

Abstract
AIM: Activated pancreatic stellate cells (PSCs) are implicated in the pathogenesis of pancreatic inflammation and fibrosis, where oxidative stress is thought to play a key role. 4-hydroxy-2,3-nonenal (HNE) is generated endogenously during the process of lipid peroxidation, and has been accepted as a mediator of oxidative stress. The aim of this study was to clarify the effects of HNE on the activation of signal transduction pathways and cellular functions in PSCs.
METHODS: PSCs were isolated from the pancreas of male Wistar rats after perfusion with collagenase P, and used in their culture-activated, myofibroblast-like phenotype unless otherwise stated. PSCs were treated with physiologically relevant and non-cytotoxic concentrations (up to 5 μmol/L) of HNE. Activation of transcription factors was examined by electrophoretic mobility shift assay and luciferase assay. Activation of mitogen-activated protein (MAP) kinases was assessed by Western blotting using anti-phosphospecific antibodies. Cell proliferation was assessed by measuring the incorporation of 5-bromo-2’-deoxyuridine. Production of type I collagen and monocyte chemoattractant protein-1 was determined by enzyme-linked immunosorbent assay. The effect of HNE on the transformation of freshly isolated PSCs in culture was also assessed.
RESULTS: HNE activated activator protein-1, but not nuclear factor κB. In addition, HNE activated three classes of MAP kinases: extracellular signal-regulated kinase, c-Jun N-terminal kinase, and p38 MAP kinase. HNE increased type I collagen production through the activation of p38 MAP kinase and c-Jun N-terminal kinase. HNE did not alter the proliferation, or monocyte chemoattractant protein-1 production. HNE did not initiate the transformation of freshly isolated PSCs to myofibroblast-like phenotype.
CONCLUSION: Specific activation of these signal transduction pathways and altered cell functions such as collagen production by HNE may play a role in the pathogenesis of pancreatic disorders.
INTRODUCTION
In 1998, star-shaped cells in the pancreas, namely pancreatic stellate cells (PSCs), were identified and characterized[1,2]. They are morphologically similar to the hepatic stellate cells that play a central role in inflammation and fibrogenesis of the liver[3]. In normal pancreas, stellate cells are quiescent and can be identified by the presence of vitamin A-containing lipid droplets in the cytoplasm. In response to pancreatic injury or inflammation, they are transformed (“activated”) from their quiescent phenotype into highly proliferative myofibroblast-like cells which express the cytoskeletal protein α -smooth muscle actin (α -SMA), and produce type I collagen and other extracellular matrix components. Many of the morphological and metabolic changes associated with the activation of PSCs in animal models of fibrosis also occur when these cells are grown in serum-containing medium in culture on plastic. There is accumulating evidence that PSCs, like hepatic stellate cells, are responsible for the development of pancreatic fibrosis[1,2,4]. It has also been suggested that PSCs may participate in the pathogenesis of acute pancreatitis[4,5]. In view of their importance in pancreatic fibrosis and inflammation, it is of particular importance to elucidate the molecular mechanisms underlying their activation. The activation of signaling pathways such as p38 mitogen-activated protein (MAP) kinase[6] and Rho-Rho kinase pathway[7] is likely to play a central role in PSC activation. However, the precise intracellular signaling pathways in PSCs are largely unknown.
The role of oxidative stress in the development of acute and chronic pancreatitis has been clarified[8,9]. Reactive oxygen species and aldehydic end-products of lipid peroxidation, such as 4-hydroxy-2,3-nonenal (HNE), could act as mediators affecting signal transduction pathways, proliferation, and functional responses of target cells[10-12]. HNE is a specific and stable end product of lipid peroxidation. It could be produced in response to oxidative insults[13], and has been regarded to be responsible for many of the effects during oxidative stress in vivo[13,14]. It reacts with DNA and proteins, generating various forms of adducts (cysteine, lysine, and histidine residues) that are capable of inducing cellular stress responses such as cell signaling and apoptosis[14]. It has been shown that alcoholic chronic pancreatitis is characterized by the co-localization of evident lipid peroxidation, as assessed by immunoreactivity of HNE inside acinar cells, and the deposition of collagen type I in the fibrous septa and fibrotic stroma surrounding the pancreatic acini[15]. The stronger HNE immunoreactivity at the periphery of acini raised the possibility that HNE might affect the cell functions of PSCs, but the effects of HNE on the signaling pathways and the regulation of cell functions in PSCs are unknown. We here report that HNE at physiologically relevant and non-cytotoxic concentrations activated activator protein-1 (AP-1) and three classes of MAP kinases in PSCs. In addition, HNE increased type I collagen production from PSCs. Specific activation of these signal transduction pathways and altered cell functions such as collagen production by HNE may play a role in the pathogenesis of pancreatic disorders.
MATERIALS AND METHODS
Materials
3-(4, 5-dimethylthiazole-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was obtained from Dojindo (Kumamoto, Japan). Poly (dI.dC)-poly (dI.dC) and [γ -32P] ATP were purchased from Amersham Biosciences UK, Ltd. (Buckinghamshire, England). Collagenase P and recombinant human interleukin (IL)-1β were from Roche Applied Science (Mannheim, Germany). Double-stranded oligonucleotide probes for AP-1 and nuclear factor κB (NF-κB) were purchased from Promega (Madison WI). Rat recombinant platelet-derived growth factor (PDGF)-BB was from R&D Systems (Minneapolis, MN). Rabbit antibodies against phosphospecific MAP kinases, total MAP kinases, and inhibitor of NF-κB (IκB-α ) were purchased from Cell Signaling Technologies (Beverly, MA). Rabbit antibody against glyceraldehyde-3-phosphate dehydrogenase (G3PDH) was from Trevigen (Gaithersburg, MD). HNE, SB203580, SP600125, and U0126 were from Calbiochem (La Jolla, CA). All other reagents were from Sigma-Aldrich (St. Louis, MO) unless specifically described.
