

Abstract
Malaria and HIV infection are coendemic in a large portion of the world and remain a major cause of morbidity and mortality. Growing resistance of Plasmodium species to existing therapies has increased the need for new therapeutic approaches. The Plasmodium glucose transporter PfHT is known to be essential for parasite growth and survival. We have previously shown that HIV protease inhibitors (PIs) act as antagonists of mammalian glucose transporters. While the PI lopinavir is known to have antimalarial activity, the mechanism of action is unknown. We report here that lopinavir blocks glucose uptake into isolated malaria parasites at therapeutically relevant drug levels. Malaria parasites depend on a constant supply of glucose as their primary source of energy, and decreasing the available concentration of glucose leads to parasite death. We identified the malarial glucose transporter PfHT as a target for inhibition by lopinavir that leads to parasite death. This discovery provides a mechanistic basis for the antimalarial effect of lopinavir and provides a direct target for novel drug design with utility beyond the HIV-infected population.
INTRODUCTION
Despite aggressive worldwide efforts to eradicate malaria, this life-threatening disease continues to affect over 200 million people per year, resulting in an annual death toll exceeding half a million, mostly among African children (1). Currently, vaccination against malaria is not available, while resistance against all known therapeutics is spreading (1). As a result, newer antimalarial agents with novel mechanisms of action are urgently needed.
The global prevalence of malaria and that of HIV infection largely overlap geographically. A combination antiviral therapy that includes the HIV protease inhibitor (PI) lopinavir has been found to dramatically decrease malaria incidence in a pediatric clinical population, by 41%, suggesting a direct effect of PIs on parasite replication (2). Indeed, lopinavir has demonstrated in vitro activity (3) against Plasmodium falciparum, the protozoan species responsible for most malaria deaths (1, 4). In addition, lopinavir reduces the malaria liver stage burden in infected rhesus monkeys in vivo at clinically relevant concentrations (5).
Despite ongoing efforts, the direct cellular target(s) of lopinavir responsible for its antimalarial properties against P. falciparum remains unclear. PIs were originally designed as antagonists of the viral aspartyl protease (6). The malaria parasite requires a class of aspartyl proteases called plasmepsins, which are necessary to degrade host hemoglobin (7) and direct export of malaria export proteins (8); however, the antimalarial activity of PIs does not appear to be mediated through plasmepsin inhibition (9, 10). Identifying the antimalarial mechanism of action of PIs is imperative for finding a novel, clinically proven drug target and developing a new class of lopinavir-like antimalarial drugs.
In clinical populations, prolonged use of PIs is associated with insulin resistance. Recent studies have identified the molecular mechanism of this effect, which is mediated by direct binding of PIs to the insulin-responsive facilitative glucose transporter GLUT4 (11–13). The human glucose transporters share sequence homology with the essential P. falciparum glucose transporter PfHT. Similar to GLUT1 and GLUT4, the predicted topology of PfHT comprises 12 transmembrane helices, forming a central glucose permeation path. Key residues that are involved in glucose binding and transport are preserved between the human and malaria glucose transporters (14, 15).
Intraerythrocytic malaria parasites depend on a constant supply of glucose as their primary source of energy (16). Not surprisingly, infected erythrocytes show an ∼100-fold increase in glucose consumption compared to uninfected erythrocytes (17). PfHT (PF3D7_0204700) is the principal glucose transporter, transcribed from a single-copy gene with no close paralogue (14). PfHT has been genetically validated as essential in Plasmodium parasites (18) and has been independently chemically validated as a novel drug target against malaria (14, 19).
Here we show that lopinavir inhibits glucose uptake into the P. falciparum parasite by blocking PfHT at therapeutically relevant concentrations. This establishes a direct molecular target for the antimalarial activity of lopinavir and validates the utility of targeting PfHT in novel drug development.
MATERIALS AND METHODS
Materials.
[14C]2-deoxyglucose ([14C]2DOG) was purchased from PerkinElmer. [3H]2DOG was purchased from American Radiolabels Inc. PfHT DNA was codon optimized and synthesized by Life Technologies (Grand Island, NY). GLUT1 short hairpin RNA (shRNA) was obtained through the RNA interference (RNAi) core at Washington University, School of Medicine. HEK293 cells were acquired from the American Type Culture Collection. HIV protease inhibitors were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. Compound 3361 was kindly donated by Sanjeev Krishna (Centre for Infection, Division of Cellular and Molecular Medicine, St. George's, University of London, London, United Kingdom).
Malaria tissue culture.
