

Abstract
Capnocytophaga canimorsus are gram-negative bacteria living as commensals in the mouth of dogs and cats. C. canimorsus cause rare but life-threatening generalized infections in humans that have been in contact with a dog or a cat. Over the last years we collected 105 C. canimorsus strains from different geographical origins and from severe human infections or healthy dogs. All these strains were analyzed by 16S rDNA sequencing and a phylogenetic tree revealed two main groups of bacteria instead of one with no relation to the geographical origin. This branching was confirmed by the whole-genome sequencing of 10 strains, supporting the evidence of a new Capnocytophaga species in dogs. Interestingly, 19 out of 19 C. canimorsus strains isolated from human infections belonged to the same species. Furthermore, most strains from this species could grow in heat-inactivated human serum (HIHS) (40/46 tested), deglycosylate IgM (48/66) and were cytochrome-oxidase positive (60/66) while most strains from the other species could not grow in HIHS (22/23 tested), could not deglycosylate IgM (33/34) and were cytochrome-oxidase negative (33/34). Here, we propose to call Capnocytophaga canis (Latin: dog) the novel, presumably less virulent dog-hosted Capnocytophaga species and to keep the name C. canimorsus for the species including human pathogens.
Keywords: ANI, bacterial taxonomy, commensalism, genome comparison, pathogenesis
Introduction
The genus Capnocytophaga, in the Bacteroidetes phylum, comprises capnophilic gram-negative bacteria that live as host-specific commensals in the oral cavities of mammals.1,2 Several species like Capnocytophaga gingivalis, Capnocytophaga ochracea, and Capnocytophaga sputigena are human hosted while others, like Capnocytophaga canimorsus and Capnocytophaga cynodegmi are commensals of dogs and cats.3 Recent studies have shown a prevalence of C. canimorsus of up to 74% in dogs.4,5,6,7,8,9 Although C. canimorsus are not reported to cause infections in dogs, they cause rare but life-threatening infections in humans that are in contact with dogs.1,4,7,9,10,11,12 The usual syndrome is septicaemia with mortality in the range of 50%. The patients are generally older than 40 years old and roughly half of them had splenectomy or a damaged liver but the other half had no medical history (for review, see Refs. 2, 13,14,15). A few elements contributing to the pathogenesis of C. canimorsus were unraveled recently. First, because of their lipopolysaccharide, C. canimorsus are not very sensitive to complement killing and phagocytosis by human polymorphonuclear leukocytes.10 Because of a low-inflammatory, penta-acylated lipid A, they also escape detection and phagocytosis by macrophages, which results in a low release of pro-inflammatory cytokines in vitro.16,17,18 C. canimorsus 5 also have the unusual capacity to harvest amino sugars from glycan chains of host cell surface and plasma N-glycoproteins like IgGs and transferrin.19,20 This capacity, which contributes to their persistence in a tissue-cage mouse model, is due to the joint action of a sialidase21 and the surface-exposed Gpd (glycoprotein deglycosylation) complex19,22 a feeding complex related to the starch utilization system (Sus) of Bacteroides thetaiotaomicron.23 The capacity to harvest amino sugars is essential during infection because C. canimorsus is a natural auxotroph for N-acetylglucosamine (GlcNAc).24 In its natural niche, C. canimorsus can also retrieve GlcNAc from salivary mucin, through the Muc complex, another Sus-like complex. A third Sus-like complex, called ICS allows C. canimorsus 5 to capture iron from transferrin.20 The three complexes, Gpd, Muc, and ICS are encoded by typical polysaccharide utilization loci (PUL), a hallmark of Bacteroidetes.25
Here we provide genetic and molecular evidences that the C. canimorsus species gathers two main distinct groups of strains and that 19 out of 19 strains isolated from severe human infections worldwide belong to only one of these groups. Since this clustering in two groups is supported by the complete sequencing of the genomes of 10 strains,26,27,28,29 we propose to create a new Capnocytophaga species, for the group of strains that does not include strains isolated from human infections. We propose the name Capnocytophaga canis for this new species.
Materials and methods
Bacterial strains
All bacterial strains used in this study are listed in Supplementary Table S1.
Isolation of Capnocytophaga from dog's saliva and bacterial growth conditions
The saliva from dogs was collected using a swab, which was rubbed against the gingival and cheek mucosa. Within 24 hours, swabs were used to inoculate Heart Infusion Agar plates (Difco, Becton, Dickinson & Co Franklin Lakes, NJ, USA) supplemented with 5% sheep blood (Oxoid, Altrincham, UK) and 20 mg mL−1 gentamicin, which is not active against bacteria from the genus Capnocytophaga. Plates were incubated for 2 days at 37 °C with 5% CO2. Single colonies resembling the ATCC35979 type strain of C. canimorsus, with regard to colony shape, color, and smell, were further analyzed by 16S rRNA sequencing.
16S rDNA sequencing and analysis
A single colony was resuspended in 100 μL ddH2O and heated for 15 minutes at 98 °C. One microliter was used as template for amplification of 1.07 kb of the 16S rDNA. Primers 27F (5′-agagtttgatcctggctcag-3′) and 1100R (5′-gggttgcgctcgttg-3′), binding in the conserved region,30,31 were used at 0.4 mM concentration with 200 mM dNTP and 1 U Taq polymerase (NEB, Ipswich, MD, USA). PCR was carried out for five initial cycles (94 °C for 30 s, 60 °C for 2 minutes, 72 °C for 3 minutes) in which the annealing temperature was reduced by 1.5 °C/cycle, followed by 30 cycles (94 °C for 30 s, 52 °C for 90 s, 72 °C for 3 minutes) and final elongation for 10 minutes at 72 °C. The 1.1 kb PCR product was extracted from a 1.2% agarose gel by NucleoSpin (Macherey Nagel, Düren, Germany). The cleaned PCR products were sequenced by Beckman Coulter genomics with primers 27F, 685R (5′-tctacgcatttcaccgctac-3′)30,31 and 1100R. The consensus sequences were obtained using the Bioedit software and then used to build the 16S phylogeny tree using the Ribosomal Database Project Tree Builder tool (http://rdp.cme.msu.edu/).32
Specific detection of the 16S rRNA gene of Capnocytophaga spp. by PCR
PCR specific detection of 16S rRNA gene was performed as reported by Suzuki et al.9 using primer sets: CaL2-AS1, CaL2-CaR, and CaL2-CyR. A single colony was resuspended in 100 μL ddH2O and heated for 15 minutes at 98 °C. One microliter was used as template for amplification using the Promega Go Taq® G2 polymerase.