Cell culture
All animal procedures were performed in accordance with the National Institutes of Health Animal Care and Use Guidelines. Rat PSCs were prepared from the pancreas tissues of male Wistar rats (Japan SLC Inc., Hamamatsu, Japan) weighing 200-250 g as previously described using the Nycodenz solution (Nycomed Pharma, Oslo, Norway) after perfusion with 0.3 g/L collagenase P[16]. The cells were resuspended in Ham's F-12 containing 100 mL/L heat-inactivated fetal bovine serum (ICN Biomedicals, Aurora, OH), penicillin sodium, and streptomycin sulfate. Cell purity was always more than 90% as assessed by a typical star-like configuration and by detecting vitamin A autofluorescence. All experiments were performed using cells between passages two and five except for those using freshly isolated PSCs. Unless otherwise stated, we incubated PSCs in serum-free medium for 24 h before the addition of experimental reagents.
Cell viability assay
Cell viability was assessed by the MTT assay as previously described[17]. After treatment with HNE for 24 h, MTT solution was added to the cells at a final concentration of 500 μg/mL and the incubation continued at 37 °C for 4 h. After the incubation period, the medium was aspirated and the formazan product was solubilized with dimethylsulfoxide. Cell viability was determined by the differences in absorbance at wavelengths of 570 min 690 nm.
Intracellular peroxide production assay
Intracellular peroxide production was examined as previously described[14]. Briefly, cells were incubated with 2’,7’-dichlorofluorescin diacetate (DCF-DA) at 50 μmol/L for 30 min at 37 °C, and then treated with experimental reagents for an additional 30 min at 37 °C. After chilled on ice, cells were washed with ice-cold PBS, scraped from the plate, and resuspended at 1 × 10 6 cells/mL in PBS containing 10 mmol/L EDTA. The fluorescence intensities of 2’, 7’-dichlorofluorescin formed by the reaction of DCF-DA with peroxides of more than 10000 viable cells from each sample were analyzed by FACSCaliber flow cytometer (Becton Dickinson Co. Ltd., Tokyo, Japan) with excitation and emission settings of 488 and 525 nm, respectively. Prior to data collection, propidium iodide was added to the sample to gate out dead cells. Experiments were repeated at least twice with similar results. The data are expressed as one representative histogram.
Cell proliferation assay
Serum-starved PSCs (approximately 80% density) were treated with HNE at the indicated concentrations in the absence or presence of PDGF-BB (25 ng/mL). Cell proliferation was assessed using a commercial kit (Cell proliferation ELISA, BrdU; Roche Applied Science) according to the manufacturer’s instruction. This is colorimetric immunoassay based on the measurement of 5-bromo-2’-deoxyuridine (BrdU) incorporation during DNA synthesis[18]. After 24-hour-incubation with experimental reagents, cells were labeled with BrdU for 3 h at 37 °C, fixed, and incubated with peroxidase-conjugated anti-BrdU antibody. Then the peroxidase substrate 3,3’,5,5’-tetramethylbenzidine was added, and BrdU incorporation was quantitated by differences in absorbance at wavelength 370 min 492 nm.
Nuclear extracts preparation and electrophoretic mobility shift assay
Nuclear extracts were prepared and electrophoretic mobility shift assay was performed as previously described[19]. Double-stranded oligonucleotide probes for AP-1 (5’-CGCTTGATGAGT CAGCCGGAA-3’) and NF-κB (5’-AGTTGAGGGGACTT TCCCAGGC-3’) were endlabeled with [γ -32P]ATP. Nuclear extracts (approximately 5 μg) were incubated with the labeled oligonucleotide probe for 20 min at 22 °C, and electrophoresed through a 40 g/L polyacrylamide gel. Gels were dried, and autoradiographed at -80 °C overnight. A 100-fold excess of unlabeled oligonucleotide was incubated with nuclear extracts for 10 min prior to the addition of the radiolabeled probe in the competition experiments.
Luciferase assay
Luciferase assays were performed as previously described[20] using luciferase expression vectors containing two consensus AP-1 binding sites (TGACTCA), or two consensus NF-κB binding sites (GGGACTTTCC), which were kindly provided by Dr. Naofumi Mukaida (Kanazawa University, Japan). Approximately 1 × 10 6 PSCs were transfected with 2 μg of each luciferase expression vector, along with 40 ng of pRL-TK vector (Promega) as an internal control, using the FuGENE6 reagent (Roche Applied Science). After 24 h, the transfected cells were treated with HNE at 1 μmol/L for an additional 24 h. At the end of the incubation, cell lysates were prepared using Pica Gene kit (Toyo Ink Co., Tokyo, Japan), and the light intensities were measured using a model Lumat LB9507 Luminescence Reader (EG&G Berthold, Bad Wildbad, Germany).
Western blot analysis
Activation of MAP kinases was examined by Western blot analysis using anti-phosphospecific MAP kinase antibodies as previously described[21]. Cells were treated with HNE, and lysed in sodium dodecyl sulfate buffer (62.5 mmol/L Tris-HCl at pH 6.8, 20 g/L sodium dodecyl sulfate, 100 mL/L glycerol, 50 mmol/L dithiothreitol, 1 g/L bromphenol blue) for 15 min on ice. The samples were then sonicated for 2 s, heated for 5 min, and centrifuged at 12000 g for 5 min to remove insoluble cell debris. Whole cell extracts (approximately 100 μg) were fractionated on a 100 g/L sodium dodecyl sulfate-polyacrylamide gel. They were transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA), and the membrane was incubated overnight at 4 °C with rabbit antibodies against phosphorylated MAP kinases (extracellular signal-regulated kinase (ERK)1/2, c-Jun N-terminal kinase (JNK), or p38 MAP kinase). After incubation with peroxidase-conjugated goat anti-rabbit secondary antibody for 1 h, proteins were visualized by using an ECL kit (Amersham Biosciences UK, Ltd.). Levels of total MAP kinases, IκB-α , α -SMA, and G3PDH were examined in a similar manner.