P. falciparum strain 3D7 was obtained from the Malaria Research and Reference Reagent Resource Center (MR4, ATCC, Manassas, VA). Unless otherwise stated, P. falciparum strains were cultured at 37°C in a 2% suspension of human erythrocytes in RPMI 1640 medium (SKU R4130; Sigma-Aldrich) supplemented with 27 mM sodium bicarbonate, 11 mM glucose, 5 mM HEPES, 1 mM sodium pyruvate, 0.37 mM hypoxanthine, 0.01 mM thymidine, 10 μg/ml gentamicin, and 0.5% Albumax (Life Technologies) in a 5% O2–5% CO2–90% N2 atmosphere, as previously described (20, 21). Culture growth was monitored by microscopic analysis of Giemsa-stained blood smears.
Drug and glucose sensitivity of P. falciparum.
Asynchronous P. falciparum cultures were diluted to 1% parasitemia and were treated at a range of concentrations of inhibitor or glucose. Growth inhibition assays were performed in opaque 96-well plates at 100-μl culture volume. After 3 days, parasite growth was quantified by measuring DNA content using Picogreen (Life Technologies), as previously described (22). Picogreen fluorescence was measured on a FLUOstar Omega microplate reader (BMG Labtech) at 485-nm excitation and 528-nm emission. IC50s (50% inhibitory concentrations) were calculated by nonlinear regression analysis using GraphPad Prism software.
Stable expression of PfHT.
HEK293 cells were stably transfected with codon-optimized PfHT DNA in the pcDNA3.1(−)/hygro plasmid (Life Technologies) as described earlier (23). After selection with hygromycin, 10 colonies were grown to near confluence in 4-cm tissue culture dishes, and the highest expresser of PfHT was selected using [3H]2DOG uptake and quantitative reverse transcription (RT)-PCR (as described below).
Short hairpin RNA lentiviruses for GLUT1 knockdown.
293T packaging cells were cotransfected with vesicular stomatitis virus glycoprotein (VSVg) and Delta 8.9 packaging plasmids plus shRNA lentiviral plasmids targeting GLUT1 (NM_006516.1-2310s1c1) using Optifect transfection reagent (Life Technologies). The medium was changed 12 h after transfection, and supernatants (10 ml) were harvested every 24 h for 72 h and kept at 4°C until they were pooled, filtered through 0.45-μm syringe filters, aliquoted, and stored at −80°C until use. HEK293 wild-type cells or HEK293 cells stably expressing PfHT were infected by exposure to viral supernatant for 48 h and then were placed in selection medium (containing 5 μg/ml puromycin).
Quantitative PCR analysis.
Total RNA was isolated using the TRIzol Plus RNA Purification System (Life Technologies). RNA was reverse transcribed using the qScript cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD). Quantitative RT-PCR was performed using the Power SYBR green PCR master mix (Applied Biosystems) as described previously (24).
2DOG uptake into P. falciparum and HEK293 cells.
Uptake of [14C]2DOG into isolated parasites was determined at room temperature using the methods described previously (25). Lopinavir and compound 3361 were added 5 min prior to the addition of [14C]2DOG (final concentration, 0.2 μCi/ml). Uptake of [3H]2DOG into HEK293 cells was measured in phosphate buffer at 37°C for 5 min as described previously (26).
Statistical analyses.
The data are reported as means ± standard errors of the means (SEM). Differences between control and experimental values were determined by one-way analysis of variance (ANOVA) analysis.
Modeling of PfHT and docking of lopinavir.
The PfHT structure was predicted using the I-TASSER webserver (27–29). The webserver was among top scorers of the Server Section of the 7th (2006), 8th (2008), 9th (2010), 10th (2012), and 11th (2014) CASPs, a community-wide experiment for testing the state of the art of protein structure predictions. We chose a model that resembled the inward open conformation. Docking was performed using AutoDock Vina (30). The receptor was prepared using MGLTools version 1.5.6, and the ligand lopinavir was prepared using ArgusLab. The docking process was repeated with number of modes of 50 and increasing exhaustiveness of 25, 50, and 100. The results in each attempt were 20 similar docked poses. Docked poses were displayed using PyMOL version 1.7.4.0.
