

Abstract
Pharmacovigilance (PV) plays a key role in the healthcare system through assessment, monitoring and discovery of interactions amongst drugs and their effects in human. Pharmaceutical and biotechnological medicines are designed to cure, prevent or treat diseases; however, there are also risks particularly adverse drug reactions (ADRs) can cause serious harm to patients. Thus, for safety medication ADRs monitoring required for each medicine throughout its life cycle, during development of drug such as pre-marketing including early stages of drug design, clinical trials, and post-marketing surveillance. PV is concerns with the detection, assessment, understanding and prevention of ADRs. Pharmacogenetics and pharmacogenomics are an indispensable part of the clinical research. Variation in the human genome is a cause of variable response to drugs and susceptibility to diseases are determined, which is important for early drug discovery to PV. Moreover, PV has traditionally involved in mining spontaneous reports submitted to national surveillance systems. The research focus is shifting toward the use of data generated from platforms outside the conventional framework such as electronic medical records, biomedical literature, and patient-reported data in health forums. The emerging trend in PV is to link premarketing data with human safety information observed in the post-marketing phase. The PV system team obtains valuable additional information, building up the scientific data contained in the original report and making it more informative. This necessitates an utmost requirement for effective regulations of the drug approval process and conscious pre and post approval vigilance of the undesired effects, especially in India. Adverse events reported by PV system potentially benefit to the community due to their proximity to both population and public health practitioners, in terms of language and knowledge, enables easy contact with reporters by electronically. Hence, PV helps to the patients get well and to manage optimally or ideally, avoid illness is a collective responsibility of industry, drug regulators, clinicians and other healthcare professionals to enhance their contribution to public health. This review summarized objectives and methodologies used in PV with critical overview of existing PV in India, challenges to overcome and future prospects with respect to Indian context.
Keywords: Pharmacovigilance, Adverse drug reaction, Clinical trials, Pharmacogenomics, Data mining, Indian Pharmacopoeia Commission
1. Introduction
Pharmacovigilance (PV) was officially introduced in December 1961 with the publication of a letter (case report) in the Lancet by W. McBride, the Australian doctor who first suspected a causal link between serious fetal deformities (phocomelia) and thalidomide, a drug used during pregnancy: Thalidomide was used as an antiemetic and sedative agent in pregnant women [1]. In 1968, the World Health Organization (WHO) promoted the “Programme for International Drug Monitoring”, a pilot project aimed to centralize world data on adverse drug reactions (ADRs). In particular, the main aim of the “WHO Programme” was to identify the earliest possible PV signals. The term PV was proposed in the mid-70s by a French group of pharmacologists and toxicologists to define the activities promoting “The assessment of the risks of side effects potentially associated with drug treatment” [2].
PV is the science of collecting, monitoring, researching, assessing and evaluating information from healthcare providers and patients on the adverse effects of medications, biological products, blood products, herbals, vaccines, medical device, traditional and complementary medicines with a view to identifying new information about hazards associated with products and preventing harm to patients. The challenge of maximizing drug safety and maintaining public confidence has become increasingly complex. Pharmaceutical and biotechnology companies must not only monitor, but also proactively estimate and manage drug risk throughout a product’s lifecycle, from development to post-market [3].
PV is particularly concerned with ADRs, which are drug responses that are noxious and unintended, and which occur at doses normally used for the prophylaxis, diagnosis or therapy of disease, or for the modification of physiological function [4]. Continuous monitoring of drug effects, side effects, contraindications and outright harmful effects which could result in a high degree of morbidity, and in some cases, even mortality, are essential to maximize benefits and minimize risks. No degree of care and caution at the pre-clinical and clinical testing stages can guarantee absolute safety, when a drug is marketed and prescribed to large populations across the country and outside. Because clinical trials involve several thousands of patients at most, less common side effects and ADRs are often unknown at the time a drug enters the market. Post marketing PV uses tools such as data mining and investigation of case reports to identify the relationships between drugs and ADRs. The drug regulatory agencies have the responsibility of having a well-established PV system to monitor ADRs during the drug development phase and later during the life time of a marketed drug [5]. A complex and vital relationship exists between wide ranges of partners in the practice of drug safety monitoring such as government, industry, health care centers, hospitals, academia, medical and pharmaceutical associations, poisons information centers, health professionals, patients, consumers and media [6-8]. Sustained collaboration and commitment are vital if future challenges in PV are to be met in order to develop and flourish.
Since very few new drugs were discovered in India and hardly any new drug was launched for the first time in India in the past, there was no major compulsion to have a strong PV system to detect ADRs of marketed products. The experience from the markets where the drug was in use for several years before its introduction in India, was used by the companies and the regulatory agencies to assess the safety parameters and take corrective actions, such as the withdrawal or banning of the drug in question. The evolution of a new patent regime in the Indian pharmaceutical and biotechnology industries as a Trade Related Intellectual Property Rights and Services (TRIPS) makes it incumbent upon India to no longer copy patented products and market them without license from the innovator company. The leading Indian companies, realizing the compulsions of the new regime, have already initiated investments of substantial resources for the discovery and development of new drugs needed for both Indian and International markets. This in turn means that during the coming year, research and development by the Indian pharmaceutical and biotech companies will hopefully lead to new drugs based on pre-clinical and clinical data generated mostly in India. In such cases, the Indian regulatory agencies cannot count on the experience of other markets to assess the incidence and prevalence of importance of a properly designed PV system in India. With the Indian companies’ capacity to develop and market new drugs out of their own research efforts, it is important that adequate PV standards are introduced to monitor ADRs of products first launched in India.
1.1. Scope of PV
The discipline of PV has developed considerably since the 1972 WHO technical report, and it remains a dynamic clinical and scientific discipline. It has been essential to meet the challenges of the increasing range and potency of pharmaceutical and biological medicines including vaccines, which carry with them an inevitable and sometimes unpredictable potential for harm. The risk of harm, however, is less when medicines are used by an informed health profession and by patients who themselves understand and share responsibility for their drugs. When adverse effects and toxicity appear, particularly when previously unknown in association with the medicine, it is essential that they are analyzed and communicated effectively to an audience that has the knowledge to interpret the information. This is the role of PV, of which much has already been achieved. But more is required for the integration of the discipline into clinical practice and public policy. To fulfill the PV obligations for its marketed products as per regulations, a pharmaceutical company in India has to essentially carry out activities such as collection, and expedited reporting of serious unexpected ADRs [9]. A typical setup for PV studies, including people involved on various levels, organizational units and their functions are shown in Figure 1.
Figure 1.
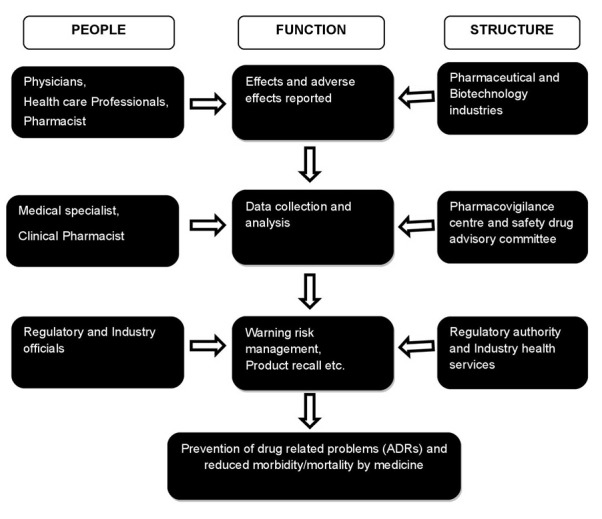
A typical pharmacovigilance setup
1.2. Vaccines and biological medicines
Vaccines and biological medicines require modified systems of safety monitoring. They are often administered to healthy children. This applies particularly to vaccines used within a national immunization program. In many countries, those exposed to a particular vaccine represent the entire birth cohort and therefore a sizeable part of the entire population. People’s expectations of safety are high, and they are reluctant to countenance even a small risk of adverse events. Concerns regarding vaccine safety, real or imagined, may result in loss of confidence in the entire vaccine programs. This can result in poor compliance and a consequent resurgence in morbidity and mortality of vaccine-preventable disease. The difficulties in monitoring and dealing with vaccine safety are complicated by the problems inherent in determining the causal link between an adverse event following immunization and a vaccine [10,11]. For example, information on dechallenge and rechallenge is often missing, and vaccines are given to most of the country’s birth cohort at an age when coincidental disease is likely. Several vaccines are likely to be administered concurrently. The possibility of programmatic errors should never be overlooked. A programmatic error is a medical incident that is caused by errors in the transportation, storage, handling or administration of vaccines. However, the responsibility of the regulatory authority is by no means limited to the safety of vaccines used in immunization programs.
Several biological products are used in specific patient populations as preventive or curative measures. The efficient regulation of these products is crucial in order to avoid potential harm to the public as a result of substandard manufacture or improper transportation and storage of imported vaccines and biological medicines. In recent years, the safety of biological products and blood products has come under public scrutiny. Concerns about the safety of medicinal products of animal origin have been raised in connection with variant Creutzfeldt-Jacob disease (vCJD) and with contamination of blood and blood products by infectious organisms such as HIV and hepatitis B [12]. The quality of screening and sterilization procedures and appropriate selection of donors are linked to the risks of contamination. Such safety issues related to the use of plasma-derived medicinal products should fall under the aegis of PV programs. For that to happen, PV centers would have to consider the special issues related to safety of these products. Expertise in biological products, virology and medical microbiology would be required. Clinical trials in large patient populations are being considered for testing the efficacy and safety of biological medicine.
2. Clinical Trials in India
Global pharmaceutical companies have found India to be a preferred destination for clinical trials because India's clinical research space and opportunities are very attractive [13]. Some of the advantages for clinical trials that India has as are as follows:
High degree of compliance to international guidelines such as the International Conference on Harmonisation (ICH) / WHO Good Clinical Practice (ICH-GCP) and the regulations lay down by the US Food and Drug Administration.
