

Abstract
DNA repair, particularly DNA double strand breaks (DSBs) repair, is essential for the survival of both normal and cancer cells. An elaborate repair mechanism has been developed in cells in order to efficiently repair the damaged DNA. The pathways that are predominately involved in DSBs repair are homologous recombination (HR) and classical nonhomologous end-joining (cNHEJ) although alternative NHEJ (aNHEJ), a third DSBs repair pathway, may also be important in certain contexts. The protein of BRCA1 encoded by the tumor suppressor gene BRCA1 regulates all DSBs repair pathways. Given the fact that DSBs represent the most biologically significant lesions induced by ionizing radiation (IR) and impaired DSBs repair leads to radiation sensitivity it has been expected that cancer patients with BRCA1 mutations should benefit from radiation therapy (RT). However, the clinical data are conflicting and inconclusive. Here, we provide an overview about the current status of the data regarding BRCA1 deficiency and RT sensitivity in both experimental models and clinical investigations. In addition, we will discuss a strategy to potentiate the effects of RT by poly(ADP ribose) polymerase (PARP) inhibitors, the pharmacologic drugs that are being investigated as a monotherapy for the treatment of patients with BRCA 1/2 mutations.
Background
DNA double-strand breaks (DSBs) are the most cytotoxic forms of DNA damage (1,2). DSBs can not only be caused by replication stress but also by the principle cytotoxic lesion from ionizing radiation (IR) and radiomimetic chemicals. Eukaryotic cells have evolved a sophisticated DNA damage response (DDR) to efficiently detect and repair these lesions, including activation of direct repair, cell cycle checkpoints, transcriptional alteration and in some types of cells induction of cell death by apoptosis (3). The importance of the response to DNA-damage, particularly DSBs, is reflected by the fact that defects in repair and signaling of DNA damage are causally linked with development of cancer due to increased genomic instability. For example, the cancer-prone syndrome ataxia telangiectasia is caused by mutation of the ATM kinase, a central protein to cell cycle checkpoint activation after DSBs (4). The genomic instability syndromes ataxia-telangiectasia-like-disorder and Nijmegen Breakage syndrome are caused by mutation of Mre11 or NBS1, components of the DSB-sensing Mre11 complex comprised of Mre11, Rad50, and NBS1 (5). Moreover, the hereditary breast and ovarian cancer (HBOC) syndrome which is related to a high risk of developing breast cancer and ovarian cancer are due to mutation in BRCA1/BRCA2, two of the most important proteins required for homologous recombination (HR)- mediated DSBs repair (6).
DNA DSB Repair Pathways and IR
The administration of IR, radiation therapy (RT) has been widely used in the medical field for the treatment of cancer patients. IR causes damage directly through energy deposition and indirectly by attacking the DNA through ionization of water molecules to produce hydroxyl radicals. Multiple forms of DNA damage are induced by IR, including damage to the bases, and cleavage of the DNA backbone to form DNA single strand breaks and DSBs (7). Compared to other types of DNA damage induced by IR, DSBs are determinant of cellular radiosensitivity (8).
Generally speaking, the pathways that are predominately involved in DSBs repair are classical nonhomologous end-joining (cNHEJ) and HR, although alternative NHEJ (aNHEJ), a third DSBs repair pathway, may also contributes to repair of DSBs. In mammalian cells, the majority of IR-induced DSBs are repaired by cNHEJ. cNHEJ occurs mainly in the G0 and G1 phase of the cell cycle. Although cNHEJ involves rejoining the two broken ends of the DNA without DNA resection, cNHEJ is generally viewed as a more error prone method of DSB repair as genetic information can be lost before the two ends are rejoined. A central component of cNHEJ is the DNA–PK complex, composed of a heterodimer of the Ku proteins, Ku70 and Ku86, and a catalytic sub-unit, DNA–PKcs. Initial binding of Ku to DSBs is important to protect the ends of DSBs from degradation. The Ku protein can then recruit DNA–PKcs, which phosphorylate itself and other enzymes involved in repair (7). Re-ligation of the strands is achieved by the XRCC4–ligase IV complex. The recently identified cNHEJ component, XRCC4-like factor, stimulates the activity of the XRCC4-DNA ligase IV complex toward noncompatible DNA ends (9). DNA end-processing enzymes such as Artemis are also required for the processing of a subset of IR-induced DSBs in vivo (10)
HR is the second major pathway for the repair of DSBs induced by IR in mammalian cells. However, HR is the predominant repair pathway for endogenous DSBs produced when replication fork collapse occurs (11). HR functions only during the late S and G2 phases of the cell cycle, when a homologous region of DNA is available. Cells that are deficient in HR are less sensitive to IR-induced damage than NHEJ deficient cells. In addition, there is evidence that cells generally demonstrate increased radio-resistance during late-S/G2-phase (12) while HR mutants exhibit the greatest radiosensitivity in late S/G2 (13). Early studies suggested that HR proteins might play a minimal role in repair of IR-induced DSBs repair (14). Recent studies demonstrate that HR repairs both direct and secondary, replication-induced, DSBs after IR (15,16). In addition, it has been demonstrated that HR repair promoted by ATM and Artemis contributes to the slowly repairing sub-fraction of IR-induced breaks in G2 phase (17). HR uses the homologous template to faithfully repair the DNA, and consequently, it is a more accurate form of DSB repair. Although, chromosome rearrangement can also occur if the template is the homologous chromosome or an ectopic repeat is located in the same or different chromosome (18). A key step in HR is initiation of exonucleolytic resection, which generates long single-stranded tails, critical intermediates for initiating homologous pairing by strand exchange. The formed RAD51 nucleoprotein filament then facilitates DNA strand invasion and exchange steps (19) leading to formation of a Holliday junction (HJ). Either gene conversion (GC) or crossovers (CO) are generated depending on how the HJs are resolved (20).
The third and most recently discovered mechanism for DSB repair is aNHEJ. Initially, aNHEJ was viewed as a backup system capable of repairing DSBs in cells with deficiencies of cNHEJ (21, 64, 73). The subsequent discovery of aNHEJ in cNHEJ-proficient cells indicates that it is not simply a backup pathway used by cells to allow survival in the absence of cNHEJ (21). aNHEJ also plays an important physiological role in repairing DSBs to maintain cell viability, especially under genomic stress (21). Early studies suggested that aNHEJ is a repair process with slow kinetics compared to rapid repair by c-NHEJ (14,22). However, studies later suggested that DNA end complexity also contributes to the repair kinetics (23,24). Consistent with this concept, the cNHEJ protein, DNA-Pkcs, was found to contribute to the repair of IR-induced DSBs repair with slower kinetics (25). Several features of aNHEJ have been characterized although the molecular mechanisms controlling it are less well delineated compared to cNHEJ. First, it has been found that aNHEJ products have a great dependence on short patches of perfectly matched sequences known as microhomologies (26,27). Frequently, aNHEJ is referred to as microhomology-mediated end joining (MMEJ) or backup-NHEJ (B-NHEJ) although not all aNHEJ events involve a microhomology sequence. Second, longer deletions or resections may occur prior to end joining. Mechanistically, aNHEJ is associated with the generation of 3’ single strand overhang, and this process involves the MRE11/RAD50/NBS1 complex and CtIP (25,28–33). Last, aNHEJ/MMEJ is a distinct DSB repair pathways compared to c-NHEJ (32). aNHEJ/MMEJ is suppressed by the core cNHEJ protein Ku but promoted by CtIP, perhaps in G1 phase cells (29,34,35). In mammalian cells, in addition to the MRE11/RAD50/NBS1 complex and CtIP, XRCC1/DNA ligase III has a critical role in aNHEJ (29,30,36–38). Additional work suggests an important role of poly(ADP-ribose) polymerase (PARP) 1 in the aNHEJ pathway (37,39–42). Our recent publication demonstrates that 53BP1 promotes aNHEJ/MMEJ in G1 phase cells (43). In yeast, MMEJ appears to be required for radioresistance. Studies suggest that a defect in PolQ-mediated aNHEJ/MMEJ in mammalian cells confer radiation sensitivity (44,45). These results indicate a critical role of aNHEJ in repair of IR-induced DSBs.
In summary, cNHEJ and HR are two major mechanisms required for repair of DSBs induced by IR. Cells with decreased ability of repair DSBs are more sensitive to IR. The role of aNHEJ pathway in the repair of IR- induced DSBs in mammalian cells needs to be further investigated and established.
BRCA1 and Double Strand Breaks Repair
BRCA1 or BRCA2 are two important tumor suppressor genes. Mutation in one copy of either gene in the germline results in HBOC syndrome, which accounts for 5–7% of all cases of breast cancer. Individuals with HBOC syndrome have a 50–80% lifetime risk of developing breast cancer, and 30–50% risk for ovarian cancer. HBOC is inherited in an autosomal-dominant manner (6). In addition to breast and ovarian cancer, BRCA1 mutation is also associated with an increased risk of pancreatic, stomach, laryngeal, fallopian tube and prostate cancer (6). BRCA1 is involved in a wide spectrum of cellular processes such as cell-cycle regulation, transcriptional regulation, chromatin remodeling, DNA DSBs and apoptosis. In contrast, the functions of BRCA2 are largely limited to DSBs repair through promoting HR (6). Loss of function of these two proteins leading to inability to undergo HR is believed to be a key step in tumorigenesis in individuals with BRCA1/2 mutations (46). Although cell cycle checkpoint and programmed cell death (apoptosis) also determine radiation sensitivity, they are beyond the scope of this review. We will focus on the roles of BRCA1 in DNA DSBs repair and its implications in RT.
BRCA1 has a well-established role in promoting HR mediated DSBs repair; (47–51) although the actual functional role of BRCA1 in HR and its regulations remain largely unknown. BRCA1 promotes single stranded DNA resection and/or acts as a recombination mediator/comediator (20). The interaction of BRCA1 with CtIP may be especially critical for regulating the processing of DNA ends at DSBs during HR (52). In addition, BRCA1 also interacts with PALB2 linking it functionally to BRCA2 and its role in RAD51 loading during HR (53,54).