RESULTS
We compared the effect of currently FDA-approved HIV protease inhibitors on the replication and survival of P. falciparum parasites in asexual erythrocyte cell culture. Consistent with prior reports (3, 9), all tested compounds inhibited parasite growth. Lopinavir was the most potent inhibitor with an IC50 of 1.9 μM (Fig. 1a). We previously determined the inhibitory effects of several HIV protease inhibitors on human glucose transport (11, 13, 26). Since HIV PIs inhibit human glucose transport, we evaluated whether lopinavir might inhibit glucose uptake in isolated P. falciparum parasites. We compared the uptake of radiolabeled 2DOG in live parasites isolated from erythrocytes treated with vehicle, lopinavir, or the chemically unrelated PfHT inhibitor compound 3361 (Fig. 2a) (19). The O-3-hexose derivative compound 3361 [3-O-((undec-10-en)-yl)-d-glucose)] has previously been shown to selectively inhibit PfHT relative to several mammalian sugar transporters and inhibit glucose uptake by the parasite, leading to inhibition of growth and proliferation in vitro and in vivo (19, 25). We found that high concentrations of lopinavir potently inhibited uptake of radiolabeled glucose by more than 70% compared to vehicle-treated control, similar to the effect observed by the known PfHT inhibitor compound 3361. In dose-response studies (Fig. 2b), we found that the half-maximal inhibitory concentration (IC50) of lopinavir was 16 ± 4.2 μM, which is within the range of average peak levels in sera of both adult and pediatric patients treated with lopinavir/ritonavir (31–34).
FIG 1.

Comparison of HIV protease inhibitors' effects on P. falciparum growth inhibition. (a) Comparison of IC50s of HIV protease inhibitors on growth of P. falciparum in blood culture. IC50s were calculated using nonlinear regression analysis and are expressed as means with 95% confidence intervals. (b) Chemical structures of the three most potent HIV protease inhibitors of PfHT-mediated glucose uptake.
FIG 2.

(a) Uptake of [14C]2DOG by isolated P. falciparum trophozoites in the presence of lopinavir (100 μM), compound 3361 (200 μM), or vehicle. The parasites were suspended in HEPES-buffered saline containing 200 μM glucose. (b) Uptake of [14C]2DOG by isolated P. falciparum trophozoites at increasing concentrations of lopinavir. The uptake was quenched after 2 min. IC50s were calculated using nonlinear regression analysis. Uptake data are expressed as means ± SEM. A 30-s or 1-min time point would have been a more accurate measurement of initial rates; however, we chose the longer time point to enhance the uptake signal, as the degree of nonlinearity will probably not greatly affect the calculated IC50 value.
To confirm that inhibition of malaria glucose transport was mediated through PfHT inhibition, we developed and tested the effect of this PI on glucose transport in a PfHT-overexpressing HEK293 cell line. HEK293 cells predominantly transport glucose using the facilitative transport protein GLUT1. Using a PfHT sequence codon optimized for mammalian cell expression, we overexpressed the malarial hexose transporter protein and knocked down the expression of GLUT1 using shRNA. In this engineered cell line, PfHT was the predominant hexose transporter expressed (Fig. 3a). Glucose uptake in the PfHT-overexpressing cell line was >5 times higher than in background cells that did not overexpress PfHT but were transduced with GLUT1 shRNA (Fig. 3b). We quantified the amount of [3H]2DOG accumulated in PfHT-overexpressing HEK293 cells in the presence of various concentrations of lopinavir and determined an IC50 of 14 ± 2 μM. The IC50s for the effect of lopinavir on glucose uptake in both P. falciparum parasites and PfHT-overexpressing HEK293 cells are closely correlated, consistent with a model in which inhibition of PfHT is responsible for decreased glucose uptake and reduced parasite growth (Fig. 4). Supporting this model, blood-stage P. falciparum is acutely sensitive to reduced extracellular glucose concentrations (17). Since the measurement of 2DOG uptake involves both the transport of this sugar and phosphorylation to 2DOG-6-P, we cannot fully exclude the possibility that lopinavir also inhibits the malarial hexokinase. However, the observation that lopinavir inhibits 2DOG uptake with different IC50s when measured in HEK293 cells overexpressing different GLUTs (ranging from 3 μM for GLUT4 to 32 μM for GLUT1) suggests that hexokinase is not directly targeted.
FIG 3.

(a) mRNA expression levels of glucose transporter proteins in HEK293 wild-type (WT), GLUT1 knockdown (KD), and GLUT1 KD + PfHT-overexpressing (OE) cells. mRNA levels were determined via quantitative PCR, and levels are displayed in copy number per nanogram of cDNA. Data are expressed as means ± SEM. (b) Comparing specific activities of glucose uptake in HEK293 cells expressing GLUT1 shRNA and overexpression of PfHT. Data are expressed as means ± SEM (*, P < 0.05; ***, P < 0.001; one-way ANOVA analysis).
FIG 4.

Specific activity of glucose uptake in HEK293 cells stably expressing PfHT and GLUT1 shRNA at different lopinavir concentrations. Glucose uptake was determined by quantifying the amount of 2DOG accumulated in HEK293 cells and normalizing to time and total protein. The experiment was repeated in an identical background cell line that does not overexpress PfHT. Normalized specific activity of PfHT-overexpressing cells was adjusted for non-PfHT-specific glucose uptake by subtracting background uptake. Data are expressed as means ± SEM. IC50 was calculated using nonlinear regression analysis.