Average nucleotide identity (ANI) calculation
ANI was calculated using the ANI calculator from the Kostas Konstantinidis laboratory (Georgia Institute of Technology) Lab Tools (http://enve-omics.ce.gatech.edu/ani/)33 with default settings.
Genome to genome distance (GGD) and DNA–DNA hybridization (DDH) calculations
GGD and DDH estimates were calculated using the GGDC 2.0 calculator (http://ggdc.dsmz.de)34 with default settings. Values considered were the ones obtained using formula 2 (identities/high-scoring segment pairs HSP length). Formula 2 is recommended by the developers because it is independent of genome length and is thus robust against the use of incomplete draft genomes.
Core genome and synteny analysis
The genome comparison between group I, group II C. canimorsus and C. cynodegmi genomes was performed using the MicroScope Comparative Genomics Pan/core-genome tool (https://www.genoscope.cns.fr/agc/microscope/compgenomics/pancoreTool.php?)35 using a cutoff of 50% amino-acid identity and 80% of amino-acid alignment coverage. Synteny statistics were obtained using the MicroScope PkGDB synteny statitistics tool35 (https://www.genoscope.cns.fr/agc/microscope/compgenomics/GOstats.php?). Putative orthologous relations based on the bi-directional best hit (BBH) criterion were considered for at least 35% of sequence identity on 80% of the length of the smallest protein. For the synteny analysis, all possible kinds of chromosomal rearrangements are allowed (inversion, insertion/deletion) and the gap parameter, representing the maximum number of consecutive genes which are not involved in a synteny group, is set to five genes.
Determination of genes unique to clinical isolates of C. canimorsus
Clinical C. canimorsus unique genes were found using the MicroScope Comparative genomics Gene phyloprofile tool (https://www.genoscope.cns.fr/agc/microscope/compgenomics/phyloprofil.php?) searching for homologs in all the four C. canimorsus clinical isolates genomes (Cc2, Cc5, Cc11, and Cc12) excluding all homologs in the three C. canis genomes (CcD38, CcD93, CcD95) choosing as homology constraints parameters: minLrap ≥ 0.8; maxLrap ≥ 0; Identity ≥ 30%.
Phylogenetics of 15 Capnocytophaga genomes
Clusters of orthologs were computed with orthoMCL (PMID: 12952885) from the genomes of C. ochracea F0287 (NC_013162), C. ochracea DSM7271 (CP001632), Cc5 (NC_015846), Cc2 (CDOJ00000000), Cc11 (NZ_CDOI00000000), Cc12 (NZ_CDOE00000000), C. sputigena ATCC 33612 (NZ_ABZV00000000), C. gingivalis ATCC 33624 (NZ_ACLQ00000000), Capnocytophaga sp. oral taxon 338 str. F0234 (NZ_AEXX00000000), Ccyn 2B (NZ_CDOD00000000), C. cynodegmi ATCC 49044 (NZ_CDOF00000000), Ccyn 74 (NZ_CDOG00000000), CcD38, CcD93 (NZ_CDOL00000000), and CcD95 (NZ_CDOH00000000).27,28,29 Orthologous group including paralogs or displaying high length variability (below or above 80% length discrepancy) were discarded from the analysis. Single cluster phylogenies were computed with PROML (http://evolution.genetics.washington.edu/phylip/doc/proml.html) with a Henikoff/Tillier probability matrix from blocks (PMB) distance model, Gamma distributed variation rates and default options. 771 trees were generated and used to compute a consensus tree with CONSENSE (PHYLIP version 3.6. (http://evolution.genetics.washington.edu/phylip/doc/consense.html)) using the Extended majority rule.36
Growth in heat-inactivated human serum (HIHS)
Growth assays were performed in 96-well plates. Inocula were prepared from cultures grown on sheep blood plates, set to an OD600 of 0.2, and serially diluted 1:10 four times. 22 μL of bacterial suspensions were used to inoculate 200 μL of HIHS.
HIHS was supplemented with iron (III) citrate (FeC6H5O7; 0.25 mM) if required.
Equivalent volumes of inocula were also plated in order to precisely determine bacterial concentrations by colony-forming unit (CFU) counting at the inoculation time point. Infected HIHS were then incubated statically for 23 hours at 37 °C in the presence of 5% CO2. Serial dilutions were plated on sheep blood plates, and CFU were determined. The number of generations was calculated according to the following formula: CFU in the well = inoculum × 2Number of generations.
Catalase and oxidase activity tests
Bacteria were grown for 2 days on blood agar plates and colonies were resuspended in a drop of H2O2. The formation of bubbles was considered as indication of catalase activity. For monitoring the cytochrome-oxidase activity, bacterial colonies were spread on oxidase test discs (Sigma, Saint-Louis, MO, USA). The formation of indophenol blue, observed within 2 minutes at 25–30 °C, was considered as indication of oxidase activity.
