

Abstract
We introduce a web portal that employs network theory for the analysis of trajectories from molecular dynamics simulations. Users can create protein energy networks following methodology previously introduced by our group, and can identify residues that are important for signal propagation, as well as measure the efficiency of signal propagation by calculating the network coupling. This tool, called MDN, was used to characterize signal propagation in Escherichia coli heat-shock protein 70-kDa. Two variants of this protein experimentally shown to be allosterically active exhibit higher network coupling relative to that of two inactive variants. In addition, calculations of partial coupling suggest that this quantity could be used as part of the criteria to determine pocket druggability in drug discovery studies.
Introduction
Computer simulations have become a widely used tool to explore the behavior of molecular systems. Great advances have been achieved in the last decade, and improvements in both software and hardware allow for calculations of ever-increasing length and complexity to be performed (1,2). However, performing a simulation is arguably the least interesting and complex part of a computational study of a molecular system. The raw output of such simulations is typically a trajectory file, which contains 3D coordinates for all the atoms in the simulation box, for different conformations explored by the system during the course of the simulation. For simulations of biomolecular systems, the number of atoms (∼105 − 107) coupled to the significant sampling requirements (∼107 − 1010 simulation steps) lead to an enormous amount of high-dimensional data output that needs to be analyzed. This analysis step is thus the core of the investigation itself. Provided the simulations were carried out appropriately, it is the analysis of these data that will determine the quality of the resulting scientific conclusions.
A number of software packages are available for performing molecular dynamics (MD) simulations of biomolecules. The most well known are arguably AMBER (3), CHARMM (4), GROMACS (5), GROMOS (6), and NAMD (7). These have slightly different functionality and provide their own set of analysis tools, ranging from simple calculations of average atomic positions to more complex tasks such as clustering and principal-component analysis (8).
A particular application that computational studies have frequently pursued is the investigation of allostery and protein signaling at the microscopic level (9). A number of specialized methods have been proposed to address this issue (10–15). Another approach that has been widely used in recent years is to analyze standard MD simulations with network theory (16–22). This approach involves modeling the protein as a graph, with residues mapped to network nodes, thus yielding a coarse-grained representation of the system. The most important choice that needs to be made is how to assign the network connections (edges) and the corresponding weights. This is usually done based on interresidue distances and correlation coefficients extracted from MD simulations. We have recently shown that this approach introduces significant statistical errors in the analysis, and networks constructed in this way were not able to identify residues important for signal propagation in the protein imidazole glycerol phosphate synthase (21) or discriminate allosterically active mutants of the lactose repressor protein (22). On the other hand, we showed that assigning network edges based on interaction energies extracted from MD simulations can give an accurate description of signal propagation in proteins (21). In addition, we introduced a network coupling measure that, when applied to protein energy networks, was able to discriminate allosterically active mutants of the lac repressor protein (22). These analyses were done with a customized version of the GROMACS software (5), along with several programs written entirely by our group. Here, we introduce a web portal that enables the performance of this type of network analysis in a user-friendly manner. The portal, called MDN, is available at http://mdn.cheme.columbia.edu, and it can currently be used to analyze output from simulations performed with the GROMACS and NAMD packages. In addition, we present results obtained by using the server to analyze molecular dynamics simulations of four variants of the Escherichia coli heat-shock protein 70 kDa (Hsp70) (DnaK). We show that the energetic coupling analysis is able to discriminate allosterically active from inactive mutants, in accordance with previous experimental results. Finally, we calculated partial coupling values and showed that this quantity can help predict the response of a protein to drug binding to a specific pocket in the molecule.
Materials and Methods
Software routines for network analysis and server details
The protocol for the construction of protein energy networks involves the calculation of a matrix of interresidue interaction energies. This is accomplished by reading the trajectory file with the corresponding simulation program and calculating the energies without integrating the equations of motion. The protocol is currently available for the programs GROMACS (5) and NAMD (7). Here, we will focus on analyzing a simulation performed with GROMACS. Instructions for NAMD users can be found on the MDN web site. To setup the GROMACS simulation for calculating interaction energies, energy groups need to be defined and explicitly included in the GROMACS simulation parameter file (.mdp). However, the official GROMACS release limits the number of energy groups used in a single simulation. We made small modifications to the GROMACS source code to circumvent this limitation, and to only write to disk the final (average) energies, thus avoiding unnecessary file input/output during postprocessing steps. We made similar modifications to the NAMD source code.
