

Abstract
Study Objectives:
Obstructive sleep apnea (OSA) induces cognitive impairment that involves intermittent hypoxia (IH). Because OSA is recognized as a low-grade systemic inflammatory disease and only some patients develop cognitive deficits, we investigated whether IH-related brain consequences shared similar pathophysiology and required additional factors such as systemic inflammation to develop.
Design:
Nine-week-old male C57BL/6J mice were exposed to 1 day, 6 or 24 w of IH (alternating 21–5% FiO2 every 30 sec, 8 h/day) or normoxia. Microglial changes were assessed in the functionally distinct dorsal (dH) and ventral (vH) regions of the hippocampus using Iba1 immunolabeling. Then the study concerned dH, as vH only tended to be lately affected. Seven proinflammatory and anti-inflammatory cytokine messenger RNA (mRNA) were assessed at all time points using semiquantitative real-time reverse transcription polymerase chain reaction (RT-PCR). Similar mRNA analysis was performed after 6 w IH or normoxia associated for the past 3 w with repeated intraperitoneal low-dose lipopolysaccharide or saline.
Measurements and Results:
Chronic (6, 24 w) but not acute IH induced significant microglial changes in dH only, including increased density and morphological features of microglia priming. In dH, acute but not chronic IH increased IL-1β and RANTES/CCL5 mRNA, whereas the other cytokines remained unchanged. In contrast, chronic IH plus lipopolysaccharide increased interleukin (IL)-6 and IL10 mRNA whereas lipopolysaccharide alone did not affect these cytokines.
Conclusion:
The obstructive sleep apnea component intermittent hypoxia (IH) causes low-grade neuroinflammation in the dorsal hippocampus of mice, including early but transient cytokine elevations, delayed but long-term microglial changes, and cytokine response alterations to lipopolysaccharide inflammatory challenge. These changes may contribute to IH-induced cognitive impairment and pathological brain aging.
Citation:
Sapin E, Peyron C, Roche F, Gay N, Carcenac C, Savasta M, Levy P, Dematteis M. Chronic intermittent hypoxia induces chronic low-grade neuroinflammation in the dorsal hippocampus of mice. SLEEP 2015;38(10):1537–1546.
Keywords: hippocampus, inflammation, intermittent hypoxia, mice, microglia
INTRODUCTION
Obstructive sleep apnea syndrome (OSA) is characterized by recurrent episodes of pharyngeal collapse during sleep resulting in intermittent hypoxia (IH) due to repetitive oxygen desaturation-reoxygenation sequences. OSA affects 5% to 25% of the general population.1,2 Currently, the gold standard therapy is continuous positive airway pressure (CPAP), which alleviates upper airway obstruction. However, its limited compliance3–5 counterbalances the therapeutic benefits.6
Several studies have demonstrated daytime sleepiness and cognitive deficits in memory, attention, executive functions, and motor abilities in patients with sleep apnea patients.7–9 There is accumulating evidence that OSA may represent an aggravating factor for neurodegenerative processes such as Alzheimer disease.10 OSA-related cognitive deficits may result from various brain alterations, as IH induces functional and structural changes in rodents: impairment of synaptic plasticity,11,12 enhanced vulnerability to middle cerebral artery occlusion,13 increased apoptosis in the cortex14 and in the CA1 region of the hippocampus,15–17 and facilitation of amyloid-beta (Aß42) generation in the brain.18
OSA is also recognized as a chronic low-grade systemic inflammatory disease, particularly through its hypoxic component.19–21 It is associated with elevated plasma levels of C-reactive protein (CRP) and proinflammatory factors such as interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) that could contribute to excessive daytime sleepiness.22 Indeed, TNF-α is involved in sleep regulation, promoting nonrapid eye movement sleep under physiological and inflammatory conditions.23 In patients with OSA, the TNF-α inhibitor etanercept can decrease sleepiness even more than CPAP.24 As neuroinflammation can affect cognitive processes,25 it could be an additional mechanism of IH-induced cognitive impairment. A few animal studies support this hypothesis. Indeed, increased levels of cyclooxygenase-2 and TNF-α messenger RNA (mRNA) have been described in brains of rats and mice exposed to IH.26,27 In addition, Aviles-Reyes et al.15 have observed significant astrogliosis in the cortex and hippocampus of rats exposed to IH. However, these studies looked at neuroinflammatory changes within the first 6 w of exposure. To our knowledge, no data are available from brain exposed to longer periods of IH that could be more representative of the human situation, because OSA is a long-lasting condition.
We hypothesize that long-term exposure to IH induces neuroinflammation that contributes to OSA-related cerebral impairment. We exposed C57BL/6J mice to IH (8 h/day) up to 24 w to reproduce the cumulative long-term effects of OSA, and studied neuroinflammatory responses over time. We focused on the hippocampus, a brain region involved in learning and memory and susceptible to IH, and evaluated dorsal and ventral hippo-campus, two functionally distinct brain structures with a different susceptibility to various pathological conditions.28–30 First, we assessed the morphology and density of microglia, which are resident immune cells of the central nervous system. Then, we completed the analysis by measuring mRNA expression of inflammatory cytokines in the dorsal hippocampal region that exhibited significant microglial alterations. Because cytokine alterations were observed after acute IH only, and because we used the C57BL/6J mouse strain that is relatively resistant to various consequences, we hypothesized that additional factors such as systemic inflammation could enhance the neuroinflammatory response. This could explain the development of cognitive deficits only in a subset of patients with OSA. Therefore, similar mRNA analyzes were performed after 6 w IH or normoxia (N) associated for the past 3 w with repeated intraperito-neal low-dose lipopolysaccharide (LPS) or saline injections.
MATERIALS AND METHODS
All experiments were conducted in accordance to the French and European Community guidelines for the use of animals in research. Mice were housed under a constant light/dark cycle (light on from 7:00 to 19:00). The room temperature was maintained at 22 ± 2°C and standard rodent food and water were available ad libitum throughout the experiment. The protocol was approved by the institutional committee of Grenoble for animal care and use (Protocol number: 56_UHTA-HP2-MD-01).
Animals
Intermittent Hypoxia Exposure
Male C57BL6J mice (9 w old) were housed in commercially available ventilated cages (eight animals per cage). Animals were randomly divided into six groups exposed either to IH or N conditions for 1 day, 6 w, or 24 w. The IH stimulus was applied automatically 8 h/day between 6:00 and 14:00, as rodents preferentially sleep during this time window. The animals were exposed to IH (60-sec cycle of normoxic (21% FiO2) and hypoxic (5% FiO2) phases of 30 sec each). FiO2 was measured throughout the experiment with a gas analyzer (ML206, ADInstruments Ltd, Paris, France). Control animals were placed in the same environment but exposed to normoxic air stimulus at similar flows to ensure similar levels of noise and turbulence related to gas circulation. Eighteen hours after the last exposure period, animals exposed to IH or N were deeply anesthetized with sodium pentobarbital (Ceva santé animale, 120 mg/kg, intraperitoneal injection).
Brain Collection and Preparation
To study the distribution and morphology of microglia, four mice per group were transcardially perfused with lactated Ringer's solution containing 0.1% heparin followed by 75 mL of a fixative solution composed of 4% paraformaldehyde in 0.1 M phosphate buffer (PB, pH 7.4). Brains were removed, post-fixed overnight at 4°C in the fixative solution, and immerged in 30% sucrose in 0.1 M PB for 3 days. Then brains were rapidly frozen in methylbutane, cooled with dry ice to −30°C, and cut in 25-μm-thick coronal sections on a cryostat. The free-floating sections were collected and stored at −20°C in a cryoprotective solution (0.05% diethyl pyrocarbonate, 20% glycerol, 30% ethylene glycol in 50 mM PB, pH 7.4).
To evaluate the level of gene expression involved in the neuroinflammation processes, five to six mice per group were used. Whole brains were rapidly removed on ice and rapidly frozen on dry ice and stored at −80°C for later sectioning and dissection for mRNA analysis.
