

Abstract
Topoisomerase functions are required in all organisms for many vital cellular processes, including transcription elongation. The C terminus domains (CTD) of Escherichia coli topoisomerase I interact directly with RNA polymerase to remove transcription-driven negative supercoiling behind the RNA polymerase complex. This interaction prevents inhibition of transcription elongation from hypernegative supercoiling and R-loop accumulation. The physiological function of bacterial topoisomerase I in transcription is especially important for a rapid network response to an antibiotic challenge. In this study, Escherichia coli with a topA66 single nucleotide deletion mutation, which results in a frameshift in the TopA CTD, was shown to exhibit increased sensitivity to trimethoprim and quinolone antimicrobials. The topoisomerase I-RNA polymerase interaction and the SOS response to the antimicrobial agents were found to be significantly reduced by this topA66 mutation. Consequently, the mutation frequency measured by rifampin selection following SOS induction was diminished in the topA66 mutant. The increased antibiotic sensitivity for the topA66 mutant can be reversed by the expression of recombinant E. coli topoisomerase I but not by the expression of recombinant Mycobacterium tuberculosis topoisomerase I that has a nonhomologous CTD even though the recombinant M. tuberculosis topoisomerase I can restore most of the plasmid DNA linking number deficiency caused by the topA66 mutation. Direct interactions of E. coli topoisomerase I as part of transcription complexes are likely to be required for the rapid network response to an antibiotic challenge. Inhibitors of bacterial topoisomerase I functions and interactions may sensitize pathogens to antibiotic treatment and limit the mutagenic response.
INTRODUCTION
Microbial pathogens resistant to current antibiotics are becoming an increasingly urgent public health crisis. For example, quinolones are widely used broad-spectrum antimicrobial agents that are highly effective for rapid bactericidal outcomes (1), but quinolone resistance can be acquired rapidly by the bacterial pathogens via a number of mechanisms (2, 3). The bacterial SOS response system has been shown to contribute to the increase in antibiotic resistance (4). There are many factors that can potentially lead to the increased resistance in response to the SOS-inducing antimicrobials. These factors include elevations in the expression levels of error-prone DNA polymerases (5) or plasmid-mediated antibiotic resistance determinants (6, 7), higher rates of transfer of resistance determinants (8, 9), and increases in the numbers of persistors in the bacterial population (10).
The transcriptome in the bacterial cell must adjust rapidly to the antibiotic challenge for survival. Due to resistance to the rotational motion of the transcription ensemble around the DNA during transcription, the transcription loci can accumulate positive supercoils ahead of the RNA polymerase (RNAP)-nascent RNA complex and negative supercoils behind the complex (11). In bacteria, DNA gyrase is responsible for removal of the positive supercoils, while topoisomerase I function is used for removal of the negative supercoils. Accumulation of hypernegative supercoils and inhibition of transcription elongation account, at least in part, for the growth defect of Escherichia coli topA null mutants without any topoisomerase I activity (12). Multiple promoters, including σ32 and σs, have been shown to be utilized for transcription initiation of the E. coli topA gene (13, 14). This suggests that the function of topoisomerase I may be especially important during a stress response. E. coli strains with topA deletion have been shown to have decreased survival rates when challenged with high temperatures or oxidative stress (15, 16).
The E. coli topA gene encodes 865 amino acids. The first 595 amino acids of topoisomerase I form the N-terminal domains that are responsible for cutting and rejoining of a single strand of DNA during the catalytic cycle. There are three tightly bound Zn(II) ions, each coordinated by a tetracysteine motif present in the region between residues 598 and 737 (17). The last 122 residues of the enzyme do not bind Zn(II) but also fold into a zinc ribbon structure (18). The C-terminal domains (CTD) between residues 598 and 865 contribute to the protein-protein interaction with RNA polymerase for relief of transcription-driven supercoiling during transcription elongation (19). This direct interaction with RNA polymerase may be critical for topoisomerase I function during a stress response when high rates of transcription of certain genes are needed for adaptation and survival. The topA66 mutation in E. coli strain DPB636 is a single nucleotide deletion changing the reading frame to −1 for the last 100 amino acids and terminating one codon beyond the normal stop codon (20). Previously reported phenotypes associated with the topA66 mutation are linked to suppression of various replication defects, including an integration host factor requirement for pSC101 plasmid replication (21), a segregation defect of muk mutants (22), and Tus-mediated replication arrest (20). There is some disagreement in the literature (23) on whether the E. coli topA deletion mutants are viable without compensatory mutations in other genes such as the gyrase genes (24, 25), but it can be stated that E. coli topA deletion mutants are likely to acquire a compensatory mutation. In contrast, there does not appear to be any selective pressure under standard laboratory growth conditions for compensatory mutations in other genes for the topA66 mutant, and the topoisomerase relaxation activity in the cell extract of the topA66 mutant was found to be significantly higher than that for other topA mutants that have been isolated and characterized (21). Previous studies have shown that the last 126 amino acids at the C terminus of E. coli topoisomerase I are not required for the enzyme to exhibit in vitro or in vivo relaxation activity (26). Although the growth rate under normal laboratory conditions is not affected significantly by this topA66 CTD mutation, the experiments described here showed that the topA66 CTD mutation can nevertheless result in a severe disadvantage due to a deficiency in the SOS response to an antibiotic challenge. Our results demonstrate that growth inhibition and loss of viability following treatment with certain antimicrobials are more severe for the topA66 CTD mutant, along with a reduced SOS response and a diminished elevation in the mutation frequency, following SOS induction when measured by rifampin resistance.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The E. coli strains and plasmids used this study are listed in Table 1. The frameshift mutation in the topA66 gene of strain DPB636 was introduced into the expression plasmid pLIC-ETOP by the QuikChange method of site-directed mutagenesis using PfuUltra II fusion DNA polymerase (with oligonucleotide primer 5′-GCCGTGCGAAAAACAGATGCTTATTTC-3′ and its complement). Plasmid pBAD/Thio was obtained from Invitrogen. Plasmids pETOP (27) and pMTOP (28) for expression of recombinant E. coli and Mycobacterium tuberculosis topoisomerase I from the BAD promoter were described previously.
