

Abstract
Candida parapsilosis is the second most prevalent fungal agent causing bloodstream infections. Nevertheless, there is little information about the molecular mechanisms underlying azole resistance in this species. Mutations (G1747A, A2619C, and A3191C) in the MRR1 transcription factor gene were identified in fluconazole- and voriconazole-resistant strains. Independent expression of MRR1 genes harboring these mutations showed that G1747A (G583R) and A2619C (K873N) are gain-of-function mutations responsible for azole resistance, the first described in C. parapsilosis.
TEXT
Candida parapsilosis is a human fungal pathogen causing infections that range from mucocutaneous to systemic. Interestingly, C. parapsilosis infections display an unusual prevalence in terms of age and geographical distribution. Invasive fungal infections caused by C. parapsilosis afflict mainly young patients (neonates and children), surpassing in number those caused by Candida albicans. Geographically, C. parapsilosis is the second most common agent of fungal infection in Mediterranean, South American, and Asian countries (1–3). Studies regarding the drug susceptibility profile of C. parapsilosis isolates have been carried out worldwide, and apart from their reduced susceptibility to echinocandins (4), their level of resistance to azoles has not been alarming so far (5–7). However, with the extensive use of azoles as therapeutic and prophylactic drugs prescribed in C. parapsilosis infections, it is expected that azole resistance will emerge more frequently. This concern was recently the focus in a study of C. parapsilosis fluconazole surveillance carried out in clinical isolates from several hospitals in the United States (6). Fluconazole susceptibility was assessed in 706 C. parapsilosis isolates, collected in 80 hospitals. Interestingly, fluconazole resistance rates vary from 70%, in isolates found in Atlanta hospitals, to 0%, in isolates coming from Portland, OR. The study of the molecular mechanisms underlying fluconazole resistance revealed the first mutations described in clinical isolates, either in the MRR1 transcription factor or in the azole target, the ERG11 gene (6).
We have demonstrated that in vitro exposure to azoles, namely, fluconazole, voriconazole, and posaconazole, triggers a stable development of azole resistance in C. parapsilosis (8). Interestingly, exposure to posaconazole induced cross-resistance to other azoles, while fluconazole and voriconazole induced only cross-resistance to each other (9). Azoles act by inhibiting an enzyme involved with the ergosterol biosynthesis pathway, the enzyme 14-α-demethylase, encoded by ERG11. The lack of ergosterol production and/or the production of toxic metabolites as a result of Erg11p inactivation seriously disrupts fungal growth. Fungal cells can overcome these effects by overexpressing ERG11, reducing azole affinity to its target molecule through mutation, or expelling the azole from the intracellular to the extracellular environment (10). The efficiency of this last strategy requires overexpression of efflux pumps, encoded by the ABC transporter superfamily (CDR1, CDR2) or the major facilitator superfamily (MDR1) (11).
The fact that fluconazole and voriconazole induce cross-resistance in C. parapsilosis suggests that the underlying mechanisms are similar. This is supported by comparison of the transcriptional profiles of fluconazole- and voriconazole-resistant strains, which showed that two genes associated with acquisition of resistance were overexpressed, MDR1 (multidrug resistance) and its regulator, the transcription factor MRR1 (multiresistance regulator) (9). Two different missense mutations (G1747A and A2619C) were identified in the MRR1 genes from fluconazole- and voriconazole-resistant strains, leading to alterations in the Mrr1p polypeptide chain from glycine to arginine (G583R) and from lysine to asparagine (K873N), respectively (9). Subsequent analysis of MRR1 from the voriconazole-resistant isolate detected an additional mutation, A3191C, causing a change from glutamine to a proline (Q1064P) in Mrr1p. Other MRR1 mutations were recently described in fluconazole-resistant clinical isolates; however, their impact in fluconazole resistance was not conclusive, since the same polymorphisms were also found in susceptible clinical isolates, with the one exception being the G2337T mutation (6).
Extensive characterization in C. albicans and C. glabrata has shown that gain-of-function mutations in transcription factors, like MRR1, TAC1, UPC2, or PDR1, are involved in the development of azole resistance in these species (12–19). These mutations activate the transcription factors, which constitutively upregulate their targets: efflux pumps or ergosterol biosynthesis enzymes.