To further investigate the mode of action of lopinavir on PfHT, we determined several parameters by Michaelis-Menten kinetics. As expected, uptake of glucose is saturated with increasing substrate concentration. The observed increase in Km with a constant maximum rate of metabolism (Vmax) indicates that lopinavir acts as a competitive inhibitor of glucose uptake (Fig. 5).
FIG 5.

Kinetic analysis of inhibition of glucose uptake by PfHT by lopinavir. [3H]2DOG uptake was determined in the presence of various concentrations of lopinavir and substrate. Uptake was quenched after 2 min. Data were fitted to the Michaelis-Menten equation by nonlinear regression using GraphPad Prism 6 to obtain Km and Vmax values and were transformed and plotted as a double-reciprocal Lineweaver-Burk plot using GraphPad Prism 6. Uptake data are expressed as means ± SEM (n = 3).
To investigate the binding site of lopinavir on PfHT, we generated a model of the transporter based on several crystal structures of homologous proteins using the I-TASSER Web server (Table 1). We selected a model with high cluster density in an inward conformation to determine the lopinavir binding site by docking analysis. We created a docking volume covering the entire protein to obtain unbiased results. Lopinavir was docked to the PfHT model using AutoDock VINA via rigid-receptor, flexible-ligand docking. All docked poses of lopinavir were bound to a single binding pocket on the intracellular side of the glucose permeation path, preventing glucose from entering and reaching its binding site (Fig. 6a). These results were replicated in several docking experiments using random seeds and different degrees of exhaustiveness, strongly supporting this position as the lowest energy binding site. We compared the PfHT-lopinavir putative binding site with the binding site of the structurally related protease inhibitor indinavir in the PfHT homologue GLUT4, published by Hresko et al. (Fig. 6b) (26). The docking experiments suggest that both indinavir and lopinavir bind to a structurally similar binding pocket in their respective glucose transporters. Lopinavir binding within the glucose permeation pathway is consistent with this drug acting as a competitive inhibitor of zero-trans glucose uptake (Fig. 5). Although inhibition of GLUT4 by indinavir requires drug binding from the intracellular side of the transporter (11), lopinavir appears to have access to this domain from the exofacial side of PfHT. Similar to the observed differences in isoform selectivity of HIV protease inhibitors for mammalian GLUTs (26), it is possible that steric influences of amino acid side chains lining the glucose permeation pathway contribute to the ability of these drugs to target PfHT.
TABLE 1.
Top 10 templates used by I-TASSERa
| Rank | PDB hit | Ident1 | Ident2 | Cov. | Norm. Z-score |
|---|---|---|---|---|---|
| 1 | 4pypA | 0.28 | 0.26 | 0.87 | 3.85 |
| 2 | 4gbyA | 0.26 | 0.26 | 0.88 | 5.79 |
| 3 | 4pypA | 0.27 | 0.26 | 0.87 | 5.39 |
| 4 | 4gc0A | 0.25 | 0.26 | 0.88 | 3.91 |
| 5 | 4gc0A | 0.26 | 0.26 | 0.89 | 3.65 |
| 6 | 4pypA | 0.28 | 0.26 | 0.87 | 5.35 |
| 7 | 4gc0A | 0.26 | 0.26 | 0.89 | 4.83 |
| 8 | 4pypA | 0.29 | 0.26 | 0.87 | 6.61 |
| 9 | 4pypA | 0.28 | 0.26 | 0.87 | 4.68 |
| 10 | 4pypA | 0.28 | 0.26 | 0.87 | 4.82 |
Rank of templates represents the top 10 threading templates used by I-TASSER; PDB hit, Protein Data Bank template hit; Ident1 is the percent sequence identity of the templates in the threading aligned region with the query sequence; Ident2 is the percent sequence identity of the whole template chains with the query sequence; Cov. represents the coverage of the threading alignment and is equal to the number of aligned residues divided by the length of query protein; Norm. Z-score is the normalized Z-score of the threading alignments; alignment with a normalized Z-score of >1 means a good alignment and vice versa.
FIG 6.
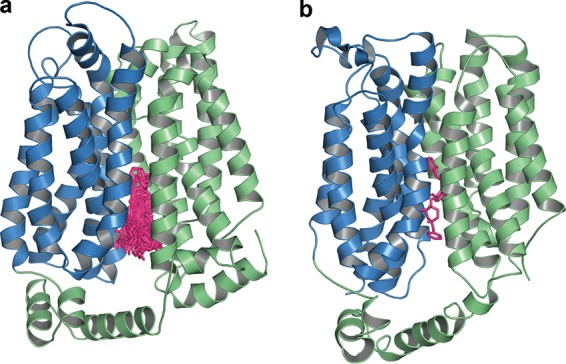
(a) Lopinavir docked within the glucose permeation channel of PfHT. Lopinavir (pink) is docked in several poses to the inward open conformation of PfHT. The amino-terminal half of PfHT is colored blue, and the carboxyl-terminal half is green. The depicted 40 poses occupy a single binding pocket and are representative of the results of several rounds of docking analysis. (b) Indinavir docked within the glucose permeation channel of GLUT4. Indinavir (pink) is docked to the inward open conformation of GLUT4 (26). The amino-terminal half of GLUT4 is colored blue, and the carboxyl-terminal half is green.
