

Abstract
Analysis of data pooled from multiple phase 2 (SILEN-C1 to 3) and phase 3 studies (STARTVerso1 to 4) of the hepatitis C virus (HCV) nonstructural protein 3/4A (NS3/4A) protease inhibitor faldaprevir plus pegylated interferon alpha/ribavirin (PR) provides a comprehensive evaluation of baseline and treatment-emergent NS3/4A amino acid variants among HCV genotype-1 (GT-1)-infected patients. Pooled analyses of GT-1a and GT-1b NS3 population-based pretreatment sequences (n = 3,124) showed that faldaprevir resistance-associated variants (RAVs) at NS3 R155 and D168 were rare (<1%). No single, noncanonical NS3 protease or NS4A cofactor baseline polymorphism was associated with a reduced sustained virologic response (SVR) to faldaprevir plus PR, including Q80K. The GT-1b NS3 helicase polymorphism T344I was associated with reduced SVR to faldaprevir plus PR (P < 0.0001) but was not faldaprevir specific, as reduced SVR was also observed with placebo plus PR. Among patients who did not achieve SVR and had available NS3 population sequences (n = 507 GT-1a; n = 349 GT-1b), 94% of GT-1a and 83% of GT-1b encoded faldaprevir treatment-emergent RAVs. The predominant GT-1a RAV was R155K (88%), whereas GT-1b encoded D168 substitutions (78%) in which D168V was predominant (67%). The novel GT-1b NS3 S61L substitution emerged in 7% of virologic failures as a covariant with D168V, most often among the faldaprevir breakthroughs; S61L in combination with D168V had a minimal impact on faldaprevir susceptibility compared with that for D168V alone (1.5-fold difference in vitro). The median time to loss of D168 RAVs among GT-1b-infected patients who did not have a sustained virologic response at 12 weeks posttreatment (non-SVR12) after virologic failure was 5 months, which was shorter than the 14 months for R155 RAVs among GT-1a-infected non-SVR12 patients, suggesting that D168V is less fit than R155K in the absence of faldaprevir selective pressure.
INTRODUCTION
The treatment of chronic hepatitis C infection has changed considerably over the past 10 years, shifting from intravenously administered pegylated interferon plus oral ribavirin (PR) to combinations of direct-acting antivirals (DAAs) with PR and, more recently, all-oral, interferon-free combinations of DAAs with or without ribavirin (RBV) (1, 2). The hepatitis C virus (HCV) nonstructural protein 3/4A (NS3/4A) protease inhibitors (PIs) telaprevir and boceprevir represented the first significant advance in the treatment of HCV (3–6); however, these drugs are associated with limitations, including potentially serious side effects, inconvenient dosing, and extensive drug-drug interactions (7, 8). More recently, the PIs simeprevir and faldaprevir have demonstrated better efficacy and tolerability in combination with PR than telaprevir and boceprevir (9–13).
Faldaprevir is a reversible, noncovalently binding, linear HCV NS3/4A PI with a pharmacokinetic profile conducive to once-daily (QD) dosing (14). Faldaprevir has been investigated in phase 1b, phase 2, and phase 3 clinical studies for the treatment of patients chronically infected with HCV genotype-1 (GT-1) (10–14). In a phase 2 study in treatment-naive patients infected with HCV GT-1, sustained virologic response (SVR) rates of up to 84% were achieved with faldaprevir plus PR (10). In a phase 3 study in treatment-naive patients, a large proportion (88%) of patients who received faldaprevir plus PR were able to stop all treatment at week 24; 88% of these achieved SVR at 12 weeks posttreatment (SVR12) (13).
The HCV RNA polymerase has low fidelity, and this, coupled with a high viral replication rate, results in the generation of highly variable viral populations within HCV-infected individuals (15). Among the viral quasispecies that make up these populations, there are likely to be variants with some degree of reduced susceptibility to antiviral agents. Under the selective pressure of antiviral treatment, these variants can rapidly become the majority population and lead to virologic failure (16, 17). HCV variants with reduced susceptibility to antiviral agents typically also have a lower replicative capacity, or “fitness,” than wild-type virus and are rapidly lost once antiviral therapy is stopped and the selective pressure is thus removed (16, 18–20). For any new antiviral agent, it is important to identify which mutations in the viral genome may emerge in the case of virologic failure and to understand the dynamics of their selection during treatment and potential deselection once treatment is stopped. This knowledge can help to select the optimal treatment combinations to avoid the emergence of resistance during treatment and to effectively treat patients who have failed to respond to first-line therapy.
The comprehensive analysis of resistance data provided here from phase 2 and phase 3 studies of faldaprevir in combination with PR includes an evaluation of the impact of baseline NS3/4A polymorphisms on treatment response, identification, and characterization of treatment-emergent resistance-associated variants (RAVs) among virologic failures and assessment of the persistence of RAVs during posttreatment follow-up.
MATERIALS AND METHODS
Study design and patient virology samples.
The analyses were performed on plasma samples derived from patients infected with HCV GT-1 and treated with faldaprevir plus PR or placebo plus PR in three phase 2 clinical studies and four phase 3 clinical studies (see Table S1 in the supplemental material), all of which have been described in detail elsewhere (10–13, 21–24).
In the SILEN-C1 (ClinicalTrials registration no. NCT00774397) phase 2 study, 429 treatment-naive patients received faldaprevir (120 mg or 240 mg QD) and PR or placebo plus PR for 24 weeks (10). In the SILEN-C3 (NCT00984620) phase 2 study, 159 treatment-naive patients received faldaprevir (120 mg QD) and PR for 12 or 24 weeks (21). In the SILEN-C2 (NCT00774397) phase 2 study, 288 treatment-experienced patients received faldaprevir (240 mg QD or twice daily [BID]) and PR for 24 weeks (11). Phase 3 studies with resistance data included STARTVerso1 (NCT01343888), STARTVerso2 (NCT01297270), STARTVerso3 (NCT01358864), and STARTVerso4 (NCT01399619). These were multicenter, randomized, double-blind, placebo-controlled, parallel-group studies. STARTVerso1 (652 patients) and STARTVerso2 (657 patients) included treatment-naive patients who received 120 mg faldaprevir QD (12 or 24 weeks) or 240 mg faldaprevir QD (12 weeks), all with PR (24 to 48 weeks), or placebo plus PR for 24 weeks followed by PR alone up to 48 weeks (13, 24). STARTVerso3 enrolled 677 treatment-experienced patients (prior PR therapy null responders, partial responders, or relapsers); treatment was with 240 mg faldaprevir QD for 12 or 24 weeks with PR (24 to 48 weeks) or placebo plus PR (partial responders or relapsers) for 24 weeks followed by PR alone up to 48 weeks (22). In STARTVerso4, 308 patients coinfected with HIV-1 and HCV GT-1 (treatment-naive or prior PR relapsers) received 120 mg or 240 mg faldaprevir and PR (12). The total treatment duration of PR was 24 or 48 weeks in all of these phase 2 and phase 3 studies.
The documentation for each study, including protocol amendments, was approved by the appropriate institutional review board, and the studies were carried out in accordance with the Declaration of Helsinki and the International Conference on Harmonisation guidelines. All patients provided written informed consent before enrollment.
Efficacy and virologic responses.