Chronic LPS Treatment of Hypoxic and Normoxic Mice
Male C57BL6J mice were randomized to IH or N conditions as previously described (10 animals per cage) and were exposed for 6 w. During the past 3 w of IH/N exposure, low dose of LPS (0.5 mg/kg) (from Escherichia coli serotype 055:B5; Sigma-Aldrich, St Louis, MO, USA) or saline 0.9% was administered intraperitoneally (10 mL/kg) at 15:00, twice a week (n = 5 in each group) as described by Sy et al.31 Mice were quickly euthanized at 11:00, 20–22 h after the last LPS injection. Brains were removed, rapidly frozen on dry ice, and stored at −80°C until semiquantitative polymerase chain reaction (qPCR) analysis.
Immunohistochemical Analysis of Microglial Cells
Brain sections were successively incubated at room temperature in a solution containing anti-Iba1 antibody. In brain, Iba1 (ionized calcium-binding adaptor molecule 1) is a specific marker of microglial cells. The antibody (1:2500; Wako Chemicals GmbH, Neuss, Germany) was diluted in a 10 mM PB solution also containing 0.9% NaCl and 0.3% Triton-100X (PBST) for 18 h, followed by a biotinylated donkey antirabbit immunoglobulin G (IgG) solution (1:1000; Rockland Immunohistochemicals Inc., Gilbertville, PA, USA) and then an avidinbiotin complex marked by horse-radish peroxidase solution (1:1000; Elite kit, Vector Laboratories, Burlingame, CA, USA) both for 90 min. Finally, the sections were immersed in a 0.05 M Tris-HCl buffer (pH 7.6) containing 0.025% 3.3′-diaminobenzidine-4 HCl (Sigma-Aldrich, St. Louis, MO, USA), 0.5% nickel ammonium sulfate and 0.003% H2O2. Three washes of 10 min in PBST were performed between each step. The sections were then rinsed two times in PBST for 10 min and then mounted on glass slides. Sections were counterstained with neutral red, dehydrated and coverslipped with Depex (Vector Laboratories). Controls were run in the absence of primary antibodies to ensure the absence of nonspecific labeling.
The labeled sections were digitized at ×40 magnification through an Axioscope microscope (Zeiss, Germany) equipped with a motorized X–Y-sensitive stage and a video camera connected to a computerized image analysis system (ICS Framework; TRIBVN, Châtillon, France). Iba1 immunoreactive (Iba1+) cells were manually plotted from hemisections of the hippocampus taken at 400 μm intervals (7 sections between −1.06 and −3.28 anteroposterior from Bregma). Hippo-campus surfaces were automatically calculated by tracing their contours with ICS Framework software and the atlas of Paxinos and Franklin.32 The microglial cell density was calculated as the number of Iba1+ cells over the surface area of the counted region. The dorsal hippocampus was present in sections 1 to 4 whereas the ventral hippocampus was in sections 5 to 7.
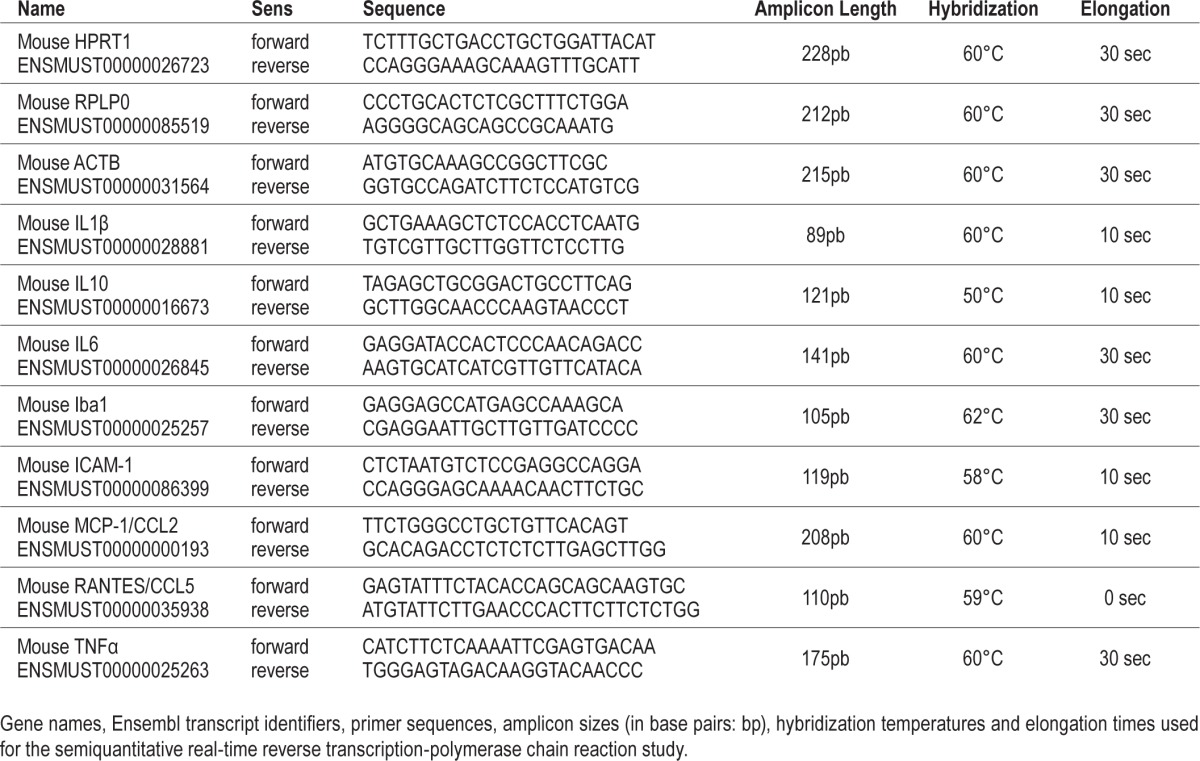
Semiquantitative Real-Time RT-PCR
Among the inflammatory mediators, we focused on the key proinflammatory chemokines RANTES (regulated upon activation, normal T-cell expressed and secreted) also known as CCL5 (Chemokine (C-C motif) ligand 5), MCP-1 (monocyte chemoattractant protein-1)/CCL2 (Chemokine (C-C motif) ligand 2), and on the intercellular adhesion molecule-1 (ICAM-1), as well as on the proinflammatory and anti-inflammatory cytokines (TNF-α; IL-1β, IL-6, and IL-10 interleukins) that are important mediators in neuroinflammation with linked activities. Brains were sliced in 400-μm-thick frontal sections at −12°C on a cryostat. The dorsal hippocampus was dissected bilaterally using a stereomicroscope and a scalpel blade (between −1.34 and −2.46 anteroposterior from Bregma). Tissue samples were kept at −80°C until their use. Total RNA was extracted using the RNeasy mini kit (QIAGEN, Courtaboeuf, France) following manufacturer's protocol with minor modification. An additional step was performed using the RNase-Free DNase Set (QIAGEN) to remove any residual DNA. The quality and quantity of total RNA were assessed with the bioanalyzer 2100 (Agilent Technologies, Inc) and NanoDrop 2000c (Thermo Scientific, Wilmington, DE, USA). A 260/280 ratio of 1.8 to 2.1 with the values of the ribosomal RNA 28S/18S ratio and RNA integrity numbers indicated the high quality of extracted RNA.