DISCUSSION
There is a compelling need for novel therapies for treatment of malaria. In particular, the emergence and spread of resistance to artemisinin-based compounds threatens malaria control efforts worldwide. Protease inhibitors, such as lopinavir, represent promising therapeutic leads for new antimalarial development. Lopinavir has potent antimalarial activity in vitro and in animal models (4, 5), and human studies of lopinavir-treated individuals in areas of endemicity indicate antimalarial potency at clinically relevant doses (2). Importantly, many of the pharmacologic challenges that slow antimalarial development have already been overcome with lopinavir, which is orally bioavailable, available in suspension form (for pediatric use), and suitable for once-daily dosing in combination with ritonavir.
Our studies provide substantial evidence that inhibition of the malaria hexose transporter, PfHT, is responsible for the antiparasitic effects of lopinavir. PfHT hexose transporter function is known to be required for Plasmodium parasite development in culture and in animal models (14, 18, 19, 25, 35). We find that not only does lopinavir directly block glucose uptake into P. falciparum parasites but it also inhibits glucose uptake in human cells engineered such that the majority of hexose transport is through heterologously expressed PfHT. Importantly, profound reductions of glucose uptake into parasites and into PfHT-expressing human cells are achieved at lopinavir concentrations well below therapeutically achieved drug levels (31–34).
Of note, even lower concentrations of both lopinavir and compound 3361 are required to inhibit parasite replication than are required to inhibit glucose transport (19). This difference may be explained by the reliance of parasites on a constant glucose supply. Plasmodium infection increases cellular glucose consumption in erythrocytes nearly 100-fold, while parasite survival is highly sensitive to reduced extracellular glucose concentrations (17). Thus, even partial inhibition of Plasmodium glucose transporters may lead to growth inhibition and eventually death of the parasite. PfHT may therefore represent a promising, parasite-specific “Achilles heel” for target-based drug discovery efforts. We cannot rule out the possibility that there are additional targets of lopinavir in P. falciparum other than PfHT. The existence of additional drug targets, if present, could delay the development of drug resistance, a major problem for malaria drugs. However, observing this difference for both lopinavir and compound 3361, two structurally unrelated drugs that inhibit PfHT-mediated glucose transport, decreases the likelihood that they would both also inhibit another secondary target.
As agents designed for long-term suppression of HIV replication, lopinavir and other protease inhibitors are well tolerated, with minor adverse events (mostly gastrointestinal) associated with short-term use (36). During chronic HIV treatment regimens that include PIs, disturbances in glucose homeostasis and other metabolic changes that increase cardiovascular risk have been reported (37). In this HIV-infected population, it has been challenging to dissociate the direct contributions of PIs from the influences of viral infection, other drug exposures (e.g., nucleoside reverse transcriptase inhibitors), and associated environmental factors. Nevertheless, the in vitro effects of PIs on the insulin-responsive facilitative glucose transporter GLUT4 correlate directly with in vivo changes in insulin sensitivity, both in rodent models (38) and in humans (39). Fortunately, the effect of PIs on glucose tolerance is readily reversible following short-term drug exposure, as required for treatment of malaria (38, 39). However, the full spectrum of PI-mediated effects on the remaining GLUT isoforms remains unknown, and not all PIs appear to have the same effect on GLUT4 activity (40). Ideally, the development of drugs that selectively target PfHT over mammalian GLUTs would provide highly selective inhibition of PfHT over the mammalian GLUTs, resulting in a superior safety profile.