To determine if G583R, K873N, and Q1064P in MRR1 are gain-of-function mutations associated with fluconazole and voriconazole resistance in C. parapsilosis sensu stricto, a set of constructs was generated using the SAT1 flipper cassette (Fig. 1A) (20). Five cassettes were constructed, representing MRR1wt (wild-type gene) (GenBank accession number KT160017), MRR1FLC (with the G583R mutation) (GenBank accession number KT160018), MRR1VRC (with K873N and Q1064P mutations) (GenBank accession number KT160019), MRR1K873N (with the K873N mutation only), and MRR1Q1064P (with the Q1064P mutation only). These were introduced into a C. parapsilosis strain with the two copies of the endogenous mrr1Δ gene deleted (21). After electroporation (20), transformants were selected in yeast extract-peptone-dextrose (YPD) agar plates containing 200 μg/ml nourseothricin. Integration of the cassettes at the native MRR1 locus was confirmed by PCR (Fig. 1B). All primer sequences are listed in Table 1. After recycling of the SAT1 flipper (Fig. 1C), the azole susceptibility profile and expression of MRR1 and MDR1 in each strain was determined.
FIG 1.

Genomic integration of the MRR1 transcription factor gene variants in the C. parapsilosis homozygous disruptant mrr1Δ strain. (A) MRR1 complementation cassettes were constructed using the SAT1 flipper cassette (20) and primers MRR1_F1, MRR1_R1, MRR1_F2, and MRR1_R2 (Table 1). MRR1wt, MRRFLC (containing mutation G1747A), MRRVRC (containing mutations A2619C and A3191C), MRR1K873N (containing mutation A2619C), and MRR1Q1064P (containing mutation A3191C) genes were integrated in the C. parapsilosis mrr1Δ double mutant, generating the JB8, JB14, JB4, JB12, and JB6 strains, respectively. Each mutation in MRRVRC was independently silenced by site-directed mutagenesis (Table 1) to originate MRR1K873N and MRR1Q1064P. (B) Complementation cassettes were integrated at the MRR1 native genomic locus and confirmed by PCR using the following pairs of primers: (i) MRR1_F3 and FLP_R, which amplified a 3-kb fragment (lanes 2 to 6; absent in lane 7), and (ii) MRR1_F4 and MRR1_R3, resulting in a 1.3-kb PCR product (lanes 9 to 13). Lanes 14 and 15 show a 1.3-kb fragment present in CLIB214 and absent in the mrr1Δ strain, respectively. Lanes 1 and 8 represent the molecular size marker (NZYDNA Ladder III). (C) Confirmation of the successful recycling of the MRR1 complementation cassettes by PCR using primers MRR1_F3 and MRR1_R4, resulting in the amplification of a 2.1-kb fragment (lanes 2 to 6), CLIB214 was used as control amplifying a 1.7-kb fragment (lane 7). The discrepancy between size fragments amplified in the transformants and the one amplified in CLIB214 is due to the presence of two promoters regions upstream of the MRR1 gene in the transformants compared with only one in the CLIB214 strain. Lane 1 represents the molecular size marker (NZYDNA Ladder III).
TABLE 1.
Primers used in this study
| Primer name | Primer sequence (5′ to 3′) |
|---|---|
| Construction of cassettes | |
| MRR1_F1 | GGGGGTACCCTACTGATATGCCTGACGCCAC |
| MRR1_R1 | GGGGGGCCCTCTCTCTTATTGAAAACAAGAAAGC |
| MRR1_F2 | TCCCCGCGGCTACTGATATGCCTGAGGCCAC |
| MRR1_R2 | GGGGAGCTCTCTCTCTTATTGAAAACAAGAAAGC |
| Site-directed mutagenesis | |
| MRR1VSDM_F1 | CGAGGTATTTTTACGCATGGAAATTGACAAAGAGTCATTCTTATT |
| MRR1VSDM_R1 | AATAAGAATGACTCTTTGTCAATTTCCATGCGTAAAAATACCTCG |
| MRR1VSDM_F2 | GCGGCCCCAGCAACAACAGCCTATAGGG |
| MRR1VSDM_R2 | CCCTATAGGCTGTTGTTGCTGGGGCCGC |
| PCR confirmation | |
| MRR1_F3 | GAAAACAAGTAATCAAAACACGGGG |
| FLP_R | TTTATGATGGAATGAATGGGATG |
| MRR1_F4 | CGGCATCTCGCAGCAACAA |
| MRR1_R3 | GTCTGTAAAGGGGGGGGTTGGA |
| MRR1_R4 | ACTTGAACGAAATGGAGACC |
| qRT-PCR | |
| TUB4_F | TGTATTCCACAATGATGCCT |
| TUB4_R | TGCCTTGAAACGAAGTAGC |
| MRR1_F | ACAATGGTCTGAGCAATGAA |
| MRR1_R | GGCAATACTGGTGATGGAA |
| MDR1_F | TTCGTGATAGTTTTGGTGGTAG |
| MDR1_R | TGAACCTGGAGTGAATCTTGT |
Azole susceptibility profiles of the strains expressing MRR1wt, MRR1FLC, MRR1VRC, MRR1K873N, MRR1Q1064P, the mrr1Δ strain, and the parent of the mrr1Δ mutant (C. parapsilosis CLIB214) were characterized according to CLSI protocols (M27-S4 and M27-A3) (22, 23). The expression of MRR1FLC (strain JB14), MRR1VRC (strain JB4), and MRR1K873N (strain JB12) conferred resistance to fluconazole and voriconazole (Table 2). In contrast, strains expressing MRR1Q1064P (JB6) or MRR1wt (JB8) and the parent CLIB214 and mrr1Δ strains are susceptible to the same antifungal drugs (Table 2). This demonstrates that missense mutations G1747A (G583R) and A2619C (K873N) in MRR1 determine resistance to fluconazole and voriconazole in C. parapsilosis, whereas Q1064P has no effect. As expected, no alteration of susceptibility phenotype was found for posaconazole (Table 2).