TABLE 1.
E. coli strains and plasmids used in this study
| Strain or plasmid | Genotype or description | Source or reference |
|---|---|---|
| Strain | ||
| DPB635 | λ−, zci-2250::mini-kan, rph-1 | Yale CGSC, 21 |
| DPB636 | λ−, topA66 zci-2250::mini-kan, rph-1 | Yale CGSC, 21 |
| Plasmid | ||
| pBAD/Thio | Protein expression vector with BAD promoter | Invitrogen |
| pETOP | pBAD/Thio-derived expression plasmid for E. coli topoisomerase I | 27 |
| pMTOP | pBAD/Thio-derived expression plasmid for M. tuberculosis topoisomerase I | 28 |
| pLIC-ETOP | T7 promoter-driven expression plasmid for E. coli topoisomerase I | 29 |
| pLIC-ETOP66 | pLIC-ETOP mutagenized for expression of topoisomerase I with topA66 mutation | This study |
| pDinlux | SOS reporter plasmid with dinD1::luxCADBE fusion | 30 |
Topoisomerase enzymes.
Wild-type E. coli topoisomerase I (ETOP) and mutant topoisomerase I (ETOP66) encoded by a gene with the topA66 frameshift mutation were expressed from pLIC-ETOP (29) and pLIC-ETOP66 in E. coli BL21 AI (Invitrogen). Protein purification to >90% homogeneity was carried out using nickel-nitrilotriacetic acid (Ni-NTA) agarose and single-stranded DNA cellulose affinity chromatography as described previously (29). The relaxation activity of purified wild-type and mutant topoisomerase I was assayed with supercoiled pBAD/Thio plasmid DNA under standard assay conditions (27).
Assays for activity of antimicrobial agents.
Activities of antimicrobials against different bacterial strains in this study were measured using (i) a spotting assay, (ii) MIC determination, and (iii) a viability assay. For spotting assays, norfloxacin, ciprofloxacin, and trimethoprim were added to Mueller-Hinton agar plates at the specified concentrations. Overnight cultures were adjusted to an optical density at 600 nm (OD600) value of 0.1, and 10-fold serial dilutions of the OD-adjusted cultures of DPB635, DPB636, and their transformants in lysogeny broth (LB) were spotted on Mueller-Hinton agar plates with or without antimicrobials and incubated at 37°C overnight to compare growth inhibition. MIC values were determined by the broth microdilution method with Mueller-Hinton medium as described by the Clinical and Laboratory Standards Institute (CLSI) approved standards. To measure the viability following treatment with the bactericidal antibiotic norfloxacin, DPB635 and DPB636 were diluted 1:100 from overnight cultures and grown in Mueller-Hinton medium for 4 h before treatment with 100 to 250 ng/ml of norfloxacin. Following incubation with shaking at 37°C for the indicated length of time, serial dilutions of the culture were plated on LB plates and incubated at 37°C for determination of the number of viable colonies. Experiments were repeated at least three times.
Pulldown of RNA polymerase by His-tagged topoisomerase I.
The same amount (8.3 nmol) of recombinant ETOP or ETOP66 protein with an N-terminal 6-histidine tag was mixed with various quantities (0 to 33.2 nmol) of E. coli RNA polymerase holoenzyme (Epicentre) in a buffer of 20 mM potassium phosphate (pH 7.5), 0.1 M KCl, and 0.1% NP-40 at 4°C for 2 h. The mixtures were added to 60 μl of HisPur Cobalt agarose resin (Thermo Fisher). After overnight mixing at 4°C, the resin was centrifuged and washed twice with the same buffer. Proteins bound to the resin were eluted with buffer containing 400 mM imidazole. Following SDS-PAGE, the β′ subunit of E. coli RNA polymerase was detected by Western blotting using NT73 monoclonal antibodies from NeoClone. The chemiluminescent Western blot signal was detected by the Li-Cor C-DiGit blot scanner and quantitated with Image Studio software.