The development of high-throughput screening protocols targeting the Plasmodium glucose transporter will help identify additional classes of PfHT antagonists. Several assays have been developed by expressing PfHT in yeast or Leishmania (14, 18, 41). In combination with these prior studies, our current findings confirm that PfHT is an attractive, druggable target for malaria. Our development of a PfHT-overexpressing HEK293 cell line and demonstration that this system can reliably test for drug-mediated inhibition of the malarial glucose transporter can significantly aid candidate drug screening. Complementing a high-throughput approach is the opportunity for structure-based design. This can allow for higher affinity and selectivity to the plasmodium transporter while minimizing effects on mammalian glucose transporters (GLUTs). GLUT1 is the most extensively characterized of the human GLUTs, and a crystal structure of this protein was recently reported (42). In the absence of structural data for PfHT and the other known GLUTs, it has been necessary to rely upon molecular modeling based on the structures of related bacterial and mammalian transporters. Although our docking results are consistent with the previously identified HIV protease inhibitor binding site in the closely related transporter GLUT4 (26), we cannot exclude the possibility that lopinavir binds to a distinct binding site accessible only in an outward open conformation of PfHT. Testing this possibility will generate valuable data once a homologue transporter is crystallized in the outward open conformation. Future efforts to establish a comprehensive structure-activity profile for the inhibition of PfHT and the human GLUTs may provide valuable information about the feasibility of this approach. Investigation of potential additive or synergistic effects of PfHT inhibitors with existing antimalarial agents or other compounds that inhibit parasite metabolism will also be of great interest.
In summary, the identification of lopinavir as a clinically meaningful inhibitor of the essential malaria hexose transporter PfHT provides a strong rationale and molecular basis for future antimalarial development efforts targeting this protein. Because PfHT inhibition retards both the liver and mosquito stage development of rodent malaria parasites (43), novel PfHT inhibitors hold great promise for use in malaria treatment and as transmission-blocking agents. The existing pharmacokinetic and safety data for lopinavir should allow rapid assessment of the suitability and efficacy of this agent in non-HIV-infected patient populations. Understanding the molecular target of lopinavir in P. falciparum will be key to any necessary medicinal chemical optimization of lopinavir and related protease inhibitors, repurposed for use as antimalarials.
ACKNOWLEDGMENTS
This work was supported by the Children's Discovery Institute of Washington University and St. Louis Children's Hospital (MD-LI-2011-171 to A.R.O.), NIH/NIAID R01AI103280 (to A.R.O.), a March of Dimes Basil O'Connor Starter Scholar Research Award (to A.R.O.), and a Doris Duke Charitable Foundation Clinical Scientist Development award (to A.R.O.).
We thank Maria Payne for conducting quantitative PCR experiments and generating plasmids.
Author contributions: T.E.K. and P.W.H. conceived the initial idea of the study; T.E.K., C.A., and M.R.H. performed the experiments; T.E.K. created the model and performed the docking; T.E.K., A.R.O., and P.W.H. contributed to the experimental design and data analysis; T.E.K., A.R.O., and P.W.H. wrote the manuscript, with feedback from M.R.H.
We declare that we have no competing financial interests.
REFERENCES
- 1.World Health Organization. 2014. World malaria report 2014, p 165–176. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Achan J, Kakuru A, Ikilezi G, Ruel T, Clark TD, Nsanzabana C, Charlebois E, Aweeka F, Dorsey G, Rosenthal PJ, Havlir D, Kamya MR. 2012. Antiretroviral agents and prevention of malaria in HIV-infected Ugandan children. N Engl J Med 367:2110–2118. doi: 10.1056/NEJMoa1200501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nsanzabana C, Rosenthal PJ. 2011. In vitro activity of antiretroviral drugs against Plasmodium falciparum. Antimicrob Agents Chemother 55:5073–5077. doi: 10.1128/AAC.05130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hobbs CV, Voza T, Coppi A, Kirmse B, Marsh K, Borkowsky W, Sinnis P. 2009. HIV protease inhibitors inhibit the development of preerythrocytic-stage plasmodium parasites. J Infect Dis 199:134–141. doi: 10.1086/594369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hobbs CV, Neal J, Conteh S, Donnelly L, Chen J, Marsh K, Lambert L, Orr-Gonzalez S, Hinderer J, Healy S, Borkowsky W, Penzak SR, Chakravarty S, Hoffman SL, Duffy PE. 2014. HIV treatments reduce malaria liver stage burden in a non-human primate model of malaria infection at clinically relevant concentrations in vivo. PLoS One 9:e100138. doi: 10.1371/journal.pone.0100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nalam MNL, Schiffer CA. 2008. New approaches to HIV protease inhibitor drug design II: testing the substrate envelope hypothesis to avoid drug resistance and discover robust inhibitors. Curr Opin HIV AIDS 3:642–646. doi: 10.1097/COH.0b013e3283136cee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banerjee R, Liu J, Beatty W, Pelosof L, Klemba M, Goldberg DE. 2002. Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc Natl Acad Sci U S A 99:990–995. doi: 10.1073/pnas.022630099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boddey JA, Hodder AN, Günther S, Gilson PR, Patsiouras H, Kapp EA, Pearce JA, de Koning-Ward TF, Simpson RJ, Crabb BS, Cowman AF. 2010. An aspartyl protease directs malaria effector proteins to the host cell. Nature 463:627–631. doi: 10.1038/nature08728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parikh S, Liu J, Sijwali P, Gut J, Goldberg DE, Rosenthal PJ. 2006. Antimalarial effects of human immunodeficiency virus type 1 protease inhibitors differ from those of the aspartic protease inhibitor pepstatin. Antimicrob Agents Chemother 50:2207–2209. doi: 10.1128/AAC.00022-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hobbs CV, Tanaka TQ, Muratova O, Van Vliet J, Borkowsky W, Williamson KC, Duffy PE. 2013. HIV treatments have malaria gametocyte killing and transmission blocking activity. J Infect Dis 208:139–148. doi: 10.1093/infdis/jit132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hresko RC, Hruz PW. 2011. HIV protease inhibitors act as competitive inhibitors of the cytoplasmic glucose binding site of GLUTs with differing affinities for GLUT1 and GLUT4. PLoS One 6:e25237. doi: 10.1371/journal.pone.0025237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murata H, Hruz PW, Mueckler M. 2000. The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J Biol Chem 275:20251–20254. doi: 10.1074/jbc.C000228200. [DOI] [PubMed] [Google Scholar]
- 13.Murata H, Hruz PW, Mueckler M. 2002. Indinavir inhibits the glucose transporter isoform Glut4 at physiologic concentrations. AIDS 16:859–863. doi: 10.1097/00002030-200204120-00005. [DOI] [PubMed] [Google Scholar]
- 14.Slavic K, Krishna S, Derbyshire ET, Staines HM. 2011. Plasmodial sugar transporters as anti-malarial drug targets and comparisons with other protozoa. Malar J 10:165. doi: 10.1186/1475-2875-10-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woodrow CJ, Penny JI, Krishna S. 1999. Intraerythrocytic Plasmodium falciparum expresses a high affinity facilitative hexose transporter. J Biol Chem 274:7272–7277. doi: 10.1074/jbc.274.11.7272. [DOI] [PubMed] [Google Scholar]
- 16.Kirk K. 2001. Membrane transport in the malaria-infected erythrocyte. Physiol Rev 81:495–537. [DOI] [PubMed] [Google Scholar]
- 17.Mehta M, Sonawat HM, Sharma S. 2006. Glycolysis in Plasmodium falciparum results in modulation of host enzyme activities. J Vector Borne Dis 43:95–103. [PubMed] [Google Scholar]
- 18.Blume M, Hliscs M, Rodriguez-Contreras D, Sanchez M, Landfear S, Lucius R, Matuschewski K, Gupta N. 2011. A constitutive pan-hexose permease for the Plasmodium life cycle and transgenic models for screening of antimalarial sugar analogs. FASEB J 25:1218–1229. doi: 10.1096/fj.10-173278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joet T, Eckstein-Ludwig U, Morin C, Krishna S. 2003. Validation of the hexose transporter of Plasmodium falciparum as a novel drug target. Proc Natl Acad Sci U S A 100:7476–7479. doi: 10.1073/pnas.1330865100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang B, Watts KM, Hodge D, Kemp LM, Hunstad DA, Hicks LM, Odom AR. 2011. A second target of the antimalarial and antibacterial agent fosmidomycin revealed by cellular metabolic profiling. Biochemistry 50:3570–3577. doi: 10.1021/bi200113y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 22.Corbett Y, Herrera L, Gonzalez J, Cubilla L, Capson TL, Coley PD, Kursar TA, Romero LI, Ortega-Barria E. 2004. A novel DNA-based microfluorimetric method to evaluate antimalarial drug activity. Am J Trop Med Hyg 70:119–124. [PubMed] [Google Scholar]