TABLE 2.
MIC values and susceptibility phenotypes of C. parapsilosis strains
| Strain | MIC (μg/ml)/phenotypea for: |
||
|---|---|---|---|
| Fluconazole | Voriconazole | Posaconazole | |
| CLIB214 | 1/S | 0.03/S | 0.06/WT |
| mrr1Δ mutant | 1/S | 0.03/S | 0.06/WT |
| MRR1wt (JB8) | 1/S | 0.03/S | 0.06/WT |
| MRR1FLC (JB14) | 64/R | 2/R | 0.12/WT |
| MRR1VRC (JB4) | 64/R | 2/R | 0.12/WT |
| MRR1K873N (JB12) | 64/R | 2/R | 0.12/WT |
| MRR1Q1064P (JB6) | 1/S | 0.03/S | 0.06/WT |
S, susceptible; R, resistant; WT, wild type.
To elucidate the mechanism of action, expression of MRR1 and MDR1 was determined by quantitative real-time PCR (qRT-PCR) (Table 1). The expression levels of the MRR1FLC, MRR1VRC, and MRR1K873N alleles were 3- to 4-fold higher than those of the MRR1Q1064P and MRR1wt alleles (Fig. 2). The effect on MDR1 expression is more dramatic; expression was up to 70 times higher in strains expressing MRR1FLC (strain JB14), MRR1VRC (JB4), and MRR1K873N (JB12) than in CLIB214 or strains expressing MRR1Q1064P (JB6) or MRR1wt alleles (JB8) (Fig. 2). Therefore, only G1747A and A2619C, but not A3191C, confer hyperactivity to the MRR1 transcription factor, which consequently upregulates the MDR1 multidrug transporter. Thus, G583R and K873N mutations are the first gain-of-function mutations described in the C. parapsilosis MRR1 transcription factor.
FIG 2.
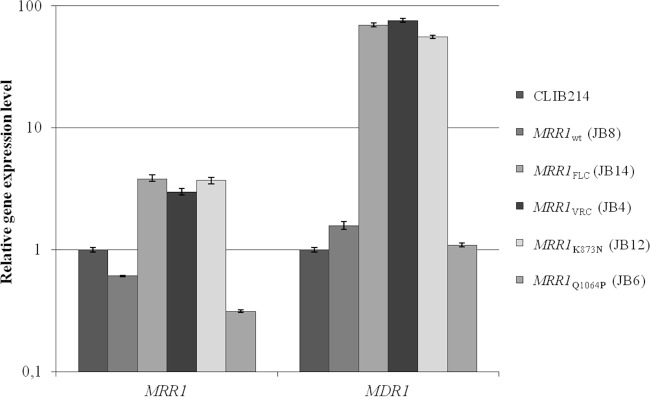
Mutations G583R and K873N induce overexpression of MRR1 and MDR1 genes. Quantitative real-time PCR (qRT-PCR) analysis of the MRR1 and MDR1 gene expression in the JB8, JB14, JB4, JB12, and JB6 strains, using the CLIB214 strain as a control. Due to the large range in gene expression levels, the y axis scale is logarithmic. The expression values displayed in the graph represent the variation of MRR1 and MDR1 gene expression relative to the CLIB214 strain and are expressed as the mean of five independent experiments, with the respective standard deviation. Amplification efficiency and the amount of TUB4 (endogenous reference gene) were used to normalize each mean value.