Availability of well qualified, English speaking research professionals including physicians.
Ongoing support and cooperation from the government.
Lower cost compared to the west [14].
Increasing prevalence of illnesses common to both developed and developing countries.
Availability of good infrastructure.
Changes in Patent Laws since January 2005.
As per a recent report from Federation of Indian Chambers of Commerce and Industry (FICCI), scientific feasibility, medical infrastructure, clinical trial experience, regulations, commercialization potential and cost competitiveness are some of the growth drivers responsible for the metamorphosis of Indian clinical research in the recent past [15]. Indian-born contract research organizations (CROs) were able to offer the advantages of understanding the Indian scenario better, provide services at more competitive costs, and having better knowledge of Investigator sites in the country compared to the newer entrants in the market. India’s existing favorable regulatory framework and regulations with international standards, increasing awareness of good clinical practice guidelines and its implementation by clinicians are some of the main reasons propelling the growth of clinical research in India [16,17]. The therapeutic area wise distribution of clinical trials and availability of diverse patient population across major therapeutic segments in India is shown in Figure 2 [18].
Figure 2.
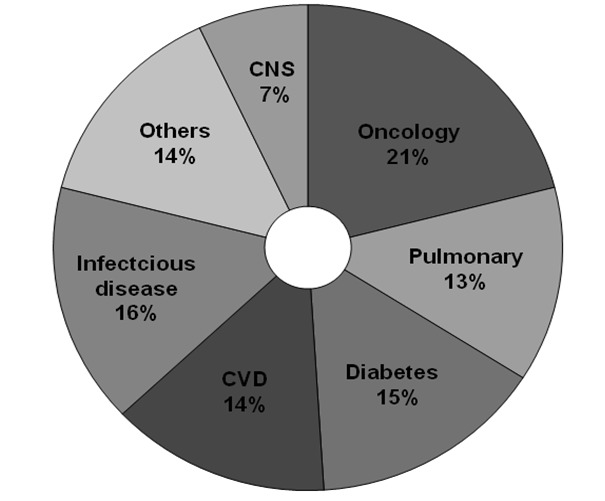
Therapeutic area wise distribution of clinical trials outsourced to India.
2.1. SWOT Analysis of Indian Clinical Trial Sector
-
Strengths
Large population of over 1.2 billion, about 16% of the world’s population.
Huge pharmaceutical and biotech industry base with availability of skilled persons.
Third largest players in the world with 500 different active pharmaceutical ingredients [19].
Currently accounts for 8% of global pharmaceutical production, being fourth in the world [20].
Conducive initiatives to harness India's innovation capability by the government.
Huge data mining related to safety profile of drugs possibilities due to large population [21].
-
Weaknesses
As per 2009-10 estimates, expenditure on health sector was 2.1% of the total budget and 0.35% of the Gross domestic product (GDP) of India [22].
Developed countries like United States, France, Switzerland and Germany, spent around 16%, 11%, 10.8% and 10.4% of their GDP respectively.
Less funding available for implementation of programs and issues of national importance such as PV [23].
-
Opportunities
The Indian population is the largest source of human biodiversity.
Consists of 4635 culturally and anthropologically well-defined populations, representing a perfect model to study efficacy, disease susceptibility, etiology, molecular pathology, and safety profile of drugs with respect to genetic diversity.
Excellent potential for skilled human resources required for an effective PV system due to >300 medical, >230 dental, >830 pharmacy and >650 recognized nursing colleges in India [24].
-
Threats
Under reporting of ADRs.
Low availability of funds.
Less ADRs monitoring centers.
2.2. Agencies Involved for Clinical Research Regulation In India
Various regulatory agencies of India and their prominent roles in overseeing clinical trial along with Ethics committee are shown in Table 1 [25].
Table 1. Roles of various regulatory agencies.
| Agencies | Role of agencies |
| Drug Controller General of India (DCGI) |
Implementation the National Pharmacovigilance |
| Central Drugs Standard Control Organization (CDSCO) |
Operate under the supervision of the National Pharmacovigilance Advisory Committee to recommend. |
| Department of Biotechnology (DBT) | Provides product evaluation and validation through support for limited and large scale field trials for agriculture products and clinical trials for health care products. |
| Ministry of Environment & Forests
(MOEF) |
Project advisory committee approves guidelines for making data entries of the information provided by the environmental experts through the field trials for agriculture products and clinical trials for health care products. |
| Indian Council of Medical Research (ICMR) |
Brought
out the 'Policy Statement on Ethical Considerations involved in
Research on Human Subjects' in 1980 and revised these guidelines in 2000 as the 'Ethical guidelines for Biomedical Research on Human Subjects'. |
| Central Bureau of Narcotics (CBN) |
Closely monitors all clinical trials, which require additional narcotics compliances relating to storage, import-export quotas and movement of the investigational drug. |
| Ministry of Health and Family Welfare (MHFW) | An autonomous body for setting of standards for drugs, pharmaceuticals and healthcare devices and technologies in India. |
| National Pharmacovigilance Advisory (NPAC) |
Collates,
analyzes and archives adverse drug Committee reaction data for creating healthy environment for the regulatory authorities to analyze the drug to be marketed in India. |
3. Role of pharmacogenomics in PV
Pharmacogenomics (PGx) combines traditional pharmaceutical sciences such as biochemistry with annotated knowledge of genes, proteins, and single nucleotide polymorphisms (SNP). It is the technology that deals with the influence of genetic variation on drug response in patients by correlating gene expression or single-nucleotide polymorphisms (SNP) with a drug's efficacy or toxicity. By doing so, (PGx) aims to develop rational means to optimize drug therapy, with respect to the patients' genotype, to ensure maximum efficacy with minimal adverse effects [26]. Such approaches promise the advent of "personalized medicine"; in which drugs and drug combinations are optimized for each individual's unique genetic makeup. The science of pharmacogenetics (PG) originated from the analysis of a few rare and sometimes serendipitously found extreme reactions (phenotypes) observed in some humans. These phenotypes were either inherited diseases or abnormal reactions to drugs or other environmental factors [27]. PG and PGx research remain iterative processes and there is more room for opportunities for improvement in each of the approaches. Figure 3 shows that multiple approaches have to be combined to obtain PGx knowledge that is of value for the development of new therapeutics or for the improvement of existing therapies.
Figure 3.
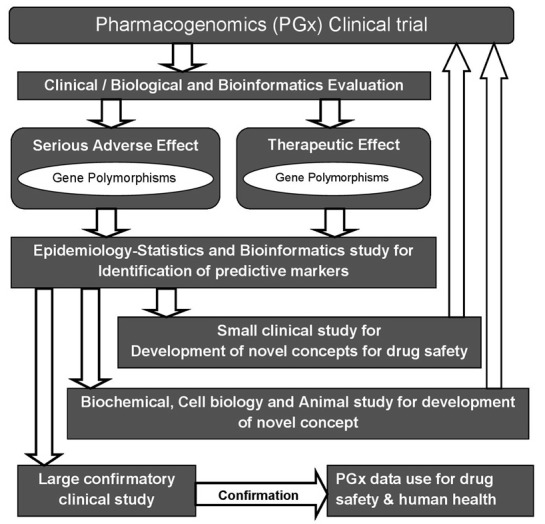
The pathways of pharmacogenomic research in clinical
3.1. Find the gene for disease risk
PG primarily deals with the therapeutic effects and the adverse effects of humans to drugs, poisons and other types of chemicals and environmental factors. However, soon after the field of PG emerged, this scope was broadened and the genetic polymorphisms were extensively studied, not only in relation to known specified exposures but also as susceptibility factors for diseases in general. In many of these studies the disease initiating conditions were not known. In parallel, the functional nature of the genes considered in PG research broadened from drug-metabolizing enzymes to almost all other classes, such as drug transport, DNA-repair, cell-cycle regulation and signal transduction [28]. The overall number of publications studying PG polymorphisms in relation to disease risk exceeds, several-fold, those studying the polymorphisms in relation to response to drugs, and it is not possible to give a meaningful summary of that research on the hundreds of candidate genes as disease susceptibility factors. All recent genome-wide screens did not focus on the response to drugs but on the identification of genotypes predisposing to certain multi factorial and polygenic diseases. Some of the basic techniques of the wet and dry laboratories methods of PG and genomics are shown in Table 2.
Table 2. Techniques for genotype analysis in pharmacogenetics and genomics.
| Methods | Short description and purpose |
| PCR | PCR, basic technique in almost all current pharmacogenetic and genomic analysis. |
| PCR-RFLP | The polymorphic genomic region is amplified by PCR and cut by sequence-specific enzymes (restriction endonucleases). The resulting fragments are analyzed by electrophoresis and are indicative of the genotypes. |
| qPCR (real-time PCR) | Detection of the PCR product formation while PCR reaction proceeds using various fluorescence quenching or fluorescence energy transfer techniques for genotyping of single SNPs in many samples. |
| qRT-PCR (quantitative reverse transcriptase PCR) | Used to quantify amounts of transcripts in a sample after a reverse transcription reaction. Useful for quantification of RNAs in big numbers of samples. |
| Denaturing high performance liquid chromatography (DHPLC) | Variant and wild-type DNA forms differently shaped hybrid molecules (homoduplex versus heteroduplex) which can be separated by ion-pair reverse phase HPLC to identify polymorphisms. |
| Sanger di-deoxy (end terminal) sequencing | Reading of DNA sequences, identification of new polymorphisms. |
| Single-base (primer) extension (known as mini-sequencing) | Short oligonucleotides are annealed so that their 3’-end directly upstream the polymorphic site. Elongation of only one single base is performed by using a mixture of (fluorescently labeled) ddNTPs without dNTPs. The products can be detected either with sequencing or using MALDI-TOF detection system. |
| DNA microarrays | Microarray
solid-phase bound DNA molecules to simultaneously genotype large numbers of SNPs (up to more than a million) in a single sample. Used in the genome-wide association studies. |
| Pyrosequencing | A method of DNA sequencing based on the sequencing by synthesis principle. Used in SNP genotyping and DNA methylation analyses. The principle behind this method is also the basis of the current large-scale DNA sequencing known as 454 “next generation” capable of sequencing more than 100 million basepairs per day. |
3.2. Statistics, bioinformatics and systems biology
Previously reported by Brockmöller and Tzvetkov, it is mentioned that there are almost 12 million SNPs in the human genome [exactly 11,811,594 SNPs, of which 5,689,286 are validated SNPs, according to the last release of the database dbSNP, build 127 (accessed March 2007): http://www.ncbi. nlm.nih.gov/SNP/Notes/build127_announce.txt]. In the near future, a clinical scientist will have to deal not only with clinical and laboratory data on his/her volunteers or patients but also with data on the patient’s 500,000 or 1 million SNPs. SNPs are DNA sequence variations that occur when a single nucleotide (A, T, C, or G) in the genome sequence is altered. SNPs occur every 100 to 300 bases along the 3-billion-base human genome, therefore millions of SNPs must be identified and analyzed to determine their involvement (if any) in drug response. Further complicating the process is our limited knowledge of which genes are involved with each drug response. Since many genes are likely to influence responses, obtaining the big picture on the impact of gene variations is highly time-consuming and complicated. Knowing one's genetic code will allow a person to make adequate lifestyle and environmental changes at an early age so as to avoid or lessen the severity of a genetic disease. Likewise, advance knowledge of particular disease susceptibility will allow careful monitoring, and treatments can be introduced at the most appropriate stage to maximize their therapy.