Enzyme-linked immunosorbent assay
After a 24-h incubation, cell culture supernatants were harvested and stored at -80 °C until the measurement. Monocyte chemoattractant protein-1 (MCP-1) levels in the culture supernatants were measured by enzyme-linked immunosorbent assay (Pierce Biotechnology, Rockford, IL) according to the manufacturer's instructions.
Collagen assay
PSCs were incubated with HNE in serum-free medium for 48 h. Type I collagen released into the culture supernatant was quantified by enzyme-linked immunosorbent assay, as previously described[22]. Briefly, immunoassay plates (Becton Dickinson, Franklin Lakes, NJ) were coated with diluted samples overnight at 4 °C. After blocking with 50 g/L dry milk in PBS, plates were incubated with goat anti-rat type I collagen antibody (SouthernBiotech, Birmingham, AL). After washing of the plate, rabbit anti-goat IgG antibody conjugated with alkaline phosphatase was added, and incubated. Finally, P-nitrophenylphosphate was added as a substrate, and the collagen levels were determined by differences in absorbance at wavelength 405 min 690 nm. Rat tail collagen type I was used as a standard. The collagen levels in each sample were normalized to the cellular DNA content, which was determined by a fluorometric assay, according to the method of Brunk et al[23]. The results are expressed as a percentage of the untreated control.
Activating effect of HNE on PSCs in culture
Freshly isolated PSCs were incubated with HNE (at 1 μmol/L ) in serum-free medium or 50 mL/L fetal bovine serum-containing medium. After 7-d incubation, morphological changes characteristic of PSC activation were assessed after staining with glial acidic fibrillary protein (GFAP) as previously described[24] using a streptavidin-biotin-peroxidase complex detection kit (Histofine Kit; Nichirei, Tokyo, Japan). Briefly, cells were fixed with 100 mL/L methanol for 5 min at -20 °C, and then endogenous peroxidase activity was blocked by incubation in methanol with 3 mL/L hydrogen peroxide for 5 min. After immersion in normal goat serum for 1 h, the slides were incubated with rabbit anti-GFAP antibody at a dilution of 1:100 overnight at 4 °C. The slides were incubated with biotinylated goat anti-rabbit antibody for 45 min, followed by peroxidase-conjugated streptavidin for 30 min. Finally, color was developed by incubating the slides for several minutes with diaminobenzidine (Dojindo, Kumamoto, Japan). In addition, total cellular proteins (approximately 25 μg) were prepared, and the levels of α -SMA and G3PDH were determined by Western blotting.
Statistical analysis
The results were expressed as mean ± SD. Luminograms and autoradiograms are representative of at least three experiments. Differences between experimental groups were evaluated by the two-tailed unpaired Student’s t test. A P-value less than 0.05 was considered statistically significant.
RESULTS
HNE was cytotoxic at high concentrations
Because HNE has been shown to be cytotoxic at high concentrations in several types of cells[25], we first examined the effect of HNE on the cell viability of PSCs. PSCs were incubated with increasing concentrations of HNE for 24 h, and the cell viability was assessed by MTT assay. HNE up to 5 μmol/L did not alter the cell viability, but above 5 μmol/L, HNE was cytotoxic (Figure 1). The results were also confirmed by trypan blue dye exclusion test (data not shown). Based on these results, we used HNE up to 5 μmol/L in the subsequent experiments.
Figure 1.

PSCs were incubated with HNE at the indicated con-centrations in serum-free medium for 24 h. Cell viability was determined by the MTT assay, and the absorbance at 570 min 690 nm (“A.”) of the sample is given. Data are shown as mean ± SD (n = 6). aP < 0.05 vs control, bP < 0.01 vs control.
HNE was a potential inducer of oxidative stress
We examined the production of pro-oxidants using a peroxide-sensitive fluorescent probe, DCF-DA. This chemical is freely permeable to cells. Once inside the cells, it is hydrolyzed to DCF and trapped intracellularly. In the presence of peroxides, especially hydrogen peroxide, DCF is oxidized to fluorescent 2’,7’-dichlorofluorescin, which can then be readily detected by flow cytometry[14]. Treatment with HNE (at 5 μmol/L) increased the peroxide-activated fluorescence intensity, indicating that HNE was a potential inducer of intracellular oxidative stress in PSCs (Figure 2).
Figure 2.

PSCs were incubated with DCF-DA (at 50 μmol/L ) for 30 min and then treated with HNE (at 1 or 5 μmol/L) for 30 min. The fluorescence intensity of more than 10000 cells was analyzed using a flow cytometer.
HNE increased AP-1, but not NF-κB binding activity
We next examined the effects of HNE on the activation of the transcription factors NF-κB and AP-1. These transcription factors are important regulators of gene expression in response to many stimuli including proinflammatory cytokines and growth factors[26,27]. PSCs were incubated with HNE, or IL-1β for 1 h. Nuclear extracts were prepared, and the specific binding activities of AP-1 and NF-κB were examined by electrophoretic mobility shift assay. HNE, as well as IL-1β , increased the AP-1 binding activity (Figure 3A). The specificity of AP-1-specific DNA binding activity was demonstrated by the addition of a 100-fold molar excess of unlabeled AP-1 oligonucleotide, but not by unrelated NF-κB oligonucleotide in competition assays (data not shown). In contrast, NF-κB binding activity was induced by IL-1β , but not by HNE (Figure 3B).
Figure 3.