- 23.Chaudhary S, Pak JE, Gruswitz F, Sharma V, Stroud RM. 2012. Overexpressing human membrane proteins in stably transfected and clonal human embryonic kidney 293S cells. Nat Protoc 7:453–466. doi: 10.1038/nprot.2011.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aerni-Flessner L, Abi-Jaoude M, Koenig A, Payne M, Hruz PW. 2012. GLUT4, GLUT1, and GLUT8 are the dominant GLUT transcripts expressed in the murine left ventricle. Cardiovasc Diabetol 11:63. doi: 10.1186/1475-2840-11-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saliba KJ, Krishna S, Kirk K. 2004. Inhibition of hexose transport and abrogation of pH homeostasis in the intraerythrocytic malaria parasite by an O-3-hexose derivative. FEBS Lett 570:93–96. doi: 10.1016/j.febslet.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 26.Hresko RC, Kraft TE, Tzekov A, Wildman SA, Hruz PW. 2014. Isoform-selective inhibition of facilitative glucose transporters: elucidation of the molecular mechanism of HIV protease inhibitor binding. J Biol Chem 289:16100–16113. doi: 10.1074/jbc.M113.528430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. 2015. The I-TASSER Suite: protein structure and function prediction. Nat Methods 12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trott O, Olson AJ. 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergshoeff AS, Fraaij PL, Ndagijimana J, Verweel G, Hartwig NG, Niehues T, De Groot R, Burger DM. 2005. Increased dose of lopinavir/ritonavir compensates for efavirenz-induced drug-drug interaction in HIV-1-infected children. J Acquir Immune Defic Syndr 39:63–68. doi: 10.1097/01.qai.0000155203.89350.85. [DOI] [PubMed] [Google Scholar]
- 32.Rosso R, Di Biagio A, Dentone C, Gattinara GC, Martino AM, Viganò A, Merlo M, Giaquinto C, Rampon O, Bassetti M, Gatti G, Viscoli C. 2006. Lopinavir/ritonavir exposure in treatment-naive HIV-infected children following twice or once daily administration. J Antimicrob Chemother 57:1168–1171. doi: 10.1093/jac/dkl136. [DOI] [PubMed] [Google Scholar]
- 33.Best BM, Stek AM, Mirochnick M, Hu C, Li H, Burchett SK, Rossi SS, Smith E, Read JS, Capparelli EV. 2010. Lopinavir tablet pharmacokinetics with an increased dose during pregnancy. J Acquir Immune Defic Syndr 54:381–388. doi: 10.1097/QAI.0b013e3181d6c9ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy RL, Brun S, Hicks C, Eron JJ, Gulick R, King M, White A, Benson CC, Thompson M, Kessler HA, Hammer S, Bertz R, Hsu A, Japour A, Sun E. 2001. ABT-378/ritonavir plus stavudine and lamivudine for the treatment of antiretroviral-naive adults with HIV-1 infection: 48-week results. AIDS 15:F1–F9. doi: 10.1097/00002030-200101050-00002. [DOI] [PubMed] [Google Scholar]
- 35.Slavic K, Straschil U, Reininger L, Doerig C, Morin C, Tewari R, Krishna S. 2010. Life cycle studies of the hexose transporter of Plasmodium species and genetic validation of their essentiality. Mol Microbiol 75:1402–1413. doi: 10.1111/j.1365-2958.2010.07060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chandwani A, Shuter J. 2008. Lopinavir/ritonavir in the treatment of HIV-1 infection: a review. Ther Clin Risk Manag 4:1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grunfeld C, Kotler DP, Arnett DK, Falutz JM, Haffner SM, Hruz P, Masur H, Meigs JB, Mulligan K, Reiss P, Samaras K. 2008. Contribution of metabolic and anthropometric abnormalities to cardiovascular disease risk factors. Circulation 118:e20–e28. doi: 10.1161/CIRCULATIONAHA.107.189623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hruz PW, Murata H, Qiu H, Mueckler M. 2002. Indinavir induces acute and reversible peripheral insulin resistance in rats. Diabetes 51:937–942. doi: 10.2337/diabetes.51.4.937. [DOI] [PubMed] [Google Scholar]
- 39.Noor MA, Lo JC, Mulligan K, Schwarz JM, Halvorsen RA, Schambelan M, Grunfeld C. 2001. Metabolic effects of indinavir in healthy HIV-seronegative men. AIDS 15:F11–F18. doi: 10.1097/00002030-200105040-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noor MA, Flint OP, Maa J-F, Parker RA. 2006. Effects of atazanavir/ritonavir and lopinavir/ritonavir on glucose uptake and insulin sensitivity: demonstrable differences in vitro and clinically. AIDS 20:1813–1821. doi: 10.1097/01.aids.0000244200.11006.55. [DOI] [PubMed] [Google Scholar]
- 41.Ortiz D, Guiguemde WA, Johnson A, Elya C, Anderson J, Clark J, Connelly M, Yang L, Min J, Sato Y, Guy RK, Landfear SM. 2015. Identification of selective inhibitors of the Plasmodium falciparum hexose transporter PfHT by screening focused libraries of anti-malarial compounds. PLoS One 10:e0123598. doi: 10.1371/journal.pone.0123598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. 2014. Crystal structure of the human glucose transporter GLUT1. Nature 510:121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- 43.Slavic K, Delves MJ, Prudêncio M, Talman AM, Straschil U, Derbyshire ET, Xu Z, Sinden RE, Mota MM, Morin C, Tewari R, Krishna S, Staines HM. 2011. Use of a selective inhibitor to define the chemotherapeutic potential of the plasmodial hexose transporter in different stages of the parasite's life cycle. Antimicrob Agents Chemother 55:2824–2830. doi: 10.1128/AAC.01739-10. [DOI] [PMC free article] [PubMed] [Google Scholar]