Human IgM deglycosylation analysis by lectin staining
Bacteria were collected from blood agar plates and resuspended in PBS at OD600 = 1. 100 µL of bacterial suspensions, corresponding to 5 × 107 bacteria, were then incubated with 100 µL of a purified human IgM (I8260 Sigma) solution (0.05 mg mL−1) for 4 hours at 37 °C. As negative control, 200 µL of 1/2 diluted IgM solution alone was incubated for 4 hours at 37 °C. Samples were then centrifuged for 5 minutes at 13 000g, supernatant collected and loaded in a 12% sodium dodecyl sulfate gel. Samples were analyzed by coomassie staining, western-blot using goat anti human IgM antibodies (AbD Serotec, STAR145, Hercules, CA, USA) and lectin staining with SNA according to manufacturer recommendations (DIG Glycan Differentiation Kit, 11210238001, Roche, Basel, Switzerland).
Ethics statement
Human blood was collected from healthy volunteers who had signed a written informed consent. The experiments were approved by the Comité d'éthique médicale from the CHU Mont-Godinne/Université de Louvain (no: B039201316262).
The sampling of dog's saliva was done by the dogs' owners themselves, after they have been duly informed about the goal of the experiment. The sampling of dogs saliva was approved by the “Commission d'éthique en expérimentation animale, CEEXPANI” of the University of Namur under the N° 13/205 and title C. canimorsus.
The CEEXPANI committee works according to the Belgian law AR 29/05/13, which is the national implementation of the EU Directive 2010/63.
Results
16S rDNA phylogeny identifies two main groups of C. canimorsus
In a previous work,8 we isolated 62 C. canimorsus strains from dogs in Switzerland (areas of Basel, Lausanne, and Valais) irrespective of race, age, and sex of the animal. For the present study, we isolated 23 new C. canimorsus strains from dogs in Belgium (areas of Namur, Liege, and Charleroi) and one strain from a dog in Italy (Sardinia). We also collected 19 strains of C. canimorsus isolated from severe human infections worldwide (Supplementary Table S1), including the C. canimorsus ATCC35979 type strain. We sequenced the 16S rDNA from the new isolates and we generated a phylogeny tree of the 105 strains (86 isolated from dogs and 19 isolated from patients) (Figure 1). The phylogeny tree clearly revealed the presence of two main C. canimorsus groups as already observed with a smaller set of dog strains isolated in Switzerland8 and with a set of strains isolated in Japan.37 Interestingly the new data confirmed that all the strains isolated from patients localized in the same group (group I). Thus, one group included 19 clinical isolates and 47 strains from dogs while the other (group II) contained 34 strains, all from dogs (Figure 1). Five dog isolates (group III) localized separately from group I and group II C. canimorsus (Figure 1) but were not further analyzed in this study.
Figure 1.

16S rDNA majority consensus tree. The C. canimorsus human isolates are indicated by *. The strains whose genome has been sequenced are indicated by #. Flavobacterium johnsoniae (accession number M59051) was selected as an outgroup (software: RDP Tree Builder).
Since the 16S rRNA tree topology strongly suggested that the two main phylogenetic groups of C. canimorsus strains could represent two different species, with all the clinical isolates belonging to the same one, we decided to further investigate this hypothesis.
Whole-genome analyses confirm the evidence of a novel Capnocytophaga species
Since the species concept for bacteria and Archaea is based on DDH we took advantage of the recent sequencing of 10 Capnocytophaga genomes,26,27,28,29 and we predicted in silico DDH scores for genome-to-genome comparisons. The 10 genomes were from four clinical isolates of C. canimorsus (Cc2, Cc5, Cc11, and Cc12 (ATCC35979)) (group I), three C. canimorsus dog strains from group II (CcD38, CcD93, and CcD95), and three strains of C. cynodegmi (Ccyn 2B, Ccyn ATCC49044, and Ccy74).26,27,28,29 We first determined the ANI, a similarity index between a given pair of genomes that can be applicable to prokaryotic organisms independently of their GC content.33 The average pairwise similarity between coding sequences (CDS) conserved between the compared genomes was calculated using the ANI calculator from the Kostas Konstantinidis laboratory (Georgia Institute of Technology) Lab Tools (http://enve-omics.ce.gatech.edu/ani/).33 With this approach, two genomes with an ANI > 95% are considered as from the same species, while those with ANI below this threshold, belong to separate species.38,39 As shown in Table 1, all ANIs between the three group II strains (CcD38, CcD93, and CcD95) and the group I Cc12 C. canimorsus type strain (ATCC35979) were below 95% suggesting that indeed CcD38, CcD93, and CcD95 belong to a different species than the C. canimorsus type strain. The ANIs between CcD38, CcD93, and CcD95 were all above 98% (Table 1), suggesting that the three group II strains belong to the same species. The ANIs between all the four group I C. canimorsus strains (Cc2, Cc5, Cc11, Cc12 ATCC35979) were between 97.74% and 98.50% indicating that all these strains belong to the same species (Table 1). Furthermore, the ANIs between the three group II strains and the C. cynodegmi type strain ATCC49044 (Ccyn ATCC49044) were all below 88% suggesting that group II bacteria belong to a species different from C. cynodegmi. Finally, the ANIs between the two C. cynodegmi strains, Ccy74 and Ccyn 2B, and the type strain Ccyn ATCC49044 were 99.48% and 96.71% respectively, confirming the homogeneity of the C. cynodegmi species. In conclusion, according to the ANI comparison shown in Table 1, group II bacteria belong to a species different from C. canimorsus and C. cynodegmi.