Table 3. Bioinformatics databases and software tools for pharmacogenetics and genomics.
| Aim | Computer solution | Website |
| Databases: | ||
| Human genome |
National Center for Biotechnology Information in USA (NCBI) | www.ncbi.nlm.nih.gov/genome/guide/human/ |
| SNP databases | dbSNP at NCBI | www.ncbi.nlm.nih.gov/SNP/ |
| Pairwise linkage disequilibrium and haplotypes |
HapMap project | www.hapmap.org |
| Gene expression analysis | Gene Expression Omnibus (GEO) By NCBI | www.ncbi.nlm.nih.gov/geo/ |
| Metabolic pathways | Kyoto Encyclopedia of Genes and Genomes (KEGG) | www.genome.jp/kegg/ |
| Software: | ||
| Homology search | BLAST at NCBI | www.ncbi.nlm.nih.gov/BLAST/ |
| Sequence alignment and identification of new SNPs |
Gap4 (part of Staden package) | http://staden.sourceforge.net/ |
| Haplotype mapping | Phase, Fastphase | http://stephenslab.uchicago.edu/software.html |
| Pairwise linkage disequilibrium and visualization of Haplotype blocks |
Haploview | www.broad.mit.edu/mpg/haploview/ |
| Extended haplotype homozygosity (EHH) | Sweep | www.broad.mit.edu/mpg/sweep/ |
| Analysis of SNPs affecting promoter function | TRANSFAC | http://transfac.bioinf.med.uni-goettingen.de/ |
| Analysis of SNPs affecting splice sites | Automated Splice Site Analyses | https://splice.cmh.edu/ |
The cytochrome P450 (CYP) family of liver enzymes is responsible for breaking down more than 30 different classes of drugs. DNA variations in genes that code for these enzymes can influence their ability to metabolize certain drugs. Less active or inactive forms of CYP enzymes that are unable to break down and efficiently eliminate drugs from the body can cause drug overdose in patients. Today, clinical trials researchers use genetic tests for variations in cytochrome P450 genes to screen and monitor patients. In addition, many pharmaceutical companies screen their chemical compounds to see how well they are broken down by variant forms of CYP enzymes [29]. Thus, the development of bioinformatics and genetic statistics plays a crucial role in the further development of PG and genomics (Table 3). Since the first interspecies comparisons of haemoglobin, protein sequences homology search has been an important tool to identify those segments in the genome that are particularly crucial for the biological function of a certain protein. Linkage of genetic variants on the same chromosome is a central basis of methods in genetic statistics. If a person has a certain variant at a certain position, the same person will probably also have other linked variants 10,000 or 50,000 bp nearby, and the same constellation will be found in several relatives of this person. Hence, PG and PGx are the most important reasons behind interethnic differences in drug effects which are studied in worldwide marketing of drugs and in PV shown in the Table 4.
Figure 4.
Adverse drug reactions online information tracking and yellow card system Sources of data
Table 4. Summarizing the Role of PGx in PV.
| Pharmacogenomics (PGx) and Pharmacogenetics (PG) Drug effects |
| 1. If the safety or efficacy of a drug may depend on genetic polymorphisms, the best choices are either to delete the drug from the market or to analyse and consider the genetic polymorphism in therapy. |
| 2. PGx and genomics are the most important reasons behind interethnic differences in drug effects. Thus, PG has to be carefully studied in worldwide marketing of drugs and in PV. |
| 3. Many PG polymorphisms may have both positive and negative consequences for human health depending on the context and exposures. |
| 4. Applying PG knowledge in the clinics should not always mean applying genotyping, often some type of phenotyping may be superior. |
| 5. Clinical study should know about the background and clinical consequences of genetic polymorphisms in G6PDH, BCHE, NAT2, CYP2D6, CYP2C19, CYP2C9, TPMT, DPD, UGT1A1, VKORC1 and factor V. Very soon that list may have to be updated. |
| 6. As with many complex and new technologies, there are problems and delays in the transfer of scientific PG knowledge to the bedside. Specific translational research has to be supported. |
| 7. The PG diagnostics will have to be based on scientifically valid mechanistic reasoning and appropriate clinical monitoring. |
| 8. In addition to concentrating on single genes, PG and genomic pathway research is required to understand the causes behind pharmacokinetic and pharmacodynamic inter-individual variation. |
| 9. Because of the mass of PG information, medical information technologies including bioinformatics are essential in the future of clinical PG. |
| 10. Besides the traditional axis between genes - mRNAs - proteins and functions, other mechanisms such as epigenetics and RNAi, are apparently relevant for understanding of interindividual variation in drug effects and adverse effects. |
| 11. The future of PG and genomic research will be a mixture of genome-wide SNP and expression analysis in appropriately designed clinical studies, and this will have to be combined with in vitro and ex vivo PGx research with human cells and model organisms and with human clinical research. Finding the right combination of research tools may be the most important demand. |
It is immediately evident that disease risk and response to drugs may depend on the combinations of several genes, and years ago scientists and companies emerged with the concept to sell predictive marker combinations. However, it is all too easy to identify in each study predictive marker combinations which are more predictive than the single markers alone. The aim, however, is to identify predictive marker combinations which remain predictive beyond the study in which they were identified. Predictive marker combinations may be identified as interaction terms in cross-tabulations, analyses of variance or logistic regression analyses, and data mining tools, such as recursive partitioning, are particularly helpful for this. The situations in which such recursive partitioning should be performed in whole genome scans is not yet clear: in the entire data sets or, as some authors have performed, in subsets identified in a monofactorial analysis as possible risk factors [30].
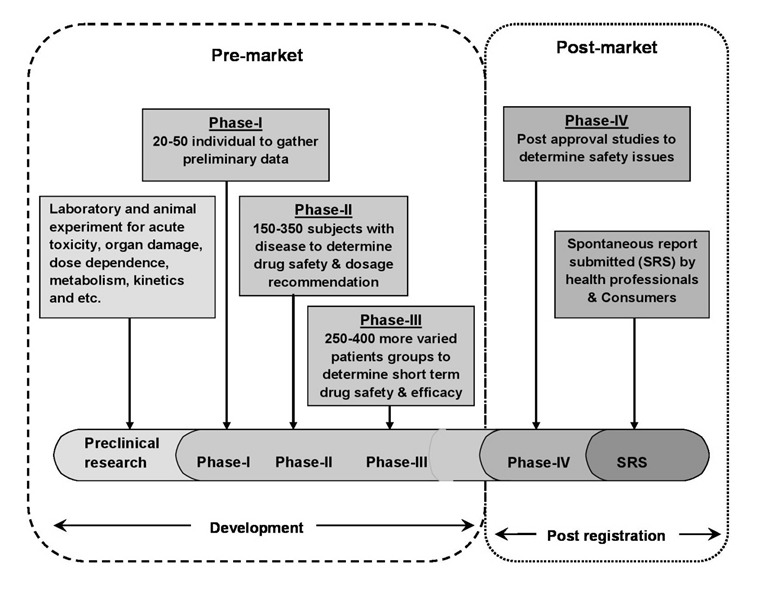
4. Data Mining for PV
PV, also known as drug safety surveillance, is the science of enhancing patient care and patient safety regarding the use of medicines by collecting, monitoring, assessing, and evaluating information from healthcare providers and patients. In that view, PV can be divided into two stages such as premarketing surveillance – information regarding ADRs is collected from pre-clinical screening and phases I to III clinical trials; and post-marketing surveillance – data accumulated in the post approval stage and throughout a drug’s market life shown in Figure 4 [31].
PV has relied on biological experiments or manual review of case reports; however, due to the vast quantities and complexity of data to be analyzed, computational methods that can accurately detect ADRs in a timely fashion have become a critical component in PV. Large-scale compound databases containing structure, bioassay, and genomic information, as well as comprehensive clinical data sets such as electronic medical record (EMR) databases, have become the enabling resources for computerized ADR detection methods [32].
4.1. Premarketing surveillance
PV at the pre-marketing stage has been devoted to predict or assess potential ADRs early in the drug development pipeline. One of the fundamental methods is the application of preclinical in vitro Safety Pharmacology Profiling (SPP) by testing compounds with biochemical and cellular assays. The hypothesis is that if a compound binds to a certain target, then its effect may translate into possible occurrence of an ADR in humans. However, experimental detection of ADRs remains challenging in terms of cost and efficiency. There have been numerous research activities devoted to developing computational approaches to predict potential ADRs using preclinical characteristics of the compounds or screening data. Most of the existing research can be categorized into protein target-based and chemical structure-based approaches. Others have also explored integrative approach [33].