Technical details on the implementation of the analysis tool (programming languages used, data management, etc.) are described in the MDN documentation, and are available to the public on the MDN web site.
Hsp70 simulations
We performed MD simulations of several variants of DnaK. The crystal structure of DnaK (23) (Protein Data Bank (PDB) entry 2KHO) was used as the starting point for the wild-type (WT), D388R, D393R, and K70E simulations. The mutants were prepared with the software UCSF Chimera (24). Structures were then solvated with TIP3P water molecules (25) in a truncated octahedral box with minimum distance between the box edges and any protein atoms set to 10 Å. The AMBER-03 force field was used to describe the protein systems (26). Long-range electrostatic interactions were treated with the particle-mesh Ewald method, with a cut-off of 12 Å, whereas van der Waals interactions were switched off between 10 and 12 Å. The systems were minimized with the steepest-descents algorithm, followed by constant-temperature MD simulations at 300 K for 500 ps and equilibration runs at T = 300 K and p = 1 bar for a further nanosecond. Finally, production runs of 200 ns were performed. Version 4.6.3 of the GROMACS simulation package (5) was used to perform the simulations. The thermostat of Bussi and co-workers (27) was used to maintain the temperature of the systems, and pressure control was achieved with the Parrinello-Rahman algorithm (28). An integration time step of 2 fs was used in the MD simulations, with bond lengths of protein and water molecules constrained with the LINCS (29) and SETTLE (30) algorithms, respectively.
Results and Discussion
Available types of analysis
The basic input for MDN consists of a trajectory file, as well as the files used for generating the simulation input. These are processed to construct the network topology and extract the interaction energies to assign network edges, following methodology previously introduced (21). Briefly, weights ωij are assigned to different residue pairs as follows:
| (1) |
The function χij is evaluated by calculating average interaction energies, ϵij. We then define , where ϵav and ϵrmsd are the average and root mean-square deviation of the distribution of interaction energies. The parameter ωb is set to 0.99, the maximum weight allowed. The resulting network is analyzed to determine nodes important for signaling and the efficiency of signal propagation.
The node betweenness is a measure of network centrality suitable for determining important nodes for signaling (21,31). This quantity measures the number of pathways that go through each node in the network. The Floyd-Warshall algorithm (32) is used to calculate the shortest pathways between all pairs of nodes specified by the user, and the resulting node betweenness and normalized node betweenness, η, are calculated according to the methods of Ribeiro and Ortiz (21).
The network coupling, recently introduced by our group (22), is a measure of the efficiency of signal propagation through the network. It uses a network efficiency measure previously introduced by Latora and Marchiori (33):
| (2) |
where N is the number of nodes and dij is the minimum-path distance between nodes i and j. The network coupling is then determined by using Eq. 2 to calculate the efficiencies of the constructed network and of the corresponding ideal network (22):
| (3) |
The ideal network uses the pathways calculated for the analyzed network but gives maximum weight to all edges along these pathways (22). We note that Eq. 3 can also be used to calculate partial energetic coupling values between different regions of the network by summing only over the pathways connecting the corresponding groups of nodes.
Job registration and file upload
The start page of MDN exhibits a registration form in which the name and email of the user should be provided. A verification email is sent to the user, who can then choose the simulation package and start to set up the analysis job. Before network construction can be attempted, a number of files need to be uploaded to the server.
The files needed for analyzing a simulation performed with GROMACS are the trajectory file (.xtc, .trr, .gro, or .pdb), and all files used to set up the simulation with the grompp tool. These include the input configuration file (.gro), the simulation parameter file (.mdp), the index file (.ndx), and the system topology file (.top). The topology file usually reads auxiliary .itp files if there is more than one biomolecule in the system. MDN reads the topology file and asks for the upload of the corresponding auxiliary files. After all necessary files are uploaded, the server checks them for consistency and provides a short summary of the system, as can be seen in Fig. 1 for the DnaK test case. We note that MDN enforces a maximum size for file uploads, currently set to 100 MB. Users may remove solvent molecules from their trajectories before upload, resulting in significantly reduced file sizes. Instructions on how to accomplish this task can be found in the MDN documentation. We also note that this method was found to be very robust with regard to structural fluctuations (21), and the user should be able to upload a trajectory file containing a relatively small number of frames, provided no major conformational changes are observed.
