

Abstract
Because no drug exists that halts or even slows any neurodegenerative disease, developing effective therapeutics for any prion disorder is urgent. We recently reported two compounds (IND24 and IND81) with the 2-aminothiazole (2-AMT) chemical scaffold that almost doubled the incubation times in scrapie prion-infected, wild-type (wt) FVB mice when given in a liquid diet. Remarkably, oral prophylactic treatment with IND24 beginning 14 days prior to intracerebral prion inoculation extended survival from ∼120 days to over 450 days. In addition to IND24, we evaluated the pharmacokinetics and efficacy of five additional 2-AMTs; one was not followed further because its brain penetration was poor. Of the remaining four new 2-AMTs, IND114338 doubled and IND125 tripled the incubation times of RML-inoculated wt and Tg4053 mice overexpressing wt mouse prion protein (PrP), respectively. Neuropathological examination of the brains from untreated controls showed a widespread deposition of self-propagating, β-sheet-rich “scrapie” isoform (PrPSc) prions accompanied by a profound astrocytic gliosis. In contrast, mice treated with 2-AMTs had lower levels of PrPSc and associated astrocytic gliosis, with each compound resulting in a distinct pattern of deposition. Notably, IND125 prevented both PrPSc accumulation and astrocytic gliosis in the cerebrum. Progressive central nervous system dysfunction in the IND125-treated mice was presumably due to the PrPSc that accumulated in their brainstems. Disappointingly, none of the four new 2-AMTs prolonged the lives of mice expressing a chimeric human/mouse PrP transgene inoculated with Creutzfeldt-Jakob disease prions.
Introduction
As the population ages, neurodegenerative diseases (NDs), including Alzheimer’s (AD) and Parkinson’s disease (PD), are becoming an increasing societal challenge and economic burden. However, there are currently no drugs that can halt or even slow the progressive neurodegeneration that attends both of these illnesses. An impressive convergence of data argues that both AD and PD are caused by pathogenic proteins that adopt alternative conformations and become self-propagating (Prusiner, 2014). These replicating conformers, i.e., prions, spread throughout the brain and produce neuronal dysfunction. In AD and PD, the specific proteins becoming prions differ, but the same fundamental mechanism appears to be operative. This observation has led to a growing consensus that these NDs are all prion disorders.
In developing therapeutics for NDs, we initially focused on the prototypical prion disease, in which the endogenous prion protein (PrP) is converted from a largely α-helical cellular isoform (PrPC) into a self-propagating, β-sheet-rich “scrapie” isoform (PrPSc). Scrapie is the classic PrP prion disease that occurs naturally in sheep and goats. Although the most common human PrP prion disease, Creutzfeldt-Jakob disease (CJD), has an incidence of only one per million per year, PrP prions offer robust cell and animal models and therefore a platform for drug discovery. Moreover, PrP prions occur in multiple “strains,” which differ in their PrPSc conformations as well as their biochemical properties and neuropathological lesions (Bessen and Marsh, 1994; Telling et al., 1996; Collinge and Clarke, 2007). The phenomenon of strains is not limited to PrP prions and has recently been recognized in the more common NDs, both in synthetic preparations (Frost et al., 2009; Guo et al., 2013; Stöhr et al., 2014) and in brains from patients (Clavaguera et al., 2013; Lu et al., 2013b; Watts et al., 2013, 2014; Sanders et al., 2014).
The relative ease of transmissibility of PrP prions has led to the development of multiple rodent models, most notably, with mouse-passaged sheep scrapie. The RML and 22L strains are derived from the same original sheep scrapie pool but with different transmission histories. After multiple passages in sheep, one lineage was passaged through goats and then mice, resulting in the “Chandler isolate,” which was later transferred to the Rocky Mountain Laboratories and has become referred to by the acronym RML. A second lineage underwent additional serial passage through sheep before direct infection into mice, resulting in the 22L strain (Dickinson, 1976; Oelschlegel et al., 2012). The ME7 strain was derived from an independent transmission of sheep scrapie to mice (Zlotnik and Rennie, 1963). These mouse-passaged scrapie strains can be successfully propagated in the mouse CAD5 cell line (Mahal et al., 2007), enabling comparison of efficacy in vitro and in vivo.
Our initial PrP drug discovery studies were based on screening chemical libraries in N2a cells propagating the RML prion strain, where we identified the 2-aminothiazole (2-AMT) scaffold (Ghaemmaghami et al., 2010), which was subsequently optimized by medicinal chemistry complemented by pharmacokinetic studies (Gallardo-Godoy et al., 2011; Li et al., 2013; Silber et al., 2013). Two compounds from this series, IND24 and IND81, were shown to almost double the incubation period of mice inoculated with RML prions (Berry et al., 2013). To date, only two other compounds have been reported to give a similar extension in survival in prion-infected mice: Compound B (Kawasaki et al., 2007; Lu et al., 2013a) and Anle138b (Wagner et al., 2013).