Studies on the role of BRCA1 in cNHEJ have yielded conflicting results, ranging from suppression of NHEJ to no effect or promotion (51,55–58), which could be due to different reporters and cell lines. BRCA1 was found to inhibit random integration; (47,51) however, there is plenty of data, including ours, which suggest that BRCA1 is important for some subtypes of cNHEJ pathway. It has been suggested that the cNHEJ pathway is impaired in BRCA1 −/− mouse embryonic fibroblasts (MEFs) (59,60) and in the human breast cancer cell line, HCC1937, which carries a homozygous mutation in the BRCA1 gene (56,57,61). The fidelity of NHEJ is severely decreased in lymphoblastoid cell lines established from women with a truncating BRCA1 mutation or missense BRCA1 mutations (56,62,63). Our previous work demonstrated a lower frequency of error-free end joining in HCC1937 cells compared to control cells with intact BRCA1 (57), which is supported by the results obtained from several cancer cell lines with BRCA1 depletion (64). All these data suggest a role for BRCA1 in cNHEJ pathway. Interestingly, ATM/CHK2 pathway-mediated BRCA1 phosphorylation at S988 promotes the fidelity of DSB repair by NHEJ (57,64). A favored model is that BRCA1 may only be involved in this particular subpathway of precise NHEJ (51,56,61,62,65). In support of this hypothesis, a very recent study demonstrated that BRCA1 binds and stabilizes Ku80 at DSBs, promotes precise DSB rejoining, and increases cellular resistance to radiation-induced DNA damage in a G1 phase-specific manner (66).
The role of BRCA1 in aNHEJ/MMEJ pathway is debatable and conflicting. It has been demonstrated that Brca1(−/−) MEFs exhibited a deficiency in MMEJ using a defined chromosomal DNA DSB introduced by a rare cutting endonuclease I-SceI (60). In this reporter, the CATG MMEJ resulted in repair products that could be cleaved by restoring the original NcoI site. Using this reporter, Brca1 was found to be important for MMEJ activity. Interestingly, our previous work suggested an inhibitive role in MMEJ using the chromatinized pEPI-HDV6 substrate and extra-chromosomal reporter detecting various stretches of microhomology around the cleavage site. We found that the proportion of rejoining events using any microhomology length (1–8 bp) was significantly elevated in cells with BRCA1 deficiency (57). The lack of consistency between these results may be at least partly explained by the use of different assays, which may measure different subtypes of aNHEJ/MMEJ, and different cell lines.
BRCA1 Deficiency and Radiation Sensitivity in Experimental Models
A number of known inherited syndromes caused by mutations in genes required for the response to DNA damage, such as ataxia telangiectasia, ataxia-telangiectasia like-disorder, radiosensitive severe combined immunodeficiency, Nijmegen breakage syndrome, and LIG4 deficiency are associated with increased radiosensitivity (67). The radiation sensitivity of cells with BRCA1 mutation has been tested in a variety of experimental models. BRCA1 −/− MEFs are highly sensitive to IR (68,69). A targeted disruption in exon 11 of the murine Brca1 gene causes IR hypersensitivity and genetic instability. Consistent with the results observed in MEFs, radiation sensitivity was also observed in human cancer cells HCC1937, a cell line with truncated BRCA1 expression (69). Human cells carrying mutations of BRCA1, BRCA2, or heterozygous BRCA1 and BRCA2 mutations also led to enhanced radiosensitivity with an impaired proliferative capacity after irradiation (69). Retrovirally expressed wild-type BRCA1 decreased the IR sensitivity and increased the efficiency of DSBs repair of the BRCA1 −/− human breast cancer line, HCC1937. It also reduced its susceptibility to DSB generation by IR (70). An increased sensitivity to IR has additionally been observed in an ovarian cancer cell line carrying defective BRCA1 (71). Work by Cortez et al. demonstrated that ATM –dependent phosphorylation on BRCA1 is important for the resistance to IR (72). Lastly, a study by Li et al. demonstrates with an animal model that BRCA1 is critical for cancer cell resistance to IR, mediated by FOXP3 deficiency (73). Tumor cell lines with or without 15 Gy irradiation were injected into the mammary fat-pad of syngeneic mice and monitored for tumor growth. It was found that the impact of Foxp3 silencing on tumor growth after radiation was abrogated by concurrent Brca1 silencing, suggesting a critical role of BRCA1 in Foxp3 mediated radiation resistance (73).
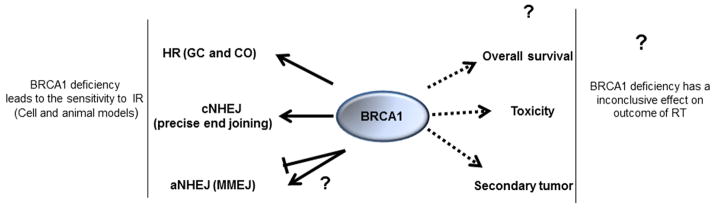
In summary, the evidence obtained from cell and animal models have provided strong evidence supporting a role of BRCA1 in radioresistance. The impaired ability for DSBs repair contributes to radiation sensitivity when BRCA1 is deficient. However, which DSBs repair pathways controlled by BRCA1 play a more important role in this regulation remains unknown. Interestingly, BRCA1 expression restores radiation resistance in BRCA1-defective cancer cells through enhancement of transcription-coupled DNA repair (71). In addition, our previous study suggested that ATM dependent phosphorylation on BRCA1 is not important for HR mediated repair (51) although it is critical for radiation resistance (72). These results may indicate that the role of BRCA1 in HR play a minor role or no role on radiation sensitivity. This hypothesis needs to be tested in the future.
BRCA1 and Radiation Therapy in Clinical Settings
Theoretically, tumors with mutated BRCA1 may be more sensitive to treatments due to their inherent inability to repair DNA DSBs caused by radiation and/or chemotherapeutic agents. On the other hand, in patients with mutant BRCA1 there is the concern that RT may increase toxicity or the risk of secondary malignancy. Many studies have sought to determine whether these theoretical benefits or risks are indeed present. The majority of these studies include patients with BRCA1 and/or BRCA2 germline mutations as they both promote HR, and the number of patients with these mutations is small.
In a study of a small number of patients, a lower rate of subsequent ipsilateral breast tumors in BRCA1/2 carriers treated for their cancers by lumpectomy and breast RT was observed compared with noncarriers treated similarly at a median follow-up time of 63 months (74). However, in the same study, the authors found that women with germline BRCA mutations were significantly more likely to develop contralateral breast cancer at 5 years. Contralateral breast cancer developed in 12 of 30 (40.0%) women with germline BRCA mutations, compared with five of 61 (8.2%) women without BRCA mutations (74). The reasons regarding why there might be different effects observed on occurrence of ipsilateral and contralateral breast cancer is not clear. It is possible that occult ipsilateral tumors in BRCA mutation carriers are more effectively treated by IR than those in non-carriers, due to their impaired DSBs repair pathways. However, the proposed roles of BRCA1 and BRCA2 in DDR suggest that IR may initiate tumorigenesis in those that carry a mutation in either BRCA1 or BRCA2 as a result of un-repaired DNA DSBs. Data on the incidence of subsequent contralateral breast cancers in BRCA1/2 carriers treated with breast conservation and adjuvant RT compared to those treated with mastectomy alone could help to clarify this issue. It is possible that the low dose scatter radiation to the contralateral breast induces tumorigenesis by causing DNA breaks that are less likely to be repaired in those carrying mutations in BRCA1/2. This could lead to an increase incidence of later-onset breast tumors, indicating a new primary as opposed to a clinical presentation of a previously occult cancer, in both the ipsilateral and contralateral breasts (75). The increased number of contralateral breast cancers in those with germline BRCA mutation could also be due to their increased baseline risk of developing breast cancer. A study from Yale, examined a group of young women with breast cancer treated with breast conserving therapy (BCT) and radiation. They performed BRCA1 and BRCA2 sequencing and compared those who were found to have deleterious mutations to those who did not. They noted that the rate of ipsilateral events were statistically significantly higher in the group with BRCA1/2 mutations, 49%, compared to those without deleterious mutations, 21%. Contralateral breast cancers were also significantly higher in the group with deleterious mutations 42% compared to 9% (76) . Another report by Pierce et al., a multi-national study, compared women with BRCA1 or BRCA2 mutation treated with either BCT and RT or mastectomy alone. They found that local recurrence rates were higher in those treated with BCT + RT, 30.2% at 20 years, compared to mastectomy, 5.5% at 20 years (77). However, no difference was noted in overall survival or in regional/systemic recurrence. The risk of contralateral breast cancer was high regardless of whether adjuvant RT was given, exceeding 40%, indicating that low dose scatter from radiation treatment does not seem to increase the risk of developing breast cancer.