Reverse transcription was performed from 1 μg of extracted total RNA using the iScript complementary DNA (cDNA) Synthesis Kit (Bio-Rad Laboratories, Marnes-la-Coquette, France) following manufacturer's instructions. Then qPCR was done using primers designed with the VectorNTI software (primer sequences in Table 1). HPRT1 (hypoxanthine phosphoribosyltransferase 1), ACTB (beta-actin) and RPLP0 (ribosomal protein, large, P0) were chosen as reference genes because their mRNA expression level was unchanged by the experimental conditions (Figures S1 and S2, supplemental material). qPCR was performed on a thermocycler (CXF96 Real-Time PCR Detection System, Bio-Rad, Marnes-La-Coquette, France) using the SsoFast Eva Green Supermix (Bio-Rad). Fifteen microliters of mix (10 μL of Eva Green plus the couple of primers) was added to 5 μL of diluted cDNA (1:40 except for IL-10: 1:160) in a 96-well plate. RNA-free water was used as negative control. The amplification conditions were as follows: enzyme activation (95°C for 30 sec) and 50 cycles of PCR (denaturation at 95°C for 3 sec, hybridization for 45 sec [temperatures in Table 1] and elongation at 72°C [times in Table 1]). Following qPCR, a melting curve analysis was performed to confirm that a single gene product had been amplified, by heating the sample to 95°C for 10 sec, then cooling it to 65°C for 5 sec, followed by a linear increase in temperature to 95°C. PCR amplification efficiency was of 95% to 105% for all studied genes. Gene expression level was analyzed according to the comparative Ct method where Ct (cycle threshold) is the threshold cycle value. Results were expressed using the following formula: Relative mRNA expression = 2_(Ct gene of interest − Ct gene of reference). Levels of mRNA were normalized to the three reference genes, then averaged and shown as a ratio to the level found in control animals exposed to either 1 day of N (experiments without LPS) or 6 w of N and receiving NaCl for the LPS study.
Table 1.
Parameters for the semiquantitative real-time reverse transcriptase-polymerase chain reaction study.
Statistical Analysis
All results (Iba1+ cell density and relative mRNA expressions) were expressed as mean ± standard error of the mean and analyzed using two-way analysis of variance (SigmaStat 3.1, Systat Software Inc). Statistical significance was set at P < 0.05. The Grubbs test was applied to both the Iba1+ cell density and the Ct values in each group to detect significant outliers. One of the Ct values from the PCR analysis concerning RANTES gene in the control group exposed to 1-day N was removed. Degrees of freedom (df) were specified for each analysis.
RESULTS
Chronic IH induces Microglia Priming in the Dorsal Hippocampus
Quantitative Alterations
Acute 1-day exposure to IH did not alter microglia density in dorsal (dH) and ventral (vH) hippocampus compared to control animals (df = 23, P = 0.89) (Figure 1A, 1B). In contrast, chronic IH induced a significant increase in microglial density in dH, with similar elevation at 6 and 24 w IH (Figure 1A). Microglia density remained unaffected in vH after 6 w IH but tended to increase after 24 w IH (t test, P = 0.099; Figure 1B). In normoxic animals, microglial cell density did not change from 1 day to 24 w, suggesting that aging per se did not affect microglial density (Figure 1A, 1B).
Figure 1.

Quantitative and qualitative microglial alterations in the dorsal hippocampus after acute and chronic intermittent hypoxia. Density of microglial cells (Iba+ cells/mm2) in dorsal (A) and ventral (B) hippocampus of mice exposed to 1 day, 6 w, or 24 w of intermittent hypoxia (IH) or normoxia (N). Results are expressed as mean ± standard error of the mean (n = 4 for each group, *P < 0.05 and **P < 0.01 versus N group, #P < 0.05 and ##P < 0.01 versus 1-day IH). (C) Representative photographs showing microglial cells (Iba1+ cells, black cytoplasmic staining) in the CA3 area of the dorsal hippocampus (−2.00 mm from Bregma) from IH and N mice. One day of IH did not affect the morphology of microglial cells because they showed a “resting” phenotype (upper microphotographs), whereas microglial cells from mice exposed to 6 and 24 w of IH exhibited morphological features of primed microglia (middle and lower microphotographs). Scale bar: 50 μm. Py, pyramidal cell layer of the hippocampus; Rad, stratum radiatum of the hippocampus.
Qualitative Alterations
In the three normoxic groups (1 day, 6 and 24 w), and in 1-day IH-exposed animals, microglial cells in dH were “ramified” with small soma, thin cellular processes, and dense distal arborization (Figure 1C). This morphological pattern is representative of resting microglia. In contrast, after 6 and 24 w of IH exposure, microglial cells in dH showed hypertrophy of the soma with decreased ramification of distal branches and thickening of proximal processes (Figure 1C). These morphological features are typical of the activated state referred to “primed” microglia. In vH, microglia soma size and branches remained unchanged after acute or chronic exposure to IH (Figure S3, supplemental material).
Because the study did not aim at assessing neuronal consequences, no specific analysis was performed. However, gross evaluation of the pyramidal cell layer on neutral red-stained tissues did not show overt structural disorganization in dH and vH of hypoxic animals compared to normoxic animals, whatever the durations of IH exposure (Figure 1C, Figure S3).
Acute But Not Chronic IH induces Inflammatory Cytokine Alterations in the Dorsal Hippocampus
As microglia priming was observed in dH only, mRNA analysis of proinflammatory and anti-inflammatory cytokines was performed in this hippocampal region. Acute IH (1 day) induced a significant increase in RANTES/CCL5 (df = 31) and IL-1β (df = 32) mRNA levels (Figure 2), with a predominant elevation for RANTES/CCL5. In contrast, RANTES/CCL5 and IL-1β mRNA levels were not affected after 6 or 24 w of IH (Figure 2). ICAM-1, MCP-1/CCL2, TNF-α, IL-6, and IL-10 mRNA levels remained unchanged whatever the durations of IH (1 day, 6 and 24 w) (df = 32) (Figure S4, supplemental material).
Figure 2.
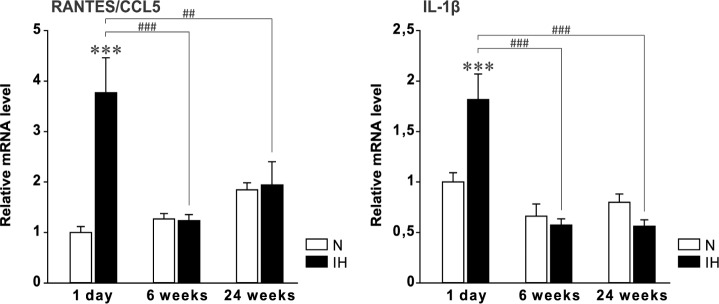
RANTES/CCL5 and IL1-β messenger RNA in the dorsal hippocampus after acute and chronic intermittent hypoxia. Levels of mRNA were normalized to the RPLP0, ACTB, and HPRT1 genes, and are shown as a ratio to the level found in control animals exposed to 1 day of normoxia (N). Results are expressed as mean ± standard error of the mean (n = 5–6 for each group, except in the 1-day normoxic group where n = 4 because one value was considered as an outlier and removed; **P < 0.01 and ***P < 0.001 versus N group, ##P < 0.01 and ###P < 0.001 versus 1-day IH). N, normoxia; IH, intermittent hypoxia.
Chronic IH Associated with Chronic Low-dose LPS induces Cytokine Alterations in the Dorsal Hippocampus
In the previous set of mice, we found that chronic IH induced microglia “priming” without cytokine alterations. Because C57BL/6J is a relatively resistant mouse strain to various consequences, we hypothesized that an additional systemic inflammation could be necessary. Thus, mRNA analyses were conducted again after 6 w IH or N associated for the past 3 w with repeated low-dose LPS or saline. Cytokine mRNA levels increased in-distinctly in both hypoxic and normoxic animals for RANTES/ CCL5, ICAM-1, TNF-α, IL-1β, and MCP-1/CCL2, suggesting that the cytokine response was due to LPS only, without an additional effect of IH (Figure 3A). These results confirmed those obtained in the previous set of experiments, i.e., that chronic IH alone was not able to increase these cytokines.
Figure 3.