In a large surveillance study of fluconazole-resistant clinical isolates of C. parapsilosis in the United States, the most common underlying molecular mechanisms were associated with polymorphisms in ERG11 and MRR1 (6). According to these authors, polymorphisms in MRR1 are common, and only some are associated with overexpression of MDR1. They suggest that there is a hot spot for gain-of-function mutations in MRR1, in the region coding from amino acids 852 to 875, which is equivalent to a similar region in MRR1 in C. albicans and C. dubliniensis (6, 14). They identified one clinical isolate with a polymorphism in this region, corresponding to L779F, which has 73-fold upregulation of MDR1 (6). Our results corroborate this finding since the K873N mutation is located in this so-called gain-of-function hot spot region but also show that mutations located in other regions (G583R) are equally important for azole resistance. Strains expressing G583R and K873N MRR1 mutants upregulate MDR1 expression to a level similar to that of the L779F isolate (around 70-fold increase compared to that of the wild type) (6), conferring resistance to fluconazole and voriconazole (MIC, ≤64 μg/ml). Grossman et al. identified four other MRR1 putative gain-of-function polymorphisms present in fluconazole-resistant clinical isolates (6). However, they did not conclusively show that polymorphisms in MRR1 cause increased expression of MDR1, as they also identified MRR1 polymorphisms in susceptible isolates.
Other mechanisms of azole resistance, such as ERG11 mutations, should also be considered. For example, the A395T (Y132F) polymorphism was found in more than 50% of 30 fluconazole-resistant clinical isolates of C. parasilosis (6).
In conclusion, we described the first gain-of-function mutations, G583R and K873N, in the C. parapsilosis MRR1 transcription factor involved in fluconazole and voriconazole resistance. Furthermore, these data confirm our previous suggestion (9) that MDR1 is a key player in fluconazole and voriconazole resistance in C. parapsilosis.
Nucleotide sequence accession numbers.
Sequences have been deposited in GenBank under the accession numbers KT160017, KT160018, and KT160019.
ACKNOWLEDGMENTS
I.M.M., J.B., and this work were supported by FEDER Funds (Programa Operacional Factores de Competitividade-COMPETE) and by National Funds within FCT (Fundação para a Ciência e Tecnologia) in the scope of the project PTDC/DTP-EPI/1660/2012 “Surveillance of Candida parapsilosis antifungal resistance.” R.M.S. is supported by the Institute for Biomedicine (no. UID/BIM/04501/2013) and the project Neuropath (no. CENTRO-07-ST24-FEDER-002034), cofunded by the QREN “Mais Centro” program and the European Union.
REFERENCES
- 1.Guinea J. 2014. Global trends in the distribution of Candida species causing candidemia. Clin Microbiol Infect 20(Suppl 6):S5–S10. doi: 10.1111/1469-0691.12539. [DOI] [PubMed] [Google Scholar]
- 2.Montagna MT, Lovero G, Borghi E, Amato G, Andreoni S, Campion L, Lo Cascio G, Lombardi G, Luzzaro F, Manso E, Mussap M, Pecile P, Perin S, Tangorra E, Tronci M, Iatta R, Morace G. 2014. Candidemia in intensive care unit: a nationwide prospective observational survey (GISIA-3 study) and review of the European literature from 2000 through 2013. Eur Rev Med Pharmacol Sci 18:661–674. [PubMed] [Google Scholar]
- 3.Wu Z, Liu Y, Feng X, Liu Y, Wang S, Zhu X, Chen Q, Pan S. 2014. Candidemia: incidence rates, type of species, and risk factors at a tertiary care academic hospital in China. Int J Infect Dis 22:4–8. doi: 10.1016/j.ijid.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Effron G, Katiyar SK, Park S, Edlind TD, Perlin DS. 2008. A naturally occurring proline-to-alanine amino acid change in Fks1p in Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis accounts for reduced echinocandin susceptibility. Antimicrob Agents Chemother 52:2305–2312. doi: 10.1128/AAC.00262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klingspor L, Tortorano AM, Peman J, Willinger B, Hamal P, Sendid B, Velegraki A, Kibbler C, Meis JF, Sabino R, Ruhnke M, Arikan-Akdagli S, Salonen J, Doczi I. 2015. Invasive Candida infections in surgical patients in intensive care units: a prospective, multicentre survey initiated by the European Confederation of Medical Mycology (ECMM) (2006-2008). Clin Microbiol Infect 21:87.e1–87.e10. [DOI] [PubMed] [Google Scholar]