PSCs were treated with IL-1β (at 2 ng/mL, lane 2) or HNE (at 5 μmol/L, lane 4) in serum-free medium for 1 h. Nuclear extracts were prepared and subjected to electro-phoretic mobility shift assay using AP-1 (panel A) or NF-κB (panel B) oligonucleotide probes. Arrows denote specific in-ducible complexes competitive with cold double-stranded oli-gonucleotide probes (lane 3). Lane 1: control (serum-free medium only). □: free probe.
HNE increased AP-1, but not NF-κB dependent transcriptional activity
To confirm that AP-1 activation observed by electrophoretic mobility shift assay was functional, the effect of HNE on AP-1 dependent transcriptional activity was examined. PSCs were transiently transfected with NF-κB- or AP-1-luciferase gene reporter constructs and assayed for the luciferase activity. As shown in Figure 4, HNE increased AP-1-dependent, but not NF-κB-dependent luciferase activity. Phosphorylation and degradation of the inhibitory protein IκB-α , and subsequent dissociation of this protein from NF-κB were thought to be necessary for the activation[26]. We also examined the effect of HNE on the degradation of IκB-α by Western blotting. IL-1β , but not HNE, induced transient degradation of IκB-α , further supporting that HNE did not activate NF-κB (Figure 4B).
Figure 4.
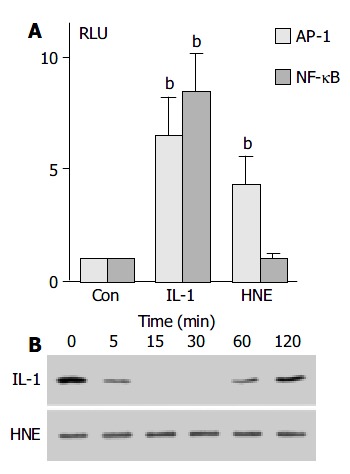
HNE increased AP-1, but not NF-κB, -dependent transcriptional activity. A: PSCs were transfected with the lu-ciferase vectors (2 X AP-1 or 2 X NF-κB) along with pRL-TK vector as an internal control. After 24 h, the transfected cells were treated with IL-1β (at 2 ng/mL) or HNE (at 1 μmol/L ). After another 24-h incubation, intracellular luciferase activities were determined. The data represent mean mean ± SD, calcu-lated from three independent experiments as fold induction compared with the activity observed in control (serum-free medium only, “Con”). bP < 0.01 vs control. RLU: relative light units. B: PSCs were treated with IL-1β (at 2 ng/mL) or HNE (at 1 μmol/L) in serum-free medium for the indicated times. Total cell lysates were prepared, and the level of IκB-α was deter-mined by Western blotting.
HNE activated MAP kinases
Induction of the expression of AP-1 components c-Fos and c-Jun by a variety of stimuli such as growth factors and cytokines is mediated by the activation of three distinct MAP kinases: ERK1/2, JNK, and p38 MAP kinase[27,28]. Activation of these kinases occurs through phosphorylation[27,28]. We determined the activation of these MAP kinases in HNE-treated cells by Western blotting using anti-phosphospecific MAP kinase antibodies. These antibodies recognize only phosphorylated forms of MAP kinases, thus allowing the assessment of activation of these kinases. HNE activated these three classes of MAP kinases in a time-dependent manner with peaking around 5 to 15 min (Figure 5).
Figure 5.
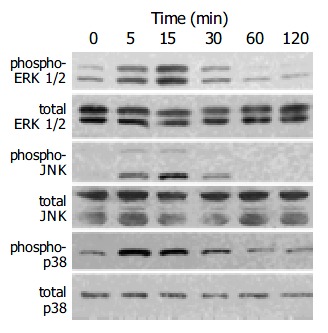
PSCs were treated with HNE (at 1 μmol/L) for the indicated times. Total cell lysates (approximately 100 μg) were prepared, and the levels of activated, phosphorylated MAP kinases were determined by Western blotting using anti- phosphospecific antibodies. Levels of total MAP kinases were also determined.
HNE did not alter basal and PDGF-BB-induced proliferation
We then examined the effects of HNE on basal and PDGF-induced proliferation. As previously reported[16], PDGF-BB significantly increased proliferation. HNE did not affect either basal or PDGF-BB-induced proliferation of PSCs (Figure 6).
Figure 6.
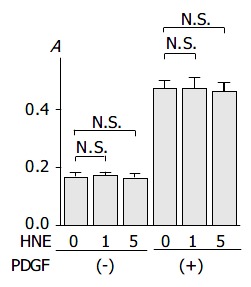
PSCs were treated with HNE (at 0, 1, or 5 μmol/L ) in the absence or presence of PDGF (at 25 ng/mL) in serum-free medium. After 24-h incubation, cells were labeled with BrdU for 3 h. Cells were fixed, and incubated with peroxidase-conju-gated anti-BrdU antibody. Then the peroxidase substrate 3,3’, 5,5’-tetramethylbenzidine was added, and BrdU incorporation was quantitated by differences in absorbance at wavelength 370 min 492 nm (“A.”, optical density). Data are shown as mean ± SD (n = 6). bP < 0.01 versus control (serum-free medium only), N.S.: not significant.
HNE did not induce MCP-1 production
Activated PSCs acquired the proinflammatory phenotype; they might modulate the recruitment and activation of inflammatory cells through the expression of MCP-1[29]. IL-1β induced MCP-1 production, but HNE did not (Figure 7).
Figure 7.