Inflammatory cytokine messenger RNA (mRNA) in the dorsal hippocampus after chronic intermittent hypoxia and chronic low-dose lipopolysaccharide. (A) RANTES/CCL5, ICAM-1, TNF-α, IL1-β, MCP-1/CCL2 and (B) IL-6 and IL-10 mRNA in the dorsal hippocampus after 6 w intermittent hypoxia (IH) or normoxia (N) associated for the past 3 w with repeated intraperitoneal low-dose lipopolysaccharide (LPS) or saline (NaCl). Levels of mRNA were normalized to the RPLP0, ACTB, and HPRT1 genes, and are shown as a ratio to the level found in control animals exposed to 6 w of N and receiving NaCl instead of LPS. Results are expressed as mean ± standard error of the mean (n = 5 for each group, *P < 0.05, **P < 0.01 and ***P < 0.001 versus N group, #P < 0.05 and ##P < 0.01 versus N/LPS).
In contrast, IL-6 and IL-10 mRNA increased in IH animals challenged with LPS, whereas the levels of these two cytokines remained unaffected in LPS-treated normoxic animals (Figure 3B). These last results suggest that LPS alone was not able to affect these two cytokines but chronic IH plus chronic LPS did.
DISCUSSION
OSA is a chronic disease that may cause cognitive impairment with varying patterns and severity.7–9,33 The mechanisms underlying these consequences are not well known. However, neuroinflammatory processes elicited by the repetitive hypoxia-reoxygenation sequence could be involved. We assessed the time course of IH-induced neuroinflammation after short- and long-term exposure to IH using a rodent model.34 We focused on the hippocampus and first assessed microglial alterations in dH and vH, two functionally distinct structures that are differentially affected by various conditions.28–30 Chronic (6, 24 w) but not acute IH (1 day) induced significant microglial changes in dH only, including increased density and morphological features of microglia priming. Then, we assessed proinflammatory and anti-inflammatory cytokine mRNA in dH only, as vH was not significantly affected. Acute but not chronic IH increased IL-1β and RANTES/CCL5 mRNA, whereas the other five cytokines remained unchanged whatever the duration of IH. In contrast, chronic IH associated for the past 3 w with repeated low-dose intraperitoneal LPS as an additional inflammatory challenge, increased IL-6 and IL10 mRNA, whereas LPS without IH did not affect these two cytokines. Collectively, our results demonstrate that IH could induce neuroinflammation in a vulnerable brain region, including an early response through transient cytokine alterations, a delayed chronic low-grade inflammation through long-term microglial changes, and a modified neuroinflammatory response to other inflammatory stimuli.
IH Induces Different Neuroinflammatory Patterns According to IH Duration
There is growing evidence that OSA is a chronic low-grade inflammatory condition at both systemic and tissue levels with evidence for an early implication in the course of the disease.19 Therefore, we assessed neuroinflammation after three durations of IH exposure using cytokine mRNA and histological analyses.
IH Induces Early But Transient Cytokine Alterations
Our data show that two of the seven investigated cytokine mRNA increased after 1-day IH. Only RANTES/CCL5 and IL-1β augmented, suggesting an early but not comprehensive immune response to IH. Moreover, these two cytokines were no longer elevated when assessed after the two longer IH exposures (6, 24 w), indicating a transient cytokine response to IH. These findings may have several meanings: (1) IH induced a particular cytokine expression profile; (2) IH elicited a time-dependent response with differential kinetics according to cytokines; (3) chronic IH may lead to some adaptations such as tolerance. This could share some patho-physiology of LPS-tolerance as our group recently demonstrated that IH-induced inflammation involved TLR4 signaling35; (4) alternatively, it could be argued that cytokine protein levels may still be increased after 6 or 24 w with no more up-regulation of mRNA expression. The correlation between mRNA and protein levels is indeed modest. Upregulated mRNA without protein changes is well known, but the opposite relationship (unaffected or downregulated mRNA with upregulated protein expression) can also occur, for example when protein half-life is increased due to various alterations (turnover, protein-protein interactions, etc.). However, here, it is quite unlikely because cytokines are short-lived proteins.
It is of interest that RANTES/CCL5 and IL-1β were rapidly expressed. Indeed, IL-1β is produced by peripheral immune cells as well as by glial cells and neurons, and is considered as a main inducer of acute-phase proteins at systemic and brain levels.36 RANTES/ CCL5, which plays an important role in recruiting inflammatory cells, was found by our group to be an early and predominant cytokine in the inflammatory response to IH.37 Because of the long intervals between time points, we do not know how long the two cytokines remained elevated, and if the other five cytokines required more than 1 day IH to increase. Although the literature brings some clues, results seem to vary according to the experimental setting. Indeed, the discrepancies may rely on numerous factors such as the type of analysis (mRNA versus proteins), the animal used (rat versus mouse, mouse strain, young versus old), the tissue analyzed (brain regions), and the IH stimulus (depth of oxygen desaturation, duration of hypoxic phases, number of hours per day, duration of IH exposure). IH-induced elevation of TNF-α has been found after 2 and 6 w IH,26,27 whereas this cytokine was unaffected in our study even after 6 w. Although MCP-1/CCL2 and ICAM-1 mRNA were found elevated in rat carotid body after 7 days IH,38 these cytokines were unaffected after 1 day or longer exposures in our study. It is likely that some cytokines may require different times of IH exposure to increase. Indeed, in a previous study from our group, RANTES/CCL5 mRNA was found elevated in mouse spleen after 5 days IH whereas MCP-1/CCL2 tended to increase after 14 days IH.37
Whether the increase of proinflammatory cytokines alone is indicative of an inflammatory reaction is debatable. The term “immune signaling” has been suggested to describe the isolated release of immune-relevant molecules without any concomitant expression of other signs of neuroinflammation.39 However, the early changes observed here have similarities to those previously observed in other tissues and that had clear patterns of inflammation.37 Moreover, the secondary appearance of primed microglia is suggestive of a dynamic process exhibiting different patterns of neuroinflammation according to the IH duration. However, IH appears to be a moderate inflammatory stimulus, especially when comparing with the inflammatory response induced by low-dose LPS. Indeed, 3 w LPS induced long-term elevation of several cytokines in both normoxic and IH animals. Therefore, the study brings evidence that IH induced an early but moderate and transient neuroinflammation through cytokine alterations. Whereas inflammation is first of all a protective mechanism, it is usually seen as a detrimental process, which is true when the inflammatory response is exaggerated in intensity and/or duration. Whether the cytokine response found here is detrimental remains to be determined, but there are clues in the literature for this hypothesis. Indeed, both RANTES/CCL5 and IL-1β may have contributed to the development of the delayed and long-lasting microglial changes. Brain injection of IL-1β increases proinflammatory cytokine expression in rodent brain40–42 and RANTES/CCL5 induces a proinflammatory microglial profile.43 In contrast, mice lacking type-1 IL-1 receptor have attenuated response to brain injury (microglial and astrocytic activation, IL-6 levels).44 In addition to neuroinflammation, the increased levels of the two cytokines could also impact cognition, as IL-1β can disrupt long-term potentiation45 and hippocampal-dependent memory consolidation.46
Chronic IH Induces Long-Term Microglial Alterations
One of the most important findings of this study is microglia involvement in mice chronically exposed to IH. This included quantitative and qualitative changes as shown by increased density of microglial cells and morphological features of microglia priming. Microglia represent innate immune cells of the brain. Under physiological conditions, microglia are in a “resting” state with highly ramified and motile processes, continuously surveying the surrounding microenvironment. Microglia respond to injuries and other endogenous and exogenous stimuli through the release of proinflammatory and anti-inflammatory cytokines. Microglia have essential physiological functions such as monitoring synapses, clearing up apoptotic debris and synaptic pruning,47 and are involved in cognition as they modulate learning and memory through their action on neuronal activity and synaptic plasticity.48 In response to noxious stimuli, microglia shift from (1) resting to (2) primed microglia with specific morphological changes that include enlargement of the cell body and shortening of cellular processes, and (3) activated microglia.49 As for other processes (e.g., free radicals), microglial activation has either beneficial (e.g., neurotrophic effects) or detrimental effects depending on the intensity of the activation, microglia overactivation often leading to neurotoxic and other deleterious consequences.50 Because IH induces cognitive deficits in rodents,16,51 we speculate that microglial priming described here is likely detrimental and could contribute to IH-related cognitive alterations (see next paragraphs).