The hypothesis that breast cancers in BRCA1/2 mutation carriers may be more sensitive to RT does not seem to be supported by retrospective studies. A report from the Institut Curie of matched retrospective case-control study examined ipsilateral tumor recurrence, contralateral tumor development, and overall survival in breast cancer patients that had undergone breast conserving surgery and RT. They demonstrated that with a median follow-up of 161 months there was no difference in ipsilateral breast tumor recurrence (IBTR) or overall survival (OS) between BRCA1/2 mutation carriers versus sporadic controls. As would be expected, contralateral breast cancers were significantly higher in those patients with BRCA1/2 mutations compared to controls, due to the high risk of new primary cancers inherent in this population (78). Pierce et al. also reported in a multi-institutional study that IBTR was similar between BRCA1/2 mutated patients compared to controls treated with breast conserving surgery. At 15 years there was no significant different in IBTR, 24% in mutation carriers compared to 17% in controls. However, in a subset analysis they did note a significantly higher IBTR rate in BRCA1/2 mutated patients who had not undergone oophorectomy, RR of 1.9, compared to controls (79). Consistent with this observation, in a retrospective study of Stage I/II breast cancer patients by Pierce et al., they did not note any difference in OS, Local recurrence (LR), or cancer specific survival (CSS) between 71 BRCA1/2 carriers and age/stage matched control patients with a median follow-up of 5.3 years for the BRCA1/2 patients and 4.6 years for controls. For the BRCA1/2 mutation carriers compared to controls, CSS at 5 years was 92% vs. 91%, respectively, and local control was 98% vs. 96%, respectively (80). In a study by Goodwin et al., they examined outcomes of breast cancer patients by either BRCA1 or BRCA2 compared to sporadic cases. They demonstrate that women with breast cancer and BRCA2 mutations may have worse prognosis than age-matched sporadic breast cancers. However, when adjusted for effects of tumor and treatment related variable, there was no significant difference in prognosis. This indicates that women with BRCA2 mutation may present with more aggressive breast cancers; however the prognosis would be similar to a stage-matched cancer in a non-mutation carrier patient (81). Thus it appears as if BRCA1/2 mutation carriers are not more sensitive to RT in terms of the ability to cure breast cancer, at least in early stage breast cancer patients. Additionally, there is currently no evidence to support a higher risk of RT induced secondary malignancies.
Although most of the studies looking at BRCA1 and breast cancer prognosis or response to treatment have been in the group of patients with detectable BRCA1/2 mutations, some groups have looked at protein expression levels in patients with no familial history of breast cancer because lower expression of BRCA1 is also frequently observed in spontaneous breast cancers. It is estimated that almost 33% of sporadic breast cancers display decreased BRCA1 expression (82). A Japanese study examined a group of breast cancer patients with no family history of breast cancer who underwent surgery with subcutaneous glandectomy and axillary lymph node dissection or mastectomy with axillary lymph node dissection followed by combination chemotherapy and tamoxifen. At a median follow-up of 4.4 years those with negative BRCA1 staining had statistically significant higher disease recurrence rate compared to those with positive staining, 35% vs. 7%, respectively (83). In a United Kingdom study, BRCA1 immunohistochemistry staining examined. After adjusting for grade, patients with BRCA1 negative tumors had a significantly longer disease free survival (DFS) than those with tumors expressing BRCA1. A similar trend, although non-significant, was noted with regards to OS. Before the adjustment by grade, there was no significant difference for OS and DFS between BRCA1 negative and positive tumors (84). Soderlund et al. examined local recurrences in relation to expression levels of BRCA1/BRCA2/RAD51, a complex that is important in HR DNA repair of DSBs. Patients from a prospective breast cancer trial comparing either adjuvant chemotherapy (CMF-cyclophosphamide, methotrexate, and 5-fluorouracil) or post-operative RT, 46 Gy/2 Gy per fraction to the chest wall and comprehensive nodal regions, after modified radical mastectomy. These patients all had node positive breast cancer and/or had tumors greater than or equal to 3 cm. They found that patients who received chemotherapy had a local-recurrence free survival (LRFS) of 85% if a high level of the BRCA1 complex was noted compared to a LRFS of 60% if there were low levels of BRCA1 complex. In patients with low levels of BRCA1 complex the LRFS for those receiving RT was significantly higher at 90% compared to those that received the chemotherapy, 60%, with a statistically significant RR of 0.38. Those patients with high levels of BRCA1 complex had similar LRFS rates of about 80% regardless of which adjuvant treatment was received (85). These studies demonstrate that BRCA1/2 expression level within a tumor, regardless of the patient’s germline status, has an effect on outcome. Tumor cells with low levels of functional BRCA1/2 are less effectively treated with standard treatments. Interestingly, it does appear that RT may be more effective than chemotherapy in cancers with low levels of BRCA1 complex. This could be dependent on the particular mechanism a chemotherapeutic agent utilizes to induce cell death and certain chemotherapies could be just as effective as RT. Alternatively the DNA DSBs caused by RT may be more effective in causing cell death compared to the chemotherapeutic agents used in the analysis by Soderlund.
Although results are somewhat mixed it is thought that there is a higher risk of tumor relapse in the ipsilateral breast in BRCA1/2 mutated patients. Thus, while not an absolute contraindication, general consensus would recommend mastectomy as opposed to breast conserving therapy in patients with known BRCA1/2 mutation (86). An additional limitation to these retrospective studies is the unknown effects of each particular BRCA1 or BRCA2 mutation. These proteins have multiple functions within the cell and different mutations may have variable effects on these cellular processes. Depending on the specific mutation, there could be differing effects on radiation and/or chemotherapy sensitivity and could increase or decrease the likelihood of recurrence.
Radiation sensitivity may not require loss of both alleles because abnormalities in the DDR and radiation sensitivity were observed in cells harboring a single mutated copy of BRCA1 or BRCA2 (69,87,88). In addition, a role for BRCA1 in mammary tumor formation after exposure to IR has been suggested using BRCA1+/− mouse model (89). Many groups have sought to determine whether patients with BRCA1 mutations are more sensitive to the normal tissue toxicities of RT and/or chemotherapy. Patients with lung cancer who received RT and had single photon emission computed tomography (SPECT) lung perfusion imaging before and after treatment were examined for radiation-induced changes. Dose-response curves were generated for each patient and genotyping was performed from blood samples. It was found that polymorphisms within XRCC1 and BRCA1 were significantly associated with higher sensitivity to RT (90). However, other studies, mostly in breast cancer patients, have not definitively found a correlation between BRCA1 mutations and increased toxicity. Pierce et al. examined a group of BRCA1 and BRCA2 carriers with age-matched controls and found that following RT there was no different in acute skin toxicity, pneumonitis or breast pain. With a median followup of 5.3 years in the BRCA1/2 mutated group there was also no increase in late toxicity of the skin, subcutaneous tissue, lung or bone(80). In another small retrospective study a trend toward increased acute pain during RT treatment was noted as well as an increase in chronic pain. This study did not find a difference in other late toxicities of edema, fibrosis, telangiectasia, rib fractures, lung fibrosis, or cardiac events (91). A study examining 22 patients with severe normal tissue reactions after RT were screened for mutations in BRCA1 or BRCA2. None of the commonly described BRCA1/2 mutations were found which implies that other genes may account for much of the severe unexpected toxicities that are seen in patients treated with RT (92). In a Korean study of 213 breast cancer patients who had undergone BRCA1/2 mutation testing and received RT, acute skin toxicity was evaluated. They classified radiosensitivity as RTOG Grade 2 or greater acute skin reaction. Approximately 20% of the tested patients were found to have mutations in BRCA1, BRCA2, or both. There were 27% of patients in this group who were found to have Grade 2 or greater skin toxicity. Multivariate analysis identified significant associations between mean breast volume and toxicity; however, BRCA1/2 mutation status was not associated with acute skin toxicity (93). These data suggest that patients with BRCA1/ 2 mutation do not have significantly worse acute toxicity or a higher risk of late toxicity from RT.
Huszno et al. examined 270 early stage breast cancer patients who were treated with the same protocol regardless of BRCA1/2 mutation status. In this cohort 15% were found to be BRCA1/2 carriers. The rates of chemotherapy and RT toxicities were compared between the BRCA1/2 carriers and the rest of the cohort. All of the patients received an anthracycline based chemotherapy regimen and 66% received RT. Huszno et al did not find any difference in the rates of grade 4 toxicity from chemotherapy. There was a significantly increased incidence of nausea and vomiting, grade 3–4, in non-BRCA1/2 carriers. Neutropenia however was significantly more frequent in BRCA1/2 carriers. Acute toxicity from RT was similar between the two groups (94). An interesting study was performed, where peripheral lymphocytes of patients with breast cancer and healthy controls were compared for the kinetics of DNA damage repair and the level of basal, oxidative and alkylative DNA damage before and during/after chemotherapy. It was found that the patients with breast cancer had slower DNA repair compared to the healthy controls. They also noted higher levels of basal, oxidative and alkylative DNA damage in the breast cancer patients’ peripheral lymphocytes, prior to treatment, compared to the damage that was noted in controls(95). Similar findings were noted when looking at patients with head and neck cancer before RT compared with healthy controls (96). It is perhaps this lowered capacity for DNA damage repair that is already present in cancer patients that mitigates the increase in toxicity that was expected in patients with mutated BRCA1. Although the majority of these small retrospective studies have not borne out any major differences for acute or chronic toxicities the theoretical risk is still present. There are a number of mutations that are seen in the hereditary breast/ovarian cancers and perhaps only certain BRCA1 mutants may have a propensity for increased radiation sensitivity in the normal tissues. As more clinical trials directed at BRCA1/2 mutated patients are performed, it will be important to prospectively record and analyze for acute and chronic toxicities. It may be that as Blasiak et al. and Palyvoda et al. demonstrated, cancer patients already exhibit a higher amount of DNA damage in their cells such that BRCA1 mutation does not increase the toxicity noted in normal tissues.
Secondary malignancies are a rare, but known, late complication from RT as well as chemotherapy treatments. There is concern that in patients with deficient DNA repair capabilities treatment with RT could increase the risk of RT-induced cancers. It was noted that the rate of contralateral breast cancers was 2% in the sporadic breast cancer group compared to 20% in the BRCA1/2 carrier group. This rate is consistent with the rate of breast cancer development in known carriers even without RT treatment and thus it does not appear that at this time point the risk of a second breast cancer is increased in the BRCA1/2 carrier group despite the RT and chemotherapy treatment. The rate of ovarian cancer development was also increased as expected 11% vs 0%; however, other malignancies were not significantly increased (80). A study was performed in Israel examining the rate of RT-associated sarcomas in BRCA1/2 carriers. In their population of patients, with a follow-up of 8 years, there was a 0.53% rate of RT-associated sarcoma development (97). This is approximately two-fold higher than previously reported rates in sporadic breast cancer patients; however still a very low percentage (98). Kadouri et al concluded that while mutations in BRCA1/2 may increase the risk of RT-associated sarcomas, the risk was still very small and should not play a significant role in decision-making with regards to RT.