Figure 1.

Short system summary after completed file upload for the DnaK test case.
Network setup
After completing the file upload, the user can set up the network. The standard approach is to construct the network at the residue level, with each network node representing one residue, as specified in the user-provided topology. The user may also employ custom node definitions, as outlined below.
The GROMACS index file (.ndx) uploaded by the user is used to define the network. This file lists groups of atoms, such as all protein atoms or all atoms belonging to a given domain. The user can create custom groups with the GROMACS tool make_ndx. MDN reads the index file and checks groups that can be used to create a network, with the requirement that the group contains all the atoms of a given residue if any of these atoms is listed. A typical group choice for network construction is shown in Fig. 2A, in which the entire protein for the DnaK test case is used to create the network, thus generating 600 nodes. The user is also able to construct smaller networks, including only residues belonging to a specific domain. This is shown in Fig. 2B, where a subset of protein residues (the group representing the substrate-binding domain (SBD) of DnaK) is used to construct the network, thus generating 108 nodes. We note that choosing a specific group will result in a network constructed by splitting the group over its constituent residues. The user may also choose to upload an index file with custom node definitions, in which case the only group available for selection will be named SPECIFIED_NODES. Instructions for using custom node definitions can be found in the MDN documentation.
Figure 2.

Network setup for the DnaK test case. (A) Network construction for group Protein (600 nodes). (B) Network construction for group SBD (108 nodes).
We note that calculating the interaction energies from the uploaded trajectories is the most demanding step of the entire protocol. Depending on the number of trajectory frames and the complexity of the molecular system and its interactions, this step may take 5 min or longer. The user is able to follow the overall progress.
Choosing pathways
The analysis of network properties depends on the network topology and the subset of possible pathways that is analyzed. A network containing N nodes has N(N − 1)/2 pairs of nodes, each giving rise to a pathway. MDN lets the user choose whether all these pathways should be calculated and analyzed. This is again accomplished through the index file, and a group is allowed if it is a subset of the group used for network construction. It should be noted that the same input index (.ndx) file used to define the nodes (see previous section) will be used here to define the groups of nodes that will be considered for calculation of the pathways. Therefore, the user should decide on these groups at the moment he/she is making the index file.
A typical choice for pathway calculation is shown in Fig. 3A, where all pathways for the network containing the entire protein are calculated (179,700 pathways). Fig. 3B is an example where two nonoverlapping regions of the network are selected for analysis. In this case, pathways between groups SBD (108 nodes) and the nucleotide-binding domain (NBD) (380 nodes) are calculated, yielding a total of 74,860 pathways.
Figure 3.

Selecting nodes for pathway calculation for the DnaK test case. A) Example showing group choice that leads to all pathways for the group called Protein being calculated. B) Example showing choice that leads to pathways between two non-overlapping groups (SBD and NBD) being calculated.
Output
Once all the information described above is specified, the program performs the energy network analysis and the user can download a zip file containing several analysis output files. A text file containing node betweenness and normalized node betweenness η values for each network node is given. The same information is also given in a file listing every atom belonging to residues represented in the network. A Python script, which is also included, can be used to generate a PDB file containing this information in the occupancy field. This allows the user visualization of the node betweenness using any of the biomolecular visualization programs available. Information about the global network coupling, as well as partial network coupling, values between different groups listed in the index file is given in another file. Finally, files containing information about the coupling between each node of the network and the groups listed in the index file are provided. Each of these files lists every atom belonging to residues represented in the network, along with the coupling value of the corresponding node. These files can be used to generate maps showing the energetic coupling to a specific region over the entire structure of the molecular system.
In the next sections, we outline the usage of MDN to analyze molecular dynamics simulations of the WT and three mutants of DnaK.