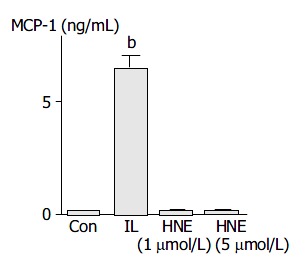
PSCs were treated with IL-1β (2 ng/mL, “IL”) or HNE (at 1 or 5 µmol/L ) in serum-free medium. After 24 h, culture supernatant was harvested, and the MCP-1 level was determined by enzyme-linked immunosorbent assay. Data shown are expressed as mean ± SD (n = 6). bP < 0.01 versus con-trol (“Con”, serum-free medium only).
HNE increased type I collagen production
It has been shown that culture-activated PSCs express α -SMA and produce type I collagen. Indeed, α -SMA expression has been accepted as a marker of PSC activation[1], and in situ hybridization techniques showed that α -SMA-positive cells were the principal source of collagen in the fibrotic pancreas[4]. HNE increased the production of type I collagen (Figure 8). Our previous finding that ethanol and acetaldehyde induced collagen production through p38 MAP kinase[30] prompted us to examine the role of MAP kinases in HNE-induced type I collagen production. Thus we employed specific inhibitors to block these pathways. A selective p38 MAP kinase inhibitor SB203580[31], and a JNK inhibitor SP600125[32] decreased type I collagen production. In contrast, U0126, a specific inhibitor of MAP kinase kinase activation and consequent ERK1/2 activation[33], did not affect the collagen production. These results suggested that HNE induced type I collagen production through the activation of p38 MAP kinase and JNK, but not ERK pathway.
Figure 8.

PSCs were treated with HNE (at 1 µmol/L , lane 2) in the absence or presence of SB203580 (at 25 mmol/L , lane 3), SP600125 (at 10 µmol/L, lane 4), or U0126 (at 5 µmol/L, lane 5). After 48-h incubation, type I collagen released into the cultute supernatant was quantified by enzyme-linked immunosorbent assay. The collagen levels in each sample were normalized to the cellular DNA content. The results are expressed as a per-centage of the untreated control (“Con”). Data are shown as mean ± SD (n = 6). bP < 0.01 vs control (serum-free medium only, lane 1). dP < 0.01 vs HNE only (lane 2).
HNE did not initiate the transformation of freshly isolated PSCs to activated phenotype
Finally, we examined whether HNE initiated the transformation of PSCs from quiescent to myofibroblast-like phenotype in culture. Freshly isolated PSCs were incubated with HNE in serum-free medium or 50 mL/L serum-containing medium for 7 d. Morphological changes characteristic of PSC activation were assessed after staining with GFAP. After 7 d, PSCs cultured with 50 mL/L serum showed transformation into cells with a myofibroblast-like phenotype (Figure 9A). In contrast, PSCs treated with HNE were small and circular, with lipid droplets presenting in many cells and slender dendritic processes (Figure 9B), as PSCs treated with serum-free medium only (Figure 9C). In agreement with the morphological changes, significant expression of α -SMA was observed on d 7 in serum-treated PSCs, whereas HNE did not induce α -SMA expression.
Figure 9.

A,B,C: Freshly isolated PSCs were incubated with 50 mL/L serum (panel A), HNE (at 1 µmol/L, panel B), or serum-free medium (panel C) for 7 d. Morphological changes characteristic of PSC activation were assessed after staining with GFAP. Original magnification: × 10 objective. D: Total cell lysates (approximately 25 µg) were prepared from cells treated with serum-free medium (“Cont”), 50 mL/L serum (“FBS”), or HNE (at 1 µmol/L) for 7 d, and the levels of α -SMA and G3PDH were determined by Western blotting.
DISCUSSION
It has been established that oxidative stress plays a critical role in the pathogenesis of both acute and chronic pancreatitis[8,9]. HNE, an aldehydic end product of lipid peroxidation, is believed to be largely responsible for pathological effects observed during oxidative stress[13]. The major effects of HNE produced during oxidative stress are believed to be damage to cellular components such as glutathione and proteins. However, recent studies have revealed that HNE at non-cytotoxic levels could potently activate a stress response mechanism[14]. In this study, we have shown for the first time that HNE, at non-cytotoxic concentrations, activated AP-1, but not NF-κB, in PSCs. This selective pattern of activation was distinct from that elicited by IL-1β and tumor necrosis factor-α , which also activated NF-κB[5]. In addition, HNE activated three classes of MAP kinases (ERK1/2, JNK, and p38 MAP kinase). HNE-induced type I collagen production was inhibited by SB203580, a p38 MAP kinase inhibitor, and by SP600125, a JNK inhibitor, suggesting a role of p38 MAP kinase and JNK in the type I collagen production. The concentrations of HNE used in this study (up to 5 μmol/L) could be easily reached in vivo in conditions of mild to moderate oxidative stress, and were considered to be physiologically relevant[13].
It has been shown that ethanol increased HNE protein adduct accumulation in the pancreas of rats[34]. During pancreatic injury in vivo, PSCs might be exposed to HNE via two pathways: (1) exogenous exposure as a result of the generation of HNE by surrounding cells (e.g. acinar cells and inflammatory cells) or (2) endogenous exposure as a result of the generation of HNE within the cells. Casini et al[15] have shown that stronger HNE immunoreactivity was observed at the periphery of acini in chronic alcoholic pancreatitis. Under such a circumstance, it is likely that PSCs exogenously expose to HNE. Our present study is exclusively concerned with the effect of exogenously-delivered HNE on cell biology, and it remains to be clarified how results of this study correlated with the potential effect of HNE formed endogenously in the cell. It is likely that PSCs were exposed to endogenous HNE during ethanol consumption, because PSCs, in addition to pancreatic acinar cells[35], had the capacity to metabolize ethanol to acetaldehyde via alcohol dehydrogenase-mediated oxidation of ethanol[36].