In summary, despite the affirmative role in radiation sensitivity when BRCA1 is deficient in experimental models, inconsistent results are obtained with clinical studies regarding breast cancer prognosis in women with BRCA mutations (Figure 1). Current studies have failed to demonstrate, for the most part, a significant overall survival difference between BRCA-associated breast cancers and sporadic breast cancers following RT. However, there is a significant increase in the risk of contralateral breast cancers in BRCA mutation carriers. In addition, the currently available data provide no evidence that a genetic alteration in BRCA1/2 has a significant role in RT-induced toxicity and secondary cancer risk (Figure.1).
Figure 1.
BRCA1 and Radiation Therapy
The Potential Factors Contributing to Inconsistences
The discrepant results from clinical data can be explained to a large extent by the underlying study methodologies used (99). In an optimal study of disease prognosis, several criteria should be met (75). These criteria included assembly of an “inception cohort”; description of the referral pattern and hence representativeness of the sample; complete follow-up; use of objective outcome criteria; “blinding” of outcome assessment; and adequate sample size. Thus, a high quality clinical study following stringent methodology is essential in standardized clinical trials. In addition, the following factors, from the point of view of molecular biology, should also be considered when we explain the inconsistence of the obvious radiosensitivity observed in BRCA1 deficient cancer cells in experimental models and the less conclusive results from clinical investigations.
Lack of functional analysis. BRCA1 is a highly polymorphic gene with more than 1,500 unique documented variants (100). The consequences on protein function due to these individual mutations are largely unknown. Cancer patients with different BRCA1 mutations may or may not have a defect in DSBs repair pathways. Thus, the effectiveness of DSBs repair pathways in these patients need to be evaluated.
Activation of alternative DSBs repair pathways. In tumor cells, a defect in one repair pathway usually activates a second pathway, which contributes to repair of DSBs. For instance, although cNHEJ machinery deficiency delays repair of IR-induced DSBs, ultimately cells will remove the majority of DSBs using the aNHEJ pathway (14,22). Given the fact that MMEJ, a subset of aNHEJ pathway, is important for the resistance to IR (44,45), it would be a possibility that the aNHEJ/MMEJ pathway is elevated in BRCA1 carriers, which can compensate for the loss of HR and precise cNHEJ pathway due to BRCA1 deficiency.
Loss of 53BP1. Loss of 53BP1 has been identified in a variety of tumors (101), particularly those with BRCA mutation (102). Studies have suggested that loss of 53BP1 is able to reverse many aspects of the phenotype associated with BRCA1 loss (102–104), including restoring HR ability and resistance to PARP inhibitors and RT (102,103,105). RT resistance was observed in breast cancer patients with low 53BP1 expression (105) despite the hypersensitivity to IR in cells with 53BP1 deficiency in numerous different cancer cell lines and animal models (106–108). Although the molecular mechanism behind this observation is not fully understood, it is likely that lower 53BP1 expression occurs in cancer patients with pre-existing BRCA1 mutations. Our recent publication suggested that 53BP1 deficiency results in an increased MMEJ frequency in BRCA1-deficient cells. Given that MMEJ is important for resistance to IR (44), increased MMEJ levels in BRCA1-deficient cells due to lower 53BP1 expression could contribute to radiation resistance.
. Second mutations in BRCA genes restore DSBs repair activity. It has been shown that a second mutation in the BRCA2 gene could “restore” the functionality lost with the initial inherited mutation (109). Such a mutation could turn a BRCA-deficient cell into a “wild-type” one with competent HR, thereby conferring resistance to PARP inhibition. Currently, it has not been reported if gene mutation in BRCA1/2 can also occur following IR. It would be interesting to test this hypothesis in the future.
. Less significant role of BRCA1 in cNHEJ. The role of BRCA1 in cNHEJ may be limited to repair of a small subset of DSBs, such as those with clear break ends, which can be repaired by a direct re-ligation. However, the dirty ends of breaks caused by IR-induced are complex and are not appropriate substrates for precise end joining mediated by BRCA1. Those DNA termini frequently contain 3’-phosphate or 3’-phosphoglycolate groups, which must be removed prior to ligation (110). Thus, the role of BRCA1 in repair of IR-induced DSBs by cNHEJ appears minimal.
. Relationship to triple negative breast cancers and p53 loss. Triple-negative breast cancer accounts for a large number of breast cancer deaths because of its poor prognosis. One of the key features of this particular subtype of breast cancer is loss of p53. Currently, the majority of studies support an association between worse survival and the presence of p53 mutations. An increased rate of p53 mutations in cancers arising in carriers of germline BRCA1 and BRCA2 mutations was observed (111).Thus, the expected improved outcome with RT due to the impaired activity of DSBs in BRCA1 carriers may be masked by a p53 deficiency.
PARP Inhibitors and Radiation Therapy
The finding that BRCA1 cells are deficient in DSBs repair and are thus more sensitive to DNA damaging agents has led to an exploration of how this pathway can be exploited for developing more effective cancer treatments. Proteins with PARP activity have been known to play important roles in a variety of cellular functions, including repair of single stranded DNA breaks. The PARP family of proteins is defined by the catalytic capacity to modify target proteins by the covalent addition of chains of poly(ADP-ribose) polymers. PARP inhibitors (PARPi) have been shown to be selectively lethal to cells deficient in BRCA1 or BRCA2 due to synthetic lethality (112–114). It is widely believed that the increased number of unrepaired endogenous single strand breaks in PARP inhibited cells result in more collapsed replication forks, which require HR for repair. BRCA1/2 deficient cells are sensitive to PARPi because the DSBs resulting from PARP inhibition fail to be repaired by HR (115). A recent study demonstrated that PARPi treatment induces phosphorylation of DNA-dependent protein kinase substrates and stimulates cNHEJ selectively in HR-deficient cells (116). They found that not only is PARP1 catalytic activity required for the regulation of cNHEJ in HR-deficient cells but that deregulated NHEJ plays a major role in generating genomic instability and cytotoxicity in HR-deficient cells treated with PARPi. However, PARPi may not be effective in patients with low 53BP1 expression due to the restoration of HR, as we discussed early.
In the past decade, PARPi as cancer therapy has been a hot topic in the field of DNA repair. Although PARPi are being explored as single agents in clinical trials for the treatment of various cancers, the knowledge that these cells are more sensitive to DNA damage has led to interest in combining PARPi with other DNA damaging agents. Many groups have shown preclinical data demonstrating mice and cells derived from mice that are PARP null are hypersensitive to DNA methylating agents, topoisomerase I poisons and IR (117). Delaney et al published that a potent PARPi, NU1085 could potentiate Temozolomide (TMZ), a DNA methylating agent, activity up to 6-fold in a variety of cancer cell lines. They also noted that the cytotoxicity of topotecan, a topoisomerase 1 poison, was also increased (118). The PARPi, ABT-888, enhanced TMZ cytotoxicity in a variety of cancer cell lines including lung, colon, cervical and prostate. In an orthotopic model of glioma as well as a mouse model of melanoma a decrease in tumor growth was noted with the combination of ABT-888 and TMZ (119,120). Tentori et al. demonstrated that cells treated with GPI 15427 had increased growth inhibition by TMZ in human glioblastoma cells (121). In pre-clinical studies concomitant use of 3-Aminobenzamide, a PARPi, in a cisplatin-resistant ovarian cancer cell line increased the cytotoxic activity of the same dose of cisplatin from the IC50 to the IC95 (122).
In vivo, GPI 15427 has been shown to delay the growth and metastatic activity of a melanoma mouse model (121). ABT-888 has also been studied in vivo and was found to increase the antitumor activity of TMZ in an orthotopic model of rat glioma (123). TMZ has been used in conjunction with CEP-6800 and demonstrated tumor regression in mice bearing human glioblastoma tumor xenografts and human colon cancer xenografts (124). The combination of GPI 15427 with TMZ and irinotecan increased antitumor activity against HT29, colon cancer, xenografts in mice (125). AG-014699, another clinical PARPi, enhanced topotecan-induced tumor growth delay in a neuroblastoma mouse model (126). PARPi combined with other classes of chemotherapies have had mixed results in increasing anti-tumor effects. There is some evidence that with platinum therapy, PARPi are more effective in cells that are HR defective (127). Multiple in vivo studies have shown that various PARPi in combination with a platinum drug cause regression of tumor xenografts which are HR deficient, for example BRCA1 or BRCA2 deficient (123,128–130).
Clinical trials using PARPi alone or in combination other chemotherapies have been undertaken in many different cancers from lung cancer to breast cancer to glioblastoma. In clinical trials, a phase I/Ib study was performed using Olaparib, a PARPi, with carboplatin in breast and ovarian cancer patients with germline BRCA1/2 mutations. The study was designed to test the safety, tolerability, and activity of Olaparib with carboplatin in patients with recurrent or refractory disease or locally advanced unresectable breast cancer. Olaparib was given orally in either a continuous daily or intermittent dosing at doses of 100 to 400 mg twice daily with carboplatin, area under the curve (AUC) 3–5, once every 21-day cycle. A maximum of eight combination cycles were given and patients who had not progressed were maintained on daily Olaparib, 400 mg every 12 hours, until disease progression. In the subset of breast cancer patients there was an 87.5% response rate with this combination treatment while platinum-sensitive ovarian cancers noted a 71% response rate. The group of platinum-resistant ovarian cancer patients still demonstrated a 25% response rate with another 45% having stable disease (131). This Phase I study shows promising results in combining PARPi with other chemotherapeutic agents to treat patients with BRCA1/2 mutations; however, the right combination of drugs and the correct patient population must be identified.