Plasmid supercoiling.
E. coli strains DPB635 and DPB636 each transformed with the respective plasmids (pBAD/Thio or pETOP or pMTOP) were grown in LB containing 100 μg/ml ampicillin. The plasmids were isolated from early stationary-phase cultures using the Qiagen Spin miniprep kit. The level of supercoiling was compared by electrophoresis in 1% agarose gel with Tris-acetate-EDTA (TAE) buffer containing a chloroquine concentration optimized for the separation of topoisomers of each of the plasmids for 16 h at 30 V. After electrophoresis, the gels were washed extensively with deionized water, stained with ethidium bromide, and photographed over UV light. Densitometry plots of the different plasmid topoisomers were generated with AlphaView software (ProteinSimple).
Viability following oxidative stress challenge from toxic electrophile.
DPB635 and DPB636 cells from cultures were challenged with the toxic electrophile N-ethylmaleimide at 0.4 to 1 mM concentrations for the indicated lengths of time at 37°C as described previously (15). Treated cells and the untreated control were diluted serially, plated on LB plates, and incubated overnight at 37°C for viable colony counts. The relative survival ratio was calculated as the ratio of the viable colony counts from treated and untreated cultures. Experiments were repeated at least three times.
Assay of SOS induction of responsive promoters.
DPB635 and DPB636 cells were transformed with the pDinlux plasmid (30) with dinD1::luxCADBE fusion as a luciferase reporter for SOS induction. The cultures in LB with 100 μg/ml ampicillin were grown to the exponential phase before treatment with inducers of SOS. Luminescence was measured at 37°C for 35 cycles at 10 min/cycle with 300 s of shaking before and during the measurements using the PerkinElmer 7000 Bio Assay reader. The luciferase response ratio was obtained as the ratio between the luciferase signal from the treated culture and the control untreated culture.
Effect of trimethoprim treatment on mutation frequency.
The increase in mutation frequency following SOS induction by trimethoprim was determined by selection of rifampin resistance with procedures similar to those previously described (31). Overnight cultures of DPB635 and DPB636 cells were diluted at a 1:10,000 ratio into LB and incubated at 37°C with shaking at 225 rpm for 3.5 h. The cultures were then diluted with fresh LB at a 1:3 ratio and either treated with 8 μg/ml of trimethoprim or left untreated as a control. Ten individual 2-ml aliquots of either treated or untreated cultures were incubated further at 37°C with shaking at 225 rpm for 20 h. The total number of viable colonies in each aliquot (CFU per milliliter) was determined by serial dilution of 30 μl of the cultures and plating on LB plates for overnight incubation. The number of rifampin-resistant colonies was determined by plating of the remainder of the cultures in aliquots on LB plates containing 100 μg/ml rifampin followed by 40 h of incubation at 37°C. The mutation frequency was calculated by the Ma-Sandri-Sarkar maximum likelihood estimator (MSS-MLE) method using the web tool FALCOR (32). The experiment on each strain was repeated three times.
RESULTS
Effect of C-terminal topA66 mutation on growth inhibition by antimicrobials.
Sequence analysis of several nondeletion topA mutant strains isolated in previous studies showed that the associated topA mutations are all found in the CTD downstream of amino acid 766 toward the C terminus (20). Biochemical activity assays have shown that the C-terminal region beyond Asp-760 is not required for the in vitro relaxation activity of the enzyme (26). The topA CTD mutations may, however, affect the previously identified interaction between RNA polymerase and topoisomerase I (19). This interaction could be more critical when a rapid cellular response to a stress challenge is required for survival. Isogenic strains of DPB635 with wild-type topA and DPB636 with topA66 CTD mutations were compared for growth inhibition by antimicrobial agents that induce the SOS response. Serial dilutions of cells from overnight culture in LB were spotted onto LB plates with and without antimicrobials. The results from 24 h of incubation of the plates (Fig. 1) showed that the topA66 mutation in strain DPB636 confers more severe growth inhibition in the presence of ciprofloxacin, norfloxacin, and trimethoprim than the topA+ isogenic strain DPB635. There is a 2-fold decrease in MIC values determined by the broth microdilution method for DPB636 with the topA66 mutation compared with that for DPB635 (Table 2).
FIG 1.