Microglia priming was found in the dorsal hippocampal region while the ventral part was not significantly affected. This finding confirms the differential vulnerability in the hippo-campal regions which has been previously observed with paradoxical (rapid eye movement) sleep deprivation,28 and other acute conditions,52 as well as in a genetic model of neurodegeneration.29 Dorsal hippocampus and vH are functionally distinct structures: dH corresponds to the posterior hippocampus in primates and performs primarily cognitive functions, whereas vH corresponds to the anterior hippocampus in primates and relates to stress, emotion, and affect.30 In agreement with this observation, dH-dependent memories such as spatial learning and verbal memory are particularly affected in IH-exposed animals16,51 and patients with OSA, respectively.53 It is difficult from imaging studies to find this differential vulnerability in patients with OSA, as conventional structural neuroimaging have reported inconclusive findings, and imaging studies using voxel-based morphometry and functional techniques reported alterations in several gray matter regions including the hippo-campus, but without details about the hippocampal regions.54 Through a gross histological analysis of pyramidal cells in dH and vH, we did not find patent differences between IH and normoxic animals whatever the exposure duration. This is not surprising as the hypoxic component of IH, even deep, is transient and appears therefore less detrimental than acute challenges such as ischemia or epileptic seizures that lead to overt neuronal loss due to severe excitotoxicity. This result does not put into question what has been previously described in the hippocampus of IH animals, as the experimental settings were clearly different (rats versus mice, different patterns of IH, time points and methods to evaluate neuronal consequences, etc.).15,16,34 Utilization of markers to assess necrosis, apoptosis, neurogenesis, or astrogliosis would have been useful but it was beyond the scope of the study. Despite no evident structural alterations of the pyramidal cells, cognitive impairment can occur due to functional impairment, for example, of cell-cell communication (e.g., glia-neuron signaling) and synaptic plasticity (long-term potentiation), which are determinant mechanisms in cognition and affected by IH.11,12 In line with other studies, our results bring further evidence that chronic IH represents a cause for pathological brain aging. In addition to the various IH-induced consequences including vascular alterations, neuronal loss and/or dysfunction, astrogliosis, and accumulation of the amyloid-beta protein, we showed that chronic IH induced long-term microglial priming. Primed microglia have been described in the aged brain55–57 and in models of neurode-generative diseases such as Prion, Alzheimer, and Parkinson diseases.58,59 This microglial activation can in turn contribute to the neurodegenerative processes.60,61 Thus, long-term IH-induced microglial changes may contribute to increased susceptibility to aging- and inflammation-induced cognitive deficits in patients with OSA.
Microglia Priming: A Vulnerable Condition for Subsequent Systemic Inflammatory Events
Chronic IH (6, 24 w) induced microglia priming but did not affect cytokine mRNA levels. These results are consistent with the fact that primed microglia do not constitutively over-produce pro-inflammatory mediators.62 In contrast, primed microglia can exacerbate neuroinflammation responses to subsequent inflammatory challenge, such as systemic inflammation.62,63 Therefore, to assess this hypothesis in our model, we repeatedly administered intraperitoneal low-dose LPS in IH-exposed animals, and assessed cytokine mRNA in dH. The low dose used in the study is able to induce neuroinflammation with moderate sickness behavior.31,64 It increased five of the seven cytokines in normoxic mice, with a predominant increase in TNF-α. These five cytokines were similarly elevated in IH mice, suggesting that there was no additional effect of IH. However, animals with IH challenged with LPS also exhibited increased IL-6 and IL-10 mRNA. Because these two cytokines were not affected by LPS alone or by 6 w IH alone (previous set of experiments), these findings suggest that the neuroinflammatory response triggered by a stimulus was altered in animals with chronic IH.
The absence of difference between IH and normoxic animals for the five cytokines may stem from the experimental setting. Peripheral LPS administration increases brain cytokine levels in a time- and dose-dependent manner.65 However the LPS response can take a number of different forms, depending on the dose and duration of exposure. Low doses can induce a state of tolerance to subsequent toxic doses of LPS, but extremely low doses have an opposite effect, priming the immune system for an even more violent response to subsequent challenge.66,67 Therefore, we cannot exclude that a milder LPS treatment would have unmasked more differences between normoxic and IH animals. Moreover, microglia are the most responsive glial cells to LPS and produce considerable amounts of TNF-α, IL-1β, and RANTES/CCL5 with smaller amounts of IL-6.68 But with repeated LPS exposure, microglia can develop LPS tolerance with reduced LPS-induced TNF-α, IL-1, and IL-6 release.66,67 In the study, although the cytokine profile induced by LPS was rather consistent with responsive microglia, LPS tolerance cannot be excluded because we did not assess cytokines after an acute LPS challenge. Nevertheless, the important finding is the LPS-induced IL-6 and IL-10 mRNA in animals exposed to IH. Because microglia are the most responsive glial cells to LPS, our results suggest that IH-induced microglia priming altered the neuroinflammatory response to LPS challenge. Whereas IL-10 is clearly a potent immune-regulating cytokine and inhibitor of inflammatory cytokine synthesis, the picture is more complex for IL-6. It has been mainly regarded as a key cytokine in acute and chronic inflammation as well as in neurodegenerative disorders. However, there is now growing evidence that IL-6 has pleiotropic actions including coordination of neuroimmune responses, anti-inflammatory activities, cell-to-cell signaling, and neuronal differentiation, growth, and survival. The dichotomic action of IL-6 may depend on the level of activation and the signaling pathway.69
Limitations of the Study
We already mentioned several limitations. We assessed the time course of IH-induced neuroinflammation in animals exposed up to 24 w to mimic chronic sleep apnea in humans. The use of only three time points have impeded a precise description of the dynamics of cytokine and histological changes. We cannot exclude the occurrence of these alterations in between the time points. Indeed, reactive astrogliosis was described in hippocampus of rats exposed to 10 days IH.15 Additional time points, between 1 day and 6 w IH, when cytokine overexpression declines and microglia priming starts, could be useful to have a better view of the mechanisms involved and eventually establish a link between the early and transient expression of proinflammatory factors and microglia priming.
We assessed cytokine alterations through their mRNA expression. However, it is well known that the correlation between mRNA and protein levels may be modest and vary according to the different cytokines as well as the experimental conditions. Therefore, mRNA results need to be confirmed by the evaluation of the corresponding proteins.
We focused the study on microglia, and did not assess specifically neuronal and astrocytic consequences as they have been investigated in several studies.14,15,70 Although gross analysis of pyramidal cells with neutral red staining did not show obvious histological differences between IH and normoxic animals even after 24 w IH, any process that could result in neuronal loss through necrosis or apoptosis cannot be excluded without histological markers.
We focused the study on the hippocampus as this brain region is involved in cognition and is susceptible to IH. However, other brain regions, either involved in cognition, affect, immunity, and wake, are affected by IH54,70 and could be investigated in a further study. We did not assess behavior because it was beyond the scope of the study, but it would be interesting to parallel behavioral findings and kinetics of cytokine mRNA and microglial alterations.
We used the rodent model of IH, which was developed more than 20 y ago and reproduces one of the major component of sleep apnea syndrome, i.e., the repetitive sequence of hypoxia-reoxygenation.34 However, the model is not “pure” because sleep is impaired, including sleep fragmentation, deficits in rapid eye movement sleep and in the delta power of nonrapid eye movement during IH exposure.71 Because sleep loss represents a cause of systemic and brain inflammation,72–74 IH-induced sleep alterations could have contributed to the inflammatory alterations observed in the study.