There has also been interest in whether PARPi could further sensitize tumors to RT because clinical fractionated RT used either as a single modality or in combination with surgery and/or chemotherapy, is a fundamental treatment modality for most locally advanced cancers, including breast cancer (132). Several combination studies of PARP inhibition and IR have been performed in human xenografts, and the promising results of these preclinical studies are summarized in a review by Chalmers in 2010 (133). Various in vivo studies utilizing PARPi in conjunction with RT have been shown to decrease tumor growth or prolong animal survival. The recent studies further support the benefit of this combination. Tuli et al. demonstrated that pancreatic xenograft tumors in mice were more responsive to RT after administration of a ABT-888. They found that at 60 days, mice treated with both ABT-888 and RT had a 40% chance of survival, compared to none that survived when treated with either ABT-888 or RT alone (134). A study using Ewing sarcoma cell lines found that Olaparib sensitized cells to RT in vitro as well as in vivo (135). Jian et al. found that RT combined with a PARP inhibitor increased DNA damage and led to an increase in apoptosis and necrosis in a xenograft model of urothelial carcinoma (136). A study of colorectal cancer cells demonstrated increased DSBs when cells were treated with ABT-888 and RT. In mouse xenograft models the combination of RT and ABT-888 led to an increased tumor growth delay compared to RT alone, and even longer growth delays when ABT-888 and RT were used in combination with irinotecan, oxaliplatin or 5-FU (137). In a study with PARP inhibition and RT, it appears that use of a PARPi with lower dose rate RT causes increased numbers of senescent cells and more extensive DNA damage compared to higher dose rates in prostate cancer cells (138). All of these studies suggested a very encouraging result of combining PARP inhibitors and RT for the treatment of cancer.
Despite the encouraging preclinical studies, very few clinical studies of PARPi in combination with RT are underway because of the difficulties of design (133). The late, irreversible effects of RT usually limit total RT dose, and these effects are not observed until months or years after treatment. Conventional drug dose-escalation studies combined with RT regimens are therefore difficult to design (133). A Phase II trial is ongoing examining whether the addition of Veliparib with whole brain RT is more effective in patients with brain metastases from non-small cell lung cancer. There are also clinical trials evaluating the feasibility and safety of a combination of Veliparib, capecitabine, and RT in patients with locally advanced rectal cancer as well as Veliparib with chest wall and nodal RT in inflammatory or loco-regionally recurrent breast cancer. The studies designed to specifically test the efficacy of the combination of PARPi and RT in cancer cells with BRCA1/2 deficiency is rare; although, PARPi were initially used for treating BRCA deficient cancer cells or cancers. A recent report from Bourton et al. suggested a hypersensitivity of BRCA1 heterozygote lymphoblastoid cells to IR and PARPi (139). In this study they found that while lymphoblastoid cells from normal individuals and those carrying a mono-allelic mutation in the BRCA2 gene did not display elevated radiosensitivity, the three lymphoblastoid cell lines with mono-allelic mutations in the BRCA1 gene exhibited a significant reduction in cell survival after IR exposure in combination with Olaparib. The reasons that BRCA1 and BRCA2 mutations display different results is not clear at this point but HR deficiency may not be a major contributing factor for the reduction in cell survival observed in BRCA1 deficient cells since both BRCA1 and BRCA2 have a critical role in HR. Interestingly, the review by Cressman et al. has discussed a variety of mechanisms that may underlie the radiosensitizing effects of PARPi in vitro and in vivo, including a decrease in HR due to hypoxia (133). In addition, B-lymphoblastoid cells heterozygous for BRCA1 show elevated cellular sensitivity to IR in the presence of Olaparib when compared to heterozygous BRCA2 B-lymphoblastoids and those from normal individuals. Thus, the authors speculate that that the clinical application of PARPi used concomitantly with RT may increase the number of individuals that experience RT-induced normal tissue toxicity. This hypothesis remains to be tested. Currently, there are no clinical trials ongoing to test if PARPi plus RT is more efficient in the context of BRCA1/2 deficiency compared to spontaneous cancer patients. In addition to PARPi, there may be other ways to increase the sensitivity of tumors to chemotherapy or RT in BRCA1 deficient cells. For instance, our recent data suggested that the function of BRCA1 in crossover- mediate HR, sister chromatid exchange, following replication fork collapse is more profound when ATR is depleted (140), this could provide another molecular mechanism to target the BRCA1 deficient cells with an ATR/Chk1 inhibitor.
Conclusions and Perspectives
The important of role of BRCA1 in DSB repair pathways in vitro has been well characterized. However, the concept that RT may be particularly effective in BRCA1-deficient tumors is still theoretical at this point. The correlation between the in vivo clinical response to RT and BRCA1 deficiency remains to be further elucidated and established through conducting well-designed and standardized studies which have adequate sample size and complete follow-up, and use objective outcome criteria and blinding of outcome assessments. In addition, as our understanding of the mechanism of biochemical details of the role of BRCA1 in DDR increases, the potential ways to target treatment for patients with BRCA1 deficiency will emerge and will have an enormous impact on future RT and chemotherapy. Success will rely on an in-depth characterization of the pathways controlled by BRCA1. It will be of great interest to observe how PARPi and other novel therapeutic approaches based on the molecular pathways controlled by BRCA1/2 impact clinical outcomes of RT, particularly in breast cancer treatment.
Acknowledgments
I apologize to colleagues whose work was not cited due to space limitations or my ignorance. Research in my laboratory is supported by American Cancer Society (IRG-58-010-51) and National Cancer Institute (grant R01CA154625) and a startup fund from the Department of Radiation Oncology, Case Western Reserve University School of Medicine (to J.Z).
The abbreviations used are
- aNHEJ
alternative NHEJ
- B-NHEJ
backup NHEJ
- cNHEJ
classical Non-homologous end joining
- CO
crossover CSS, cancer specific survival
- DDR
DNA damage response
- DFS
disease free survival
- DSBs
DNA double strand breaks
- GC
Gene conversion
- HBOC
Hereditary breast and ovarian cancer
- HJ
Holliday junction
- HR
Homologous recombination
- IBTR
ipsilateral breast tumor recurrence
- IR
Ionizing radiation
- LR
Local recurrence
- LRFS
local-recurrence free survival
- MEFs
Mouse embryonic fibroblast cells
- MMEJ
microhomology-mediated end joining
- OS
overall survival
- PARP
Poly(ADP-ribose) polymerase
- PARPi
Poly(ADP-ribose) polymerase inhibitors
- RT
Radiation Therapy
- SPECT
single photon emission computed tomography
- TMZ
Temozolomide
Footnotes
Authors’ contributions
CK and JZ drafted the manuscript. JZ approved the final manuscript.
Conflict of interest: none
References
- 1.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nature genetics. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 2.Pfeiffer P, Goedecke W, Kuhfittig-Kulle S, Obe G. Pathways of DNA double-strand break repair and their impact on the prevention and formation of chromosomal aberrations. Cytogenet Genome Res. 2004;104:7–13. doi: 10.1159/000077460. [DOI] [PubMed] [Google Scholar]
- 3.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 4.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 5.Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 6.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nature reviews. Cancer. 2012;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. The Biochemical journal. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Frontiers in oncology. 2013;3:113. doi: 10.3389/fonc.2013.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 10.Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 11.Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutation research. 2003;532:103–115. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Tamulevicius P, Wang M, Iliakis G. Homology-directed repair is required for the development of radioresistance during S phase: interplay between double-strand break repair and checkpoint response. Radiation research. 2007;167:1–11. doi: 10.1667/RR0751.1. [DOI] [PubMed] [Google Scholar]
- 13.Wilson PF, Hinz JM, Urbin SS, Nham PB, Thompson LH. Influence of homologous recombinational repair on cell survival and chromosomal aberration induction during the cell cycle in gamma-irradiated CHO cells. DNA repair. 2010;9:737–744. doi: 10.1016/j.dnarep.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iliakis G, Wang H, Perrault AR, Boecker W, Rosidi B, Windhofer F, Wu W, Guan J, Terzoudi G, Pantelias G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenetic and genome research. 2004;104:14–20. doi: 10.1159/000077461. [DOI] [PubMed] [Google Scholar]