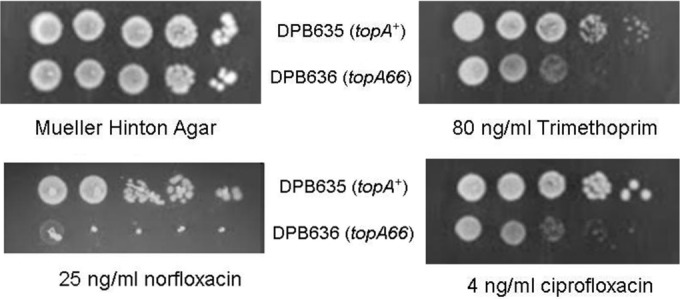
C-terminal domain mutation of topA results in more severe growth inhibition by antimicrobials. Overnight cultures of DPB635 (topA+) and DPB636 (topA66) in LB were adjusted to an OD600 value of 0.1. Volumes of 5 μl from 10-fold serial dilutions of OD600-adjusted cultures were spotted on Mueller-Hinton agar plates with and without antimicrobials. The cell growth on the plates was photographed after 24 h of incubation at 37°C.
TABLE 2.
Effect of topA66 mutation on MICs
| Drug | MIC (μg/ml) |
|
|---|---|---|
| DPB635 (topA+) | DPB636 (topA66) | |
| Norfloxacin | 0.125 | 0.0625 |
| Ciprofloxacin | 0.032 | 0.016 |
| Trimethoprim | 4 | 2 |
Relaxation activity of mutant topoisomerase I with frameshift mutation in the CTD.
The topA66 mutation was introduced by site-directed mutagenesis into the T7 promoter-driven expression plasmid for E. coli topoisomerase I enzyme (pLIC-ETOP). The resulting TopA enzyme (ETOP66) with a shift in the reading frame for the last 100 amino acids and terminating earlier by one codon was purified using the same procedures as those used for wild-type recombinant ETOP. The in vitro relaxation activity of ETOP66 protein was compared with that of wild-type ETOP and found to be decreased by 2- to 3-fold (see Fig. S1 in the supplemental material).
The topA66 CTD mutation decreases interaction between topoisomerase I and RNA polymerase.
Purified ETOP and ETOP66 proteins with N-terminal 6-histidine tags were incubated with increasing amounts of E. coli RNA polymerase prior to pulldown with Cobalt agarose resin. The amounts of RNA polymerase bound to ETOP or ETOP66 at the different RNA polymerase to topoisomerase molar ratios were analyzed by immunoblotting using antibodies against the β′ subunit of RNA polymerase. The results in Fig. 2 show that the affinity for RNA polymerase was reduced for ETOP66 as a result of the topA66 CTD mutation. The maximal level of RNA polymerase (RNAP) can be pulled down by ETOP at an ∼3-fold lower RNAP/topoisomerase ratio than by ETOP66.
FIG 2.

Effect of topA66 mutation on interaction with RNA polymerase. Pulldown of E. coli RNA polymerase at increasing RNA polymerase to topoisomerase molar ratios (RNAP:Topo) was measured by Western blotting using antibodies against the β′ subunit of RNA polymerase. The level of the β′ subunit signal relative to the maximal signal seen in each experiment was plotted as the average plus standard deviation for three independent experiments.
Recombinant Mycobacterium tuberculosis topoisomerase I can complement for DNA supercoiling but not for antibiotic hypersensitivity.
It was previously reported that plasmids extracted from E. coli with the topA66 mutation are more negatively supercoiled than plasmids extracted from E. coli with wild-type topA (20, 21). This was confirmed by electrophoretic analysis of the pBAD/Thio plasmid extracted from strains DPB635 and DPB636 in agarose gel containing chloroquine. Plasmid pBAD/Thio topoisomers from the topA66 mutant DPB636 were more negatively supercoiled and showed a decrease in the linking number difference (ΔΔLk) of −8 and a change in the superhelical density (Δσ) value of −0.0189 compared to the pBAD/Thio plasmid topoisomers from topA+ DPB635 (Fig. 3A). The basal level expression of recombinant E. coli or M. tuberculosis topoisomerase I from pETOP and pMTOP was shown previously to be sufficient for complementation of the topAts allele in E. coli AS17 for growth at nonpermissive temperatures (28). Here we found that the deficiency in the relaxation activity for DPB636 due to topA66 mutation could be complemented by the basal recombinant topoisomerase I activities contributed by pETOP completely and to a large extent by those of pMTOP. The pETOP plasmid DNA extracted from DPB635 and DPB636 had identical supercoiling levels (Fig. 3B). The pMTOP plasmid DNA extracted from DPB636 had a broader range of linking number distribution than the pMTOP plasmid DNA extracted from DPB635 (Fig. 3C) with the center of the topoisomer distribution shifted by a linking number difference of 2 (ΔΔLk = −2), corresponding to a change in the superhelical density (Δσ) value of only −0.0029.
FIG 3.

Comparison of plasmid DNA supercoiling in DPB635 and DPB636 with and without recombinant topoisomerase I. Plasmid DNA cloning vector pBAD/Thio (A), pETOP-expressing recombinant E. coli topoisomerase I (B), and pMTOP-expressing recombinant M. tuberculosis topoisomerase I (C) were extracted from early stationary-phase cultures of DPB635 (topA+) or DPB636 (topA66) transformants and analyzed by electrophoresis in agarose gel containing 4, 1.5, and 2 μg/ml of chloroquine, respectively. The chloroquine concentrations were optimized for comparison of the linking numbers of each of the plasmids.