Application to analysis of Hsp70 variants
The Hsp70 chaperones are essential for protein transport and folding and have been shown to be the most conserved proteins in all organisms (34,35). They are essential for cell survival, as they help cells withstand stresses by helping unfolded/misfolded proteins fold properly. For this reason, inhibition of Hsp70 activity has been suggested as a treatment for cancer (36) and Alzheimer’s disease (37). The DnaK structure, shown in Fig. 4, has three domains: the NBD, the SBD, and an α-helical C-terminal domain. This is an important example of an allosteric system, as ATP binding at the NBD triggers substrate release at the SBD, thus involving communication over a distance of 50 Å. In addition, motivation to understand allosteric signaling in Hsp70 comes from the idea that in designing inhibitors of Hsp70 function, allosteric drugs might offer higher selectivity than drugs designed to bind to the molecule’s primary binding sites (34).
Figure 4.
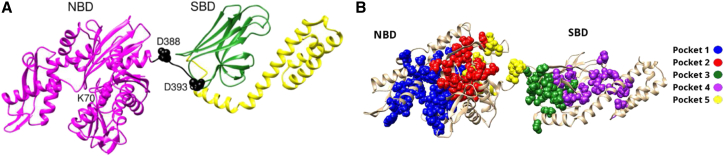
DnaK structure (PDB 2KHO). (A) Visualization of the three domains: the NBD (magenta), the SBD (green), and the C-terminal domain (yellow). The NBD-SBD linker is colored in black. Residues K70, D388, and D393 are shown as spheres. (B) Visualization of the five pockets identified by Rodina and co-workers (40). Residues lining the pockets are represented as spheres colored according to the color code. To see this figure in color, go online.
Experimental attempts to characterize the sources of Hsp70 activity have shown that certain single-point mutations can affect both ATP binding and interdomain communication, leading to inactive Hsp70 mutants. We have performed MD simulations of four experimentally studied DnaK variants: WT, D388R and D393R (38), and K70E (39). The WT and D388R variants were shown to be allosterically active, whereas the remaining variants are inactive. It is particularly interesting to note that the mutation from Asp to Arg at position 388 does not suppress allosteric activity, whereas the opposite is true at position 393. Both residues are part of the linker that connects the NBD with the SBD (Fig. 4, black).
MDN was used to calculate the partial network energetic coupling between each amino acid residue in the NBD and the entire SBD. Fig. 5 shows the results, with each residue in the NBD colored according to the corresponding coupling value. It can be seen that the coupling values are higher for the two active proteins, WT and D388R.
Figure 5.

Maps of energetic coupling for DnaK WT and for mutants experimentally found to be allosterically active (D388R) or inactive (D393R and K70E). Each amino acid in the NBD is colored according to the energetic coupling calculated between that amino acid and all amino acids forming the SBD (green). To see this figure in color, go online.
A recent publication by Rodina and co-workers (40) reported on a potential Hsp70 inhibitor found by designing a drug that would fit to a cavity found in the protein by using software called SiteMap. This software finds cavities in the molecule and uses local information (size, hydrophobicity, etc.) to determine their druggability, a measure of the probability that the drug will bind to a cavity (41). However, because it does not consider the global response of the protein to the binding of a drug to a particular cavity, the software cannot make predictions on the probability that the drug will exert the desired effect on the molecule (which, in the case of Hsp70, is to block the allosteric communication between the domains). In the work of Rodina and co-workers, Sitemap was used to identify the five most druggable pockets in Hsp70 (Fig. 4B), and the authors then picked one pocket based on considerations of size and hydrophobicity and performed experiments to check that the drug binds to Hsp70 and to test for Hsp70 inhibition.
We calculated the energetic coupling between the residues that line each of the five pockets (P.D. Pattel, Memorial Sloan-Kettering Cancer Center and St. John’s University, personal communication, 2014) and the other domain of Hsp70 (i.e., if the pocket was in the NBD, we would calculate its coupling with the SBD, and vice versa). Table 1 shows the values obtained. Pockets 1 and 4 show the best communication with the other domain. Of these, pocket 4 is a small cavity, whereas pocket 1, chosen by the investigators, shows good communication with the SBD domain (0.64) and is a large cavity, so we would also have chosen pocket 1 for the target site. This suggests that energetic coupling could offer some predictive power over the response the molecule will have to binding of a small molecule to one of its cavities, before going into the experimental validation phase.
Table 1.