Activated PSCs acquire the proinflammatory phenotype; they may modulate the recruitment and activation of inflammatory cells. We have reported that activated PSCs expressed MCP-1[29] in response to IL-1β and tumor necrosis factor-α in vitro. Indeed, MCP-1 expression by activated PSCs was shown to be increased in fibrous tissue sections from patients with chronic pancreatitis[37]. In this study, HNE failed to induce NF-κB activation and consequent MCP-1 production. The failure of HNE to induce MCP-1 expression is not surprising because the activation of NF-κB has been shown to play a central role in MCP-1 expression in PSCs[29]. This is in contrast to human endothelial cells where the AP-1 proteins c-Fos and c-Jun directly induce MCP-1 expression independently of the NF-κB pathway[38]. The effects of HNE on the activation of NF-κB and AP-1 appeared to depend on the types of cells. In a similar manner to PSCs, HNE activated AP-1, but not NF-κB, in murine and human macrophages[39] and human hepatic stellate cells[40]. On the other hand, HNE did not activate AP-1, but did attenuate IL-1β - and phorbol ester-induced NF-κB activation in human monocytic THP-1 cells[41]. The inhibition of NF-κB activation was mediated through the inhibition of IκB phosphorylation and subsequent proteolysis[41]. Recent reviews[42,43] have emphasized that redox-dependent activation of NF-κB was cell and stimulus specific as opposed to the concept that oxidative stress was a common mediator of diverse NF-κB activators. Li and Karin[42] reported that when a redox-regulated effect on NF-κB was observed, it appeared to occur downstream from the IκB kinase, at the level of ubiquitination and/or degradation of IκB.
In this study, HNE activated three classes of MAP kinases in PSCs. This is in agreement with the previous study showing that HNE activated these three classes of MAP kinases in rat liver epithelial RL34 cells[14]. In human hepatic stellate cells, Parola et al[40] reported that HNE formed adducts with the p46 and p54 isoforms of JNK, leading to the nuclear translocation and activation of these signaling molecules, and, in turn, the induction of the transcription factors c-jun and AP-1. Unlike human hepatic stellate cells, we showed that HNE induced phosphorylation of p46 and p54 JNK in rat PSCs. On the other hand, we have shown that activation of ERK played a central role in PDGF-BB-induced proliferation of PSCs[16]. In spite of the ability to activate ERK, HNE failed to alter the proliferation regardless of the presence or absence of PDGF-BB, suggesting that activation of ERK is required but not sufficient for the proliferation of PSCs. This is in agreement with the concept that activation of ERK, although necessary, may not be sufficient to justify the mitogenic properties of an agonist[44].
We here showed that HNE did not initiate the transformation of quiescent PSCs. This is in disagreement with the previous reports showing that ethanol and acetaldehyde activated quiescent PSCs through the generation of oxidative stress[36]. However, it should be noted that previous studies examining the effects of oxidative stress on the activation of hepatic stellate cells have reported conflicting results. Lee et al[45] reported that quiescent hepatic stellate cells were activated upon exposure to the pro-oxidant compound ascorbate/ferrous sulfate or the lipid peroxidation product malondialdehyde. Maher et al[46] reported that stimulatory effects of malondialdehyde only occurred after hepatic stellate cells had been preactivated by culture on plastic. Olynyk et al[47] reported that malondialdehyde and HNE decreased α -SMA expression in hepatic stellate cells at d 3 when compared to controls not incubated with the compounds. Previous studies have suggested that cytokines and growth factors such as transforming growth factor-β 1 and tumor necrosis factor-α induced the transformation of quiescent PSCs[48,49]. These mediators were released or produced from inflammatory cells, platelets, and pancreatic acinar cells, during the course of pancreatitis[48,49], leading to the activation of PSCs. Once activated, PSCs were sensitive to HNE, and showed profibrogenic responses, thus playing a role in the pathogenesis of pancreatic fibrosis.
In agreement with the previous study in human hepatic stellate cells[40], HNE increased type I collagen production in PSCs. Lee et al[45] reported that oxidative stress-induced collagen synthesis in hepatic stellate cells was associated with induction of transcription factors NF-κB and c-myb[45]. In rat PSCs, the role of NF-κB was unlikely because HNE failed to activate NF-κB. Although the molecular mechanisms by which HNE increased type I collagen production in hepatic stellate cells were not further pursued, we have shown that HNE-induced type I collagen production was mediated by the activation of JNK and p38 MAP kinase. Along this line, it has been reported that malondialdehyde increased α 2 (I) collagen gene activity through JNK activation in hepatic stellate cells[50]. It should be noted that the biological action of HNE may result in multiple effects within the complex network of events occurring in the process of fibrosis following reiterated pancreatic injury, with the resulting overall biological effects being influenced by the actual HNE concentration, the target cell or cells available, and the presence of growth factors and other mediators in the microenvironment. Nevertheless, our findings that HNE induced type I collagen production suggest possible links between oxidant stress, aldehyde-adduct formation, and pancreatic fibrosis. Very recently, it has been reported that HNE acted as a potent pro-fibrogenic stimulus in the expression of genes involved in extracellualr matrix deposition in activated hepatic stellate cells[51]. Indeed, we have found that HNE increased the transcriptional activity of prolyl 4-hydroxylase (α ), a key enzyme that catalyzes the formation of 4-hydroxyproline (Satoh et al manuscript in preparation). Elucidation of the functions of PSCs would provide better understanding and rational approaches for the control of pancreatic inflammation and fibrosis.
ACKNOWLEDGEMENT
The authors thank Dr. Naofumi Mukaida for the luciferase vectors.
Footnotes
Supported by Grant-in-Aid for Encouragement of Young Scientists from Japan Society for the Promotion of Science (to A.M.), by Pancreas Research Foundation of Japan (to A.M.), and by the Kanae Foundation for Life and Socio-Medical Science (to A. M.)