However, while the expression of recombinant M. tuberculosis topoisomerase I complemented much of the DNA supercoiling effect from the topA66 mutation in DPB636, only recombinant E. coli topoisomerase I from pETOP was able to reverse the increased growth inhibition by antimicrobials for DPB636 as shown by the growth of the serially diluted cultures on plates with 4 ng/ml of ciprofloxacin (Fig. 4). DPB636 transformed with pMTOP was as susceptible to the antibiotic as DPB636 transformed with pBAD/Thio. This pattern of reversal in antibiotic susceptibility can also be seen in MIC determination assays, wherein the DPB636 strain transformed with pETOP shows a 2-fold increase in MIC compared to that for the DPB636 strain transformed with pBAD/Thio or pMTOP (Table 3).
FIG 4.
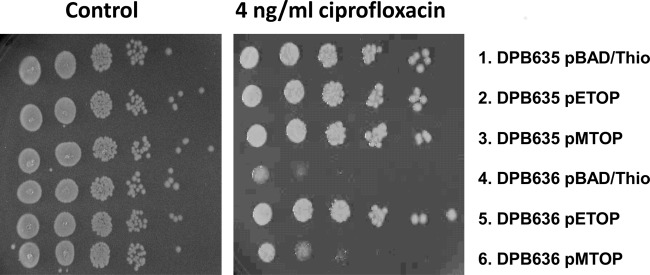
Increased sensitivity of DPB 636 to ciprofloxacin can be reversed by expression of recombinant E. coli topoisomerase I but not by recombinant M. tuberculosis topoisomerase I. Tenfold serial dilutions of DPB635 (topA+) and DPB636 (topA66) transformed with pBAD/Thio vector or pETOP- or pMTOP-expressing recombinant E. coli and M. tuberculosis topoisomerase I were spotted on control plates with or without ciprofloxacin.
TABLE 3.
Effect of recombinant topoisomerase I on ciprofloxacin MIC of DPB635 (topA+) and DPB636 (topA66)
| Strain | Plasmid | MIC (μg/ml) |
|---|---|---|
| DPB635 | pBAD/Thio vector | 0.032 |
| DPB635 | pETOP | 0.032 |
| DPB635 | pMTOP | 0.032 |
| DPB636 | pBAD/Thio vector | 0.016 |
| DPB636 | pETOP | 0.032 |
| DPB636 | pMTOP | 0.016 |
The topoisomerase I CTD mutant has decreased viability following treatment with bactericidal antibiotics and a toxic electrophile.
Norfloxacin and N-ethylmaleimide were used to determine the effect of the topA66 mutation on survival following different bactericidal challenges. Cultures of DPB635 and DPB636 cells were treated with norfloxacin at 100 to 250 ng/ml concentrations. Comparisons of the ratios of viable colony counts from treated cultures to those of untreated cultures showed that the topA66 mutation in DPB636 decreased the viability by up to 80-fold after the norfloxacin treatment (Fig. 5A and B). Survival following bactericidal stress induced by N-ethylmaleimide as a toxic nucleophile has been shown previously to depend on topA function (15). Results in Fig. 5C and D showed that DPB636 had up to a 49-fold lower survival ratio than DPB635 after treatment with 0.4 to 1 mM N-ethylmaleimide. Therefore, the topA66 CTD mutation in DPB636 not only resulted in greater bacteriostatic growth inhibition by antimicrobial agents but also resulted in decreased viability following bactericidal stress challenge by antimicrobials and a toxic electrophile.
FIG 5.

The CTD mutation resulted in decreased viability of topA66 mutant DPB636 following stress challenge. DPB635 (topA+) and DPB636 (topA66) were treated with 100 to 250 ng/ml of norfloxacin for 17 h (A), 250 ng/ml of norfloxacin for the indicated time periods (B), 0.4, 0.8, or 1 mM N-ethylmaleimide (NEM) for 1 h (C), or 0.4 mM NEM for the indicated time periods (D). Viable colony counts were determined before and after treatment by serial dilutions and plating on LB plates. The relative survival ratio after different treatments is calculated by dividing the viable colonies at the indicated time period or concentration by the viable count of respective nontreated cultures. Error bars correspond to standard deviations.
SOS response is impaired by the CTD mutation in topoisomerase I.
The luciferase signal produced by the dinD1::luxCADBE fusion present in the reporter plasmid (30) can be used to monitor the SOS response in E. coli. The topA66 CTD mutation was found to impair transcription from the dinD1 promoter during the SOS response to low concentrations of ciprofloxacin and trimethoprim. The luciferase signal did not increase relative to that in the untreated culture in DPB636 following treatment with ciprofloxacin (Fig. 6A). Transcription from the dinD1 promoter following treatment with trimethoprim is also diminished in DPB636 compared to that in DPB635 (Fig. 6B).
FIG 6.