Here, we report on the impact of multiple variables that alter the treatment efficacy of IND24, including the PrP prion strain, dose level and duration, and PrP expression level. We also report on the pharmacokinetics and efficacy of five additional 2-AMTs. Although some of these new 2-AMTs were superior therapeutics to those reported previously, none were capable of extending the survival of mice expressing a chimeric human (Hu)/mouse (Mo) PrP transgene that had been inoculated with CJD prions.
Materials and Methods
Chemical Synthesis.
The synthesis of IND24 has been reported previously (Silber et al., 2013), as have the syntheses for IND114138 and IND126461 (referenced as compounds 15 and 47, respectively) (Li et al., 2013). IND124, IND125, and IND126 were synthesized using the Hantzsch-type condensation reaction of bromomethyl ketones with thioureas. Specifically, 4-(1,3,-oxazol-5-yl)aniline was converted to the corresponding thiourea by reaction with benzoyl isothiocyanate, followed by hydrolysis in refluxing 5 N NaOH. The resulting thiourea was then reacted with the appropriate bromomethyl ketones. Detailed synthesis and characterization of these samples is included in the Supplemental Material. For efficacy studies, compounds were scaled up to 10–100 g by Chemveda (Hyderabad, India) or Sundia Ltd. (Shanghai, China).
In Vivo Studies.
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco. FVB and CD-1 mice were purchased from Charles River Laboratories (Hollister, CA). All other mice were bred in-house. Mice were 6–14 weeks old and were maintained on alternating 12-hour light/dark cycles, with free access to food and water.
Single-dose pharmacokinetic studies were performed by oral gavage in female CD-1 mice, except for IND114338 and IND124, which were performed in female FVB mice. Compounds were dosed at 10 mg/kg dissolved in 20% propylene glycol, 5% labrosol, 5% ethanol, and 70% polyethylene glycol 400 (PEG400), except IND126, which was dosed in dimethylsulfoxide delivered in a 200-µl bolus through a 22-gauge gavage needle. At specified time points after dosing (0.5, 1, 2, 4, and 6 hours), mice were euthanized by CO2, and brain and plasma samples were collected and stored at −80°C until analysis. For IND24 and IND124, samples were also collected at 0.25 and 24 hours.
Dosing for 3 days or longer was performed via a complete liquid diet as previously reported (Lu et al., 2013a). Briefly, compounds dissolved in excipient were blended with diet F1256SP and chocolate powder (BioServ, Frenchtown, NJ). For vehicle controls, an equivalent amount of excipient was used. Dosing levels of the compound were calculated based on prior studies that determined a 25-g mouse consumes an ∼20 ml liquid diet per day. The amount of liquid diet served was based on the number of mice in each cage (up to three) and was replaced with a freshly prepared batch every 2–4 days. All 3-day feeding studies were performed in female CD-1 mice. Efficacy studies were performed in FVB, FVB-Tg(Gfap-luc)+/− (Zhu et al., 2004), and FVB-Tg(Gfap-luc)+/+ (Stöhr et al., 2012) mice, which all express wild-type (wt) levels of MoPrP and are not further differentiated in the text; FVB-Tg(MoPrP)4053 mice overexpressing mouse PrP ∼4-fold (Carlson et al., 1994); or Tg(MHu2M,M111V,M165V,E167Q)1014 Prnp0/0 mice expressing chimeric Hu/Mo PrP (Giles et al., 2010). Female mice were used unless otherwise stated and were generally housed two to four per cage unless a health issue or fighting required them to be singly housed.
Quantification of Compound Levels in Tissue.
Twenty percent (w/v) brain homogenates were prepared in water by one 30-second cycle of bead beating at 5500 rpm with a Precellys 24 tissue homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France), after which tubes were placed on ice. Compounds were recovered by mixing equal parts of brain homogenate with a 50/50 (v/v) solution of water and methanol. This mixture was diluted 4-fold in acetonitrile with 0.1% formic acid. After centrifugation at 3000g at 4°C for 10 minutes, the supernatant was diluted 4-fold in water with 0.1% formic acid.
Analysis was performed using a liquid chromatography–tandem mass spectrometry system consisting of an API4000 triple quadrapole instrument (AB Sciex, Foster City, CA) interfaced with a CBM-20A controller, LC20AD 230 pumps, and a SIL-5000 auto sampler (Shimadzu Scientific, Columbia, MD). Samples were injected onto a BDS Hypersil C8 column maintained at room temperature. The amount of acetonitrile (ACN) in the gradient was increased from 75–95% ACN over 2.0 minutes, held for 1.0 minute, and then re-equilibrated to 75% ACN over 1.4 minutes. Data acquisition used multiple reaction monitoring in the positive ion mode. Specific methods were developed for each compound, enabling the determination of absolute concentrations.
Pharmacokinetic parameters, including Cmax, tmax, AUClast (the area under the concentration–time curve to the last measured point), and t1/2 were calculated with the PKSolver 2.0 plug-in for Microsoft Excel (Redmond, WA) (Zhang et al., 2010).