HCV RNA in plasma samples was quantified using the Cobas TaqMan HCV/high pure system (HPS) test v2.0 (Roche). The primary efficacy endpoint of phase 2 studies was SVR24, defined as HCV RNA of <25 IU/ml 24 weeks after the last planned dose of study drug. The primary efficacy endpoint of phase 3 studies was SVR12, defined as HCV RNA of <25 IU/ml 12 weeks after the last planned dose of study drug. Patients who did not achieve SVR were classified by the type of treatment failure and categorized as faldaprevir breakthrough, PR breakthrough, relapse, or other reasons. Faldaprevir breakthrough was defined as virologic rebound (≥1-log10 increase in HCV RNA from the nadir) during faldaprevir treatment, while PR breakthrough was defined as virologic rebound during PR treatment alone after completion of faldaprevir treatment. Relapse occurred when HCV RNA was undetected at the end of treatment, and virologic rebound occurred after completion of planned treatment with all study medications. Other reasons for not achieving SVR included a loss to follow-up or premature discontinuation (in phase 2 and phase 3 studies), an SVR viral load assessment not at <25 IU/ml (phase 2), a null response (phase 3: failure to achieve a 2-log10 reduction in HCV RNA from baseline by week 12), a partial response (phase 3: completed 24 weeks of treatment without achieving HCV RNA of <25 IU/ml), or other reasons not falling into one of the previous categories.
NS3/4A population sequencing.
Population-based sequencing was performed on all baseline virology samples. Postbaseline sequencing was performed on the first virologic rebound sample with HCV RNA of ≥1,000 IU/ml, which was the lower limit for the population sequencing assessment, or on samples in which the HCV RNA plateaued above the lower limit of quantification. For patients with a first viral load rebound sample or virologic plateau sample with detectable RAVs, subsequent plasma samples during the posttreatment follow-up period were sequenced to assess the persistence of resistance mutations during the outgrowth of wild-type virus.
NS3/4A population sequencing for phase 2 studies was performed by Boehringer Ingelheim (Canada) Ltd./Ltée, R&D (Laval, QC, Canada) as previously described (23). NS3/4A population sequencing for phase 3 studies was performed by Janssen Diagnostics (Beerse, Belgium) using subtype-specific amplification and sequencing primers; if the full NS3/4A sequence could not be obtained, sequencing of the NS3 protease (amino acids 1 to 181) was attempted. The NS3/4A nucleotide and amino acid changes were identified by comparison with GT-1 subtype-specific reference sequences (GenBank accession no.: AF009606 for GT-1a and AJ238799 for GT-1b). For subtype assignment in phase 2 studies, full-length NS3/4A population sequences were aligned with the subtype 1a and subtype 1b references in the AlignX module of Vector NTI Advance 10 software (Invitrogen), and the HCV subtype was assigned using the guide tree of the alignment. For phase 3 studies, NS3/4A (2,055 nucleotides) or NS3 (543 nucleotides) population sequences were assessed using the Oxford HCV Subtyping Tool version 2.0 (25, 26). The subtype provided by the tool was compared with those obtained with Versant HCV Genotype 2.0 assay (LiPA) and Abbott RealTime HCV Genotyping II assay reflex testing performed at the central laboratory. If results were discordant, the sequence alignment was performed as described above for the sequences processed in phase 2 studies. The rare non-GT-1a/-1b patients identified are not described here and were not included in this analysis.
NS3 protease phenotyping.
In vitro transient HCV RNA replication assays with a GT-1b bicistronic HCV subgenomic replicon phenotyping vector generated by Boehringer Ingelheim (Canada) Ltd./Ltée, R&D were used to evaluate the impact of NS3 protease mutations on drug susceptibility. Assessment of NS3 proteases in laboratory strains in vitro may reflect the reduced susceptibility of DAA treatment-emergent HCV protease mutants in patients, relative to their pretreatment virus. The phenotyping construct and drug susceptibility assay were performed as previously described (16) with the modifications below. The phenotyping vector with wild-type GT-1b NS3 (control) and the GT-1b NS3 D168V site-directed mutant were provided by Boehringer Ingelheim. DDL Diagnostic Laboratory (Rijswijk, The Netherlands) generated the GT-1b NS3 S61L and GT-1b dual NS3 S61L plus D168V site-directed mutants within the same phenotyping vector and performed in vitro drug susceptibility assays with all the constructs using a panel of HCV NS3/4A protease inhibitors. In vitro transcribed HCV RNAs were generated from linearized DNA plasmids (T7 RiboMAX kit; Promega, WI, USA) and electroporated into Huh-7.5 cells using 4-mm cuvettes and a Gene Pulser (Bio-Rad). Cells were seeded at a density of 3,100 cells per well of a 96-well plate. Luciferase activity (Luciferase 1000 assay system; Promega) was measured 96 h posttransfection as a marker for HCV RNA replication and normalized to a 4-hour measurement. Serial dilutions of inhibitors were added to the cells 24 h postelectroporation. PIs for use in these assays were synthesized by Boehringer Ingelheim (Canada) Ltd., R&D. Inhibitor stocks were dissolved in 100% dimethyl sulfoxide (DMSO), and final dilutions on cells contained 0.5% DMSO, 10% fetal bovine serum, and 1% nonessential amino acids in Dulbecco's modified Eagle's medium (Invitrogen). The concentration giving 50% inhibition of HCV RNA replication (EC50) was determined based on a four-parameter logistic nonlinear regression model (Hill equation).
Analyses.
Baseline polymorphisms were identified relative to HCV subtype-specific reference sequences. Comparisons of the percentages of patients who achieved a virologic response with and without a specific baseline amino acid variant, an amino acid mixture, or “any” variant other than wild-type reference at a particular amino acid position were carried out using a 2-sided Fisher's exact test. For many amino acid positions, there were small numbers of patients with baseline amino acid substitutions detected, which reduced the power of the 2-sided Fisher's exact test.
RAVs that emerged during treatment were characterized using pooled data from all faldaprevir treatment groups (combining results for 120-mg and 240-mg doses). Data from patients who did not achieve SVR were pooled from phase 2 (non-SVR24 patients) and phase 3 (non-SVR12 patients) studies. Amino acid changes were identified relative to each patient's respective baseline NS3/4A sequence. Non-SVR12 patients with postbaseline sequences but missing matching baseline data were excluded from analyses of treatment-emergent amino acid substitutions.
The long-term persistence and median time to loss of faldaprevir RAVs among a pooled population of all non-SVR12 patients were estimated using the Kaplan-Meier method. The detection of RAVs (any variant) at individual amino acid sites (NS3 R155 or D168) was evaluated separately, and wild type was defined as loss of RAVs at the single amino acid site of interest (e.g., NS3 S61 was not assessed in the outgrowth of wild type at NS3 D168). The time of origin was the date of virologic failure. If a variant was detected after the first date of virologic failure, the variants were imputed back to the failure date. If two consecutive visits had detectable RAVs, the RAVs were imputed for the entire time interval. An “event” was flagged at visits when RAVs were no longer detected due to outgrowth of the wild type. If a RAV was detected at the last visit and no subsequent visits with sequence data were available, then the patient was censored, and the RAV was not imputed or carried forward.
Nucleotide sequence accession numbers.
The baseline sequences from phase 2 and phase 3 study samples were submitted to GenBank under accession numbers KT232441 to KT235564 (see Table S2 in the supplemental material).
RESULTS
HCV-infected patients and virology sequence data.
The numbers of patients with HCV NS3 protease (amino acids 1 to 181) and NS3 helicase/NS4A (NS3 amino acids 182 to 631 + NS4A amino acids 1 to 54) sequences are summarized in Tables S3 and S4 in the supplemental material. Among 1,568 patients with HCV GT-1a infection, baseline NS3 protease sequences from virologic samples were available for 1,549 (99%) patients, and baseline NS3 helicase/NS4A sequences were available for 1,203 (77%). Of the 1,583 patients with HCV GT-1b infection, baseline NS3 protease sequences were available for 1,575 (99%) patients, and baseline NS3 helicase/NS4A sequences were available for 1,415 (89%). Among HCV GT-1a-infected patients who did not achieve SVR, matched baseline and postbaseline NS3 protease sequence(s) were available for 507 patients, and NS3 helicase/NS4A sequences were available for 376 patients. For HCV GT-1b-infected patients who did not achieve SVR, matched baseline and postbaseline NS3 protease sequences were available for 349 patients, and NS3 helicase/NS4A sequences were available for 307 patients.