4.2. Post-marketing surveillance
Although a drug undergoes extensive screening before its approval by the Food and Drug Administration (FDA), many ADRs may still be missed because the clinical trials are often small, short, and biased by excluding patients with comorbid diseases. Premarketing trials do not mirror actual clinical use situations for diverse (e.g. inpatient) populations, thus it is important to continue the surveillance postmarket. PV plays an essential role in the post-market analysis of newly developed drugs [34,35]. Pharmaceutical companies' competition along with rigorous regulatory evaluation procedures empowers a complex research and development process before launching a new drug into the market. Several unique data sources are available for postmarketing PV [36].
PV research is based on the analysis of "signals". The World Health Organization (WHO) defines signals as undisclosed assertions on direct relationships between effects on the human organism and a drug to induce adverse events [37]. To generate comprehensive signal datasets, clinicians and researchers use spontaneous reporting systems (SRS). Electronic SRSs are already in place throughout some European countries and the United States. Likewise, other solutions, such as general practitioners’ databases analysis, post market studies or prescription monitoring, among others, are being thoroughly explored. Nevertheless, the majority of data is not publicly available for researchers, which, jointly with other barriers, severely limits signal detection [38-40]. Although drug companies are required to track and manage adverse events reported by clinicians, lawyers or patients, the detection process relies mostly on the physician's ability to recognize a given trait as a drug adverse event. Whereas the problem for collecting and filtering ADR data from multiple distributed nodes has already been studied in the past, researchers continue to pursue the best strategies to delve into the wealth of collected data in conjunction with other post drug administration inputs [41]. With data and text-mining techniques scavenging millions of electronic medical records, PV researchers are now faced with the problem of delivering knowledge-oriented tools and services that exploit the scope of collected data. Ultimately, the adequate exploration of these data will pave the way for improved drug evaluations, critical for pharmaceutical companies, regulatory entities and researchers [42].
4.3. Spontaneous Reports
A spontaneous report is an unsolicited communication by health care professionals or consumers to a company, regulatory authority, or other organization that describes one or more ADRs in a patient who was given one or more medicinal products and that does not derive from a study or any organized data collection scheme. Spontaneous reports play a major role in the identification of safety signals once a drug is marketed. In many instances, a company can be alerted to rare adverse events that were not detected in earlier clinical trials or other pre-marketing studies [43,44].
Spontaneous reporting of ADRs and adverse events is an important tool for gathering the safety information for early detection. Case reports collected by such a system represent the source of information providing the lowest level of evidence and highest level of uncertainty regarding causality. Spontaneous reporting has advantages in that it is available immediately after a new product is marketed, continues indefinitely and covers all patients receiving the drug. It is the most likely method of detecting new, rare ADRs and frequently generates safety signals which need to be examined further [45]. The main limitations are the difficulty in recognizing previously unknown reactions, particularly events that are not usually thought of as being ADRs, and under-reporting, which is variable and sensitive to reporting stimuli and difficult to quantify. It usually does not confirm hypotheses; although situations exist where spontaneous reporting data alone allow conclusions that a signal indeed represents a true ADR [46].
4.4. Yellow Card Scheme
Yellow card schemes (YCS) were applied to spontaneous reporting systems. It was established in 1964 as a result of the thalidomide tragedy. Since then, the system has become one of the major international PV resources [47]. The yellow cards are classified into seven priorities by a member of the scientific staff according to the drugs and the nature of the ADR shown in Figure 5.
Figure 5.
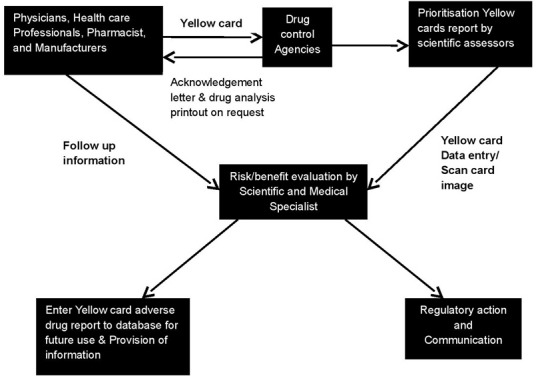
Adverse drug reactions online information tracking and yellow card system Sources of data
The YCS is run jointly by the Medicines Control Agency (MCA) which is the regulatory agency and the Committee on Safety of Medicines (CSM) which is the experts committee. Since 1991, the YCS has been enhanced by a new computer system, the ADROIT (Adverse Drug Reaction Online Information Tracking) system. ADROIT is different from other databases. Not only does it store the details of the report, but also the image of the yellow card in the optical system. Multiple users can view any yellow card on screen at the same time. The reports are prioritized so that serious adverse drug reactions receive early attention [48].
4.5. Detection and reporting
A healthcare professional or marketing authorization holder reports suspected ADRs related to one or more medicinal products, to a PV centre. Reports are made on writing report forms, by telephone, electronically, or by any other approved way [49]. Reports are collected and validated by the PV centre and are usually entered into a database. Serious reactions should be handled with the highest priority. The database is used to identify potential signals and analyze data in order to clarify risk factors, apparent changes in reporting profiles etc [50]. A typical ADR reporting form is shown Figure 6.
Figure 6.

Adverse Drug Reaction reporting form
Systematic methods for the detection of safety signals from spontaneous reports have been used. These methods include the calculation of the proportional reporting ratio, as well as the use of Bayesian and other techniques for signal detection. Data mining techniques have also been used to examine drug-drug interactions [51]. Data mining techniques should always be used in conjunction with, and not in place of, analyses of single case reports. Data mining techniques facilitate the evaluation of spontaneous reports by using statistical methods to detect potential signals for further evaluation (Figure 7).
Figure 7.
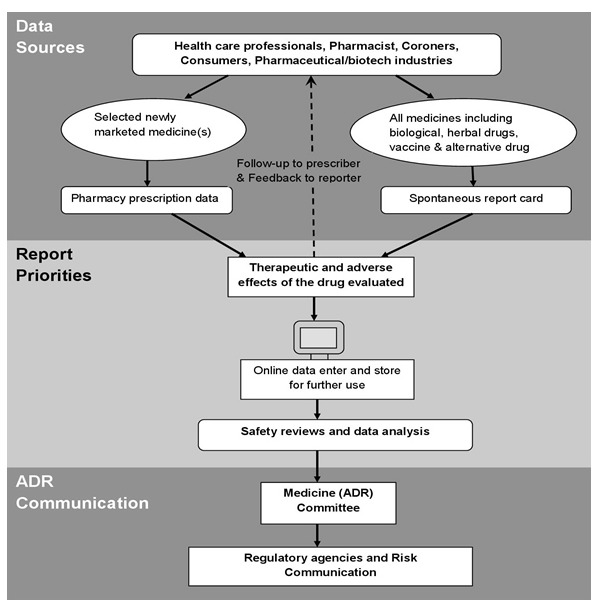
Pharmacovigilance systematic methods for the Evaluation of Spontaneous Reports collected from different data sources.
This tool does not quantify the magnitude of risk, and caution should be exercised when comparing drugs [52]. Furthermore, when using data mining techniques, consideration should be given to the threshold established for detecting signals, since this will have implications for the sensitivity and specificity of the method (a high threshold is associated with high specificity and low sensitivity). Confounding factors that influence spontaneous adverse event reporting are not removed by data mining. Results of data mining should be interpreted with the knowledge of the weaknesses of the spontaneous reporting system and, more specifically, the large differences in the ADR reporting rate among different drugs and the many potential biases inherent in spontaneous reporting. All signals should be evaluated recognizing the possibility of false positives. In addition, the absence of a signal does not mean that a problem does not exist.
5. PV in India
In India, consideration for the surveillance of ADRs developed relatively late, as traditionally there was no concept of surveillance of medicines in the country. Even though PV is still in its infancy, it is not new to India. It was not until 1986 when a few physicians, mainly from academic institutions, called for greater attention to be devoted to the potential adverse effects of prescription medicines and rational prescribing of medicines. This led to the formation of the first ADR monitoring program consisting of 12 regional centers, each covering a population of 50 million, but was unsuccessful [53]. Nothing much happened until a decade later when India joined the WHO Adverse Drug Reaction Monitoring Programme based in Uppsala, Sweden in 1997. Three centers for ADR monitoring were identified, mainly based in the teaching hospitals: A National Pharmacovigilance Center located in the Department of Pharmacology, All India Institute of Medical Sciences (AIIMS), New Delhi and two WHO special centers in Mumbai (KEM Hospital) and Aligarh (JLN Hospital, Aligarh). These centers were to report ADRs to the drug regulatory authority of India. The major role of these centers was to monitor ADRs to medicines marketed in India. However, they were non-functional as information about the need to report ADRs and about the functions of these monitoring centers never reached the prescribers and there was lack of funding from the government. This attempt was unsuccessful, and hence, again from 1 January 2005, the WHO-sponsored and World Bank-funded National Pharmacovigilance Program (NPVP) for India was formulated [54]. NPVP structure is shown in Figure 8.
Figure 8.