Partial energetic couplings between residues surrounding each pocket and the other domain of Hsp70
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Domain | SBD | SBD | NBD | NBD | SBD |
| Coupling | 0.64 | 0.42 | 0.43 | 0.66 | 0.36 |
| ±0.05 | ±0.06 | ±0.08 | ±0.05 | ±0.06 |
Conclusions
We have built an online tool, called MDN (http://mdn.cheme.columbia.edu), that allows the application of energy-based network theory to the analysis of molecular dynamics trajectories of proteins, for the characterization of allosteric signaling pathways. The tool employs a method recently published by our group (21), which assigns network edge weights based on the strength of energetic interaction between residues (used to define the network nodes). MDN provides an interface that allows the user to upload all the necessary data, as well as define the groups of residues that are to be considered in the analysis. Its output includes values for the node betweeness (21) (a measure of the importance of a residue for efficient signal propagation in a protein), and the energetic coupling (22) (a measure of efficiency of signal propagation in a protein network). MDN can be used to analyze output from simulations performed with GROMACS and NAMD, and future improvements will include support for additional simulation packages.
We used MDN to characterize allosteric signaling in the WT and three mutants of DnaK. Our calculations of energetic coupling between the NBD and the SBD in DnaK correlate well with experimental results. Two allosterically active mutants showed high values for the energetic coupling, whereas two allostericaly inactive mutants showed lower values of this quantity. In addition, a pocket recently used to design an allosteric inhibitor for Hsp70 also showed high values of energetic coupling. This suggests that energetic coupling could be used as a measure of druggability of a pocket to predict the response of the protein to drug binding before going into the (more expensive) experimental validation phase.
Author Contributions
A.A.S.T.R. and V.O. designed the research; A.A.S.T.R. designed and implemented the MDN server tool, performed the simulations, and analyzed the data; A.A.S.T.R. and V.O. wrote the manuscript.
Acknowledgments
We would like to thank Dr. Gabriela Chiosis and Dr. Pallav Patel for kindly providing information about the residues lining the five Hsp70 pockets presented in their report.
This material is based upon work supported by a Parkinson’s Disease Foundation Columbia University Center Pilot grant program.
Editor: Ivet Bahar.
References
- 1.Lindahl E. Molecular Modeling of Proteins. Springer; New York: 2015. Molecular dynamics simulations; pp. 3–26. [Google Scholar]
- 2.Bernardi R.C., Melo M.C.R., Schulten K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta. 2014;1850:872–877. doi: 10.1016/j.bbagen.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Case D.A., Cheatham T.E., 3rd, Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pronk S., Páll S., Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunz A.-P.E., Allison J.R., van Gunsteren W.F. New functionalities in the GROMOS biomolecular simulation software. J. Comput. Chem. 2012;33:340–353. doi: 10.1002/jcc.21954. [DOI] [PubMed] [Google Scholar]
- 7.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balsera M. a., Wriggers W., Schulten K. Principal component analysis and long time protein dynamics. J. Phys. Chem. 1996;100:2567–2572. [Google Scholar]
- 9.Collier G., Ortiz V. Emerging computational approaches for the study of protein allostery. Arch. Biochem. Biophys. 2013;538:6–15. doi: 10.1016/j.abb.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 10.Ota N., Agard D.A. Intramolecular signaling pathways revealed by modeling anisotropic thermal diffusion. J. Mol. Biol. 2005;351:345–354. doi: 10.1016/j.jmb.2005.05.043. [DOI] [PubMed] [Google Scholar]
- 11.Sharp K., Skinner J.J. Pump-probe molecular dynamics as a tool for studying protein motion and long range coupling. Proteins. 2006;65:347–361. doi: 10.1002/prot.21146. [DOI] [PubMed] [Google Scholar]
- 12.Ming D., Wall M.E. Quantifying allosteric effects in proteins. Proteins. 2005;59:697–707. doi: 10.1002/prot.20440. [DOI] [PubMed] [Google Scholar]
- 13.Chennubhotla C., Bahar I. Markov propagation of allosteric effects in biomolecular systems: application to GroEL-GroES. Mol. Sys. Biol. 2006;2:36. doi: 10.1038/msb4100075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chennubhotla C., Yang Z., Bahar I. Coupling between global dynamics and signal transduction pathways: a mechanism of allostery for chaperonin GroEL. Mol. Biosyst. 2008;4:287–292. doi: 10.1039/b717819k. [DOI] [PubMed] [Google Scholar]