- 6.Grossman NT, Pham CD, Cleveland AA, Lockhart SR. 2015. Molecular mechanisms of fluconazole resistance in Candida parapsilosis isolates from a U.S. surveillance system. Antimicrob Agents Chemother 59:1030–1037. doi: 10.1128/AAC.04613-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silva AP, Miranda IM, Lisboa C, Pina-Vaz C, Rodrigues AG. 2009. Prevalence, distribution, and antifungal susceptibility profiles of Candida parapsilosis, C. orthopsilosis, and C. metapsilosis in a tertiary care hospital. J Clin Microbiol 47:2392–2397. doi: 10.1128/JCM.02379-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinto e Silva AT, Costa-de-Oliveira S, Silva-Dias A, Pina-Vaz C, Rodrigues AG. 2009. Dynamics of in vitro acquisition of resistance by Candida parapsilosis to different azoles. FEMS Yeast Res 9:626–633. doi: 10.1111/j.1567-1364.2009.00508.x. [DOI] [PubMed] [Google Scholar]
- 9.Silva AP, Miranda IM, Guida A, Synnott J, Rocha R, Silva R, Amorim A, Pina-Vaz C, Butler G, Rodrigues AG. 2011. Transcriptional profiling of azole-resistant Candida parapsilosis strains. Antimicrob Agents Chemother 55:3546–3556. doi: 10.1128/AAC.01127-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuenca-Estrella M. 2014. Antifungal drug resistance mechanisms in pathogenic fungi: from bench to bedside. Clin Microbiol Infect 20(Suppl 6):S54–S59. doi: 10.1111/1469-0691.12495. [DOI] [PubMed] [Google Scholar]
- 11.Pfaller MA. 2012. Antifungal drug resistance: mechanisms, epidemiology, and consequences for treatment. Am J Med 125:S3–13. doi: 10.1016/j.amjmed.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Coste A, Turner V, Ischer F, Morschhauser J, Forche A, Selmecki A, Berman J, Bille J, Sanglard D. 2006. A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics 172:2139–2156. doi: 10.1534/genetics.105.054767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morio F, Pagniez F, Besse M, Gay-andrieu F, Miegeville M, Le Pape P. 2013. Deciphering azole resistance mechanisms with a focus on transcription factor-encoding genes TAC1, MRR1, and UPC2 in a set of fluconazole-resistant clinical isolates of Candida albicans. Int J Antimicrob Agents 42:410–415. doi: 10.1016/j.ijantimicag.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Dunkel N, Blass J, Rogers PD, Morschhauser J. 2008. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol Microbiol 69:827–840. doi: 10.1111/j.1365-2958.2008.06309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schubert S, Rogers PD, Morschhauser J. 2008. Gain-of-function mutations in the transcription factor MRR1 are responsible for overexpression of the MDR1 efflux pump in fluconazole-resistant Candida dubliniensis strains. Antimicrob Agents Chemother 52:4274–4280. doi: 10.1128/AAC.00740-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flowers SA, Barker KS, Berkow EL, Toner G, Chadwick SG, Gygax SE, Morschhauser J, Rogers PD. 2012. Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot Cell 11:1289–1299. doi: 10.1128/EC.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacPherson S, Akache B, Weber S, De Deken X, Raymond M, Turcotte B. 2005. Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob Agents Chemother 49:1745–1752. doi: 10.1128/AAC.49.5.1745-1752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunkel N, Liu TT, Barker KS, Homayouni R, Morschhauser J, Rogers PD. 2008. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot Cell 7:1180–1190. doi: 10.1128/EC.00103-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vale-Silva L, Ischer F, Leibundgut-Landmann S, Sanglard D. 2013. Gain-of-function mutations in PDR1, a regulator of antifungal drug resistance in Candida glabrata, control adherence to host cells. Infect Immun 81:1709–1720. doi: 10.1128/IAI.00074-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding C, Butler G. 2007. Development of a gene knockout system in Candida parapsilosis reveals a conserved role for BCR1 in biofilm formation. Eukaryot Cell 6:1310–1319. doi: 10.1128/EC.00136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holland LM, Schroder MS, Turner SA, Taff H, Andes D, Grozer Z, Gacser A, Ames L, Haynes K, Higgins DG, Butler G. 2014. Comparative phenotypic analysis of the major fungal pathogens Candida parapsilosis and Candida albicans. PLoS Pathog 10:e1004365. doi: 10.1371/journal.ppat.1004365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clinical and Laboratory Standards Institute. 2012. Reference method for broth dilution antifungal susceptibility testing of yeasts; 4th informational supplement. CLSI M27-S4. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 23.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard— 3rd ed CLSI document M27-A3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]