Table 1. Average nucleotide identity (ANI).
| Strain ID | CcD38 | CcD93 | CcD95 | Cc2 | Cc5 | Cc11 | Cc12 ATCC35979 | Ccyn 2B | Ccy74 | Ccyn ATCC49044 |
|---|---|---|---|---|---|---|---|---|---|---|
| CcD38 | 100% | |||||||||
| CcD93 | 98.58% | 100% | ||||||||
| CcD95 | 98.87% | 98.56% | 100% | |||||||
| Cc2 | 91.27% | 91.16% | 91.33% | 100% | ||||||
| Cc5 | 90.71% | 90.54% | 90.89% | 98.50% | 100% | |||||
| Cc11 | 89.67% | 89.59% | 89.88% | 97.75% | 97.81% | 100% | ||||
| Cc12 ATCC35979 | 89.64% | 90.05% | 90.25% | 97.79% | 97.86% | 97.74% | 100% | |||
| Ccyn 2B | 87.56% | 87.06% | 87.55% | 91.52% | 91.52% | 91.57% | 91.32% | 100% | ||
| Ccy74 | 87.12% | 86.90% | 87.31% | 91.35% | 91.45% | 91.59% | 91.83% | 96.70% | 100% | |
| Ccyn ATCC49044 | 87.40% | 86.88% | 87.62% | 91.48% | 91.56% | 91.53% | 91.63% | 96.71% | 99.48% | 100% |
We also looked at the genomic distances between our sequenced strains using the Genome to Genome Distance Calculator (GGDC) 2.0 (http://ggdc.dsmz.de/distcalc2.php).34 A practical advantage of GGDC over ANI is that it operates on the same scale as wet-lab DDH values, which facilitates comparisons. Genomes with a DDH ≥ 70% are considered to be of the same species. The results, presented in Table 2, clearly indicate that indeed CcD38, CcD93, and CcD95 belong to the same species, different from C. canimorsus and from C. cynodegmi. Similarly, all group I strains appeared as belonging to the same species, as did the C. cynodegmi strains.
Table 2. GGDC probability that DDH is ≥ 70% (i.e. same species).
| Strain ID | CcD38 | CcD93 | CcD95 | Cc2 | Cc5 | Cc11 | Cc12 ATCC35979 | Ccyn ATCC49044 | Ccy74 | Ccyn 2B |
|---|---|---|---|---|---|---|---|---|---|---|
| Cc12 ATCC35979 | 5.18% | 6.49% | 6.29% | 90.31% | 90.72% | 90.21% | 98.3% | 7.12% | 6.62% | 6.28% |
| Ccyn ATCC49044 | 1% | 0.83% | 1.06% | 9.62% | 6.31% | 7.35% | 7.12% | 98.3% | 97.31% | 81.61% |
| CcD38 | 98.3% | 94.18% | 95.48% | 10.12% | 8.96% | 7.19% | 5.18% | 1% | 1.26% | 0.94% |
| CcD93 | 94.18% | 98.3% | 93.97% | 11.19% | 9.43% | 9.57% | 6.49% | 0.83% | 0.79% | 0.76% |
We then looked at gene synteny using the MicroScope PkGDB synteny statistics tool.35 The results, reported in Supplementary Table S2, show that more than 80% of the genes of the Cc12 type strain are in synteny with genes in Cc5, Cc2, and Cc11 genomes (group I) but only less than 50% of the Cc12 genes were in synteny with group II bacteria CcD93, CcD95, and CcD38. On the contrary, around 90% of the CcD38 genes were in synteny with genes of CcD93 and CcD95 but only 50% of the CcD38 genes were in synteny with genes in C. cynodegmi or group I C. canimorsus genomes. Finally, C. cynodegmi ATTCC49044 had more than 80% synteny with Ccy74 and Ccyn 2B but less than 50% with the group I and group II C. canimorsus. Thus, group I clinical isolates of C. canimorsus share a common gene organization different from the one found in group II strains and in C. cynodegmi.
Finally, we built a consensual phylogenetic tree of the Capnocytophaga genus considering 771 groups of orthologues with a single representative in each of the 10 genomes considered before and five genomes from human Capnocytophaga species (Figure 2). Clinical isolates (group I C. canimorsus), group II C. canimorsus isolates and C. cynodegmi isolates separately formed three clades in the consensual phylogeny and these three clades were supported by a majority of tree topologies (677, 729, and 686 respectively) (Figure 2). Interestingly, group II isolates and clinical isolates did not cluster together in a majority of trees.
Figure 2.

Consensus phylogeny tree of Capnocytophaga spp. based on the 771 most conserved orthologs. Extended majority rule consensus tree derived from trees of 771 clusters of orthlogs exhibiting a single representative protein per genome (unrooted). The number on the branches indicate the number of times the partition of the species into the two sets which are separated by that branch occurred among the trees, out of the 771 trees. This also corresponds to the number of protein trees that followed the topology displayed here. Consensus majority values higher and lower than 80% are displayed in black and gray font respectively.
Finally, the GC content of the C. canimorsus ATCC35979 type strain Cc12 is 36.23% and the other group I strains have a GC content ranging from 36.08% to 36.23%. The GC content of the three group II strains ranges from 35.48% to 35.61%. The three C. cynodegmi strains have a GC content between 34.49% and 34.42%. The GC content of the two C. canimorsus groups is thus different from each other and different from that of C. cynodegmi, although the differences are not striking (Supplementary Table S3).
Taken all together, our results indicate that group II C. canimorsus strains belong to a novel Capnocytophaga species for which we propose the name C. canis (canis: Latin = dog). Since group I includes the ATCC C. canimorsus-type strain, it keeps the C. canimorsus name.
C. canis is taxonomically closer from C. cynodegmi than from C. canimorsus
As shown in Figure 3, the three C. canis strains share 1875 gene families (C. canis core genome) corresponding to 75–85% of the CDS while the three C. cynodegmi strains share 1910 gene families (C. cynodegmi core genome) corresponding to 80% of the CDS of Ccyn ATCC49044. In contrast, the four C. canimorsus strains share only 1294 gene families (C. canimorsus core genome) corresponding to 50% of the CDS of the type strain Cc12. These results indicate that the C. cynodegmi and C. canis species are more homogeneous than the C. canimorsus species. We then looked at the number of shared gene families between the three dog Capnocytophaga species. C. canimorsus and C. canis share 1020 gene families while C. canimorsus and C. cynodegmi share 1047 gene families and finally, C. canis and C. cynodegmi share 1404 CDS. Thus, C. canis appears to be taxonomically closer to C. cynodegmi than to C. canimorsus.