Effect of topA66 mutation on transcription from the SOS response promoter. The luciferase signal from plasmid-encoded dinD1::luxCADBE fusion in DPB635 (topA+) and DPB636 (topA66) was monitored after treatment with 25 ng/ml norfloxacin (A) or 10 ng/ml ciprofloxacin (B). The response ratio was calculated as the ratio of luciferase from a treated culture versus the luciferase signal from an untreated culture.
Mutation frequency increase following trimethoprim treatment is diminished by the CTD mutation in topoisomerase I.
The SOS response from treatment with sublethal levels of antimicrobial agents has been shown to increase the mutation frequency in E. coli, which can be measured by the selection of rifampin resistance (4). Because the topA66 mutation reduced the number of viable colonies following treatment with bactericidal quinolones, the bacteriostatic antibiotic trimethoprim was used to induce the SOS response in DPB635 and DPB636. The trimethoprim-treated cultures were spread on plates containing rifampin to determine the mutation frequency. The results from three independent sets of experiments showed that the increase in mutation frequency following trimethoprim pretreatment (0.25 and 0.5 μg/ml) was diminished by >75% as a result of the topA66 mutation in DPB636 (Fig. 7).
FIG 7.

Decreased mutation frequency due to the topA66 mutation. Control and trimethoprim (TMP)-treated cultures of DPB635 (topA+) and DPB636 (topA66) were plated on LB plates with or without rifampin to determine the mutation frequency. The plot shows the averages and standard deviations of the mutation frequencies from three experiments. The fold increase in mutation frequency was calculated as the ratio of the mutation frequency for TMP-treated cultures versus the mutation frequency for the untreated control cultures.
DISCUSSION
Mutant topoisomerase I with CTD mutations retains a partial level of relaxation activity that appears sufficient for viability of the organism under normal laboratory growth conditions (20, 21). The results reported here demonstrated that the topA66 CTD mutation resulted in increased bacteriostatic and bactericidal activity of antimicrobials such as fluoroquinolones and trimethoprim. A number of suppression phenotypes associated with DNA replication were observed for the topA CTD mutants (20–22). These suppression phenotypes have been attributed to the increase in negative DNA supercoiling observed in the topA CTD mutants. However, while recombinant M. tuberculosis topoisomerase I restores nearly all of the relaxation activity, it did not alleviate the increased antibiotic sensitivity observed for strain DPB636 due to the topA66 CTD mutation. The CTD sequences of E. coli and M. tuberculosis topoisomerase I have diverged in evolution even though they appear to play similar roles of interaction with a passing strand of DNA in the mechanism of relaxation of negative DNA supercoiling (33, 34). This suggests that the protein-protein interaction between the CTD of topoisomerase I and RNA polymerase (19) may be the basis for the increased bacteriostatic and bactericidal effect of antimicrobials observed for the topA66 mutant.
In order to compete and survive in the host environment, bacterial pathogens are likely to require the activity of topoisomerase I to be tightly associated with the RNA polymerase complex for relief of transcription-driven supercoiling (11) generated during rapid transcription at gene loci associated with the stress response. Hypernegative supercoiling leads to R-loop accumulation and inhibition of transcription elongation (12). The bacterial SOS response induces a set of genes that counteract the stress challenge from DNA damage and improves bacterial survival. The SOS response has also been shown to increase the rate of mutations that can lead to increased frequency of resistance to antibiotics (4, 5). The topA66 CTD mutation resulted in a diminished SOS response. This correlated with decreased viability following the stress challenge and also affected the increase in the frequency of antibiotic resistance development that would have been a result of the SOS response.
E. coli topoisomerase I has been shown recently to have a role in genome maintenance by preventing overreplication originating from oriC and R loops (35). In addition to a diminished SOS response, the topA mutation may also be expected to result in overreplication that could potentially increase the sensitivity to the antimicrobials tested here. Gyrase and topoisomerase IV targeted by the fluoroquinolones tested here are required for movement of the DNA replication fork and resolution of the replication intermediates.
Topoisomerase I function has been shown to be essential for viability even under nonselective growth conditions for certain bacterial pathogens, including M. tuberculosis and Helicobacter pylori (36, 37). The presence of topoisomerase I in every bacterial pathogen as a potential target for a topoisomerase I poison inhibitor supports the targeting of topoisomerase I for discovery of novel antimicrobial agents, since the bactericidal action of the topoisomerase poison inhibitors does not require the topoisomerase function to be essential (38). The results reported here suggest that catalytic inhibitors of bacterial topoisomerase I could also be very useful if used along with other antibiotics. Perturbation of the function of bacterial topoisomerase I from the action of catalytic inhibitors might affect sensitivity to other antibiotics and might also decrease the frequency of general antibiotic resistance that can be developed.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the technical assistance of Angelo Andres, Maria Abrenica, Priyanka Bansod, and Sherin Thomas.
This work was supported by grants R01GM054226 and R01AI069313 from the National Institutes of Health to Y.-C.T.-D.