Frequency of baseline polymorphisms.
The frequencies of baseline polymorphisms detected at NS3 amino acid positions 61, 80, 155, 156, 168, and 344 are shown in Table 1. NS3 Q80K was a common polymorphism among GT-1a baseline sequences, detected (alone or in a mixture with other variants) in 28.8% of samples. Wild-type amino acid at position 61 was less conserved among GT-1b sequences (86% S61) than among GT-1a sequences (93.5% T61), and the T/S61L substitution was not observed in any baseline sample across all the phase 2 and phase 3 studies. R155, A156, and D168 polymorphisms were rare, occurring in <1% of baseline samples.
TABLE 1.
Baseline frequency of HCV NS3 polymorphismsa
| Enzyme | Amino acid position | Genotype-1a |
Genotype-1b |
||
|---|---|---|---|---|---|
| Amino acid(s) | % | Amino acid(s) | % | ||
| NS3 proteaseb | 61d | ||||
| ≥1% variant | T | 93.8 | S | 86.0 | |
| S, S/T | 4.1 | T, S/T | 7.9 | ||
| A, A/T | 1.7 | A, A/S, A/V | 3.9 | ||
| P, P/S | 1.7 | ||||
| <1% variant | G, P, N/T, A/V | <1 | N, C, G, N/S/T/Y | <1 | |
| 80 | |||||
| ≥1% variant | Q | 68.9 | Q | 94.3 | |
| K, K/Q, K/R | 28.8 | L, L/Q | 4.8 | ||
| L, L/Q | 1.3 | ||||
| <1% variant | R, Q/R, N | <1 | K, K/Q R, Q/R, G, H, N | <1 | |
| 156 | |||||
| ≥1% variant | A | 100 | A | 99.9 | |
| <1% variant | None | NAe | A/T | <1 | |
| 155 | |||||
| ≥1% variant | R | 99.5 | R | 100.0 | |
| <1% variant | K, K/R | <1 | |||
| 168 | |||||
| ≥1% variant | D | 99.6 | D | 99.2 | |
| <1% variant | E, D/E | <1 | E, D/E, D/N | <1 | |
| NS3 helicasec | 344 | ||||
| ≥1% variant | T | 98.5 | T | 69.1 | |
| I, I/T | 20.9 | ||||
| V | 5.2 | ||||
| M, M/T | 1.1 | ||||
| <1% variant | A, A/T, S, N, N/T, A/V, I/T, D | <1 | D, D/V, I/V, I/S, S, S/T, A, A/T, A/I/T/V, I/M, I/M/T, I/M/V, N, N/T, Q | <1 | |
Data were pooled from phase 2 (SILEN-C1 to -3) and phase 3 (STARTVerso1 to -4) studies.
For NS3 protease, 1,549 patients with genotype-1a infection and 1,575 patients with genotype-1b infection.
For NS3 helicase, 1,203 patients with genotype-1a infection and 1,415 patients with genotype-1b infection.
NS3 T/S61L was not detected in any baseline samples.
NA, not applicable.
In the NS3 helicase region, threonine (T) was highly conserved at amino acid 344 among GT-1a sequences (98.5%); the corresponding T344I polymorphism, commonly detected in around 21% of GT-1b samples overall, was prevalent in only <1% of GT-1a sequences (Table 1). Furthermore, this polymorphism was more frequent among GT-1b samples from PR treatment-experienced patients (24.1%) than among samples from treatment-naive patients (15%) (see Table S5 in the supplemental material).
Baseline polymorphisms and faldaprevir efficacy.
In a comprehensive analysis of all baseline NS3/4A polymorphisms, the NS3 helicase polymorphism T344I detected in HCV GT-1b sequences was the only noncanonical variant associated with reduced SVR to faldaprevir plus PR treatment that was significant (P < 0.0001) in both pooled phase 3 analysis (64/134 [47.8%] compared with 491/652 [75.3%] patients without the T344I variant) (Fig. 1) and pooled phase 2 analysis (see Table S6 in the supplemental material). However, this observation was not associated with faldaprevir treatment only. Notably, the SVR12 rate following treatment with placebo plus PR in phase 3 studies was also lower among treatment-naive patients in whom the HCV GT-1b T344I variant was detected at baseline (7/18 [39%] compared with 60/101 [60%] patients without the T344I variant; P = 0.1263).
FIG 1.
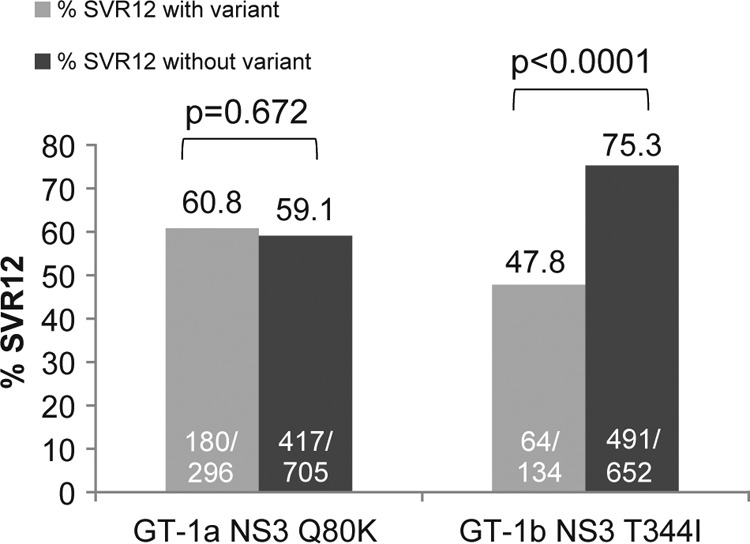
Impact of baseline NS3 polymorphisms on response to faldaprevir plus PR in phase 3 studies. Data pooled from phase 3 studies (STARTVerso1 to -4) including faldaprevir plus PR-treated patients with SVR12 and those without SVR12 for any reason (breakthrough during faldaprevir plus PR treatment or PR only or relapse or experiencing virologic failure for other reasons). “With variant” includes only the single amino acid variant of interest and does not include the wild type, other variants, or mixtures of the variant of interest with wild-type or other amino acids. “Without variant” includes the wild type and all other amino acid variants or mixtures detected. P values were determined using Fisher's exact test. GT, genotype; PR, pegylated interferon/ribavirin; SVR12, sustained virologic response at 12 weeks posttreatment.
Pooled phase 3 analysis of treatment-naive and treatment-experienced patients infected with HCV GT-1a showed that SVR12 rates were comparable among patients with and without the Q80K polymorphism detected at baseline (180/296 [60.8%] and 417/705 [59.1%] patients, respectively) (Fig. 1). Similarly, there was no significant impact of the Q80K substitution on the rates of SVR24 among treatment-naive or treatment-experienced patients infected with HCV GT-1a across the three phase 2 studies (23).
Treatment-emergent NS3/4A variants in patients without SVR.
RAVs at NS3 155 and/or 168 were detected in the majority of patients who did not achieve SVR following treatment with faldaprevir plus PR; R155 RAVs were most frequently detected in HCV GT-1a and D168 RAVs were most frequently detected in HCV GT-1b (Table 2 and Fig. 2). The predominant NS3 variant that emerged in GT-1a was R155K (88% [446/507]) and, to a lesser extent, D168V (10.3% [52/507]). D168V was the predominant amino acid substitution in GT-1b (67.3% [235/349]), and in GT-1b the R155Q and R155K variants were detected in fewer cases (14.6% [51/349] and 8.6% [30/349], respectively). NS3 T/S61L was a novel variant that was not detected among any baseline isolates, but S61L emerged at a low frequency (6.9% [24/349]) in HCV GT-1b consistently with amino acid substitutions that included the predominant D168V variant.
