal DR, Millar EV, Blackbourne LH, Dunne JR, Craft D, Mende K, Wortmann GW, Herlihy R, McDonald J, Murray CK. 2011. Infection-associated clinical outcomes in hospitalized medical evacuees after traumatic injury: trauma infectious disease outcome study. J Trauma 71:S33–S42. doi: 10.1097/TA.0b013e318221162e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA, Wolcott RD. 2008. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol 8:43. doi: 10.1186/1471-2180-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arivett BA, Ream DC, Fiester SE, Mende K, Murray CK, Thompson MG, Kanduru S, Summers AM, Roth AL, Zurawski DV, Actis LA. 2015. Draft genome sequences of Klebsiella pneumoniae clinical type strain ATCC 13883 and three multidrug-resistant clinical isolates. Genome Announce 3(1):e01385-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thompson MG, Black CC, Pavlicek RL, Honnold CL, Wise MC, Alamneh YA, Moon JK, Kessler JL, Si Y, Williams R, Yildirim S, Kirkup BC Jr, Green RK, Hall ER, Palys TJ, Zurawski DV. 2014. Validation of a novel murine wound model of Acinetobacter baumannii infection. Antimicrob Agents Chemother 58:1332–1342. doi: 10.1128/AAC.01944-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goncalves J, Wasif N, Esposito D, Coico JM, Schwartz B, Higgins PJ, Bockman RS, Staiano-Coico L. 2002. Gallium nitrate accelerates partial thickness wound repair and alters keratinocyte integrin expression to favor a motile phenotype. J Surg Res 103:134–140. doi: 10.1006/jsre.2001.6347. [DOI] [PubMed] [Google Scholar]
- 63.Collery P, Keppler B, Madoulet C, Desoize B. 2002. Gallium in cancer treatment. Crit Rev Oncol Hematol 42:283–296. doi: 10.1016/S1040-8428(01)00225-6. [DOI] [PubMed] [Google Scholar]
- 64.Choi JH, Lee JH, Roh KH, Seo SK, Choi IW, Park SG, Lim JG, Lee WJ, Kim MH, Cho KR, Kim YJ. 2014. Gallium nitrate ameliorates type II collagen-induced arthritis in mice. Int Immunopharmacol 20:269–275. doi: 10.1016/j.intimp.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 65.Eby G. 2005. Elimination of arthritis pain and inflammation for over 2 years with a single 90 min, topical 14% gallium nitrate treatment: case reports and review of actions of gallium III. Med Hypotheses 65:1136–1141. doi: 10.1016/j.mehy.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 66.Harrington JR, Martens RJ, Cohen ND, Bernstein LR. 2006. Antimicrobial activity of gallium against virulent Rhodococcus equi in vitro and in vivo. J Vet Pharmacol Ther 29:121–127. doi: 10.1111/j.1365-2885.2006.00723.x. [DOI] [PubMed] [Google Scholar]
- 67.Arnold CE, Bordin A, Lawhon SD, Libal MC, Bernstein LR, Cohen ND. 2012. Antimicrobial activity of gallium maltolate against Staphylococcus aureus and methicillin-resistant S. aureus and Staphylococcus pseudintermedius: an in vitro study. Vet Microbiol 155:389–394. doi: 10.1016/j.vetmic.2011.09.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.