We declare no conflicts of interest.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00855-15.
REFERENCES
- 1.Collignon P, Powers JH, Chiller TM, Aidara-Kane A, Aarestrup FM. 2009. World Health Organization ranking of antimicrobials according to their importance in human medicine: a critical step for developing risk management strategies for the use of antimicrobials in food production animals. Clin Infect Dis 49:132–141. doi: 10.1086/599374. [DOI] [PubMed] [Google Scholar]
- 2.Redgrave LS, Sutton SB, Webber MA, Piddock LJ. 2014. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol 22:438–445. doi: 10.1016/j.tim.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Aldred KJ, Kerns RJ, Osheroff N. 2014. Mechanism of quinolone action and resistance. Biochemistry. doi: 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baharoglu Z, Mazel D. 2014. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol Rev. doi: 10.1111/1574-6976.12077. [DOI] [PubMed] [Google Scholar]
- 5.Blázquez J, Couce A, Rodriguez-Beltran J, Rodriguez-Rojas A. 2012. Antimicrobials as promoters of genetic variation. Curr Opin Microbiol 15:561–569. doi: 10.1016/j.mib.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Wang M, Jacoby GA, Mills DM, Hooper DC. 2009. SOS regulation of qnrB expression. Antimicrob Agents Chemother 53:821–823. doi: 10.1128/AAC.00132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briales A, Rodriguez-Martinez JM, Velasco C, Machuca J, Diaz de Alba P, Blazquez J, Pascual A. 2012. Exposure to diverse antimicrobials induces the expression of qnrB1, qnrD and smaqnr genes by SOS-dependent regulation. J Antimicrob Chemother 67:2854–2859. doi: 10.1093/jac/dks326. [DOI] [PubMed] [Google Scholar]
- 8.Beaber JW, Hochhut B, Waldor MK. 2004. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427:72–74. doi: 10.1038/nature02241. [DOI] [PubMed] [Google Scholar]
- 9.Hastings PJ, Rosenberg SM, Slack A. 2004. Antibiotic-induced lateral transfer of antibiotic resistance. Trends Microbiol 12:401–404. doi: 10.1016/j.tim.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Dörr T, Lewis K, Vulic M. 2009. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet 5:e1000760. doi: 10.1371/journal.pgen.1000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu LF, Wang JC. 1987. Supercoiling of the DNA template during transcription. Proc Natl Acad Sci U S A 84:7024–7027. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massé E, Drolet M. 1999. Relaxation of transcription-induced negative supercoiling is an essential function of Escherichia coli DNA topoisomerase I. J Biol Chem 274:16654–16658. doi: 10.1074/jbc.274.23.16654. [DOI] [PubMed] [Google Scholar]
- 13.Lesley SA, Jovanovich SB, Tse-Dinh YC, Burgess RR. 1990. Identification of a heat shock promoter in the topA gene of Escherichia coli. J Bacteriol 172:6871–6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qi H, Menzel R, Tse-Dinh YC. 1997. Regulation of Escherichia coli topA gene transcription: involvement of a sigmaS-dependent promoter. J Mol Biol. 267:481–489. doi: 10.1006/jmbi.1997.0901. [DOI] [PubMed] [Google Scholar]
- 15.Tse-Dinh YC. 2000. Increased sensitivity to oxidative challenges associated with topA deletion in Escherichia coli. J Bacteriol 182:829–832. doi: 10.1128/JB.182.3.829-832.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qi H, Menzel R, Tse-Dinh YC. 1999. Increased thermosensitivity associated with topoisomerase I deletion and promoter mutations in Escherichia coli. FEMS Microbiol Lett 178:141–146. doi: 10.1111/j.1574-6968.1999.tb13770.x. [DOI] [PubMed] [Google Scholar]
- 17.Tse-Dinh YC, Beran-Steed RK. 1988. Escherichia coli DNA topoisomerase I is a zinc metalloprotein with three repetitive zinc-binding domains. J Biol Chem 263:15857–15859. [PubMed] [Google Scholar]
- 18.Grishin NV. 2000. C-terminal domains of Escherichia coli topoisomerase I belong to the zinc-ribbon superfamily. J Mol Biol. 299:1165–1177. doi: 10.1006/jmbi.2000.3841. [DOI] [PubMed] [Google Scholar]
- 19.Cheng B, Zhu CX, Ji C, Ahumada A, Tse-Dinh YC. 2003. Direct interaction between Escherichia coli RNA polymerase and the zinc ribbon domains of DNA topoisomerase I. J Biol Chem 278:30705–30710. doi: 10.1074/jbc.M303403200. [DOI] [PubMed] [Google Scholar]
- 20.Valjavec-Gratian M. 2005. Tus-mediated arrest of DNA replication in is modulated by DNA supercoiling influences Tus activity. Mol Microbiol 58:758–773. doi: 10.1111/j.1365-2958.2005.04860.x. [DOI] [PubMed] [Google Scholar]