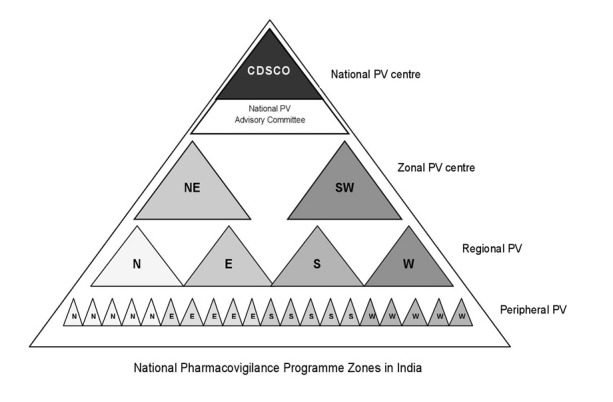
National Pharmacovigilance Program zone structure
The NPVP, established in January 2005, was to be overseen by the National Pharmacovigilance Advisory Committee based at the Central Drugs Standard Control Organization (CDSCO). Two zonal centers, the South-West (SW) zonal center (located in the Department of Clinical Pharmacology, Seth GS Medical College and KEM Hospital, Mumbai) and the North-East (NE) zonal center (located in the Department of Pharmacology, AIIMS, New Delhi) were to collect the information from all over the country and send it to the committee as well as to the Uppsala Monitoring Centre (UMC) in Sweden [55]. Three regional centers would report to the Mumbai center and two to the New Delhi one. Each regional center, in turn, would have several peripheral centers (24 in total) reporting to it. The program had three broad objectives. The short-term objective was to foster a reporting culture, the intermediate objective was to involve large number of healthcare professionals in the system in information dissemination, and the long-term objective was for the program to be a benchmark for global drug monitoring. However, this program also failed [56].
5.1. Current PV program in India
Recognizing the need to restart the NPVP, in a brainstorming workshop jointly organized by the Department of Pharmacology, AIIMS and CDSCO in late 2009, the framework of the new and current program was formulated. The program, now rechristened as the Pharmacovigilance Programme of India (PVPI) was initiated by the Government of India on 14th July 2010 with the AIIMS, New Delhi as the National Coordination Centre (NCC) for monitoring ADRs in the country for safe-guarding public health. In the year 2010, 22 ADR monitoring centers including AIIMS, New Delhi was set up under this programme [57]. To ensure implementation of this programme in a more effective way, the NCC was shifted from the AIIMS, New Delhi to the Indian Pharmacopoeia Commission (IPC), Ghaziabad, Uttar Pradesh on 15th April 2011 [58]. The main aim of the NCC at IPC is to generate an independent data on the safety of medicines, which will be at par with global drug safety monitoring standards. Year-wise target phases of PVPI is shown in Figure 9.
Figure 9.
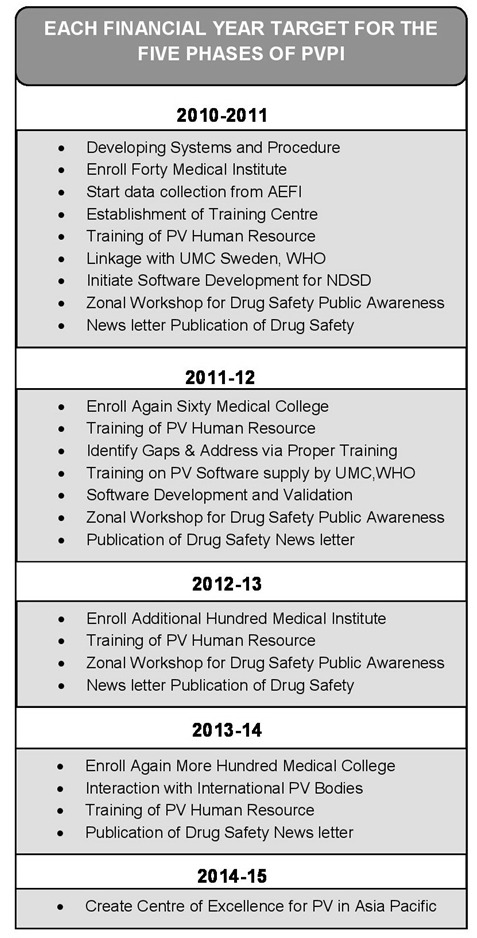
Targets for the Pharmacovigilance Program in India
The recent report of the AMC’s under the PVPI has been reported based on the entry of ADR’s in the VigiFlow. In total, NCC received 3,537 Individual Case Safety Reports (ICSRs) and 1,948 Adverse Events Following Immunization (AEFI) from the AMCs in the month of May 2014. Seven more centres have been given access to VigiFlow by UMC, Sweden. Out of 97 AMC’s where the Vigiflow is functional, 82 centres have provided the ADR’s reports via VigiFlow. It was observed that PGIMER, Chandigarh entered the highest number of ADR’s reports (311 reports in May 2014) followed by 225 reports from MMC, Chennai; 216 reports from JSS, Mysore; 184 reports from UCMS-GTBH, Delhi; 167 reports from LHMC, New Delhi. These reports are under (quality and medical) assessment at NCC [59]. Current status of NCC-PVPI published on May 31, 2014 by IPC that is shown in Table 5.
Table 5.
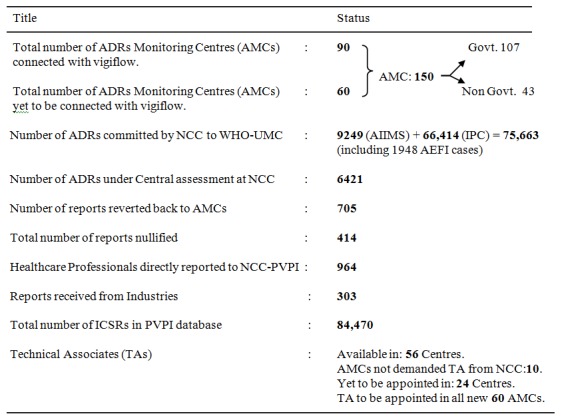
Current Status of NCC-PVPI
5.2. Framework of the new program
The center at IPC focuses on developing India’s own database on drug information and ADRs, so that India will not have to be dependent on data from other countries to take decisions relating to the banning and suspension of drugs. At present, India does not have a strong database on ADRs and has to depend on information from Western countries. So far, only 2823 ADRs have been reported since September 2010 under the current PVPI, which is very small to draw any meaningful conclusion implicated for any particular signal [60].
Table 6.
| Year | Event |
| 1747 | First reported clinical trials by James Lind, proving the effectiveness of lemon juice in preventing scurvy. |
| 1937 | Death of 107 children due to sulfanilamide toxicity. |
| 1950 | Apalstic anemia reported due to chloramphenicol |
| 1961 | Global disaster due to thalidomide toxicity |
| 1963 | 16th World Health Assembly recognize to rapid action on ADR’s |
| 1968 | WHO pilot research project for international drug monitoring |
| 1996 | Clinical trials of global standards started in India |
| 1997 | India joined WHO Adverse Drug Reaction Monitoring Program |
| 1998 | Pharmacovigilance initiated in India |
| 2002 | 67th National Pharmacovigilance Center established in India. |
| 2004-05 | National Pharmacovigilance Program launched in India |
| 2005 | Conduct of structured clinical trials in India |
| 2009-10 | PVPI initiated |
It is being envisaged that all the medical institutions, hospitals, colleges, and public health programs in the country, both government as well as private, will take part in the PVPI and report ADRs to IPC, so that all the data generated will be collated and analyzed at one place. The chronological developments in the field of PV with special reference to India are given in Table 6 [61]. PVPI administered governing body and monitored centers are shown in Figure 10.
Figure 10.
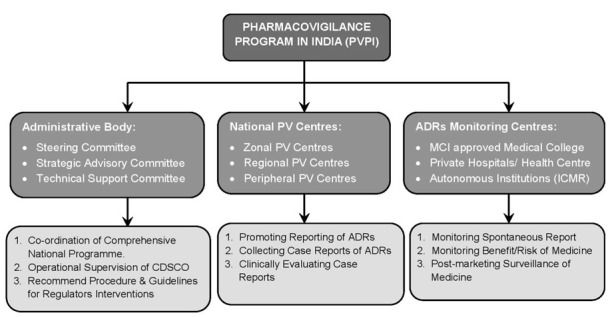
Pharmacovigilance program in India and responsibilities.
The program was envisaged to be rolled out in three phases. Phase I would include 40 ADR monitoring centers (AMC) to be rolled out in 2010. The program would be expanded in phase II to include up to 140 MCI recognized medical colleges by 2011. Until the end of 2011, a total of only 60 AMCs have been included. Phase III would ultimately cover the entire healthcare system by 2013 [62]. The AMCs get operational and logistic support from the respective zonal CDSCO centers situated at Ghaziabad, Kolkata, Mumbai, and Chennai. The zonal CDSCO centers will be under administrative control of the CDSCO headquarters at New Delhi. Organizational structure of PVPI and respective responsibilities of are shown in Figure 11 [63].
Figure 11.
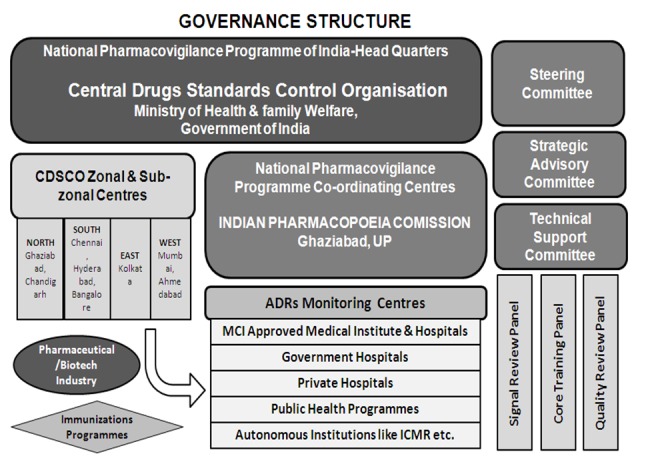
Governance structure
5.3. ADR data flow
ADR reports were collected at the AMC by the PV staff who checked for validity of the report and conducted provisional causality assessment. The ADR forms are then dispatched to the coordinating center. The AMC staff maintains a log of all the activities of the center and the selected AMCs also carry out focused ADR monitoring of drugs as per the watch list. The coordinating centers are conducting causality assessment and upload the reports into the PV database. The coordinating center also prepare a consolidated report of ADRs collected at defined time intervals and then implement and integrate PV activities into public health programs involving mass usage of drugs [64]. Finally, the integrated ADR data is then transmitted through VigiFlow interface into the UMC adverse reaction database where signal processing can be carried out [65] (Lindquist 2008). Programme communication of ADR data flow is shown in the Figure 12. There is a quality review panel which ensures the quality of ADR data that has been constituted for maintaining quality assurance in the program. All the centers will be assessed based on performance metrics criteria, completeness of reports, training imparted, and other parameters mentioned in the PV program protocol.