- 15.Groth P, Orta ML, Elvers I, Majumder MM, Lagerqvist A, Helleday T. Homologous recombination repairs secondary replication induced DNA double-strand breaks after ionizing radiation. Nucleic acids research. 2012;40:6585–6594. doi: 10.1093/nar/gks315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeggo PA, Geuting V, Lobrich M. The role of homologous recombination in radiation-induced double-strand break repair. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2011;101:7–12. doi: 10.1016/j.radonc.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 17.Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Lobrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. The EMBO journal. 2009;28:3413–3427. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nature reviews. Molecular cell biology. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sung P, Krejci L, Van Komen S, Sehorn MG. Rad51 recombinase and recombination mediators. The Journal of biological chemistry. 2003;278:42729–42732. doi: 10.1074/jbc.R300027200. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J. The role of BRCA1 in homologous recombination repair in response to replication stress: significance in tumorigenesis and cancer therapy. Cell & bioscience. 2013;3:11. doi: 10.1186/2045-3701-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, Razavian N, Berns MW, Wu X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:7720–7725. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DiBiase SJ, Zeng ZC, Chen R, Hyslop T, Curran WJ, Jr, Iliakis G. DNA-dependent protein kinase stimulates an independently active, nonhomologous, end-joining apparatus. Cancer research. 2000;60:1245–1253. [PubMed] [Google Scholar]
- 23.Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Lobrich M, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. The EMBO journal. 2011;30:1079–1092. doi: 10.1038/emboj.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ritter S, Durante M. Heavy-ion induced chromosomal aberrations: a review. Mutation research. 2010;701:38–46. doi: 10.1016/j.mrgentox.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Molecular cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 26.Boulton SJ, Jackson SP. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. The EMBO journal. 1996;15:5093–5103. [PMC free article] [PubMed] [Google Scholar]
- 27.Liang F, Jasin M. Ku80-deficient cells exhibit excess degradation of extrachromosomal DNA. The Journal of biological chemistry. 1996;271:14405–14411. doi: 10.1074/jbc.271.24.14405. [DOI] [PubMed] [Google Scholar]
- 28.Verkaik NS, Esveldt-van Lange RE, van Heemst D, Bruggenwirth HT, Hoeijmakers JH, Zdzienicka MZ, van Gent DC. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. European journal of immunology. 2002;32:701–709. doi: 10.1002/1521-4141(200203)32:3<701::AID-IMMU701>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 29.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS genetics. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nature structural & molecular biology. 2009;16:814–818. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–463. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends in genetics : TIG. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deriano L, Stracker TH, Baker A, Petrini JH, Roth DB. Roles for NBS1 in alternative nonhomologous end-joining of V(D)J recombination intermediates. Molecular cell. 2009;34:13–25. doi: 10.1016/j.molcel.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nature structural & molecular biology. 2011;18:80–84. doi: 10.1038/nsmb.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Advances in immunology. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 36.Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nature structural & molecular biology. 2009;16:819–824. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- 37.Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. The Journal of biological chemistry. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 38.Wang H, Rosidi B, Perrault R, Wang M, Zhang L, Windhofer F, Iliakis G. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer research. 2005;65:4020–4030. doi: 10.1158/0008-5472.CAN-04-3055. [DOI] [PubMed] [Google Scholar]
- 39.Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic acids research. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wray J, Williamson EA, Singh SB, Wu Y, Cogle CR, Weinstock DM, Zhang Y, Lee SH, Zhou D, Shao L, et al. PARP1 is required for chromosomal translocations. Blood. 2013;121:4359–4365. doi: 10.1182/blood-2012-10-460527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robert I, Dantzer F, Reina-San-Martin B. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. The Journal of experimental medicine. 2009;206:1047–1056. doi: 10.1084/jem.20082468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng Q, Barboule N, Frit P, Gomez D, Bombarde O, Couderc B, Ren GS, Salles B, Calsou P. Ku counteracts mobilization of PARP1 and MRN in chromatin damaged with DNA double-strand breaks. Nucleic acids research. 2011;39:9605–9619. doi: 10.1093/nar/gkr656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiong X, Du Z, Wang Y, Feng Z, Fan P, Yan C, Willers H, Zhang J. 53BP1 promotes microhomology-mediated end-joining in G1-phase cells. Nucleic acids research. 2015;43:1659–1670. doi: 10.1093/nar/gku1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma JL, Kim EM, Haber JE, Lee SE. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Molecular and cellular biology. 2003;23:8820–8828. doi: 10.1128/MCB.23.23.8820-8828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yousefzadeh MJ, Wyatt DW, Takata K, Mu Y, Hensley SC, Tomida J, Bylund GO, Doublie S, Johansson E, Ramsden DA, et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS genetics. 2014;10:e1004654. doi: 10.1371/journal.pgen.1004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jasin M. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 2002;21:8981–8993. doi: 10.1038/sj.onc.1206176. [DOI] [PubMed] [Google Scholar]
- 47.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Molecular cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 48.Snouwaert JN, Gowen LC, Latour AM, Mohn AR, Xiao A, DiBiase L, Koller BH. BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of a brca1 transgene. Oncogene. 1999;18:7900–7907. doi: 10.1038/sj.onc.1203334. [DOI] [PubMed] [Google Scholar]
- 49.Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer research. 2001;61:4842–4850. [PubMed] [Google Scholar]
- 50.Westermark UK, Reyngold M, Olshen AB, Baer R, Jasin M, Moynahan ME. BARD1 participates with BRCA1 in homology-directed repair of chromosome breaks. Molecular and cellular biology. 2003;23:7926–7936. doi: 10.1128/MCB.23.21.7926-7936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, Chung JH, Powell SN, Xia F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Molecular and cellular biology. 2004;24:708–718. doi: 10.1128/MCB.24.2.708-718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Molecular cancer research : MCR. 2009;7:1110–1118. doi: 10.1158/1541-7786.MCR-09-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang H, Zeng ZC, Bui TA, DiBiase SJ, Qin W, Xia F, Powell SN, Iliakis G. Nonhomologous end-joining of ionizing radiation-induced DNA double-stranded breaks in human tumor cells deficient in BRCA1 or BRCA2. Cancer research. 2001;61:270–277. [PubMed] [Google Scholar]
- 56.Bau DT, Mau YC, Shen CY. The role of BRCA1 in non-homologous end-joining. Cancer letters. 2006;240:1–8. doi: 10.1016/j.canlet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 57.Zhuang J, Zhang J, Willers H, Wang H, Chung JH, van Gent DC, Hallahan DE, Powell SN, Xia F. Checkpoint kinase 2-mediated phosphorylation of BRCA1 regulates the fidelity of nonhomologous end-joining. Cancer research. 2006;66:1401–1408. doi: 10.1158/0008-5472.CAN-05-3278. [DOI] [PubMed] [Google Scholar]
- 58.Al-Hakim A, Escribano-Diaz C, Landry MC, O’Donnell L, Panier S, Szilard RK, Durocher D. The ubiquitous role of ubiquitin in the DNA damage response. DNA repair. 2010;9:1229–1240. doi: 10.1016/j.dnarep.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhong Q, Boyer TG, Chen PL, Lee WH. Deficient nonhomologous end-joining activity in cell-free extracts from Brca1-null fibroblasts. Cancer research. 2002;62:3966–3970. [PubMed] [Google Scholar]
- 60.Zhong Q, Chen CF, Chen PL, Lee WH. BRCA1 facilitates microhomology-mediated end joining of DNA double strand breaks. The Journal of biological chemistry. 2002;277:28641–28647. doi: 10.1074/jbc.M200748200. [DOI] [PubMed] [Google Scholar]
- 61.Bau DT, Fu YP, Chen ST, Cheng TC, Yu JC, Wu PE, Shen CY. Breast cancer risk and the DNA double-strand break end-joining capacity of nonhomologous end-joining genes are affected by BRCA1. Cancer research. 2004;64:5013–5019. doi: 10.1158/0008-5472.CAN-04-0403. [DOI] [PubMed] [Google Scholar]
- 62.Coupier I, Baldeyron C, Rousseau A, Mosseri V, Pages-Berhouet S, Caux-Moncoutier V, Papadopoulo D, Stoppa-Lyonnet D. Fidelity of DNA double-strand break repair in heterozygous cell lines harbouring BRCA1 missense mutations. Oncogene. 2004;23:914–919. doi: 10.1038/sj.onc.1207191. [DOI] [PubMed] [Google Scholar]
- 63.Baldeyron C, Jacquemin E, Smith J, Jacquemont C, De Oliveira I, Gad S, Feunteun J, Stoppa-Lyonnet D, Papadopoulo D. A single mutated BRCA1 allele leads to impaired fidelity of double strand break end-joining. Oncogene. 2002;21:1401–1410. doi: 10.1038/sj.onc.1205200. [DOI] [PubMed] [Google Scholar]
- 64.Wang HC, Chou WC, Shieh SY, Shen CY. Ataxia telangiectasia mutated and checkpoint kinase 2 regulate BRCA1 to promote the fidelity of DNA end-joining. Cancer research. 2006;66:1391–1400. doi: 10.1158/0008-5472.CAN-05-3270. [DOI] [PubMed] [Google Scholar]
- 65.Zhang J, Powell SN. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Molecular cancer research : MCR. 2005;3:531–539. doi: 10.1158/1541-7786.MCR-05-0192. [DOI] [PubMed] [Google Scholar]