TABLE 2.
Treatment-emergent NS3 substitutions in patients who did not achieve SVR in phase 2 and phase 3 studiesa
| Frequency of NS3 amino acid variants in non-SVR patientsb | Amino acid variant (%) after treatment with FDV plus PRc |
|
|---|---|---|
| HCV GT-1a (507 patients) | HCV GT-1b (349 patients) | |
| ≥10% | R155K (88.0) | D168V (67.3) |
| D168V (10.3) | R155Qd (14.6) | |
| 1% to <10% | D168E (3.4) | R155K (8.6) |
| D168A/T (7.4) | ||
| D168Ne (7.2) | ||
| S61Lf (6.9) | ||
| D168I (5.7) | ||
| D168E (4.9) | ||
| D168H (2.6) | ||
| D168Y (1.4) | ||
| <1% | D168A/Ne/Y/Ig/T | |
| R155T/S | R155C/Rh or G/Rh | |
| D168 ambiguous mixtures with F | D168 ambiguous mixtures with L/F/K or P | |
Pooled data for the SVR24 endpoint were from phase 2 studies (SILEN-C1 to -3) and those for the SVR12 endpoint were from phase 3 studies (STARTVerso1 to -4). SVR, sustained virologic response.
Amino acids reported are those at positions associated with resistance to faldaprevir (NS3 155 or 168), detected alone or in combination with other substitutions (includes mixtures) among any post-baseline virology sample that was sequenced per patient including, but not limited to, the first virologic failure sample. Substitutions are listed in the order of their prevalence.
FDV, faldaprevir; PR, pegylated interferon/ribavirin; GT, genotype.
R155Q was detected only as a predominant variant with D168V/N/T/I substitutions and otherwise detected in R155K/Q/R or Q/R mixtures.
D168N was detected only as a predominant variant with R155Q and otherwise in D168D/N or complex D168 mixtures.
Only detected as a mixture with D168V variants.
Only detected in complex mixtures of D/I/N/V or A/I/T/V.
Only detected as a mixture with wild-type amino acid in one patient.
FIG 2.
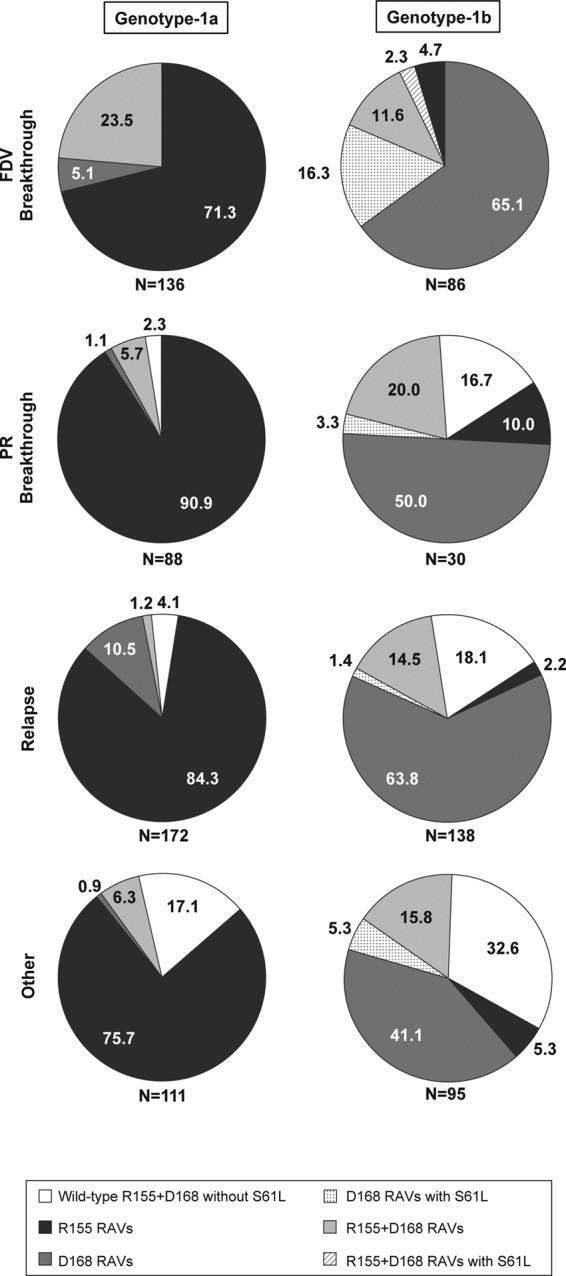
Emergence of resistance-associated variants at one or multiple NS3 codons in faldaprevir plus PR-treated patients who did not achieve SVR in phase 2 and phase 3 studies. Data were pooled for the SVR24 endpoint from phase 2 studies (SILEN-C1 to -3) and for the SVR12 endpoint from phase 3 studies (STARTVerso1 to -4). Numbers on the figures represent the percentages of patients in whom the amino acid substitutions were detected. Per patient analyses, data from all emerging variants detected among all postbaseline virology samples that were sequenced, including but not limited to the first virologic failure sample, were pooled; thus, dual RAVs in this figure represent variants detected together from the same sample or each detected individually from temporally different samples. The denominator (N) is the number of patients for whom matched baseline and virologic failure NS3 sequence data were available. FDV, faldaprevir; PR, pegylated interferon/ribavirin; RAV, resistance-associated variant; SVR, sustained virologic response.
All patients who experienced virologic breakthrough during faldaprevir plus PR treatment and had NS3 protease sequences available had faldaprevir RAVs detected at amino acid position 155 or 168, or, less frequently, both (Fig. 2). The combination of GT-1b NS3 S61L and D168 amino acid substitutions (with or without R155 RAVs) was more commonly detected among patients with virologic breakthroughs during faldaprevir plus PR treatment (18.6% [16/86]). Some patients with breakthroughs during PR treatment, some who relapsed, or some who experienced virologic failure for other reasons had no detectable treatment-emergent RAVs at positions 155 or 168: a lack of RAVs overall was more common among non-SVR patients infected with GT-1b (17.5% [61/349]) than among those infected with GT-1a (5.5% [28/507]) (Fig. 2). Among relapsers specifically, a lack of NS3 R155 and D168 RAVs was observed in 18.1% (25/138) of GT-1b-infected patients, which was less than that in GT-1a-infected patients (4.1% [7/172]).
NS3 A156 variants, which conferred reduced susceptibility to faldaprevir in preclinical studies (27), were rarely detected in faldaprevir plus PR-treated patients (<1% [3/349 GT-1b and 1/507 GT-1a]) and only as mixed amino acid substitutions with the wild type (A/T or A/G). GT-1b A156A/G emerged transiently with the D168V RAV in only one case during faldaprevir plus PR treatment (phase 3) and, in the remaining three cases, only in posttreatment plasma samples from virologic failures with predominant R155K and/or D168V/E RAVs (data not shown).
Persistence of NS3 variants in posttreatment follow-up.
The persistence of NS3 variants (by population-based sequencing) during posttreatment follow-up was evaluated in the phase 3 studies, using Kaplan-Meier analysis, among patients with virologic failure (breakthrough on faldaprevir or PR, relapse, or other reasons) and at least one postbaseline NS3 sequence and included 353 GT-1a-infected patients and 228 GT-1b-infected patients. The number of non-SVR12 patients who had additional follow-up virology sequences after the first virologic failure sequence (i.e., >1 postbaseline sequence) included 92% (326/353) of GT-1a patients and 86% (197/228) of GT-1b patients. The number of postbaseline sequences per non-SVR12 patient in the persistence analysis ranged from 1 to 8 sequences (median, 3 per patient). The median follow-up time after the day of virologic failure to the last NS3 sequence available for GT-1a patients with >1 postbaseline sequence was 373 days (range, 1 to 826 days) and for GT-1b patients was 256 days (range, 8 to 791 days).