Figure 12.
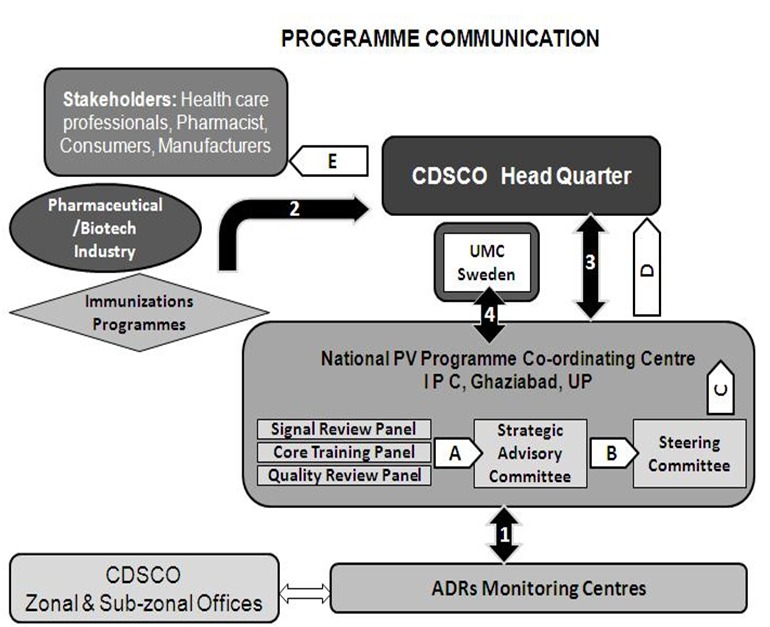
ADR data Program Communications
5.4. Haemovigilance Programme
Haemovigilance is a continuous process of data collection and analysis of transfusion-related adverse reactions in order to investigate their causes and outcomes and prevent their occurrence or recurrence. IPC, in collaboration with the National Institute of Biologicals (NIB) at Noida, has launched a Haemovigilance Programme of India (HVPI) across the country under its PVPI with two main objectives. First to track adverse reactions/events and incidence associated with blood transfusion and blood product administration, and second to help identify trends, recommend best practices and interventions required to improve patient care and safety, while reducing overall cost of the healthcare system [66].
The recognition and management of transfusion reactions (TRs) are critical to ensure patient safety during and after a blood transfusion. Transfusion reactions are classified into acute transfusion reactions (ATRs) or delayed transfusion reactions, and each category includes different subtypes. Different ATRs share common signs and symptoms which can make categorization difficult at the beginning of the reaction. Moreover, TRs are often under-recognized and under-reported. To ensure uniform practice and safety, it is necessary to implement a national haemovigilance system and a set of national guidelines establishing policies for blood transfusion and for the detection and management of TRs [67]. In this context the haemovigilance programme was launched on 10th Dec 2012 and has already enrolled 90 Medical Colleges of India under PVPI as an integral part of PVPI. NIB is the coordinating centre for HVPI to collect and collate data pertaining to Haemovigilance coming from medical institutions all over the country. A software “Haemo-Vigil” has been developed by IT Division of the NIB [68].
5.5. Visibility, communication, and feedback of PVPI
A website created by CDSCO is dedicated to PVPI. In phase II of the program, there is a provision for online reporting of ADRs by healthcare professionals who are not covered under the program. The CDSCO headquarters, in collaboration with NCC, published a quarterly “Medicine Safety Newsletter” comprising 4-16 pages. Approximately 3000 copies will be printed for circulation to healthcare institutions across the nation. A Medicine Safety Card has been included in the Medicine Safety Newsletter, and in national medical journals, to ensure that healthcare professionals not covered under the program can report ADRs directly to any of the centers. These are creating awareness about the program and ensure reporters get adequate feedback and remain motivated. In addition, to enhance the awareness and visibility of the programme, focused workshops, symposia, and group meetings on ADR reporting and causality assessment are carried out at regular intervals by all the centers [69].
5.6. Guideline of PVPI
Globally, many countries have formulated their own PV guidelines with the aim of having a systematic process of safety reporting. The ICH has six guidelines pertaining to various aspects of drug safety [70,71]. E2A- Clinical Safety Data Management: Definitions and standards for expedited reporting, E2B- Clinical Safety Data Management: Data elements for transmission of individual case safety reports, E2C- Clinical Safety Data Management: Periodic safety update reports for marketed drugs, E2D- Post-approval Safety Data Management: Definitions and standards for expedited reporting, E2E-Pharmacovigilanve planning, and E2F- Development Safety Update Report. Hence, legislative requirements of PV in India are guided by specifications of Schedule Y of the Drugs and Cosmetics Act 1945. Schedule Y also deals with regulations relating to pre-clinical and clinical studies for development of a new drug, as well as clinical trial requirements for import, manufacture, and obtaining marketing approval for a new drug in India. Schedule Y was revised and amended on 20 January 2005 as a continued commitment of the drugs controller general of India (DCGI) to ensure adequate compliance of PV obligations of pharmaceutical companies. An attempt has been made in the amended Schedule Y to better define the roles and responsibilities of pharmaceutical companies for their products, as well as relating to reporting of AEs from clinical trials [72].
India has only a small section of Schedule Y dedicated to drug safety, which when viewed in light of contemporary global practice, seems to have many lacunae. There is thus a felt need that CDSCO must formulate detailed PV guidelines. Such guidelines shall incorporate all relevant areas of pre and post marketing safety, address the current lacunae and bring about clarity on issues as discussed above. Most importantly, the guidelines shall be in tune with the current international norms, so as to support India’s growth like any participant in multinational clinical trials [73].
5.7. The challenges of PV in India
The biggest challenge facing the PVPI is the gross underreporting of adverse effects. There are many reasons for this, including lack of medical expertise in drug administration and adequate skilled resources in PV, and inadequate nationwide awareness of PV. The other challenges are infrastructure which are still conservative, wide time interval between guidelines and laws, orthodox attitude to new drug research, and PV and regulatory inspections that are almost non-existent. The system needs to be refined with the help of PV experts in collaboration with information technology (IT) because India boasts of a highly developed IT sector. Since PV deals with large numbers of ADRs, it would be wise for PV experts to collaborate with software professionals to develop and build a robust system. Software programs developed can be used for collection and analyses of data sets, determining trends of drug usage in various disease areas, compliance, medication errors and drug interactions leading to ADRs. Moreover, with more clinical research and PV outsourcing work now being conducted in India, it has been worthwhile for the DCGI to invest in a robust PV system to enable assessors and decision makers to analyze safety data and take regulatory decisions without the need to depend on other countries [74].
However, sometimes ADRs are not recognized by the physicians on admission and ADRs may be responsible for the death of many patients. Furthermore, the financial cost of ADRs to the healthcare system is also vast [75]. In the market, when new medicines are launched without long term safety studies by the regulatory authorities, patients self-medicate and switch from prescription-only medicines (POM) to over-the-counter (OTC) more widely, and this is the main reason of exposing itself to ADRs. In the earlier period, India's regulatory agencies and drug companies based their safety assessments on experiences derived from long-term use [76]. In recent years, many Indian companies are increasing their investment in research and development and are enhancing their capacity to develop and market new drugs with their own research efforts. Once a product is marketed, new information will be generated, which may have an impact on the benefit-risk profile of the product. The detailed evaluation of the new information generated through PV activities is important for all products to ensure their safe use. Hence, DCGI should take some tough decisions and make commitments to make PV mandatory and start the culture of PV inspections.
5.8. Future prospects
As future prospects increase, PV systems capable to detect new ADRs, and taking regulatory actions are needed to protect public health. Little emphasis has been put into generating information that can assist a healthcare professional or a patient in the decision-making process. The gathering and communication of this information is an important goal of PV [77]. Information about the safety of drug active surveillance is necessary. When develop new methods for active post-marketing surveillance, one has to keep in mind that the important to collect complete and accurate data on every serious reported event. Spontaneous reporting is a useful tool in generating signals, but the relatively low number of reports received for a specific association makes it less useful in identifying patient characteristics and risk factors [78]. PV methods must also be able to describe which patients are at risk of developing an ADR. As a source of information, the PV approach would be consistent with the growing patient involvement in drug safety. The PG could play a role in identifying individual risk factors for the occurrence of certain ADRs. In the future, PV has to concentrate on the patients as a source of information in addition to the more traditional groups, such as the health professionals.
At present, the DCGI should act quickly to improve PV so as to integrate Good Pharmacovigilance Practice (GPP) into the processes and procedures to help ensure regulatory compliance and enhance clinical trial safety and post marketing surveillance. An appropriately working PV system is essential if medicines are to be used carefully. It will benefit healthcare professionals, regulatory authorities, pharmaceutical companies and the consumers. It helps pharmaceutical companies to monitor their medicines for risk [79]. Post-marketing PV is currently a challenging and laborious process, not only industry-wide, but also for regulatory agencies.
The aim of the PV is to receive the information, documentation of the work and knowledge online while giving priority to the new and important safety issues. Non-serious events have less priority than serious events but important in comparing the changes in health, although they are also screened routinely [80]. In present time, GlaxoSmithKline has created a powerful new approach to PV, integrating traditional, case-based PV methods with disproportionality and data visualization tools. These tools exist within a system framework that facilitates in-stream review, tracking of safety issues and knowledge management [81]. This very innovative tool and the processes will help to advance PV by improving efficiency and providing new analytical capabilities. Similar approach may be adopted by pharmaceutical companies for prompt detection and analysis of ADRs. Transparency and communication would strengthen consumer reporting, which are positive steps towards involving consumers more in PV.