Edited by Zhu LH and Xu FM
References
- 1.Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43:128–133. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grünert A, Adler G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115:421–432. doi: 10.1016/s0016-5085(98)70209-4. [DOI] [PubMed] [Google Scholar]
- 3.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 4.Haber PS, Keogh GW, Apte MV, Moran CS, Stewart NL, Crawford DH, Pirola RC, McCaughan GW, Ramm GA, Wilson JS. Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am J Pathol. 1999;155:1087–1095. doi: 10.1016/S0002-9440(10)65211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masamune A, Sakai Y, Kikuta K, Satoh M, Satoh A, Shimosegawa T. Activated rat pancreatic stellate cells express intercellular adhesion molecule-1 (ICAM-1) in vitro. Pancreas. 2002;25:78–85. doi: 10.1097/00006676-200207000-00018. [DOI] [PubMed] [Google Scholar]
- 6.Masamune A, Satoh M, Kikuta K, Sakai Y, Satoh A, Shimosegawa T. Inhibition of p38 mitogen-activated protein kinase blocks activation of rat pancreatic stellate cells. J Pharmacol Exp Ther. 2003;304:8–14. doi: 10.1124/jpet.102.040287. [DOI] [PubMed] [Google Scholar]
- 7.Masamune A, Kikuta K, Satoh M, Satoh K, Shimosegawa T. Rho kinase inhibitors block activation of pancreatic stellate cells. Br J Pharmacol. 2003;140:1292–1302. doi: 10.1038/sj.bjp.0705551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanfey H, Bulkley GB, Cameron JL. The pathogenesis of acute pancreatitis. The source and role of oxygen-derived free radicals in three different experimental models. Ann Surg. 1985;201:633–639. doi: 10.1097/00000658-198505000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braganza JM. The pathogenesis of chronic pancreatitis. QJM. 1996;89:243–250. doi: 10.1093/qjmed/89.4.243. [DOI] [PubMed] [Google Scholar]
- 10.Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 11.Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA. Reactive oxygen species, cell signaling, and cell injury. Free Radic Biol Med. 2000;28:1456–1462. doi: 10.1016/s0891-5849(00)00252-5. [DOI] [PubMed] [Google Scholar]
- 12.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 13.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 14.Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J Biol Chem. 1999;274:2234–2242. doi: 10.1074/jbc.274.4.2234. [DOI] [PubMed] [Google Scholar]
- 15.Casini A, Galli A, Pignalosa P, Frulloni L, Grappone C, Milani S, Pederzoli P, Cavallini G, Surrenti C. Collagen type I synthesized by pancreatic periacinar stellate cells (PSC) co-localizes with lipid peroxidation-derived aldehydes in chronic alcoholic pancreatitis. J Pathol. 2000;192:81–89. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH675>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 16.Masamune A, Kikuta K, Satoh M, Kume K, Shimosegawa T. Differential roles of signaling pathways for proliferation and migration of rat pancreatic stellate cells. Tohoku J Exp Med. 2003;199:69–84. doi: 10.1620/tjem.199.69. [DOI] [PubMed] [Google Scholar]
- 17.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 18.Porstmann T, Ternynck T, Avrameas S. Quantitation of 5-bromo-2-deoxyuridine incorporation into DNA: an enzyme immunoassay for the assessment of the lymphoid cell proliferative response. J Immunol Methods. 1985;82:169–179. doi: 10.1016/0022-1759(85)90236-4. [DOI] [PubMed] [Google Scholar]
- 19.Masamune A, Igarashi Y, Hakomori S. Regulatory role of ceramide in interleukin (IL)-1 beta-induced E-selectin expression in human umbilical vein endothelial cells. Ceramide enhances IL-1 beta action, but is not sufficient for E-selectin expression. J Biol Chem. 1996;271:9368–9375. doi: 10.1074/jbc.271.16.9368. [DOI] [PubMed] [Google Scholar]
- 20.Masamune A, Shimosegawa T, Masamune O, Mukaida N, Koizumi M, Toyota T. Helicobacter pylori-dependent ceramide production may mediate increased interleukin 8 expression in human gastric cancer cell lines. Gastroenterology. 1999;116:1330–1341. doi: 10.1016/s0016-5085(99)70497-x. [DOI] [PubMed] [Google Scholar]
- 21.Masamune A, Satoh K, Sakai Y, Yoshida M, Satoh A, Shimosegawa T. Ligands of peroxisome proliferator-activated receptor-gamma induce apoptosis in AR42J cells. Pancreas. 2002;24:130–138. doi: 10.1097/00006676-200203000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Moshage H, Casini A, Lieber CS. Acetaldehyde selectively stimulates collagen production in cultured rat liver fat-storing cells but not in hepatocytes. Hepatology. 1990;12:511–518. doi: 10.1002/hep.1840120311. [DOI] [PubMed] [Google Scholar]
- 23.Brunk CF, Jones KC, James TW. Assay for nanogram quantities of DNA in cellular homogenates. Anal Biochem. 1979;92:497–500. doi: 10.1016/0003-2697(79)90690-0. [DOI] [PubMed] [Google Scholar]
- 24.Masamune A, Satoh M, Kikuta K, Suzuki N, Shimosegawa T. Establishment and characterization of a rat pancreatic stellate cell line by spontaneous immortalization. World J Gastroenterol. 2003;9:2751–2758. doi: 10.3748/wjg.v9.i12.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grilli M, Chiu JJ, Lenardo MJ. NF-kappa B and Rel: participants in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- 27.Karin M, Liu Zg, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 28.Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 29.Masamune A, Kikuta K, Satoh M, Sakai Y, Satoh A, Shimosegawa T. Ligands of peroxisome proliferator-activated receptor-gamma block activation of pancreatic stellate cells. J Biol Chem. 2002;277:141–147. doi: 10.1074/jbc.M107582200. [DOI] [PubMed] [Google Scholar]