- 66.Jiang G, Plo I, Wang T, Rahman M, Cho JH, Yang E, Lopez BS, Xia F. BRCA1-Ku80 protein interaction enhances end-joining fidelity of chromosomal double-strand breaks in the G1 phase of the cell cycle. The Journal of biological chemistry. 2013;288:8966–8976. doi: 10.1074/jbc.M112.412650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chistiakov DA, Voronova NV, Chistiakov PA. Genetic variations in DNA repair genes, radiosensitivity to cancer and susceptibility to acute tissue reactions in radiotherapy-treated cancer patients. Acta Oncol. 2008;47:809–824. doi: 10.1080/02841860801885969. [DOI] [PubMed] [Google Scholar]
- 68.Shen SX, Weaver Z, Xu X, Li C, Weinstein M, Chen L, Guan XY, Ried T, Deng CX. A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene. 1998;17:3115–3124. doi: 10.1038/sj.onc.1202243. [DOI] [PubMed] [Google Scholar]
- 69.Foray N, Randrianarison V, Marot D, Perricaudet M, Lenoir G, Feunteun J. Gamma-rays-induced death of human cells carrying mutations of BRCA1 or BRCA2. Oncogene. 1999;18:7334–7342. doi: 10.1038/sj.onc.1203165. [DOI] [PubMed] [Google Scholar]
- 70.Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Genetic analysis of BRCA1 function in a defined tumor cell line. Molecular cell. 1999;4:1093–1099. doi: 10.1016/s1097-2765(00)80238-5. [DOI] [PubMed] [Google Scholar]
- 71.Abbott DW, Thompson ME, Robinson-Benion C, Tomlinson G, Jensen RA, Holt JT. BRCA1 expression restores radiation resistance in BRCA1-defective cancer cells through enhancement of transcription-coupled DNA repair. The Journal of biological chemistry. 1999;274:18808–18812. doi: 10.1074/jbc.274.26.18808. [DOI] [PubMed] [Google Scholar]
- 72.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 73.Li W, Katoh H, Wang L, Yu X, Du Z, Yan X, Zheng P, Liu Y. FOXP3 regulates sensitivity of cancer cells to irradiation by transcriptional repression of BRCA1. Cancer research. 2013;73:2170–2180. doi: 10.1158/0008-5472.CAN-12-2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Robson M, Gilewski T, Haas B, Levin D, Borgen P, Rajan P, Hirschaut Y, Pressman P, Rosen PP, Lesser ML, et al. BRCA-associated breast cancer in young women. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1998;16:1642–1649. doi: 10.1200/JCO.1998.16.5.1642. [DOI] [PubMed] [Google Scholar]
- 75.Phillips KA, Andrulis IL, Goodwin PJ. Breast carcinomas arising in carriers of mutations in BRCA1 or BRCA2: are they prognostically different? Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1999;17:3653–3663. doi: 10.1200/JCO.1999.17.11.3653. [DOI] [PubMed] [Google Scholar]
- 76.Haffty BG, Harrold E, Khan AJ, Pathare P, Smith TE, Turner BC, Glazer PM, Ward B, Carter D, Matloff E, et al. Outcome of conservatively managed early-onset breast cancer by BRCA1/2 status. Lancet. 2002;359:1471–1477. doi: 10.1016/S0140-6736(02)08434-9. [DOI] [PubMed] [Google Scholar]
- 77.Pierce LJ, Phillips KA, Griffith KA, Buys S, Gaffney DK, Moran MS, Haffty BG, Ben-David M, Kaufman B, Garber JE, et al. Local therapy in BRCA1 and BRCA2 mutation carriers with operable breast cancer: comparison of breast conservation and mastectomy. Breast cancer research and treatment. 2010;121:389–398. doi: 10.1007/s10549-010-0894-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kirova YM, Savignoni A, Sigal-Zafrani B, de La Rochefordiere A, Salmon RJ, This P, Asselain B, Stoppa-Lyonnet D, Fourquet A. Is the breast-conserving treatment with radiotherapy appropriate in BRCA1/2 mutation carriers? Long-term results and review of the literature. Breast cancer research and treatment. 2010;120:119–126. doi: 10.1007/s10549-009-0685-6. [DOI] [PubMed] [Google Scholar]
- 79.Pierce LJ, Levin AM, Rebbeck TR, Ben-David MA, Friedman E, Solin LJ, Harris EE, Gaffney DK, Haffty BG, Dawson LA, et al. Ten-year multi-institutional results of breast-conserving surgery and radiotherapy in BRCA1/2-associated stage I/II breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:2437–2443. doi: 10.1200/JCO.2005.02.7888. [DOI] [PubMed] [Google Scholar]
- 80.Pierce LJ, Strawderman M, Narod SA, Oliviotto I, Eisen A, Dawson L, Gaffney D, Solin LJ, Nixon A, Garber J, et al. Effect of radiotherapy after breast-conserving treatment in women with breast cancer and germline BRCA1/2 mutations. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2000;18:3360–3369. doi: 10.1200/JCO.2000.18.19.3360. [DOI] [PubMed] [Google Scholar]
- 81.Goodwin PJ, Phillips KA, West DW, Ennis M, Hopper JL, John EM, O’Malley FP, Milne RL, Andrulis IL, Friedlander ML, et al. Breast cancer prognosis in BRCA1 and BRCA2 mutation carriers: an International Prospective Breast Cancer Family Registry population-based cohort study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:19–26. doi: 10.1200/JCO.2010.33.0068. [DOI] [PubMed] [Google Scholar]
- 82.Kennedy RD, Quinn JE, Mullan PB, Johnston PG, Harkin DP. The role of BRCA1 in the cellular response to chemotherapy. Journal of the National Cancer Institute. 2004;96:1659–1668. doi: 10.1093/jnci/djh312. [DOI] [PubMed] [Google Scholar]
- 83.Yang Q, Sakurai T, Mori I, Yoshimura G, Nakamura M, Nakamura Y, Suzuma T, Tamaki T, Umemura T, Kakudo K. Prognostic significance of BRCA1 expression in Japanese sporadic breast carcinomas. Cancer. 2001;92:54–60. doi: 10.1002/1097-0142(20010701)92:1<54::aid-cncr1291>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 84.Lambie H, Miremadi A, Pinder SE, Bell JA, Wencyk P, Paish EC, Macmillan RD, Ellis IO. Prognostic significance of BRCA1 expression in sporadic breast carcinomas. The Journal of pathology. 2003;200:207–213. doi: 10.1002/path.1348. [DOI] [PubMed] [Google Scholar]
- 85.Soderlund K, Skoog L, Fornander T, Askmalm MS. The BRCA1/BRCA2/Rad51 complex is a prognostic and predictive factor in early breast cancer. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2007;84:242–251. doi: 10.1016/j.radonc.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 86.Goldhirsch A, Winer EP, Coates AS, Gelber RD, Piccart-Gebhart M, Thurlimann B, Senn HJ. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2013;24:2206–2223. doi: 10.1093/annonc/mdt303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Buchholz TA, Wu X, Hussain A, Tucker SL, Mills GB, Haffty B, Bergh S, Story M, Geara FB, Brock WA. Evidence of haplotype insufficiency in human cells containing a germline mutation in BRCA1 or BRCA2. International journal of cancer. Journal international du cancer. 2002;97:557–561. doi: 10.1002/ijc.10109. [DOI] [PubMed] [Google Scholar]
- 88.Shorrocks J, Tobi SE, Latham H, Peacock JH, Eeles R, Eccles D, McMillan TJ. Primary fibroblasts from BRCA1 heterozygotes display an abnormal G1/S cell cycle checkpoint following UVA irradiation but show normal levels of micronuclei following oxidative stress or mitomycin C treatment. International journal of radiation oncology, biology, physics. 2004;58:470–478. doi: 10.1016/j.ijrobp.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 89.Cressman VL, Backlund DC, Hicks EM, Gowen LC, Godfrey V, Koller BH. Mammary tumor formation in p53- and BRCA1-deficient mice. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research. 1999;10:1–10. [PubMed] [Google Scholar]
- 90.Kelsey CR, Jackson IL, Langdon S, Owzar K, Hubbs J, Vujaskovic Z, Das S, Marks LB. Analysis of single nucleotide polymorphisms and radiation sensitivity of the lung assessed with an objective radiologic endpoin. Clinical lung cancer. 2013;14:267–274. doi: 10.1016/j.cllc.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shanley S, McReynolds K, Ardern-Jones A, Ahern R, Fernando I, Yarnold J, Evans G, Eccles D, Hodgson S, Ashley S, et al. Late toxicity is not increased in BRCA1/BRCA2 mutation carriers undergoing breast radiotherapy in the United Kingdom. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:7025–7032. doi: 10.1158/1078-0432.CCR-06-1244. [DOI] [PubMed] [Google Scholar]
- 92.Leong T, Whitty J, Keilar M, Mifsud S, Ramsay J, Birrell G, Venter D, Southey M, McKay M. Mutation analysis of BRCA1 and BRCA2 cancer predisposition genes in radiation hypersensitive cancer patients. International journal of radiation oncology, biology, physics. 2000;48:959–965. doi: 10.1016/s0360-3016(00)00728-8. [DOI] [PubMed] [Google Scholar]
- 93.Park H, Choi DH, Noh JM, Huh SJ, Park W, Nam SJ, Lee JE. Acute skin toxicity in Korean breast cancer patients carrying BRCA mutations. International journal of radiation biology. 2014;90:90–94. doi: 10.3109/09553002.2013.835504. [DOI] [PubMed] [Google Scholar]
- 94.Huszno J, Budryk M, Kolosza Z, Nowara E. The influence of BRCA1/BRCA2 mutations on toxicity related to chemotherapy and radiotherapy in early breast cancer patients. Oncology. 2013;85:278–282. doi: 10.1159/000354834. [DOI] [PubMed] [Google Scholar]
- 95.Blasiak J, Arabski M, Krupa R, Wozniak K, Rykala J, Kolacinska A, Morawiec Z, Drzewoski J, Zadrozny M. Basal, oxidative and alkylative DNA damage, DNA repair efficacy and mutagen sensitivity in breast cancer. Mutation research. 2004;554:139–148. doi: 10.1016/j.mrfmmm.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 96.Palyvoda O, Polanska J, Wygoda A, Rzeszowska-Wolny J. DNA damage and repair in lymphocytes of normal individuals and cancer patients: studies by the comet assay and micronucleus tests. Acta biochimica Polonica. 2003;50:181–190. [PubMed] [Google Scholar]
- 97.Kadouri L, Sagi M, Goldberg Y, Lerer I, Hamburger T, Peretz T. Genetic predisposition to radiation induced sarcoma: possible role for BRCA and p53 mutations. Breast cancer research and treatment. 2013;140:207–211. doi: 10.1007/s10549-013-2621-z. [DOI] [PubMed] [Google Scholar]
- 98.Kirova YM, Vilcoq JR, Asselain B, Sastre-Garau X, Fourquet A. Radiation-induced sarcomas after radiotherapy for breast carcinoma: a large-scale single-institution review. Cancer. 2005;104:856–863. doi: 10.1002/cncr.21223. [DOI] [PubMed] [Google Scholar]
- 99.Bordeleau L, Panchal S, Goodwin P. Prognosis of BRCA-associated breast cancer: a summary of evidence. Breast cancer research and treatment. 2010;119:13–24. doi: 10.1007/s10549-009-0566-z. [DOI] [PubMed] [Google Scholar]