The C57BL/6J mouse strain is classically used in hypoxia studies, but this strain has been shown to be relatively more resistant than other strains to several deleterious stimuli such as the excitotoxicity due to kainate-induced seizures.75,76 In addition, young adults are also more resistant than aged mice to inflammatory stress.77 It is possible, however, that using older mice or a more vulnerable strain such as NMRI mice78 would have shown more pronounced alterations than those found in the study. In humans, age is indeed a condition for cognitive decline because of various causes including physiological alterations, neurodegeneration, vascular disease, and increased prevalence of OSA.
In conclusion, we showed that IH induced different patterns of neuroinflammation including early but transient cytokine alterations, then delayed but long-term low-grade inflammation involving microglia. Moreover, chronic IH altered the response to LPS inflammatory challenge. As observed in the aged brain, our results suggest that chronic IH induced a shift from resting microglia to primed microglia, resulting in altered inflammatory response when primed microglia is activated by a triggering stimulus. All these alterations may affect cognitive functions and aggravate neurodegenerative processes. Therefore, in light of these findings and in addition to other IH-related consequences such as vascular, neuronal, astroglial, and sleep alterations, IH contributes to pathological brain aging and is essential to detect and treat.
DISCLOSURE STATEMENT
This was not an industry supported study. The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by AGIR@dom and the French Society for Sleep Research and Sleep Medicine. Emilie Sapin received fellowships from the French-language Society of Pneumology and the French Society for Sleep Research and Sleep Medicine. Emilie Sapin thanks Dr Corinne Loeuillet (CNRS UMR 5163, Université Joseph Fourier Grenoble 1, Grenoble, F-38042, France) for her help in the semiquantitative real-time RT-PCR experiments.
SUPPLEMENTAL MATERIAL
Validation of reference genes for semiquantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis of RANTES/CCL5, ICAM-1, IL-1β, MCP-1/CCL2, TNF-α, IL-6 mRNA. Semiquantitative real-time RT-PCR cycle threshold numbers (Cq values) for the three reference genes RPLP0, ACTB, and HPRT1 were unaffected by the experimental conditions: 1 day, 6 and 24 w of intermittent hypoxia (IH) or normoxia (N) and 6 w IH or N with 3 w intraperitoneal low-dose lipopolysaccharide (LPS) or saline (NaCl). Complementary DNA were diluted to 1/40. Results are expressed as mean ± standard error of the mean (n = 5–6 for each group).
Validation of reference genes for semiquantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis of IL-10 cytokine messenger RNA (mRNA). Semiquantitative real-time RT-PCR cycle threshold numbers (Cq values) for the three reference genes RPLP0, ACTB, and HPRT1 were unaffected by the experimental conditions: 1 day, 6 and 24 w of intermittent hypoxia (IH) or normoxia (N) and 6 w IH or N with 3 w intraperitoneal low-dose lipopolysaccharide (LPS) or saline (NaCl). Complementary DNA were diluted to 1/160. Results are expressed as mean ± standard error of the mean (n = 5–6 for each group).
Acute and chronic intermittent hypoxia do not induce qualitative alterations of microglia in the ventral hippocampus. Representative photographs showing microglial cells (Iba1+ cells, black cytoplasmic staining) in CA3 area of the ventral hippocampus (−3.00 mm from Bregma) from intermittent hypoxia (IH) and normoxia (N) mice after 1 day, 6 and 24 w of exposure. Scale bar: 50 μm. Py, pyramidal cell layer of the hippocampus; Rad, stratum radiatum of the hippocampus.
Inflammatory cytokine messenger RNA (mRNA) in the dorsal hippocampus after acute and chronic intermittent hypoxia. Levels of ICAM-1, MCP-1/CCL2, TNF-α, IL-6, and IL-10 mRNA were normalized to the RPLP0, ACTB, and HPRT1 genes, and are shown as a ratio to the level found in control animals exposed to 1 day of normoxia. Results are expressed as mean ± standard error of the mean (n = 5–6 for each group). IH, intermittent hypoxia; N, normoxia.
REFERENCES
- 1.Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5:136–43. doi: 10.1513/pats.200709-155MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328:1230–5. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 3.Engleman HM, Wild MR. Improving CPAP use by patients with the sleep apnoea/hypopnoea syndrome (SAHS) Sleep Med Rev. 2003;7:81–99. doi: 10.1053/smrv.2001.0197. [DOI] [PubMed] [Google Scholar]
- 4.Olsen S, Smith S, Oei TP. Adherence to continuous positive airway pressure therapy in obstructive sleep apnoea sufferers: a theoretical approach to treatment adherence and intervention. Clin Psychol Rev. 2008;28:1355–71. doi: 10.1016/j.cpr.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Zozula R, Rosen R. Compliance with continuous positive airway pressure therapy: assessing and improving treatment outcomes. Curr Opin Pulm Med. 2001;7:391–8. doi: 10.1097/00063198-200111000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Dorkova Z, Petrasova D, Molcanyiova A, Popovnakova M, Tkacova R. Effects of continuous positive airway pressure on cardiovascular risk profile in patients with severe obstructive sleep apnea and metabolic syndrome. Chest. 2008;134:686–92. doi: 10.1378/chest.08-0556. [DOI] [PubMed] [Google Scholar]
- 7.Bedard MA, Montplaisir J, Richer F, Rouleau I, Malo J. Obstructive sleep apnea syndrome: pathogenesis of neuropsychological deficits. J Clin Exp Neuropsychol. 1991;13:950–64. doi: 10.1080/01688639108405110. [DOI] [PubMed] [Google Scholar]
- 8.Beebe DW, Groesz L, Wells C, Nichols A, McGee K. The neuropsychological effects of obstructive sleep apnea: a meta-analysis of norm-referenced and case-controlled data. Sleep. 2003;26:298–307. doi: 10.1093/sleep/26.3.298. [DOI] [PubMed] [Google Scholar]
- 9.Lal C, Strange C, Bachman D. Neurocognitive impairment in obstructive sleep apnea. Chest. 2012;141:1601–10. doi: 10.1378/chest.11-2214. [DOI] [PubMed] [Google Scholar]
- 10.Buratti L, Viticchi G, Falsetti L, et al. Vascular impairment in Alzheimer's disease: the role of obstructive sleep apnea. J Alzheimers Dis. 2014;38:445–53. doi: 10.3233/JAD-131046. [DOI] [PubMed] [Google Scholar]
- 11.Gu XQ, Haddad GG. Decreased neuronal excitability in hippocampal neurons of mice exposed to cyclic hypoxia. J Appl Physiol. 2001;91:1245–50. doi: 10.1152/jappl.2001.91.3.1245. [DOI] [PubMed] [Google Scholar]
- 12.Payne RS, Goldbart A, Gozal D, Schurr A. Effect of intermittent hypoxia on long-term potentiation in rat hippocampal slices. Brain Res. 2004;1029:195–9. doi: 10.1016/j.brainres.2004.09.045. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Guo SZ, Bonen A, et al. Monocarboxylate transporter 2 and stroke severity in a rodent model of sleep apnea. J Neurosci. 2011;31:10241–8. doi: 10.1523/JNEUROSCI.1462-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu W, Chi L, Row BW, et al. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience. 2004;126:313–23. doi: 10.1016/j.neuroscience.2004.03.055. [DOI] [PubMed] [Google Scholar]
- 15.Aviles-Reyes RX, Angelo MF, Villarreal A, Rios H, Lazarowski A, Ramos AJ. Intermittent hypoxia during sleep induces reactive gliosis and limited neuronal death in rats: implications for sleep apnea. J Neurochem. 2010;112:854–69. doi: 10.1111/j.1471-4159.2009.06535.x. [DOI] [PubMed] [Google Scholar]
- 16.Gozal D, Daniel JM, Dohanich GP. Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci. 2001;21:2442–50. doi: 10.1523/JNEUROSCI.21-07-02442.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gozal D, Row BW, Kheirandish L, et al. Increased susceptibility to intermittent hypoxia in aging rats: changes in proteasomal activity, neuronal apoptosis and spatial function. J Neurochem. 2003;86:1545–52. doi: 10.1046/j.1471-4159.2003.01973.x. [DOI] [PubMed] [Google Scholar]