Among the 351 GT-1a-infected patients with virologic failure (two patients with baseline R155K variants were excluded), the assessed duration was 428 days (approximately 14 months) from the first day of virologic failure to the point when 50% of patients had HCV that encoded wild-type NS3 R155 and concomitant loss of R155 RAVs (any variant) (Fig. 3). The elapsed time to when 50% of the 228 GT-1b non-SVR12 patients encoded wild-type HCV NS3 D168 with concomitant loss of D168 RAVs (any variant) was 147 days (approximately 5 months) after virologic failure.
FIG 3.
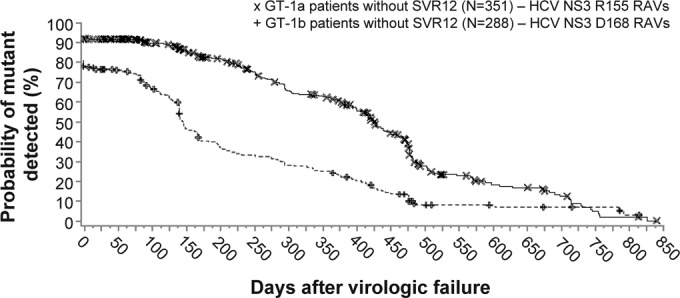
Persistence of NS3 R155 and D168 amino acid substitutions after virologic failure in pooled phase 3 studies. Kaplan-Meier estimations of the time to loss of RAVs (population-based sequencing) after the time of virologic failure (plot origin) among all non-SVR12 patients treated with faldaprevir plus PR in phase 3 studies (STARTVerso1 to -4). Analysis included 351 GT-1a- and 228 GT-1b-infected patients without SVR12 for whom baseline and at least one or more postbaseline sequences were available (with or without RAVs) and excluded patients with RAVs detected at baseline. Symbols (× and +) represent time points where patient(s) were censored, i.e., RAVs were detected but a subsequent follow-up sequence was not available. GT, genotype; PR, pegylated interferon/ribavirin; RAV, resistance-associated variant; SVR12, sustained virologic response 12 weeks posttreatment.
Characterization of the NS3 S61L site-directed mutant.
The impact of the newly identified NS3 S61L treatment-emergent substitution on in vitro susceptibility to HCV PIs was assessed using site-directed mutagenesis that introduced S61L, D168V, or the combination S61L plus D168V into an HCV GT-1b subgenomic replicon used in phenotyping assays. The replication capacity of the NS3 S61L variant (25.9%) was lower than that of GT-1b wild-type NS3 but greater than that of the NS3 D168V variant (6.9%) (Table 3). The replication capacity of the NS3 S61L plus D168V dual variant (9.0%) was not substantially different from that of D168V alone. S61L alone conferred only a 2-fold reduction in susceptibility to faldaprevir and simeprevir and did not have any measurable effect on susceptibility to telaprevir. The fold changes in faldaprevir EC50 were 1,159-fold with D168V and 1,737-fold with the dual S61L plus D168V mutant relative to those for the wild type and were not substantially different (approximately 1.5-fold difference) (Table 3).
TABLE 3.
In vitro characterization of the impact of NS3 S61L site-directed mutants on HCV GT-1b drug susceptibilitya
| HCV GT-1b NS3 site-directed mutant | Mean replication capacity ± SD (n) | Mean EC50b fold change ± SD (n) |
||
|---|---|---|---|---|
| Faldaprevir | Simeprevir | Telaprevir | ||
| S61L | 25.9 ± 5.7 (4) | 1.8 ± 0.5 (4) | 2.3 ± 0.4 (4) | 1.0 ± 0.2 (3) |
| D168V | 6.9 ± 1.7 (4) | 1,159 ± 148 (4) | 1,694 ± 317 (4) | 0.4 ± 0.05 (4) |
| S61L+D168V | 9.0 ± 2.6 (3) | 1,737 ± 316 (3) | 1,243 ± 135 (3) | 0.5 ± 0.1 (3) |
Means were calculated from interexperimental values. GT, genotype.
EC50s (50% effective concentrations) are given in Table S7 in the supplemental material.
Characterization of novel NS3 R155 and D168 mutants from patient isolates.
In vitro phenotypic characterization of NS3 proteases derived from patient isolates from earlier studies encoding the single NS3 mutants R155K or D168V in GT-1a and GT-1b and D168T or D168E in GT-1b has been previously described (16). The previous report showed that the faldaprevir EC50 fold change relative to baseline for GT-1b NS3 D168V chimeric replicons is 1,700-fold and GT-1a NS3 R155K provides a 330-fold change (16). Phenotypic analyses of single or dual R155 and D168 variants from patient-derived NS3 proteases from the clinical trials described in this report and not previously characterized in earlier studies were performed. The NS3 proteases derived from patient virology samples included single R155S, D168A, D168E, and D168Y substitutions and dual R155K plus D168E, R155Q plus D168N, and R155Q plus D168V variants. These variants reduced susceptibility to faldaprevir by 24- to 1,260-fold relative to baseline (Table 4).
TABLE 4.
Characterization of novel NS3 R155 and D168 variants derived from isolates from patients in phase 2 studies
| GTa | NS3 variant | No. of samples | Mean (range) faldaprevir EC50b fold change relative to baseline |
|---|---|---|---|
| 1a | R155K + D168E | 3 | 623 (588–680) |
| D168E | 1 | 24 | |
| D168A | 1 | 360 | |
| R155S | 1 | 131 | |
| 1b | R155Q + D168N | 1 | 162 |
| R155Q + D168V | 1 | 38 | |
| D168Y | 1 | 1,261 |
GT, genotype.
EC50, 50% effective concentration.
DISCUSSION
The baseline frequency of polymorphisms identified in this analysis of NS3/4A protease sequences from phase 2 and 3 studies of faldaprevir plus PR was generally consistent with the reported results from other studies assessing the frequency of polymorphisms in this region of the HCV genome, confirming that the prevalence of faldaprevir RAVs in patients naive to HCV PIs is rare (16, 23, 28). Several baseline and host factors may confound the interpretation of whether an HCV amino acid polymorphism has any impact on virologic response, for example, small sample sizes for particular variants, baseline IL28B genotype, innate immune responses, and the degree of liver cirrhosis. Nevertheless, common viral polymorphisms that reduce response to HCV antiviral agents have been identified. One example is the reduced susceptibility and reduced virologic response to simeprevir plus PR therapy in patients infected with GT-1a virus encoding the NS3 Q80K polymorphism (9). In phase 2 and phase 3 studies of faldaprevir plus PR, a comprehensive analysis of common baseline NS3 polymorphisms in HCV GT-1a or GT-1b showed that no single NS3 protease polymorphism reduced SVR rates in patients treated with faldaprevir plus PR. This result was confirmed among treatment-naive patients infected with HCV GT-1a in an analysis of pooled data from STARTVerso1 and STARTVerso2 published elsewhere (D. M. Jensen, T. Asselah, D. Dieterich, G. R. Foster, M. S. Sulkowski, S. Zeuzem, P. Mantry, E. M. Yoshida, C. Moreno, D. Ouzan, M. Wright, L. E. Morano, R. Buynak, M. Bourlière, T. Hassanein, S. Nishiguchi, J.-H. Kao, M. Omata, S. W. Paik, D. K. Wong, E. Tam, K. Kaita, S. V. Feinman, J. O. Stern, J. Scherer, A.-M. Quinson, F. Voss, J.-P. Gallivan, W. O. Böcher, and P. Ferenci, submitted for publication). The rates of SVR12 with and without Q80K were 65% and 64%, respectively, and in both cases were higher than the response rate achieved with placebo plus PR in the same subgroups.