Figure 3.
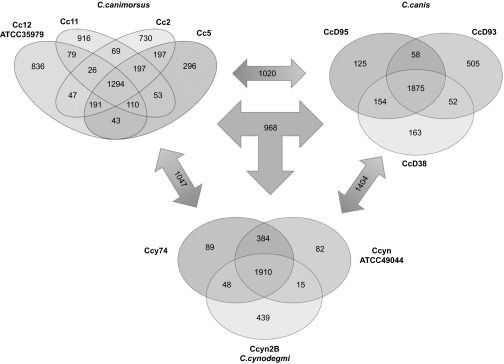
Core/Pan genome comparison between C. canimorsus, C. canis, and C. cynodegmi sequenced strains. Genome comparison was performed using the MicroScope Comparative Genomics Pan/core genome tool. Numbers correspond to gene families shared by the different genomes. Numbers in double arrows indicate families shared by two species while numbers in the triple arrow correspond to gene families found in all three groups.
Human isolates of C. canimorsus share a set of genes that are absent in C. canis
None of the 19 C. canimorsus strains isolated from human infections, worldwide, belong to the new C. canis species, suggesting that C. canis are less pathogenic than C. canimorsus. We thus questioned whether we could identify the genes underlying the capacity of C. canimorsus to infect and survive in the human host. With this aim, we compared the genomes of the four clinical C. canimorsus strains to those of the three C. canis strains searching for homologs in all the four clinical C. canimorsus genomes (Cc2, Cc5, Cc11, and Cc12) absent in the three C. canis genomes (CcD38, CcD93, CcD95). This comparative analysis identified a total of 256 genes shared only by C. canimorsus clinical isolates (Supplementary Table S4).
For only 118 genes (46%) a Clusters of Orthologous Groups functional annotation was found: metabolism (45 genes); cellular processes and signaling (21 genes); information storage and processing (18 genes); poorly characterized (34 genes). The genes involved in metabolic processes mainly belong to the classes: inorganic ions transport and metabolism; amino acid transport and metabolism; energy production and conversion. The genes involved in cellular processes and signaling belong mainly to the classes: cell cycle control, cell division, chromosome partitioning; cell wall/membrane/envelope biogenesis; posttranslational modification, protein turnover, chaperones; signal transduction mechanisms. The genes involved in information storage and processing mainly belong to the replication, recombination, and repair functional class. Interestingly among the genes, shared only by the human isolates of C. canimorsus, were 22 genes belonging to 9 of the 13 C. canimorsus 5 encoded PUL, namely: PUL2 (four genes), PUL3 (four genes), PUL4 (two genes), PUL5 (one gene), PUL6 (two genes), PUL7 (five genes), PUL9 (two genes), PUL11 (one gene), and PUL13 (one gene). PUL3 allows iron acquisition from transferrin20 and PUL5 encodes the N-glycoprotein deglycosylation system,19 two functions that contribute to survival in the host20,22 and may be considered as virulence factors. The function of the other PUL is still unknown.
C. canimorsus and C. canis have a different capacity to proliferate in human serum
Since PUL3 encodes an iron capturing system, we questioned whether the two species, C. canimorsus and C. canis, might differ in their capacity to acquire iron from human transferrin. Since 69 strains from our collection have already been tested for growth in HIHS,20 we sorted the strains between the C. canimorsus and C. canis species and we plotted the results (Figure 4 and Supplementary Table S5). Out of 46 C. canimorsus strains, 40 produced more than eight generations in HIHS. In contrast, out of 23 strains, now identified as C. canis, only one (CcD36) grew vigorously.
Figure 4.

Growth of C. canimorsus and C. canis in human serum. Number of generations achieved after 23 hours in HIHS for C. canimorsus (triangles) and C. canis (circles) strains. C. canimorsus clinical isolates are represented by gray triangles.
When free iron was supplemented to the strains that grew poorly, growth was improved but, with the exception of only one strain (CcD93), not to the level of the human isolates, indicating that it is not only their inability to obtain iron that limits their proliferation (Supplementary Figure S1). In conclusion, this in vitro experiment thus supports the hypothesis that C. canis is less virulent for humans than C. canimorsus.
C. canimorsus and C. canis differ for the cytochrome-oxidase activity
According to their initial description, C. canimorsus ferment sucrose and raffinose while C. cynodegmi do not.1 In a previous study, we extended this observation on 66 C. canimorsus strains but noticed that a few strains, isolated from dogs could ferment sucrose and raffinose.8 We now sorted the previously tested strains between the C. canimorsus and C. canis species. The results, reported in Supplementary Table S6, show that only one C. canimorsus out of 44 was able to ferment raffinose and only two were fermenting sucrose. Of the 22 C. canis, only two could ferment raffinose and nine fermented sucrose. Thus from these data, the ability to ferment sucrose and raffinose does not allow to discriminate C. canis from C. canimorsus.
The other phenotypic traits reported for C. canimorsus include positive catalase and cytochrome-oxidase reactions.1 We thus tested on our collection whether these reactions would allow discriminating C. canimorsus from C. canis.
Our data show no difference between C. canimorsus and C. canis for the catalase test (Supplementary Table S7). In contrast, 60 out of 66 (91%) of the C. canimorsus strains were cytochrome-oxidase positive while only one of the 34 C. canis (2.9%) was positive. Interestingly, all the C. canimorsus isolated from human infections were oxidase positive. This finding correlates with the genomic comparison data showing the presence of cytochrome C oxidase orthologous genes only in the genomes of C. canimorsus (Supplementary Table S4). Interestingly, the six C. canimorsus canine isolates which were oxidase negative, were previously tested for growth in human serum20 and found to be unable to proliferate (Supplementary Table S5). All together this suggests that the oxidase activity could contribute to the pathogenicity of C. canimorsus.