- 18.Shiota S, Takekawa H, Matsumoto SE, et al. Chronic intermittent hypoxia/reoxygenation facilitate amyloid-beta generation in mice. J Alzheimers Dis. 2013;37:325–33. doi: 10.3233/JAD-130419. [DOI] [PubMed] [Google Scholar]
- 19.Arnaud C, Dematteis M, Pepin JL, Baguet JP, Levy P. Obstructive sleep apnea, immuno-inflammation, and atherosclerosis. Semin Immunopathol. 2009;31:113–25. doi: 10.1007/s00281-009-0148-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malakasioti G, Alexopoulos E, Befani C, et al. Oxidative stress and inflammatory markers in the exhaled breath condensate of children with OSA. Sleep Breath. 2012;16:703–8. doi: 10.1007/s11325-011-0560-7. [DOI] [PubMed] [Google Scholar]
- 21.Tam CS, Wong M, McBain R, Bailey S, Waters KA. Inflammatory measures in children with obstructive sleep apnoea. J Paediatr Child Health. 2006;42:277–82. doi: 10.1111/j.1440-1754.2006.00854.x. [DOI] [PubMed] [Google Scholar]
- 22.Vgontzas AN, Papanicolaou DA, Bixler EO, Kales A, Tyson K, Chrousos GP. Elevation of plasma cytokines in disorders of excessive daytime sleepiness: role of sleep disturbance and obesity. J Clin Endocrinol Metab. 1997;82:1313–6. doi: 10.1210/jcem.82.5.3950. [DOI] [PubMed] [Google Scholar]
- 23.Krueger JM. The role of cytokines in sleep regulation. Curr Pharm Des. 2008;14:3408–16. doi: 10.2174/138161208786549281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vgontzas AN, Zoumakis E, Lin HM, Bixler EO, Trakada G, Chrousos GP. Marked decrease in sleepiness in patients with sleep apnea by etanercept, a tumor necrosis factor-alpha antagonist. J Clin Endocrinol Metab. 2004;89:4409–13. doi: 10.1210/jc.2003-031929. [DOI] [PubMed] [Google Scholar]
- 25.Lee JW, Lee YK, Yuk DY, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37. doi: 10.1186/1742-2094-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li RC, Row BW, Gozal E, et al. Cyclooxygenase 2 and intermittent hypoxia-induced spatial deficits in the rat. Am J Respir Crit Care Med. 2003;168:469–75. doi: 10.1164/rccm.200211-1264OC. [DOI] [PubMed] [Google Scholar]
- 27.Zhan G, Serrano F, Fenik P, et al. NADPH oxidase mediates hypersomnolence and brain oxidative injury in a murine model of sleep apnea. Am J Respir Crit Care Med. 2005;172:921–9. doi: 10.1164/rccm.200504-581OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ravassard P, Pachoud B, Comte JC, et al. Paradoxical (REM) sleep deprivation causes a large and rapidly reversible decrease in long-term potentiation, synaptic transmission, glutamate receptor protein levels, and ERK/MAPK activation in the dorsal hippocampus. Sleep. 2009;32:227–40. doi: 10.1093/sleep/32.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuster-Matanzo A, Llorens-Martin M, de Barreda EG, Avila J, Hernandez F. Different susceptibility to neurodegeneration of dorsal and ventral hippocampal dentate gyrus: a study with transgenic mice overexpressing GSK3beta. PLoS One. 2011;6:e27262. doi: 10.1371/journal.pone.0027262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7–19. doi: 10.1016/j.neuron.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sy M, Kitazawa M, Medeiros R, et al. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011;178:2811–22. doi: 10.1016/j.ajpath.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paxinos G, Franklin KBJ. Second Edition. San Diego: Academic Press; 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- 33.Vaessen TJ, Overeem S, Sitskoorn MM. Cognitive complaints in obstructive sleep apnea. Sleep Med Rev. 2015;19:51–8. doi: 10.1016/j.smrv.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 34.Dematteis M, Godin-Ribuot D, Arnaud C, et al. Cardiovascular consequences of sleep-disordered breathing: contribution of animal models to understanding of the human disease. ILAR J. 2009;50:262–81. doi: 10.1093/ilar.50.3.262. [DOI] [PubMed] [Google Scholar]
- 35.Poulain L, Richard V, Lévy P, Dematteis M, Arnaud C. Toll-Like Receptor-4 mediated inflammation is involved in the cardiometabolic alterations induced by intermittent hypoxia. Mediators Inflamm. 2015;2015:620258. doi: 10.1155/2015/620258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–18. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arnaud C, Beguin PC, Lantuejoul S, et al. The inflammatory preatherosclerotic remodeling induced by intermittent hypoxia is attenuated by RANTES/CCL5 inhibition. Am J Respir Crit Care Med. 2011;184:724–31. doi: 10.1164/rccm.201012-2033OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lam SY, Liu Y, Ng KM, et al. Chronic intermittent hypoxia induces local inflammation of the rat carotid body via functional upregulation of proinflammatory cytokine pathways. Histochem Cell Biol. 2012;137:303–17. doi: 10.1007/s00418-011-0900-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15:43–53. doi: 10.1038/nrn3617. [DOI] [PubMed] [Google Scholar]
- 40.Proescholdt MG, Chakravarty S, Foster JA, Foti SB, Briley EM, Herkenham M. Intracerebroventricular but not intravenous interleukin-1beta induces widespread vascular-mediated leukocyte infiltration and immune signal mRNA expression followed by brain-wide glial activation. Neuroscience. 2002;112:731–49. doi: 10.1016/s0306-4522(02)00048-9. [DOI] [PubMed] [Google Scholar]
- 41.Anthony D, Dempster R, Fearn S, et al. CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood-brain barrier breakdown. Curr Biol. 1998;8:923–6. doi: 10.1016/s0960-9822(07)00373-9. [DOI] [PubMed] [Google Scholar]
- 42.Moore AH, Olschowka JA, O'Banion MK. Intraparenchymal administration of interleukin-1beta induces cyclooxygenase-2-mediated expression of membrane- and cytosolic-associated prostaglandin E synthases in mouse brain. J Neuroimmunol. 2004;148:32–40. doi: 10.1016/j.jneuroim.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 43.Skuljec J, Sun H, Pul R, et al. CCL5 induces a pro-inflammatory profile in microglia in vitro. Cell Immunol. 2011;270:164–71. doi: 10.1016/j.cellimm.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 44.Basu A, Krady JK, O'Malley M, Styren SD, DeKosky ST, Levison SW. The type 1 interleukin-1 receptor is essential for the efficient activation of microglia and the induction of multiple proinflammatory mediators in response to brain injury. J Neurosci. 2002;22:6071–82. doi: 10.1523/JNEUROSCI.22-14-06071.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bellinger FP, Madamba S, Siggins GR. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–34. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- 46.Rachal Pugh C, Fleshner M, Watkins LR, Maier SF, Rudy JW. The immune system and memory consolidation: a role for the cytokine IL-1beta. Neurosci Biobehav Rev. 2001;25:29–41. doi: 10.1016/s0149-7634(00)00048-8. [DOI] [PubMed] [Google Scholar]
- 47.Yang I, Han SJ, Kaur G, Crane C, Parsa AT. The role of microglia in central nervous system immunity and glioma immunology. J Clin Neurosci. 2010;17:6–10. doi: 10.1016/j.jocn.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ben Achour S, Pascual O. Glia: the many ways to modulate synaptic plasticity. Neurochem Int. 2010;57:440–5. doi: 10.1016/j.neuint.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 49.Griffin R, Nally R, Nolan Y, McCartney Y, Linden J, Lynch MA. The age-related attenuation in long-term potentiation is associated with microglial activation. J Neurochem. 2006;99:1263–72. doi: 10.1111/j.1471-4159.2006.04165.x. [DOI] [PubMed] [Google Scholar]