The NS3 Q80K polymorphism was more prevalent among HCV GT-1a sequences (31.1% [362/1,164]) than among GT-1b sequences (0.5% [5/1,098]). Phylogenetic analyses of GT-1a isolates show their separation into two distinct clades, each with a different prevalence among geographical regions (29, 30). GT-1a clade 1 is more prevalent among North American isolates, and clade 2 is more prevalent among isolates from Europe (31). The Q80K polymorphism has been suggested to exist almost exclusively within GT-1a clade 1 and rarely within clade 2 (31).
The NS3 helicase polymorphism T344I was associated with reduced SVR in GT-1b-infected patients regardless of treatment (faldaprevir plus PR or placebo plus PR). T344I has also been reported to reduce response rates to simeprevir plus PR or placebo plus PR (32). Furthermore, assessment of the frequency of T344I in faldaprevir studies suggests that this polymorphism may be more frequent among patients previously treated with PR, suggesting potential enrichment of this variant during PR treatment. However, T344I did not emerge as a predominant variant during treatment with faldaprevir plus PR (data not shown). The impact of NS3 T344I during HCV infection remains unclear, and its association with reduced responses to antiviral therapy may not be DAA treatment specific.
The predominant RAVs that emerged in patients not achieving SVR following treatment with faldaprevir plus PR in phase 2 and phase 3 were detected at HCV NS3 codons 155 and/or 168. Faldaprevir RAVs at these positions emerged at a high frequency in non-SVR patients, with 89% (452/507) of GT-1a with R155 substitutions and 78% (273/349) of GT-1b with D168 variants (Fig. 2). Very few non-SVR patients lacked R155 or D168 RAVs and encoded wild type: 5.5% of GT-1a and 17.5% of GT-1b. NS3 R155K and NS3 D168V were the most frequently detected RAVs in non-SVR GT-1a and GT-1b patients, respectively. Other variants that emerged in >10% of non-SVR GT-1b patients included R155K and R155Q; D168V was detected in 10.3% of GT-1a non-SVR patients. D168 variants were more heterogeneous (V/A/T/N/I/E/H/Y) than R155 substitutions (K/Q/T/S), and in a small number of patients, dual R155 plus D168 RAVs emerged (R155Q plus D168V/N in GT-1b or R155K plus D168E in GT-1a) in the same clinical isolate. D168 variants typically conferred larger reductions in faldaprevir susceptibility than R155 variants in in vitro phenotyping assays (27).
The NS3 amino acid substitutions A156V and A156T emerged in GT-1b under faldaprevir selective pressure in preclinical in vitro resistance studies but were only selected with the lower faldaprevir concentration tested (0.4 μM) and were not detected with a 10-fold higher faldaprevir concentration (27). Although A156 variants have been shown to reduce faldaprevir susceptibility in vitro (up to 270-fold with A156T), A156 variants did not emerge as the predominant RAVs among virologic failures and were only detected as rare amino acid mixtures with the wild type (A/T or A/G) in phase 2 and phase 3 studies. A156 amino acid changes have been detected in posttreatment virology samples after the emergence of other variants such as R155K or D168V/E; in one case, A156A/G was detected together with the predominant D168V RAV in a faldaprevir breakthrough sample.
Although GT-1b NS3 S61L was a novel low-frequency variant that emerged only in combination with D168V and most often among faldaprevir breakthroughs, in vitro phenotyping assays showed that the S61L single mutant only modestly reduced susceptibility to faldaprevir (2-fold). Even in combination with D168V, S61L did not substantially affect the faldaprevir EC50 compared with D168V alone (only an ∼1.5-fold difference in the faldaprevir EC50 was observed). However, in HCV-infected patients, it is possible that the dual NS3 S61L plus D168V mutant replicates modestly better and confers a selective advantage in the presence of faldaprevir compared with that of D168V alone. This possibility may not have been adequately evaluated in vitro because the EC50 change conferred by D168V alone approaches the upper limit of the in vitro phenotyping assay.
The phenotypic analysis of previously uncharacterized patient-derived NS3 proteases from faldaprevir studies included the single amino acid substitutions D168E, D168A, R155S, and D168Y, which conferred faldaprevir EC50 fold changes ranging from 24- to 1,260-fold relative to baseline. These levels of resistance are similar to the levels observed with other single amino acid substitutions at positions 155 and 168 during in vitro studies (16, 27). Among these variants, the lowest fold change was observed for the single GT-1a D168E mutant (24-fold). However, this substitution resulted in a greater reduction in susceptibility to faldaprevir in a GT-1b background (180-fold) (16). The 1,260-fold change in faldaprevir EC50 observed with a single D168Y substitution in a GT-1b isolate indicates a high level of resistance that is also similar to that of previously profiled D168V in both GT-1a (1,800-fold) and GT-1b (1,700-fold) isolates (16). The three double variants tested (R155K plus D168E in GT-1a and R155Q plus D158N and R155Q plus D168V in GT-1b) all showed lower levels of reduced susceptibility to faldaprevir (38-fold to 623-fold) than a single D168V substitution (1,700-fold) as previously described (16). However, the dual GT-1a R155K plus D168E mutant reduced susceptibility to faldaprevir by more than each of the single mutants (623-fold compared with 24-fold for GT-1a D168E alone and 330-fold for R155K alone) as previously described (16). Similarly, addition of GT-1b D168N to R155Q resulted in a greater reduction in faldaprevir susceptibility (162-fold) than that with R155Q alone (60-fold) (16, 27). In contrast, mutants encoding both D168V and R155 RAVs had faldaprevir EC50s more similar to those of R155 single RAVs. In these cases, D168V did not further reduce susceptibility in combination with R155 RAVs. These trends of faldaprevir susceptibility comparing single and dual mutants from patient isolates were consistent with site-directed NS3 R155 and D168 mutants (data not shown and our unpublished data); in general, comparisons of relative faldaprevir susceptibility between different NS3 mutants in patient-derived chimeric replicons are similar among NS3 site-directed mutants (16, 27). Considering that X-ray crystallographic studies show structural compensation occurring between NS3 R155 and D168 amino acid residues (33), it is possible that faldaprevir binding to NS3 protease with R155 RAVs in combination with D168V, but not with D168E, reflects aspects of this compensation.
Analysis of the persistence of NS3 R155 and D168 RAVs by clonal sequencing in phase 1b studies, while only based on a small sample size, initially indicated longer persistence of R155K variants compared with that for D168V (16). Based on Kaplan-Meier analyses of phase 3 data used to evaluate RAVs and outgrowth of wild-type amino acid at individual NS3 codons, the median time to loss of GT-1a R155 RAVs (approximately 14 months) was longer than the median time to loss of GT-1b D168 RAVs (approximately 5 months). These findings from faldaprevir clinical studies are supportive of eventual wild-type virus outgrowth reported from non-SVR patients in boceprevir clinical trials in whom T54S and R155K variants were found in a minority (25%) of patients after 2.5 years of follow-up (7). Similarly, some telaprevir treatment failures had detectable R155K variants (>25% of viral population) at 25 months, but only 3% of patient isolates still had R155K variants at 36 months (6); the median time to loss of this variant following discontinuation of telaprevir treatment was reported to be 9.8 months based on Kaplan-Meier parameters that differ from those used in this report (34). The lower genetic barrier for emergence of R155K in GT-1a (i.e., single nucleotide transition under drug-selective pressure in GT-1a versus two in GT-1b as previously described [35]), the higher replicative capacity of R155K relative to that of D168V observed in vitro, and the apparent fitness of R155K in vitro compared to that of the wild type (36) all contribute to the higher prevalence of R155K and long-term persistence of faldaprevir resistance in GT-1a.