6. Conclusion
The PV in India has become an important public health issue as regulators, drug manufacturers, consumers, and healthcare professionals are faced with a number of challenges. The PV in India continues to grow, evolve, and improve. India is the largest producer of pharmaceuticals and now emerging as an important clinical trial hub in the world. Apparently, the requirements for professional specialization, a combined view on PGx and clinical requirements are needed. That helps to identify factors that increase the risk of unwanted outcomes from drug therapy and prior to commencing drug treatment and in tailoring drug treatment for individual patients. The PV has also involved in Data Mining Technology in spontaneous reports submit to the national surveillance systems. The PVPI is coordinated at IPC through NCC under the control of Indian Government to generate an independent data on safety of medicines, which will be at par with global drug safety monitoring standards. National and regional PV systems are well-adapted bodies, attuned to the intricate collection and analysis of ADR data that leads to timely alerts and interventions to protect population health. Furthermore, it is responsible in India of entire campaign to improve PV knowledge and increase the number of ADRs reports up to the gold standard level established by the WHO.
The adverse events reported by PV system will potentially benefit to the community due to their proximity to both the population and public health practitioners, in terms of language and knowledge of the lifestyle and habits of patients, enables easy contact with reporters, for example by telephone, Email, text massages by mobile phones. The development of new and effective medicinal products makes a positive contribution to the health and well being of individuals. However, there is a need to improve PV systems to more effectively monitor and take action on safety issues associated with medicines to enhance their contribution to public health. Hence, PV for medicinal product safety to help the patients get well and to manage optimally or ideally, avoid illness is a collective responsibility of industry, drug regulators and clinicians and other healthcare professionals. The financial support and future projects should help to achieve a more comprehensive PV activity in India.
Authors’ contribution
SGS performed literature research, drafted, edited and reviewed the manuscript and also carried out the graphic design of all figures. PK and HN read and approved the final manuscript.
Footnotes
This article received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. All authors declare that they do not have any conflicts of interest with this work.
References
- 1.McBride WG. 1961. Thalidomide and congenital abnormalities. Lancet. 278, 1358 10.1016/S0140-6736(61)90927-8 [DOI] [Google Scholar]
- 2.Bégaud B, Chaslerie A, Haramburu F. 1994. Organization and results of drug vigilance in France. Rev Epidemiol Sante Publique. 42, 416-23. [PubMed] [Google Scholar]
- 3.WHO. The Importance of Pharmacovigilance, Safety Monitoring of medicinal products. World Health Organization 2002. [Google Scholar]
- 4.Report E. Effective communications in Pharmacovigilance. International Conference on Developing Effective Communications in Pharmacovigilance, Erice, Sicily 1997; 24-27. [Google Scholar]
- 5.Shuka SS, Gidwani B, Pandey R, Rao SP, Singh V, et al. 2012. Importance of Pharmacovigilance in Indian Pharmaceutical Industry. Asian J. Res. Pharm. Sci. 2, 4-8. [Google Scholar]
- 6.Hornbuckle K, Wu H-H, Fung MC. 1999. Evaluation of spontaneous adverse event reports by primary reporter - a 15-year review (1983 to 1997). Drug Inf J. 33, 1117-24. [Google Scholar]
- 7.WHO 2000. Consumer reporting of ADRs. WHO Drug Information. 14, 211-15. [Google Scholar]
- 8.Egberts GPG, Smulderes M, De Konig FHP, et al. 1996. Can adverse drug reactions be detected earlier?: a comparison of reports by patients and professionals. BMJ. 313, 530-31. 10.1136/bmj.313.7056.530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Norwood PK, Sampson AR. 1988. A statistical methodology for postmarketing surveillance of adverse drug reaction reports. Stat Med. 7, 1023-30. 10.1002/sim.4780071004 [DOI] [PubMed] [Google Scholar]
- 10.WHO. Immunization safety surveillance: Guidelines for managers of immunization programmes on reporting and investigating adverse events following immunization. Immunization Focus: WHO Regional Office for the Western Pacific. Manila 1999. [Google Scholar]
- 11.WHO. Global programme for vaccines and immunization: Expanded Programme on Immunization. Surveillance of adverse events following immunization: Field guide formanagers of immunization programmes. Geneva 1997. [Google Scholar]
- 12.Simonsen L, Kane A, Lloyd J, Zaffran M, Kane M. 1999. Unsafe injections in the developing world and transmission of bloodborne pathogens: a review. Bull World Health Organ. 77, 789-800. [PMC free article] [PubMed] [Google Scholar]
- 13.Strom BL. Overview of automated databases in pharmacoepidemiology (ed). Pharmacoepidemiology Chichester. UK, John Wiley & Sons. 2005;p.219–22. [Google Scholar]
- 14.Butlen S, Ducuing F. 2010. European medicines agency support mechanisms fostering orphan drug development. Drug News Perspect. 23, 71-81. 10.1358/dnp.2010.23.1.1437303 [DOI] [PubMed] [Google Scholar]
- 15.Giezen TJ. 2009. Evaluation of post-authorization safety studies in the first cohort of EU risk management plans at time of regulatory approval. Drug Saf. 32, 1175-87. 10.2165/11318980-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 16.Herret E. 2010. Validation and validity of diagnoses in the general practice research database. J Clin Pharmacol. 69, 4-14. 10.1111/j.1365-2125.2009.03537.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abenhaim L. 1996. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. N Engl J Med. 335, 609-16. 10.1056/NEJM199608293350901 [DOI] [PubMed] [Google Scholar]
- 18.Kaufman DW. 2001. Signal generation and clarification: Use of case-control data. Pharmacoepidemiol Drug Saf. 10, 197-203. 10.1002/pds.571 [DOI] [PubMed] [Google Scholar]
- 19.Greene W. The Emergence of India’s Pharmaceutical Industry and Implications for the U.S. Generic Drug Market. U.S. International Trade Commission 2007. [Google Scholar]
- 20.Iskander J. 2005. Monitoring vaccine safety during an influenza pandemic. Yale J Biol Med. 78, 265-75. [PMC free article] [PubMed] [Google Scholar]
- 21.Stefano F, Tokars J. 2010. H1N1 vaccine safety monitoring: Beyond background rates. Lancet. 375, 1146-47. 10.1016/S0140-6736(09)61917-6 [DOI] [PubMed] [Google Scholar]
- 22.Chen RT. 2000. The vaccine safety datalink: Immunization research in health maintenance organizations in the USA. Bull World Health Organ. 78, 186-94. [PMC free article] [PubMed] [Google Scholar]
- 23.Vander H, Stichele R. 2004. European Surveillance of Antimicrobial Consumption (ESAC): Data collection performance and methodological approach. Br J Clin Pharmacol. 58, 419-28. 10.1111/j.1365-2125.2004.02164.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martirosyan LA. 2010. Systematic literature review: Prescribing indicators related to type 2 diabetes mellitus and cardiovascular risk management. Pharmacoepidemiol Drug Saf. 19, 319-34. [DOI] [PubMed] [Google Scholar]
- 25.Von NC, Schwappach DL, Koeck CM. 2003. The epidemiology of preventable adverse drug events: A review of literature. Wien Klin Wochenschr. 115(12), 407-15. [DOI] [PubMed] [Google Scholar]
- 26.Aneesh TP, Sonal Sekhar M, Jose A, Chandran L, Zachariaha SM. 2009. Pharmacogenomics: The Right Drug to the Right Person. J Clin Med Res. 1, 191-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brockmöller J, Tzvetkov MV. 2008. Pharmacogenetics: data, concepts and tools to improve drug discovery and drug treatment. Eur J Clin Pharmacol. 64, 133-57. 10.1007/s00228-007-0424-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalow W, Meyer U, Tyndale R. Pharmacogenomics 2nd edition, Taylor and Francis 2005;156:341–51. [Google Scholar]
- 29.Hodgson J, Marshall A. 1998. Pharmacogenomics: will the regulators approve. Nat Biotechnol. 16, 243-46. 10.1038/5143 [DOI] [PubMed] [Google Scholar]
- 30.Warren LL, Hughes AR, Lai EH, Zaykin DV, et al. 2007. Use of pairwise marker combination and recursive partitioning in a pharmacogenetic genome-wide scan. Pharmacogenomics J. 7, 180-89. 10.1038/sj.tpj.6500414 [DOI] [PubMed] [Google Scholar]
- 31.Berlin JA, Glasser SC, Ellenberg SS. 2008. Adverse Event Detection in Drug Development: Recommendations and Obligations Beyond Phase 3. Am J Public Health. 98, 1366-71. 10.2105/AJPH.2007.124537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kahn MG, Ranade D. 2010. The impact of electronic medical records data sources on an adverse drug event quality measure. J Am Med Inform Assoc. 17, 185-91. 10.1136/jamia.2009.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitebread S, Hamon J, Bojanic D, Urban L. 2005. Keynote review: in vitro safety pharmacology profiling: an essential tool for successful drug development. Drug Discov Today. 10, 1421-33. 10.1016/S1359-6446(05)03632-9 [DOI] [PubMed] [Google Scholar]
- 34.McClure DL. 2009. Improving Drug Safety: Active Surveillance Systems Should be Paramount. Pharmaceutical Medicine. 23, 127-30. 10.1007/BF03256760 [DOI] [Google Scholar]