- 100.Millot GA, Carvalho MA, Caputo SM, Vreeswijk MP, Brown MA, Webb M, Rouleau E, Neuhausen SL, Hansen T, Galli A, et al. A guide for functional analysis of BRCA1 variants of uncertain significance. Human mutation. 2012;33:1526–1537. doi: 10.1002/humu.22150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nuciforo PG, Luise C, Capra M, Pelosi G, d’Adda di Fagagna F. Complex engagement of DNA damage response pathways in human cancer and in lung tumor progression. Carcinogenesis. 2007;28:2082–2088. doi: 10.1093/carcin/bgm108. [DOI] [PubMed] [Google Scholar]
- 102.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature structural & molecular biology. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Molecular cell. 2009;35:534–541. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Neboori HJ, Haffty BG, Wu H, Yang Q, Aly A, Goyal S, Schiff D, Moran MS, Golhar R, Chen C, et al. Low p53 binding protein 1 (53BP1) expression is associated with increased local recurrence in breast cancer patients treated with breast-conserving surgery and radiotherapy. International journal of radiation oncology, biology, physics. 2012;83:e677–683. doi: 10.1016/j.ijrobp.2012.01.089. [DOI] [PubMed] [Google Scholar]
- 106.Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Molecular and cellular biology. 2003;23:2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nakamura K, Sakai W, Kawamoto T, Bree RT, Lowndes NF, Takeda S, Taniguchi Y. Genetic dissection of vertebrate 53BP1: a major role in non-homologous end joining of DNA double strand breaks. DNA repair. 2006;5:741–749. doi: 10.1016/j.dnarep.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 108.Iwabuchi K, Hashimoto M, Matsui T, Kurihara T, Shimizu H, Adachi N, Ishiai M, Yamamoto K, Tauchi H, Takata M, et al. 53BP1 contributes to survival of cells irradiated with X-ray during G1 without Ku70 or Artemis. Genes to cells : devoted to molecular & cellular mechanisms. 2006;11:935–948. doi: 10.1111/j.1365-2443.2006.00989.x. [DOI] [PubMed] [Google Scholar]
- 109.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 110.Friedberg ECWG, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM; Washington, D.C: 2005. [Google Scholar]
- 111.Gasco M, Shami S, Crook T. The p53 pathway in breast cancer. Breast cancer research : BCR. 2002;4:70–76. doi: 10.1186/bcr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 113.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 114.McCabe N, Lord CJ, Tutt AN, Martin NM, Smith GC, Ashworth A. BRCA2-deficient CAPAN-1 cells are extremely sensitive to the inhibition of Poly (ADP-Ribose) polymerase: an issue of potency. Cancer biology & therapy. 2005;4:934–936. doi: 10.4161/cbt.4.9.2141. [DOI] [PubMed] [Google Scholar]
- 115.Helleday T, Bryant HE, Schultz N. Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle. 2005;4:1176–1178. doi: 10.4161/cc.4.9.2031. [DOI] [PubMed] [Google Scholar]
- 116.Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3406–3411. doi: 10.1073/pnas.1013715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Curtin NJ, Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Molecular aspects of medicine. 2013;34:1217–1256. doi: 10.1016/j.mam.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Delaney CA, Wang LZ, Kyle S, White AW, Calvert AH, Curtin NJ, Durkacz BW, Hostomsky Z, Newell DR. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000;6:2860–2867. [PubMed] [Google Scholar]
- 119.Liu SK, Coackley C, Krause M, Jalali F, Chan N, Bristow RG. A novel poly(ADP-ribose) polymerase inhibitor, ABT-888, radiosensitizes malignant human cell lines under hypoxia. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2008;88:258–268. doi: 10.1016/j.radonc.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 120.Liu X, Shi Y, Guan R, Donawho C, Luo Y, Palma J, Zhu GD, Johnson EF, Rodriguez LE, Ghoreishi-Haack N, et al. Potentiation of temozolomide cytotoxicity by poly(ADP)ribose polymerase inhibitor ABT-888 requires a conversion of single-stranded DNA damages to double-stranded DNA breaks. Molecular cancer research : MCR. 2008;6:1621–1629. doi: 10.1158/1541-7786.MCR-08-0240. [DOI] [PubMed] [Google Scholar]
- 121.Tentori L, Leonetti C, Scarsella M, D’Amati G, Vergati M, Portarena I, Xu W, Kalish V, Zupi G, Zhang J, et al. Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2003;9:5370–5379. [PubMed] [Google Scholar]
- 122.Nguewa PA, Fuertes MA, Cepeda V, Alonso C, Quevedo C, Soto M, Perez JM. Poly(ADP-ribose) polymerase-1 inhibitor 3-aminobenzamide enhances apoptosis induction by platinum complexes in cisplatin-resistant tumor cells. Med Chem. 2006;2:47–53. doi: 10.2174/157340606775197697. [DOI] [PubMed] [Google Scholar]
- 123.Donawho CK, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE, Ferguson DC, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:2728–2737. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 124.Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S, Maegley KA, Newell DR, Skalitzky D, Wang LZ, et al. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Molecular cancer therapeutics. 2007;6:945–956. doi: 10.1158/1535-7163.MCT-06-0552. [DOI] [PubMed] [Google Scholar]
- 125.Tentori L, Leonetti C, Scarsella M, Muzi A, Mazzon E, Vergati M, Forini O, Lapidus R, Xu W, Dorio AS, et al. Inhibition of poly(ADP-ribose) polymerase prevents irinotecan-induced intestinal damage and enhances irinotecan/temozolomide efficacy against colon carcinoma. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20:1709–1711. doi: 10.1096/fj.06-5916fje. [DOI] [PubMed] [Google Scholar]
- 126.Daniel RA, Rozanska AL, Thomas HD, Mulligan EA, Drew Y, Castelbuono DJ, Hostomsky Z, Plummer ER, Boddy AV, Tweddle DA, et al. Inhibition of poly(ADP-ribose) polymerase-1 enhances temozolomide and topotecan activity against childhood neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:1241–1249. doi: 10.1158/1078-0432.CCR-08-1095. [DOI] [PubMed] [Google Scholar]
- 127.Evers B, Drost R, Schut E, de Bruin M, van der Burg E, Derksen PW, Holstege H, Liu X, van Drunen E, Beverloo HB, et al. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:3916–3925. doi: 10.1158/1078-0432.CCR-07-4953. [DOI] [PubMed] [Google Scholar]
- 128.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Drew Y, Mulligan EA, Vong WT, Thomas HD, Kahn S, Kyle S, Mukhopadhyay A, Los G, Hostomsky Z, Plummer ER, et al. Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. Journal of the National Cancer Institute. 2011;103:334–346. doi: 10.1093/jnci/djq509. [DOI] [PubMed] [Google Scholar]
- 130.Drew Y, JAL, Jones A, Hall G, Jayson GC, Highley M, Rea D, Glasspool RM, Halford SER, Crosswell G, Colebrook S, Boddy AV, Curtin NJ, Plummer ER. Phase II trial of the poly(ADP-ribose) polymerase (PARP) inhibitor AG-014699 in BRCA 1 and 2–mutated, advanced ovarian and/or locally advanced or metastatic breast cancer 2011 [Google Scholar]
- 131.Lee JM, Hays JL, Annunziata CM, Noonan AM, Minasian L, Zujewski JA, Yu M, Gordon N, Ji J, Sissung TM, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. Journal of the National Cancer Institute. 2014;106:dju089. doi: 10.1093/jnci/dju089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lawrence TSTHR, Giaccia A. Principles of Radiation Oncology. In: DeVita VT Jr, Lawrence TS, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. 8. Lippincott, Williams and Wilkins; Philadelphia: 2008. [Google Scholar]
- 133.Chalmers AJ, Lakshman M, Chan N, Bristow RG. Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Seminars in radiation oncology. 2010;20:274–281. doi: 10.1016/j.semradonc.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 134.Tuli R, Surmak AJ, Reyes J, Armour M, Hacker-Prietz A, Wong J, DeWeese TL, Herman JM. Radiosensitization of Pancreatic Cancer Cells In Vitro and In Vivo through Poly (ADP-ribose) Polymerase Inhibition with ABT-888. Translational oncology. 2014 doi: 10.1016/j.tranon.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee HJ, Yoon C, Schmidt B, Park do J, Zhang AY, Erkizan HV, Toretsky JA, Kirsch DG, Yoon SS. Combining PARP-1 inhibition and radiation in Ewing sarcoma results in lethal DNA damage. Molecular cancer therapeutics. 2013;12:2591–2600. doi: 10.1158/1535-7163.MCT-13-0338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 136.Jian W, Xu HG, Chen J, Xu ZX, Levitt JM, Stanley JA, Yang ES, Lerner SP, Sonpavde G. Activity of CEP-9722, a poly (ADP-ribose) polymerase inhibitor, in urothelial carcinoma correlates inversely with homologous recombination repair response to DNA damage. Anti-cancer drugs. 2014;25:878–886. doi: 10.1097/CAD.0000000000000114. [DOI] [PubMed] [Google Scholar]
- 137.Shelton JW, Waxweiler TV, Landry J, Gao H, Xu Y, Wang L, El-Rayes B, Shu HK. In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT-888 in colorectal cancer cells. International journal of radiation oncology, biology, physics. 2013;86:469–476. doi: 10.1016/j.ijrobp.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 138.Chatterjee P, Choudhary GS, Sharma A, Singh K, Heston WD, Ciezki J, Klein EA, Almasan A. PARP inhibition sensitizes to low dose-rate radiation TMPRSS2-ERG fusion gene-expressing and PTEN-deficient prostate cancer cells. PloS one. 2013;8:e60408. doi: 10.1371/journal.pone.0060408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bourton ECFH, Plowman PN, Harvey AJ, Parris CN. Hypersensitivity of BRCA1 Heterozygote Lymphoblastoid Cells to Gamma Radiation and PARP Inhibitors. J Genet Syndr Gene Ther 2013 [Google Scholar]
- 140.Feng Z, Zhang J. A dual role of BRCA1 in two distinct homologous recombination mediated repair in response to replication arrest. Nucleic acids research. 2012;40:726–738. doi: 10.1093/nar/gkr748. [DOI] [PMC free article] [PubMed] [Google Scholar]