Most C. canis bacteria are unable to cleave human IgM N-glycans
Thanks to the PUL5-encoded Gpd membrane complex, Cc5 bacteria cleave complex N-glycans from host cell membranes and soluble host glycoproteins such as human IgGs and transferrin.19,20 Host glycoprotein deglycosylation is crucial for the growth of Cc5 in co-culture with eukaryotic cells and for persistence in a mouse model and hence can be considered as a pathogenicity factor.22 Whether C. canimorsus can deglycosylate human IgM is not known yet. We thus monitored the capacity of C. canimorsus and C. canis strains to cleave human IgM N-glycans. To this aim we incubated human IgM with bacteria in suspension in PBS and then monitored the glycosylation state of IgM via SNA lectin staining and western blot. Incubation of IgM with wt Cc5 bacteria determined a dramatic reduction of SNA staining (Figures 5A and 5B) and a size shift of the IgM heavy chain (Figures 5C and 5D) indicating deglycosylation while incubation with PUL5 mutant bacteria did not affect the glycosylation state of IgM showing that IgM deglycosylation by Cc5 is only PUL5 dependent. Out of the 66 C. canimorsus strains of our collection, 48 strains (72.7%) cleaved human IgM glycans while 18 strains (27.3%) did not (Figure 5E). Among the 19 human isolates of C. canimorsus, 15 (79%) cleaved IgM glycans while only 4 (21%) did not. In contrast, out of the 34 C. canis strains, only one (2.9%) deglycosylated human IgM while all the other 33 strains (97.1%) did not (Figure 5F). Interestingly, the only strain of C. canis that was able to deglycosylate IgM (CcD123) was also the only one which gave a positive oxidase test suggesting that this strain is somewhat different from the other C. canis and closer to the C. canimorsus strains. Thus, among the C. canimorsus and C. canis strains, the proportion of strains able to cleave IgM N-glycans was very different, reinforcing the hypothesis that C. canis are less pathogenic for humans than C. canimorsus. This finding correlates with the genomic data which showed the absence of the PUL5 encoded endo-β-N-acetylglucosaminidase (GpdG) enzyme in the three C. canis genomes analyzed.
Figure 5.

IgM deglycosylation by C. canimorsus and C. canis. Glycosylation state of human IgM samples incubated for 4 hours in the presence of Cc5 and Cc5 ΔPUL5 bacteria monitored by staining with SNA (A). Schematic drawing of the SNA lectin target oligosaccharides (B). Coomassie staining (C) and western blot analysis with anti-IgM antibodies (D) of human IgM samples incubated in the presence of Cc5 and Cc5 ΔPUL5 bacteria. Prevalence of IgM deglycosylating strains in C. canimorsus (E) and C. canis (F).
In conclusion, our data suggest that the ability to cleave host N-glycoproteins via the Gpd complex could be considered as a pathogenicity factor, which discriminates C. canimorsus from C. canis.
16S rRNA species-specific PCR amplification does not detect C. canis
In 2010, Suzuki et al. described a rapid and sensitive species-specific PCR method that allows the detection of dog hosted Capnocytophaga and the discrimination between C. canimorsus and C. cynodegmi.9 We tested whether the three different oligonucleotides combinations of this method would also detect C. canis. As shown in Supplementary Figure S2, no amplification was detected when C. canis DNA was used as template suggesting that this method does not detect C. canis. We thus tested whether the C. canimorsus specific oligonucleotides combination (CaL2-CaR) would detect all the C. canimorsus of our collection. The results reported in Supplementary Table S8 show that out of the 66 C. canimorsus strains of our collection, 57 (86.4%) were detected by PCR and the 19 clinical isolates were all among them. Out of the 34 C canis strains, only four were detected as C. canimorsus by this PCR assay. Thus, our data indicate that this PCR method can be efficiently used for the rapid detection of C. canimorsus in clinical samples.
Discussion
C. canimorsus are part of the normal dog mouth flora and hence a large proportion of the population is in contact with this pathogen. Simple licks are sufficient to transmit the disease and, yet, the disease is very rare. Part of the answer resides in the immune status of the patient. Indeed, roughly half of them have a damaged liver or had splenectomy but half of them have no known medical history. We thus investigated whether only a subpopulation of C. canimorsus would be pathogenic for humans. Since there is, so far, no fully reliable animal model, the answer can only be brought by comparing strains isolated from human septicemia to strains isolated from healthy dogs. In 2009, we already observed that, based on 16S rRNA sequences C. canimorsus strains fall in two groups8 and this finding was confirmed more recently, by Umeda et al.37 In both reports, the few strains isolated from patients felt in the same group. These studies prompted us to analyze a large number of strains by 16S rDNA sequencing and then to analyze the full genome of representatives of the two subgroups, including the ATCC35979 type strain of C. canimorsus.