- 30.Masamune A, Kikuta K, Satoh M, Satoh A, Shimosegawa T. Alcohol activates activator protein-1 and mitogen-activated protein kinases in rat pancreatic stellate cells. J Pharmacol Exp Ther. 2002;302:36–42. doi: 10.1124/jpet.302.1.36. [DOI] [PubMed] [Google Scholar]
- 31.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 32.Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 34.McKim SE, Uesugi T, Raleigh JA, McClain CJ, Arteel GE. Chronic intragastric alcohol exposure causes hypoxia and oxidative stress in the rat pancreas. Arch Biochem Biophys. 2003;417:34–43. doi: 10.1016/s0003-9861(03)00349-7. [DOI] [PubMed] [Google Scholar]
- 35.Haber PS, Apte MV, Applegate TL, Norton ID, Korsten MA, Pirola RC, Wilson JS. Metabolism of ethanol by rat pancreatic acinar cells. J Lab Clin Med. 1998;132:294–302. doi: 10.1016/s0022-2143(98)90042-7. [DOI] [PubMed] [Google Scholar]
- 36.Apte MV, Phillips PA, Fahmy RG, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Naidoo D, Wilson JS. Does alcohol directly stimulate pancreatic fibrogenesis Studies with rat pancreatic stellate cells. Gastroenterology. 2000;118:780–794. doi: 10.1016/s0016-5085(00)70148-x. [DOI] [PubMed] [Google Scholar]
- 37.Saurer L, Reber P, Schaffner T, Büchler MW, Buri C, Kappeler A, Walz A, Friess H, Mueller C. Differential expression of chemokines in normal pancreas and in chronic pancreatitis. Gastroenterology. 2000;118:356–367. doi: 10.1016/s0016-5085(00)70218-6. [DOI] [PubMed] [Google Scholar]
- 38.Wang N, Verna L, Hardy S, Forsayeth J, Zhu Y, Stemerman MB. Adenovirus-mediated overexpression of c-Jun and c-Fos induces intercellular adhesion molecule-1 and monocyte chemoattractant protein-1 in human endothelial cells. Arterioscler. Thromb Vasc Biol. 1999;19:2078–2084. doi: 10.1161/01.atv.19.9.2078. [DOI] [PubMed] [Google Scholar]
- 39.Camandola S, Scavazza A, Leonarduzzi G, Biasi F, Chiarpotto E, Azzi A, Poli G. Biogenic 4-hydroxy-2-nonenal activates transcription factor AP-1 but not NF-kappa B in cells of the macrophage lineage. Biofactors. 1997;6:173–179. doi: 10.1002/biof.5520060211. [DOI] [PubMed] [Google Scholar]
- 40.Parola M, Robino G, Marra F, Pinzani M, Bellomo G, Leonarduzzi G, Chiarugi P, Camandola S, Poli G, Waeg G, et al. HNE interacts directly with JNK isoforms in human hepatic stellate cells. J Clin Invest. 1998;102:1942–1950. doi: 10.1172/JCI1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Page S, Fischer C, Baumgartner B, Haas M, Kreusel U, Loidl G, Hayn M, Ziegler-Heitbrock HW, Neumeier D, Brand K. 4-Hydroxynonenal prevents NF-kappaB activation and tumor necrosis factor expression by inhibiting IkappaB phosphorylation and subsequent proteolysis. J Biol Chem. 1999;274:11611–11618. doi: 10.1074/jbc.274.17.11611. [DOI] [PubMed] [Google Scholar]
- 42.Li N, Karin M. Is NF-kappaB the sensor of oxidative stress. FASEB J. 1999;13:1137–1143. [PubMed] [Google Scholar]
- 43.Bowie A, O'Neill LA. Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol. 2000;59:13–23. doi: 10.1016/s0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- 44.Blenis J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci U S A. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee KS, Buck M, Houglum K, Chojkier M. Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. J Clin Invest. 1995;96:2461–2468. doi: 10.1172/JCI118304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maher JJ, Tzagarakis C, Giménez A. Malondialdehyde stimulates collagen production by hepatic lipocytes only upon activation in primary culture. Alcohol Alcohol. 1994;29:605–610. [PubMed] [Google Scholar]
- 47.Olynyk JK, Khan NA, Ramm GA, Brown KE, O'Neill R, Britton RS, Bacon BR. Aldehydic products of lipid peroxidation do not directly activate rat hepatic stellate cells. J Gastroenterol Hepatol. 2002;17:785–790. doi: 10.1046/j.1440-1746.2002.02798.x. [DOI] [PubMed] [Google Scholar]
- 48.Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999;44:534–541. doi: 10.1136/gut.44.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneider E, Schmid-Kotsas A, Zhao J, Weidenbach H, Schmid RM, Menke A, Adler G, Waltenberger J, Grünert A, Bachem MG. Identification of mediators stimulating proliferation and matrix synthesis of rat pancreatic stellate cells. Am J Physiol Cell Physiol. 2001;281:C532–C543. doi: 10.1152/ajpcell.2001.281.2.C532. [DOI] [PubMed] [Google Scholar]
- 50.Anania FA, Womack L, Jiang M, Saxena NK. Aldehydes potentiate alpha(2)(I) collagen gene activity by JNK in hepatic stellate cells. Free Radic Biol Med. 2001;30:846–857. doi: 10.1016/s0891-5849(01)00470-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zamara E, Novo E, Marra F, Gentilini A, Romanelli RG, Caligiuri A, Robino G, Tamagno E, Aragno M, Danni O, et al. 4-Hydroxynonenal as a selective pro-fibrogenic stimulus for activated human hepatic stellate cells. J Hepatol. 2004;40:60–68. doi: 10.1016/s0168-8278(03)00480-x. [DOI] [PubMed] [Google Scholar]