- 50.Luo XG, Chen SD. The changing phenotype of microglia from homeostasis to disease. Transl Neurodegener. 2012;1:9. doi: 10.1186/2047-9158-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li RC, Guo SZ, Raccurt M, et al. Exogenous growth hormone attenuates cognitive deficits induced by intermittent hypoxia in rats. Neuroscience. 2011;196:237–50. doi: 10.1016/j.neuroscience.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashton D, Van Reempts J, Haseldonckx M, Willems R. Dorsal-ventral gradient in vulnerability of CA1 hippocampus to ischemia: a combined histological and electrophysiological study. Brain Res. 1989;487:368–72. doi: 10.1016/0006-8993(89)90842-1. [DOI] [PubMed] [Google Scholar]
- 53.Bucks RS, Olaithe M, Eastwood P. Neurocognitive function in obstructive sleep apnoea: a meta-review. Respirology. 2013;18:61–70. doi: 10.1111/j.1440-1843.2012.02255.x. [DOI] [PubMed] [Google Scholar]
- 54.Ferini-Strambi L, Marelli S, Galbiati A, Castronovo C. Effects of continuous positive airway pressure on cognitition and neuroimaging data in sleep apnea. Int J Psychophysiol. 2013;89:203–12. doi: 10.1016/j.ijpsycho.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 55.Barrientos RM, Frank MG, Watkins LR, Maier SF. Memory impairments in healthy aging: role of aging-induced microglial sensitization. Aging Dis. 2010;1:212–31. [PMC free article] [PubMed] [Google Scholar]
- 56.Dilger RN, Johnson RW. Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J Leukoc Biol. 2008;84:932–9. doi: 10.1189/jlb.0208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi JH, Won MH. Microglia in the normally aged hippocampus. Lab Anim Res. 2011;27:181–7. doi: 10.5625/lar.2011.27.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–7. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- 59.Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4:103–12. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- 60.von Bernhardi R, Tichauer JE, Eugenin J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J Neurochem. 2010;112:1099–114. doi: 10.1111/j.1471-4159.2009.06537.x. [DOI] [PubMed] [Google Scholar]
- 61.Meda L, Baron P, Scarlato G. Glial activation in Alzheimer's disease: the role of Abeta and its associated proteins. Neurobiol Aging. 2001;22:885–93. doi: 10.1016/s0197-4580(01)00307-4. [DOI] [PubMed] [Google Scholar]
- 62.Wynne AM, Henry CJ, Godbout JP. Immune and behavioral consequences of microglial reactivity in the aged brain. Integr Comp Biol. 2009;49:254–66. doi: 10.1093/icb/icp009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10:217–24. doi: 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
- 64.Ifuku M, Katafuchi T, Mawatari S, et al. Anti-inflammatory/anti-amyloidogenic effects of plasmalogens in lipopolysaccharide-induced neuroinflammation in adult mice. J Neuroinflammation. 2012;9:197. doi: 10.1186/1742-2094-9-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Biesmans S, Meert TF, Bouwknecht JA, et al. Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediators Inflamm. 2013;2013:271359. doi: 10.1155/2013/271359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morris M, Li L. Molecular mechanisms and pathological consequences of endotoxin tolerance and priming. Arch Immunol Ther Exp (Warsz) 2012;60:13–8. doi: 10.1007/s00005-011-0155-9. [DOI] [PubMed] [Google Scholar]
- 67.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–87. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 68.Rock RB, Gekker G, Hu S, et al. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17:942–64. doi: 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Spooren A, Kolmus K, Laureys G, et al. Interleukin-6, a mental cytokine. Brain Res Rev. 2011;67:157–83. doi: 10.1016/j.brainresrev.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 70.Zhu Y, Fenik P, Zhan G, et al. Selective loss of catecholaminergic wake active neurons in a murine sleep apnea model. J Neurosci. 2007;27:10060–71. doi: 10.1523/JNEUROSCI.0857-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Polotsky VY, Rubin AE, Balbir A, et al. Intermittent hypoxia causes REM sleep deficits and decreases EEG delta power in NREM sleep in the C57BL/6J mouse. Sleep Med. 2006;7:7–16. doi: 10.1016/j.sleep.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 72.Wisor JP, Clegern WC, Schmidt MA. Toll-like receptor 4 is a regulator of monocyte and electroencephalographic responses to sleep loss. Sleep. 2011;34:1335–45. doi: 10.5665/SLEEP.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ingiosi AM, Opp MR, Krueger JM. Sleep and immune function: glial contributions and consequences of aging. Curr Opin Neurobiol. 2013;23:806–11. doi: 10.1016/j.conb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zielinski MR, Kim Y, Karpova SA, McCarley RW, Strecker RE, Gerashchenko D. Chronic sleep restriction elevates brain interleukin-1 beta and tumor necrosis factor-alpha and attenuates brain-derived neurotrophic factor expression. Neurosci Lett. 2014;580:27–31. doi: 10.1016/j.neulet.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McKhann GM, 2nd, Wenzel HJ, Robbins CA, Sosunov AA, Schwartzkroin PA. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience. 2003;122:551–61. doi: 10.1016/s0306-4522(03)00562-1. [DOI] [PubMed] [Google Scholar]
- 76.Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci U S A. 1997;94:4103–8. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Starr ME, Hu Y, Stromberg AJ, et al. Gene expression profile of mouse white adipose tissue during inflammatory stress: age-dependent upregulation of major procoagulant factors. Aging Cell. 2013;12:194–206. doi: 10.1111/acel.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Babri S, Doosti MH, Salari AA. Strain-dependent effects of prenatal maternal immune activation on anxiety- and depression-like behaviors in offspring. Brain Behav Immun. 2014;37:164–76. doi: 10.1016/j.bbi.2013.12.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Validation of reference genes for semiquantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis of RANTES/CCL5, ICAM-1, IL-1β, MCP-1/CCL2, TNF-α, IL-6 mRNA. Semiquantitative real-time RT-PCR cycle threshold numbers (Cq values) for the three reference genes RPLP0, ACTB, and HPRT1 were unaffected by the experimental conditions: 1 day, 6 and 24 w of intermittent hypoxia (IH) or normoxia (N) and 6 w IH or N with 3 w intraperitoneal low-dose lipopolysaccharide (LPS) or saline (NaCl). Complementary DNA were diluted to 1/40. Results are expressed as mean ± standard error of the mean (n = 5–6 for each group).
Validation of reference genes for semiquantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis of IL-10 cytokine messenger RNA (mRNA). Semiquantitative real-time RT-PCR cycle threshold numbers (Cq values) for the three reference genes RPLP0, ACTB, and HPRT1 were unaffected by the experimental conditions: 1 day, 6 and 24 w of intermittent hypoxia (IH) or normoxia (N) and 6 w IH or N with 3 w intraperitoneal low-dose lipopolysaccharide (LPS) or saline (NaCl). Complementary DNA were diluted to 1/160. Results are expressed as mean ± standard error of the mean (n = 5–6 for each group).
Acute and chronic intermittent hypoxia do not induce qualitative alterations of microglia in the ventral hippocampus. Representative photographs showing microglial cells (Iba1+ cells, black cytoplasmic staining) in CA3 area of the ventral hippocampus (−3.00 mm from Bregma) from intermittent hypoxia (IH) and normoxia (N) mice after 1 day, 6 and 24 w of exposure. Scale bar: 50 μm. Py, pyramidal cell layer of the hippocampus; Rad, stratum radiatum of the hippocampus.
Inflammatory cytokine messenger RNA (mRNA) in the dorsal hippocampus after acute and chronic intermittent hypoxia. Levels of ICAM-1, MCP-1/CCL2, TNF-α, IL-6, and IL-10 mRNA were normalized to the RPLP0, ACTB, and HPRT1 genes, and are shown as a ratio to the level found in control animals exposed to 1 day of normoxia. Results are expressed as mean ± standard error of the mean (n = 5–6 for each group). IH, intermittent hypoxia; N, normoxia.