Our study confirmed that the sequences of 16S rDNA divide the isolates of C. canimorsus in two distinct phylogenetic groups. This division was reinforced by genome to genome comparison: (i) the computed DDH indicated a percentage of hybridization below 70% between the two groups; (ii) the ANI confirmed that strains from group II could represent a species different from that of the C. canimorsus type strain; (iii) the analysis of synteny, although not a classical taxonomy tool, reinforced the view that the C. canimorsus species could be split into two new species. Keeping the name C. canimorsus for the group that includes the type strain, we propose to call C. canis the new species. This proposal is highly relevant if one considers pathogenesis. Indeed, all the strains isolated from severe human infections are C. canimorsus, suggesting that C. canis, like C. cynodegmi, is not a dangerous pathogen for humans. This discrimination may help in the understanding of pathogenesis. Thus, in order to identify the genes underlying the capacity of C. canimorsus to infect and survive in the human host, we compared the genomes of the four clinical C. canimorsus strains to those of the three C. canis strains and we found a set of 256 genes that are unique to C. canimorsus. Interestingly, among them, 22 genes belonged to PUL and in particular we found members of the PUL3 encoded iron capturing system and the PUL5 encoded Gpd glycoprotein deglycosylation system. We showed that indeed, thanks to PUL3, C. canimorsus grow better in HIHS than C. canis and that C. canimorsus, via the Gpd system, generally deglycosylate human immunoglobulins M while very few strains of C. canis do. Although these two phenotypes are probably required to confer virulence to a strain, there must be other virulence factors and the discrimination between pathogenic C. canimorsus and non-pathogenic C. canis will help to identify these. Indeed, it drastically improves a comparative genomic analysis by offering a new closely related species that is non-pathogenic. In addition, a better identification of potentially pathogenic strains may open the door to prevention in some special situation. As an example, it would be worthwhile testing the mouth flora of dogs that are trained to help handicapped people or that are pets of people at risk. In this view, it is important to have reliable and robust methods to discriminate C. canimorsus, C. canis, and C. cynodegmi. The cytochrome-oxidase test could be one of them. We also evaluated the species-specific PCR method developed by Suzuki et al.9 This method identified correctly all the C. canimorsus human isolates, but there was a limited discrepancy in the identifications for dog isolates. Thus, we would recommend to identify potentially dangerous isolates by PCR of the 16S rDNA.
Finally, Umeda et al.37 included cat's strains in their study and they showed that these strains all belong to the group that we propose to call C. canis. This observation would tend to suggest that cat strains are not pathogenic for humans but this is contradicted by some case reports.40,41 A possible explanation is that the proportion of C. canimorsus versus C. canis is lower in cats than in dogs. This would also fit with the observation that the large majority of human infections are caused by dogs. For instance, among our 19 clinical isolates none comes from a cat transmitted infection.
Acknowledgments
This work was supported by grants 3100A0-128659 from the Swiss National Science Foundation and ERC 2011-ADG 20110310 from the European Research Council to Guy Richard Cornelis. Francesco Renzi is a research fellow (chargé de recherche) of the Belgian «Fonds de la recherche scientifique» (FNRS). We are grateful to Jean-Yves Matroule, Claire Diederich, and all the members of the URBM research unit (Université de Namur) for their help in raising the collection of dogs' strains. We also thank: G. Wauters, M. Delmée, Y. Glupczynski, D. Huang, and A. Magnette (Université Catholique de Louvain); O. Denis (Université Libre de Bruxelles); R. Jarsumbeck (Medizinisches Labor Ostsachsen, Dresden); K. Mühlemann (Universität Bern); J. Schrenzel (Université de Genève); F. S. Stals (Laurentius Ziekenhuis, Roermond) for providing us clinical strains.
Footnotes
Supplementary information of this article can be found on the Emerging Microbes & Infections' website (http://www.nature.com/emi).
Supplementary Information
References
- 1Brenner DJ, Hollis DG, Fanning GR et al. Capnocytophaga canimorsus sp. nov. (formerly CDC group DF-2), a cause of septicemia following dog bite, and C. cynodegmi sp. nov., a cause of localized wound infection following dog bite. J Clin Microbiol 1989; 27: 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2Tierney DM, Strauss LP, Sanchez JL. Capnocytophaga canimorsus mycotic abdominal aortic aneurysm: why the mailman is afraid of dogs. J Clin Microbiol 2006; 44: 649–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3Leadbetter ER, Holt SC, Socransky SS. Capnocytophaga: new genus of gram-negative gliding bacteria. I. General characteristics, taxonomic considerations and significance. Arch Microbiol 1979; 122: 9–16. [DOI] [PubMed] [Google Scholar]
- 4Dilegge SK, Edgcomb VP, Leadbetter ER. Presence of the oral bacterium Capnocytophaga canimorsus in the tooth plaque of canines. Vet Microbiol 2011; 149: 437–445. [DOI] [PubMed] [Google Scholar]
- 5Lipman L, Tienhoven N, Gaastr W. [The presence of Capnocytophaga canimorsus and Capnocytophaga cynodegmi in companion animals in the Netherlands]. Tijdschr Diergeneeskd 2011; 136: 490–492. Dutch. [PubMed] [Google Scholar]
- 6van Dam AP, van Weert A, Harmanus C et al. Molecular characterization of Capnocytophaga canimorsus and other canine Capnocytophaga spp. and assessment by PCR of their frequencies in dogs. J Clin Microbiol 2009; 47: 3218–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7Blanche P, Bloch E, Sicard D. Capnocytophaga canimorsus in the oral flora of dogs and cats. J Infect 1998; 36: 134. [DOI] [PubMed] [Google Scholar]
- 8Mally M, Paroz C, Shin H et al. Prevalence of Capnocytophaga canimorsus in dogs and occurrence of potential virulence factors. Microbes Infect 2009; 11: 509–514. [DOI] [PubMed] [Google Scholar]