- 21.Biek DP, Cohen SN. 1989. Involvement of integration host factor (IHF) in maintenance of plasmid pSC101 in Escherichia coli: mutations in the topA gene allow pSC101 replication in the absence of IHF. J Bacteriol 171:2066–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sawitzke JA, Austin S. 2000. Suppression of chromosome segregation defects of Escherichia coli muk mutants by mutations in topoisomerase I. Proc Natl Acad Sci U S A 97:1671–1676. doi: 10.1073/pnas.030528397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stockum A, Lloyd RG, Rudolph CJ. 2012. On the viability of Escherichia coli cells lacking DNA topoisomerase I. BMC Microbiol 12:26. doi: 10.1186/1471-2180-12-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiNardo S, Voelkel KA, Sternglanz R, Reynolds AE, Wright A. 1982. Escherichia coli DNA topoisomerase I mutants have compensatory mutations in DNA gyrase genes. Cell 31:43–51. doi: 10.1016/0092-8674(82)90403-2. [DOI] [PubMed] [Google Scholar]
- 25.Pruss GJ, Manes SH, Drlica K. 1982. Escherichia coli DNA topoisomerase I mutants: increased supercoiling is corrected by mutations near gyrase genes. Cell 31:35–42. doi: 10.1016/0092-8674(82)90402-0. [DOI] [PubMed] [Google Scholar]
- 26.Zumstein L, Wang JC. 1986. Probing the structural domains and function in vivo of Escherichia coli DNA topoisomerase I by mutagenesis. J Mol Biol 191:333–340. doi: 10.1016/0022-2836(86)90130-0. [DOI] [PubMed] [Google Scholar]
- 27.Cheng B, Shukla S, Vasunilashorn S, Mukhopadhyay S, Tse-Dinh YC. 2005. Bacterial cell killing mediated by topoisomerase I DNA cleavage activity. J Biol Chem 280:38489–38495. doi: 10.1074/jbc.M509722200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narula G, Becker J, Cheng B, Dani N, Abrenica MV, Tse-Dinh YC. 2010. The DNA relaxation activity and covalent complex accumulation of Mycobacterium tuberculosis topoisomerase I can be assayed in Escherichia coli: application for identification of potential FRET-dye labeling sites. BMC Biochem 11:41. doi: 10.1186/1471-2091-11-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sorokin EP, Cheng B, Rathi S, Aedo SJ, Abrenica MV, Tse-Dinh YC. 2008. Inhibition of Mg2+ binding and DNA religation by bacterial topoisomerase I via introduction of an additional positive charge into the active site region. Nucleic Acids Res 36:4788−4796. doi: 10.1093/nar/gkn460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng B, Liu I, Tse-Dinh YC. 2007. Compounds with antibacterial activity that enhance DNA cleavage by bacterial DNA topoisomerase I. J Antimicrob Chemother 59:640–645. doi: 10.1093/jac/dkl556. [DOI] [PubMed] [Google Scholar]
- 31.Kohanski MA, DePristo MA, Collins JJ. 2010. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell 37:311–320. doi: 10.1016/j.molcel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hall BM, Ma CX, Liang P, Singh KK. 2009. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 25:1564–1565. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmed W, Bhat AG, Leelaram MN, Menon S, Nagaraja V. 2013. Carboxyl terminal domain basic amino acids of mycobacterial topoisomerase I bind DNA to promote strand passage. Nucleic Acids Res 41:7462–7471. doi: 10.1093/nar/gkt506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahumada A, Tse-Dinh YC. 2002. The role of the Zn(II) binding domain in the mechanism of E. coli DNA topoisomerase I. BMC Biochem 3:13. doi: 10.1186/1471-2091-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Usongo V, Nolent F, Sanscartier P, Tanguay C, Broccoli S, Baaklini I, Drlica K, Drolet M. 2008. Depletion of RNase HI activity in Escherichia coli lacking DNA topoisomerase I leads to defects in DNA supercoiling and segregation. Mol Microbiol 69:968–981. doi: 10.1111/j.1365-2958.2008.06334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmed W, Menon S, Godbole AA, Karthik PV, Nagaraja V. 2014. Conditional silencing of topoisomerase I gene of Mycobacterium tuberculosis validates its essentiality for cell survival. FEMS Microbiol Lett 353:116–123. doi: 10.1111/1574-6968.12412. [DOI] [PubMed] [Google Scholar]
- 37.Suerbaum S, Brauer-Steppkes T, Labigne A, Cameron B, Drlica K. 1998. Topoisomerase I of Helicobacter pylori: juxtaposition with a flagellin gene (flaB) and functional requirement of a fourth zinc finger motif. Gene 210:151–161. doi: 10.1016/S0378-1119(98)00065-1. [DOI] [PubMed] [Google Scholar]
- 38.Tse-Dinh YC. 2009. Bacterial topoisomerase I as a target for discovery of antibacterial compounds. Nucleic Acids Res 37:731–737. doi: 10.1093/nar/gkn936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.