- 15.Ribeiro A.A.S.T., Ortiz V. Local elastic constants of LacI and implications for allostery. J. Mol. Graph. Model. 2015;57:106–113. doi: 10.1016/j.jmgm.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Sethi A., Eargle J., Luthey-Schulten Z. Dynamical networks in tRNA: protein complexes. Proc. Natl. Acad. Sci. USA. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gasper P.M., Fuglestad B., McCammon J.A. Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities. Proc. Natl. Acad. Sci. USA. 2012;109:21216–21222. doi: 10.1073/pnas.1218414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freddolino P.L., Gardner K.H., Schulten K. Signaling mechanisms of LOV domains: new insights from molecular dynamics studies. Photochem. Photobiol. Sci. 2013;12:1158–1170. doi: 10.1039/c3pp25400c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanwart A.T., Eargle J., Amaro R.E. Exploring residue component contributions to dynamical network models of allostery. J. Chem. Theory Comput. 2012;8:2949–2961. doi: 10.1021/ct300377a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh A., Vishveshwara S. A study of communication pathways in methionyl- tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc. Natl. Acad. Sci. USA. 2007;104:15711–15716. doi: 10.1073/pnas.0704459104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ribeiro A.A.S.T., Ortiz V. Determination of signaling pathways in proteins through network theory: importance of the topology. J. Chem. Theory Comput. 2014;10:1762–1769. doi: 10.1021/ct400977r. [DOI] [PubMed] [Google Scholar]
- 22.Ribeiro A.A.S.T., Ortiz V. Energy propagation and network energetic coupling in proteins. J. Phys. Chem. B. 2015;119:1835–1846. doi: 10.1021/jp509906m. [DOI] [PubMed] [Google Scholar]
- 23.Bertelsen E.B., Chang L., Zuiderweg E.R.P. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA. 2009;106:8471–8476. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 25.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 26.Duan Y., Wu C., Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 27.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 28.Parrinello M., Rahman A. Crystal structure and pair potentials: a molecular-dynamics study. Phys. Rev. Lett. 1980;45:1196–1199. [Google Scholar]
- 29.Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 30.Miyamoto S., Kollman P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 31.Boccaletti S., Latora V., Hwang D. Complex networks: structure and dynamics. Phys. Rep. 2006;424:175–308. [Google Scholar]
- 32.Floyd R.W. Algorithm 97: shortest path. Commun. ACM. 1962;5:345. [Google Scholar]
- 33.Latora V., Marchiori M. Efficient behavior of small-world networks. Phys. Rev. Lett. 2001;87:198701. doi: 10.1103/PhysRevLett.87.198701. [DOI] [PubMed] [Google Scholar]
- 34.Zuiderweg E.R.P., Bertelsen E.B., Ahmad A. Allostery in the Hsp70 chaperone proteins. Top. Curr. Chem. 2013;328:99–153. doi: 10.1007/128_2012_323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta R.S. Protein phylogenies and signature sequences: a reappraisal of evolutionary relationships among archaebacteria, eubacteria, and eukaryotes. Microbiol. Mol. Biol. Rev. 1998;62:1435–1491. doi: 10.1128/mmbr.62.4.1435-1491.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy M.E. The HSP70 family and cancer. Carcinogenesis. 2013;34:1181–1188. doi: 10.1093/carcin/bgt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jinwal U.K., Miyata Y., Dickey C.A. Chemical manipulation of hsp70 ATPase activity regulates τ stability. J. Neurosci. 2009;29:12079–12088. doi: 10.1523/JNEUROSCI.3345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vogel M., Mayer M.P., Bukau B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J. Biol. Chem. 2006;281:38705–38711. doi: 10.1074/jbc.M609020200. [DOI] [PubMed] [Google Scholar]
- 39.O’Brien M.C., Flaherty K.M., McKay D.B. Lysine 71 of the chaperone protein Hsc70 Is essential for ATP hydrolysis. J. Biol. Chem. 1996;271:15874–15878. doi: 10.1074/jbc.271.27.15874. [DOI] [PubMed] [Google Scholar]
- 40.Rodina A., Patel P.D., Chiosis G. Identification of an allosteric pocket on human hsp70 reveals a mode of inhibition of this therapeutically important protein. Chem. Biol. 2013;20:1469–1480. doi: 10.1016/j.chembiol.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halgren T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]