- 9Suzuki M, Kimura M, Imaoka K et al. Prevalence of Capnocytophaga canimorsus and Capnocytophaga cynodegmi in dogs and cats determined by using a newly established species-specific PCR. Vet Microbiol 2010; 144: 172–176. [DOI] [PubMed] [Google Scholar]
- 10Shin H, Mally M, Meyer S et al. Resistance of Capnocytophaga canimorsus to killing by human complement and polymorphonuclear leukocytes. Infect Immun 2009; 77: 2262–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11Bobo RA, Newton EJ. A previously undescribed gram-negative bacillus causing septicemia and meningitis. Am J Clin Pathol 1976; 65: 564–569. [DOI] [PubMed] [Google Scholar]
- 12Butler T, Weaver RE, Ramani TK et al. Unidentified gram-negative rod infection. A new disease of man. Ann Intern Med 1977; 86: 1–5. [DOI] [PubMed] [Google Scholar]
- 13Gaastra W, Lipman LJ. Capnocytophaga canimorsus. Vet Microbiol 2010; 140: 339–346. [DOI] [PubMed] [Google Scholar]
- 14Janda JM, Graves MH, Lindquist D et al. Diagnosing Capnocytophaga canimorsus infections. Emerg Infect Dis 2006; 12: 340–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15Butler T. Capnocytophaga canimorsus: an emerging cause of sepsis, meningitis, and post-splenectomy infection after dog bites. Eur J Clin Microbiol Infect Dis 2015; 34: 1271–1280. [DOI] [PubMed] [Google Scholar]
- 16Shin H, Mally M, Kuhn M et al. Escape from immune surveillance by Capnocytophaga canimorsus. J Infect Dis 2007; 195: 375–386. [DOI] [PubMed] [Google Scholar]
- 17Ittig S, Lindner B, Stenta M et al. The lipopolysaccharide from Capnocytophaga canimorsus reveals an unexpected role of the core-oligosaccharide in MD-2 binding. PLoS Pathog 2012; 8: e1002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18Zahringer U, Ittig S, Lindner B et al. NMR-based structural analysis of the complete rough-type lipopolysaccharide isolated from Capnocytophaga canimorsus. J Biol Chem 2014; 289: 23963–23976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19Renzi F, Manfredi P, Mally M et al. The N-glycan glycoprotein deglycosylation complex (Gpd) from Capnocytophaga canimorsus deglycosylates human IgG. PLoS Pathog 2011; 7: e1002118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20Manfredi P, Lauber F, Renzi F et al. New iron acquisition system in Bacteroidetes. Infect Immun 2015; 83: 300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21Mally M, Shin H, Paroz C et al. Capnocytophaga canimorsus: a human pathogen feeding at the surface of epithelial cells and phagocytes. PLoS Pathog 2008; 4: e1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22Manfredi P, Renzi F, Mally M et al. The genome and surface proteome of Capnocytophaga canimorsus reveal a key role of glycan foraging systems in host glycoproteins deglycosylation. Mol Microbiol 2011; 81: 1050–1060. [DOI] [PubMed] [Google Scholar]
- 23Shipman JA, Berleman JE, Salyers AA. Characterization of four outer membrane proteins involved in binding starch to the cell surface of Bacteroides thetaiotaomicron. J Bacteriol 2000; 182: 5365–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24Renzi F, Manfredi P, Dol M et al. Glycan-foraging systems reveal the adaptation of Capnocytophaga canimorsus to the dog mouth. MBio 2015; 6: e02507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 2008; 4: 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26Manfredi P, Pagni M, Cornelis GR. Complete genome sequence of the dog commensal and human pathogen Capnocytophaga canimorsus strain 5. J Bacteriol 2011; 193: 5558–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27Manfredi P, Renzi F, Cornelis GR. Draft genome sequences of three Capnocytophaga cynodegmi strains isolated from the oral cavity of healthy dogs. Genome Announc 2015; 3: pii: e00200–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28Manfredi P, Renzi F, Cornelis GR. Draft genome sequences of three Capnocytophaga canimorsus strains isolated from healthy canine oral cavities. Genome Announc 2015; 3: pii: e00199–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29Manfredi P, Renzi F, Cornelis GR. Draft genome sequences of three Capnocytophaga canimorsus strains isolated from septic patients. Genome Announc 2015; 3: pii: e00193–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30Lane DJ. 16S/23S rRNA sequencing. In: Stackebrandt EG, Goodfellow M (eds.), Nucleic Acid Techiques in Bacterial Systematics. New York: John Wiley & Sons, 1991: 115–174. [Google Scholar]
- 31Johnson JL. Similarity analysis of rRNAs. In: Gerhard P, Murray RGE, Wood GA, Krieg NR (eds.), Methods for General and Molecular Bacteriology. Washington, DC: American Society for Microbiology, 1994: 683–700. [Google Scholar]
- 32Cole JR Wang Q, Fish JA et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 2014; 42: D633–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33Goris J, Konstantinidis KT, Klappenbach JA et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 2007; 57: 81–91. [DOI] [PubMed] [Google Scholar]
- 34Meier-Kolthoff JP, Auch AF, Klenk HP et al. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 2013; 14: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35Vallenet D, Labarre L, Rouy Z et al. MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res 2006; 34: 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36Felsenstein J. Using the quantitative genetic threshold model for inferences between and within species. Philos Trans R Soc Lond B Biol Sci 2005; 360: 1427–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37Umeda K, Hatakeyama R, Abe T et al. Distribution of Capnocytophaga canimorsus in dogs and cats with genetic characterization of isolates. Vet Microbiol 2014; 171: 153–159. [DOI] [PubMed] [Google Scholar]
- 38Konstantinidis KT, Ramette A, Tiedje JM. The bacterial species definition in the genomic era. Philos Trans R Soc Lond B Biol Sci 2006; 361: 1929–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39Konstantinidis KT, Tiedje JM. Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr Opin Microbiol 2007; 10: 504–509. [DOI] [PubMed] [Google Scholar]
- 40Takegawa K, Sonozaki K, Nogami M et al. Four cases of Capnocytophaga canimorsus sepsis caused by dog and/or cat bites, 2007–2008—Kobe City. Infect Agent Surveill Rep 2010; 31: 109–110. [Google Scholar]
- 41Carpenter PD, Heppner BT, Gnann JW Jr. DF-2 bacteremia following cat bites. Report of two cases. Am J Med 1987; 82: 621–623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.