The pooled analysis of resistance data from phase 2 and phase 3 studies of faldaprevir shows that faldaprevir RAVs were rare at baseline but that R155 variants (in GT-1a) and D168 variants (in GT-1b) were detected in the majority of patients who experience virologic failure. These studies provide valuable information about HCV NS3 PI therapy that may be relevant to the use of other DAAs with overlapping resistance profiles.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by Boehringer Ingelheim Pharma GmbH & Co. KG. Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Esther Race of Choice Healthcare Solutions during the preparation of the manuscript.
We thank all patients and study investigators who participated in the clinical studies described here.
K.L.B., J.S., M.R., N.S., J.O.S., A.-M.Q., and G.K. contributed to data collection. J.S., M.R., and N.S. conducted statistical analyses. K.L.B., J.S., J.O.S., A.-M.Q., and G.K. contributed to data interpretation. J.S., M.R., N.S., J.O.S., A.-M.Q., and G.K. contributed to study designs. All authors contributed to the writing and review of the manuscript and approved the final version of the manuscript.
K.L.B., J.S., M.R., N.S., J.O.S., and A.-M.Q. are employees of Boehringer Ingelheim Pharmaceuticals, Inc. At the time of the study, G.K. was an employee of Boehringer Ingelheim (Canada) Ltd./Ltée and is presently employed by Gilead Sciences, Inc.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00932-15.
REFERENCES
- 1.American Association for the Study of Liver Disease, Infectious Diseases Society of America HCV Panel. 2015. HCV guidance: recommendations for testing, managing, and treating hepatitis C. http://www.hcvguidelines.org/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.European Association for Study of Liver. 2014. EASL clinical practice guidelines: management of hepatitis C virus infection. J Hepatol 60:392–420. doi: 10.1016/j.jhep.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, Poordad F, Goodman ZD, Sings HL, Boparai N, Burroughs M, Brass CA, Albrecht JK, Esteban R, HCV RESPOND-2 Investigators . 2011. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 364:1207–1217. doi: 10.1056/NEJMoa1009482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S, ADVANCE Study Team. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 364:2405–2416. doi: 10.1056/NEJMoa1012912. [DOI] [PubMed] [Google Scholar]
- 5.Poordad F, McCone J Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP, SPRINT-2 Investigators. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, Ferenci P, Nevens F, Mullhaupt B, Pockros P, Terg R, Shouval D, van Hoek B, Weiland O, Van Heeswijk R, De Meyer S, Luo D, Boogaerts G, Polo R, Picchio G, Beumont M, REALIZE Study Team. 2011. Telaprevir for retreatment of HCV infection. N Engl J Med 364:2417–2428. doi: 10.1056/NEJMoa1013086. [DOI] [PubMed] [Google Scholar]
- 7.Vertex Pharmaceuticals Incorporated. 2014. Incivek (telaprevir) prescribing information. Vertex Pharmaceuticals Incorporated, Cambridge, MA. [Google Scholar]
- 8.Merck Sharp & Dohme Corp. 2013. Victrelis (boceprevir) prescribing information. Merck Sharp & Dohme Corp, Kenilworth, NJ. [Google Scholar]
- 9.Janssen Products LP. 2014. Olysio (simeprevir) prescribing information. Janssen Products LP, Titusville, NJ. [Google Scholar]
- 10.Sulkowski MS, Asselah T, Lalezari J, Ferenci P, Fainboim H, Leggett B, Bessone F, Mauss S, Heo J, Datsenko Y, Stern JO, Kukolj G, Scherer J, Nehmiz G, Steinmann GG, Bocher WO. 2013. Faldaprevir combined with pegylated interferon alfa-2a and ribavirin in treatment-naive patients with chronic genotype-1 HCV: SILEN-C1 trial. Hepatology 57:2143–2154. doi: 10.1002/hep.26276. [DOI] [PubMed] [Google Scholar]
- 11.Sulkowski MS, Bourliere M, Bronowicki JP, Asselah T, Pawlotsky JM, Shafran SD, Pol S, Mauss S, Larrey D, Datsenko Y, Stern JO, Kukolj G, Scherer J, Nehmiz G, Steinmann GG, Bocher WO. 2013. Faldaprevir combined with peginterferon alfa-2a and ribavirin in chronic hepatitis C virus genotype-1 patients with prior nonresponse: SILEN-C2 trial. Hepatology 57:2155–2163. doi: 10.1002/hep.26386. [DOI] [PubMed] [Google Scholar]
- 12.Dieterich D, Nelson M, Soriano V, Arasteh K, Guardiola JM, Rockstroh JK, Bhagani S, Laguno M, Tural C, Ingiliz P, Jain MK, Stern JO, Manero M, Vinisko R, Kort J, STARTVerso4 Study Group . STARTVerso4: faldaprevir and pegylated interferon alpha-2a/ribavirin in individuals co-infected with hepatitis C virus genotype-1 and HIV. AIDS, in press. [DOI] [PubMed] [Google Scholar]
- 13.Ferenci P, Asselah T, Foster GR, Zeuzem S, Sarrazin C, Moreno C, Ouzan D, Maevskaya M, Calinas F, Morano LE, Crespo J, Dufour JF, Bourliere M, Agarwal K, Forton D, Schuchmann M, Zehnter E, Nishiguchi S, Omata M, Kukolj G, Datsenko Y, Garcia M, Scherer J, Quinson AM, Stern JO, STARTVerso1 Study Group . 2015. STARTVerso1: a randomized trial of faldaprevir plus pegylated interferon/ribavirin for chronic HCV genotype-1 infection. J Hepatol 62:1246−1255. doi: 10.1016/j.jhep.2014.12.024. [DOI] [PubMed] [Google Scholar]
- 14.Manns MP, Bourliere M, Benhamou Y, Pol S, Bonacini M, Trepo C, Wright D, Berg T, Calleja JL, White PW, Stern JO, Steinmann G, Yong C-L, Kukolj G, Scherer J, Boecher WO. 2011. Potency, safety, and pharmacokinetics of the NS3/4A protease inhibitor BI201335 in patients with chronic HCV genotype-1 infection. J Hepatol 54:1114–1122. doi: 10.1016/j.jhep.2010.08.040. [DOI] [PubMed] [Google Scholar]
- 15.Halfon P, Locarnini S. 2011. Hepatitis C virus resistance to protease inhibitors. J Hepatol 55:192–206. doi: 10.1016/j.jhep.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 16.Berger KL, Lagace L, Triki I, Cartier M, Marquis M, Lawetz C, Bethell R, Scherer J, Kukolj G. 2013. Viral resistance in hepatitis C virus genotype 1-infected patients receiving the NS3 protease inhibitor faldaprevir (BI 201335) in a phase 1b multiple-rising-dose study. Antimicrob Agents Chemother 57:4928–4936. doi: 10.1128/AAC.00822-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.HCV Phenotype Working Group, HCV Drug Development Advisory Group. 2012. Clinically relevant HCV drug resistance mutations figure and tables (updated). Ann Forum Collab HIV Res 14(2):1–10. [Google Scholar]
- 18.Dierynck I, Ghys A, Witek J, Luo D, Janssen K, Daems B, Picchio G, Buti M, De Meyer S. 2014. Incidence of virological failure and emergence of resistance with twice-daily vs every 8-h administration of telaprevir in the OPTIMIZE study. J Viral Hepat 21:835–842. doi: 10.1111/jvh.12347. [DOI] [PubMed] [Google Scholar]