- 35.Shibata A, Hauben M. Pharmacovigilance, signal detection and signal intelligence overview. Proceedings of the 14th International Conference on Information Fusion (FUSION-2011). Chicago, IL. 2011; pp. 1-7. [Google Scholar]
- 36.Lopes P, Nunes T, Campos D, Furlong LI, Bauer-Mehren A, et al. 2013. Gathering and Exploring Scientific Knowledge in Pharmacovigilance. PLoS ONE. 8(12), e83016. 10.1371/journal.pone.0083016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stahl M, Edwards IR, Bowring G, Kiuru A, Lindquist M. 2003. Assessing the Impact of Drug Safety Signals from the WHO Database Presented inSIGNAL': Results from a Questionnaire of National Pharmacovigilance Centres. Drug Saf. 26, 721-27. [DOI] [PubMed] [Google Scholar]
- 38.Lazarou J, Pomeranz BH, Corey PN. 1998. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 279, 1200-05. 10.1001/jama.279.15.1200 [DOI] [PubMed] [Google Scholar]
- 39.Meyboom RHB, Lindquist M, Egberts ACG, Edwards IR. 2002. Signal Selection and Follow-Up in Pharmacovigilance. Drug Saf. 25, 459-65. 10.2165/00002018-200225060-00011 [DOI] [PubMed] [Google Scholar]
- 40.De Bruin ML, Van Puijenbroek EP, Egberts AC, Hoes AW, Leufkens HG. 2002. Non-sedating antihistamine drugs and cardiac arrhythmias– biased risk estimates from spontaneous reporting systems? Br J Clin Pharmacol. 53, 370-74. 10.1046/j.1365-2125.2002.01569.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coloma PM, Schuemie MJ, Trifirò G, Gini R, Herings R, et al. 2011. Combining electronic healthcare databases in Europe to allow for largescale drug safety monitoring: the EU-ADR Project. Pharmacoepidemiol Drug Saf. 20, 1-11. 10.1002/pds.2053 [DOI] [PubMed] [Google Scholar]
- 42.Härmark L, van Grootheest AC. 2008. Pharmacovigilance: methods, recent developments and future perspectives. Eur J Clin Pharmacol. 64, 743-52. 10.1007/s00228-008-0475-9 [DOI] [PubMed] [Google Scholar]
- 43.Xu L, Anchordoquy T. 2011. Drug delivery trends in clinical trials and translational medicine: Challenges and opportunities in the delivery of nucleic acid-based therapeutics. J Pharm Sci. 100, 38-52. 10.1002/jps.22243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robb MA, Racoosin JA, Sherman RE, Gross TP, Ball R, et al. 2012. The US Food and Drug Administration's Sentinel Initiative: Expanding the horizons of medical product safety. Pharmacoepidemiol Drug Saf. 21, 9-11. 10.1002/pds.2311 [DOI] [PubMed] [Google Scholar]
- 45.Waller PC, Evans SJ. 2003. A model for the future conduct of pharmacovigilance. Pharmacoepidemiol Drug Saf. 12, 17-29. 10.1002/pds.773 [DOI] [PubMed] [Google Scholar]
- 46.Kurz X. 2010. The Eudra Vigilance database of spontaneous adverse reactions as a tool for H1N1 vaccine safety monitoring. Pharmacoepidemiol Drug Saf. 19, 330-31. [Google Scholar]
- 47.Gupta SK. Drug Discovery and Clinical Research. Jaypee Brothers Medical Pubishers (P) Ltd. First Edition 2011. [Google Scholar]
- 48.Yom-Tov E, Gabrilovich E. 2013. Postmarket Drug Surveillance Without Trial Costs: Discovery of Adverse Drug Reactions Through Large-Scale Analysis of Web Search Queries. J Med Internet Res. 15, e124. 10.2196/jmir.2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bate A, Evans SJ. 2009. Quantitative signal detection using spontaneous ADR reporting. Pharmacoepidemiol Drug Saf. 18, 427-36. 10.1002/pds.1742 [DOI] [PubMed] [Google Scholar]
- 50.Hauben M. 2004. Application of an empiric Bayesian data mining algorithm to reports of pancreatitis associated with atypical antipsychotics. Pharmacotherapy. 24, 1122-29. 10.1592/phco.24.13.1122.38098 [DOI] [PubMed] [Google Scholar]
- 51.Harpaz R, DuMouchel W, Shah NH, Madigan D, Ryan P, et al. 2012. Novel Data-Mining Methodologies for Adverse Drug Event Discovery and Analysis. Clin Pharmacol Ther. 91, 1010-21. 10.1038/clpt.2012.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edwards LD, Fox AW, Stonier PD. Principles and Practice of Pharmaceutical Medicine. 3rd Edition, Wiley-Blackwell 2011. [Google Scholar]
- 53.Kulkarni RD. 1986. Reporting system for rare side effects of non-narcotic analgesics in India: Problems and opportunities. Med Toxicol. 1, 110-13. [PubMed] [Google Scholar]
- 54.Moride Y, Haramburu F, Requejo A, Begaud B. 1997. Under-reporting of adverse drug reactions in general practice. Br J Clin Pharmacol. 43(2), 177-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adithan C. 2005. National pharmacovigilance programme. Indian J Pharmacol. 37, 347 10.4103/0253-7613.19069 [DOI] [Google Scholar]
- 56.Biswas P, Biswas AK. 2007. Setting Standards for Proactive Pharmacovigilance in India: The Way Forward. Indian J Pharmacol. 39, 124-28. 10.4103/0253-7613.33431 [DOI] [Google Scholar]
- 57.Gupta YK. Ensuring Patient Safety - Launching the New Pharmacovigilance Programme of India. Pharma Times – August 2010, Vol 42 - No. 08.
- 58.NPP. National Pharmacovigilance Protocol. Ministry of Health & Family Welfare, Government of India. Downloaded from www.cdsco.nic.in accessed on 15 October 2014.
- 59.Indian Pgarmacopoeia Comission IPC. AMC’s Progress Report – May 2014 Downloaded from www.ipc.gov.in accessed on 15 October 2014.
- 60.Biswas P. 2013. Pharmacovigilance in Asia. J Pharmacol Pharmacother. 4, S7-19. 10.4103/0976-500X.120941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar S, Baldi A. 2013. Pharmacovigilance in India: Perspectives and Prospects. Journal of Drug Delivery & Therapeutics. 3, 237-46. [Google Scholar]
- 62.Andrews EB, Moore N. Mann's Pharmacovigilance. Third Edition, Wiley Blackwell. 2014; pp 287–88. [Google Scholar]
- 63.Tripathy R, Basha S, Jain R, Shetty S, Ramachandran A. 2008. Exenatide and acute pancreatitis. J Assoc Physicians India. 56, 987-88. [PubMed] [Google Scholar]
- 64.Palanisamy S, Kottur SG, Kumaran A, Rajasekaran A. 2013. A study on assessment of knowledge about adverse drug reactions. Der Pharmacia Lettre. 5, 41-52. [Google Scholar]
- 65.Lindquist M. 2008. VigiBase, the WHO Global ICSR Database System: Basic Facts. Drug Inf J. 42, 409-19. [Google Scholar]
- 66.Francis PA. National Haemovigilance Programme. PharmaBiz-2013 October 30.
- 67.Al-Riyami AZ, Al-Hashmi S, Al-Arimi Z, et al. 2014. Recognition, Investigation and Management of Acute Transfusion Reactions: Consensus guidelines for Oman. Sultan Qaboos Univ Med J. 14, e306-18. [PMC free article] [PubMed] [Google Scholar]
- 68.IPC-NIB. Guidance document for reporting serious adverse reactions in blood transfusion service. National Institute of Biologicals documents, 2012. Downloaded from www.ipc.gov.in accessed on 15 October 2014.
- 69.CDSCO. Pharmacovigilance Programme for India (PvPI): Assuring Drug Safety. Directorate General of Health Services, Ministry of Health and Family Welfare. Downloaded from http://cdsco.nic.in/pharmacovigilance.htm accessed on 15 October 2014.
- 70.Meslin EM. 1990. Protecting human subjects from harm through improved risk judgments. IRB. 12, 7-10. 10.2307/3563683 [DOI] [PubMed] [Google Scholar]
- 71.King NM, Churchill LR. Assessing and comparing potential benefits and risks of harm, The Oxford Textbook of Clinical Research Ethics. In: Emanuel E, Grady C, Crouch RA, Lie RA, Miller FG, Wendler D editors. New York: Oxford University Press; pp. 2008;514-526. [Google Scholar]
- 72.Brahmachari B, Fernandes M, Bhatt A. 2011. Pharmacovigilance for clinical trials in India: Current practice and areas for reform. Perspect Clin Res. 2, 49-53. 10.4103/2229-3485.80366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garrard E, Dawson A. 2005. What is the role of the research ethics committee? Paternalism, inducements, and harm in research ethics. J Med Ethics. 31, 419-23. 10.1136/jme.2004.010447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Khattri S, Balamuralidhara V, Pramod Kumar TM, Valluru R, Venkatesh MP. 2012. Pharmacovigilance regulations in India: A Step forward. Clin Res Regul Aff. 29, 41-45. 10.3109/10601333.2012.692688 [DOI] [Google Scholar]
- 75.Kalantri SP. 2003. Ethics in clinical research. Indian J Anaesth. 47, 30-32. [Google Scholar]
- 76.Grady C. 2005. Payment of clinical research subjects. J Clin Invest. 115, 1681-87. 10.1172/JCI25694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.ICH. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. Guideline for Good Clinical Practice E6 1996. Available from: http:// www.ich.org/LOB/media/MEDIA482.pdf.
- 78.FDA. Food and Drug Administration. Code of Federal Regulations Title 21. Sec. 312.33 IND Annual Reports 2010. Available from: http://www.accessdata.fda.gov/scripts/cdrh/ cfdocs/cfcfr/CFRSearch.cfm?fr= 312.33.
- 79.EC. European Commission. Detailed Guidance on the Collection, Verification and Presentation of Adverse Reaction Reports Arising from Clinical Trials on Medicinal Products for Human Use 2006. Available from: http://ec.europa.eu/health/files/ eudralex/vol-10/21_ susar_rev2_2006_04_11_en.pdf.
- 80.ICH. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. Final Concept Paper- E2F-Development Safety Update Report 2006. Available from: http://www.ich.org/LOB/media/MEDIA 3302.pdf.
- 81.IOM. Institute of Medicine. Emerging safety science: Workshop summary. Washington, DC: The National Academies Press. Chapter 8: Pharmacovigilance. 2008; pp74–84. [PubMed] [Google Scholar]