- 19.Haseltine EL, De Meyer S, Dierynck I, Bartels DJ, Ghys A, Davis A, Zhang EZ, Tigges AM, Spanks J, Picchio G, Kieffer TL, Sullivan JC. 2014. Modeling viral evolutionary dynamics after telaprevir-based treatment. PLoS Comput Biol 10:e1003772. doi: 10.1371/journal.pcbi.1003772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenz O, Verbinnen T, Fevery B, Tambuyzer L, Vijgen L, Peeters M, Buelens A, Ceulemans H, Beumont M, Picchio G, Meyer SD. 2015. Virology analyses of HCV isolates from genotype 1-infected patients treated with simeprevir plus peginterferon/ribavirin in phase IIb/III studies. J Hepatol 62:1008−1014. doi: 10.1016/j.jhep.2014.11.032. [DOI] [PubMed] [Google Scholar]
- 21.Dieterich D, Asselah T, Guyader D, Berg T, Schuchmann M, Mauss S, Ratziu V, Ferenci P, Larrey D, Maieron A, Stern JO, Ozan M, Datsenko Y, Böcher YW, Steinmann G. 2014. SILEN-C3: a phase 2 randomized trial with faldaprevir plus PegIFN/ribavirin in treatment-naive HCV genotype-1-infected patients. Antimicrob Agents Chemother 58:3429–3436. doi: 10.1128/AAC.02497-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobson I, Asselah T, Ferenci P, Foster GR, Jensen D, Negro F, Mantry P, Wright D, Forns X, Garcia-Samaniego J, Oliveira C, Carvalho A, Forton D, Agarwal K, Arasteh K, Cooper C, Ghesquiere W, Dufour JF, Sakai Y, Tanaka Y, Stern JO, Sha N, Boecher WO, Steinmann G, Quinson A-M. 2013. STARTVerso3: a randomized, double-blind, placebo-controlled phase III trial of faldaprevir in combination with pegylated interferon alfa-2a and ribavirin in treatment-experienced patients with chronic HCV genotype-1 infection. Hepatology 58(Suppl):742A. doi: 10.1002/hep.26858. [DOI] [Google Scholar]
- 23.Berger KL, Triki I, Cartier M, Marquis M, Massariol MJ, Bocher WO, Datsenko Y, Steinmann G, Scherer J, Stern JO, Kukolj G. 2014. Baseline hepatitis C virus (HCV) NS3 polymorphisms and their impact on treatment response in clinical studies of the HCV NS3 protease inhibitor faldaprevir. Antimicrob Agents Chemother 58:698–705. doi: 10.1128/AAC.01976-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jensen DM, Asselah T, Dieterich D, Foster GR, Sulkowski M, Zeuzem S, Mantry P, Moreno C, Ouzan D, Wright M, Morano LE, Buynak R, Bourliere M, Hassanein T, Nishiguchi S, Kao J-H, Omata M, Paik SW, Wong DK, Tam E, Kaita K, Feinman SV, Stern JO, Garcia M, Quinson AM, Voss F, Gallivan JP, Bocher W, Ferenci P, STARTVerso1 and STARTVerso2 Study Groups . 2013. A pooled analysis of two randomized, double-blind, placebo-controlled phase III trials (STARTVerso1&2) of faldaprevir plus pegylated interferon alfa-2a and ribavirin in treatment-naive patients with chronic hepatitis C genotype-1 infection. Hepatology 58(Suppl):734A. doi: 10.1002/hep.26858. [DOI] [Google Scholar]
- 25.Alcantara LC, Cassol S, Libin P, Deforche K, Pybus OG, Van Ranst M, Galvao-Castro B, Vandamme AM, de Oliveira T. 2009. A standardized framework for accurate, high-throughput genotyping of recombinant and non-recombinant viral sequences. Nucleic Acids Res 37(Web Server issue):W634−W642. doi: 10.1093/nar/gkp455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Oliveira T, Deforche K, Cassol S, Salminen M, Paraskevis D, Seebregts C, Snoeck J, van Rensburg EJ, Wensing AM, van de Vijver DA, Boucher CA, Camacho R, Vandamme AM. 2005. An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics 21:3797–3800. doi: 10.1093/bioinformatics/bti607. [DOI] [PubMed] [Google Scholar]
- 27.Lagacé L, White PW, Bousquet C, Dansereau N, Do F, Llinas-Brunet M, Marquis M, Massariol MJ, Maurice R, Spickler C, Thibeault D, Triki I, Zhao S, Kukolj G. 2012. In vitro resistance profile of the hepatitis C virus NS3 protease inhibitor BI 201335. Antimicrob Agents Chemother 56:569–572. doi: 10.1128/AAC.05166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartels DJ, Sullivan JC, Zhang EZ, Tigges AM, Dorrian JL, De Meyer S, Takemoto D, Dondero E, Kwong AD, Picchio G, Kieffer TL. 2013. Hepatitis C virus variants with decreased sensitivity to direct-acting antivirals (DAAs) were rarely observed in DAA-naive patients prior to treatment. J Virol 87:1544–1553. doi: 10.1128/JVI.02294-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vicenti I, Rosi A, Saladini F, Meini G, Pippi F, Rossetti B, Sidella L, Di Giambenedetto S, Almi P, De Luca A, Caudai C, Zazzi M. 2012. Naturally occurring hepatitis C virus (HCV) NS3/4A protease inhibitor resistance-related mutations in HCV genotype 1-infected subjects in Italy. J Antimicrob Chemother 67:984–987. doi: 10.1093/jac/dkr581. [DOI] [PubMed] [Google Scholar]
- 30.Pickett BE, Striker R, Lefkowitz EJ. 2011. Evidence for separation of HCV subtype 1a into two distinct clades. J Viral Hepat 18:608–618. doi: 10.1111/j.1365-2893.2010.01342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Luca A, Di Gambenedetto S, Prosperi M, Lo Presti A, Caudai C, Vincenti I, Saladini F, Almi P, Grima P, Blanc P, Fabbiani M, Rossetti B, Gagliardini R, Ciccozzi M, Zazzi M. 2013. Two distinct HCV genotype 1a clades: geographical distribution and association with natural resistance mutations to HCV NS3/4A inhibitors. Antivir Ther 18:A47. [Google Scholar]
- 32.U.S. Food and Drug Administration Center for Drug Evaluation Research. 2013. Background package for NDA205123, simeprevir (TMC435). U.S. Food and Drug Administration Center for Drug Evaluation Research, Silver Spring, MD. [Google Scholar]
- 33.Lemke CT, Goudreau N, Zhao S, Hucke O, Thibeault D, Llinas-Brunet M, White PW. 2011. Combined X-ray, NMR, and kinetic analyses reveal uncommon binding characteristics of the hepatitis C virus NS3-NS4A protease inhibitor BI 201335. J Biol Chem 286:11434–11443. doi: 10.1074/jbc.M110.211417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sullivan JC, De Meyer S, Bartels DJ, Dierynck I, Zhang EZ, Spanks J, Tigges AM, Ghys A, Dorrian J, Adda N, Martin EC, Beumont M, Jacobson IM, Sherman KE, Zeuzem S, Picchio G, Kieffer TL. 2013. Evolution of treatment-emergent resistant variants in telaprevir phase 3 clinical trials. Clin Infect Dis 57:221–229. doi: 10.1093/cid/cit226. [DOI] [PubMed] [Google Scholar]
- 35.Sarrazin C, Zeuzem S. 2010. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138:447–462. doi: 10.1053/j.gastro.2009.11.055. [DOI] [PubMed] [Google Scholar]
- 36.He Y, King MS, Kempf DJ, Lu L, Lim HB, Krishnan P, Kati W, Middleton T, Molla A. 2008. Relative replication capacity and selective advantage profiles of protease inhibitor-resistant hepatitis C virus (HCV) NS3 protease mutants in the HCV genotype 1b replicon system. Antimicrob Agents Chemother 52:1101–1110. doi: 10.1128/AAC.01149-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.