Prion Strains and Inoculation.
The RML and ME7 strains have been serially passaged in CD-1 mice. The 22L strain was a generous gift from Dr. Sue Priola, Rocky Mountain Laboratories, and was passaged in FVB mice prior to the experiments reported here. All RML inocula were derived from a single pool of 50 brains, which were homogenized, aliquoted, and stored at –80°C. We have previously shown that aliquots of this preparation result in highly reproducible incubation periods (coefficient of variance of 5%) over years of independent experiments (Tamgüney et al., 2008). Two independent preparations of sporadic CJD(MM1) [sCJD(MM1)] prions were used, both of which were derived from a single previously reported case (Telling et al., 1994), that contained no mutations in the PrP sequence. Eight- to ten-week-old mice were inoculated intracerebrally into the right parietal lobe in the vicinity of the thalamus, with 30 µl of a 1% brain homogenate in a filtered solution of phosphate-buffered saline with 5% (w/v) bovine serum albumin. Mice were monitored daily and screened twice a week for neurologic dysfunction based on standard diagnostic criteria for prion disease (Carlson et al., 1986). Mice with prion disease were euthanized by CO2, and their brains were collected and frozen at −80°C or fixed in 10% buffered formalin.
Cell Culture.
CAD5 cells were infected with RML, 22L, or ME7 prions, as previously described (Butler et al., 1988), maintained in Opti-MEM supplemented with 90 units of penicillin-streptomycin and 9% fetal bovine serum at 37°C in 100-mm plates, and fed with fresh media every 2–3 days. Dividing cells were plated at 10% confluency and split 1:10 when they became confluent. Approximately 4 × 106 cells (∼40% confluency) at 10–20 passages postinfection were incubated with IND24 at the doses indicated over 5 days with refeeding, by which time they had reached confluency. IND24 was diluted in dimethylsulfoxide, with 50 µl of the final volume in 10 ml of media. At the end of the 5-day treatment period, lysis buffer (100 mM Tris⋅HCl, pH 8.0; 150 mM NaCl; 0.5% Nonidet P-40; and 0.5% sodium deoxycholate) was added to the cells, and protein concentrations were measured using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, MA). Protein extracts were normalized to 1 mg/ml total protein with lysis buffer before proteinase K (PK) digestion. Lysates were digested with 20 µg/ml PK at 37°C for 1 hour. The reaction was stopped with phenylmethylsulfonyl fluoride (2 mM final concentration), and samples were ultracentrifuged at 100,000g for 1 hour, resuspended in loading buffer, and subjected to immunoblot analysis.
Immunoblot Analysis.
Detergent-extracted brain homogenates were subjected to the same quantification and digestion process as described for the cell lysates above, but instead of ultracentrifugation, 4× loading buffer was added to the digested sample. Brain homogenate or cell lysates were heated to 100°C for 10 minutes and run in a 4–12% Tris⋅glycine SDS gel (Invitrogen, Carlsbad, CA). The gel was transferred to a polyvinyl difluoride membrane using an iBlot system (Invitrogen), and the membrane was blocked with 5% milk for 30–60 minutes. The membranes were subsequently incubated overnight with horseradish peroxidase–conjugated human-mouse chimeric (HuM) D13 or HuM-P Fabs (Williamson et al., 1998) and washed 3× with Tris-buffered saline with Tween 20 for 10 minutes before developing with an enhanced chemiluminescence reagent (Invitrogen).
Statistical Analysis of Glycoform Intensities.
PK-digested control and treated samples were run on the same gel, and isoform band intensities were quantified using ImageJ (http://imagej.nih.gov/ij). A statistical analysis was performed using Prism 6 (GraphPad Software, Inc., La Jolla, CA), and the differences among the groups were assessed by Student’s t test without correction for multiple comparisons using a significance threshold of P < 0.05.
Neuropathological Analysis.
Formalin-fixed brains were coarsely cut into four pieces using a slicing matrix and then embedded into a single paraffin block. Coronal sections (of 8 µm) were cut using a microtome at the following bregma coordinates: 1) 0.86 mm, 2) −1.82 mm, 3) −3.52 mm, and 4) −5.40 mm. Brain sections were then mounted on frosted glass slides. These were then deparaffinized and processed for immunohistochemistry or stained with hematoxylin and eosin. Endogenous tissue peroxidases were inhibited by incubating the slides in a 3% hydrogen peroxide solution in methanol for 30 minutes. Slides were then blocked with 10% (v/v) normal goat serum for 1 hour and then incubated with a primary antibody overnight at 4°C. For detection of astrocytic gliosis, the anti-GFAP rabbit polyclonal antibody Z0334 (Dako, Carpiteria, CA) was used at 1:500 dilution. To detect PrPSc deposition, sections were subjected to hydrolytic autoclaving (121°C for 10 minutes in citrate buffer), prior to blocking, and anti-PrP HuM Fab R2 (Williamson et al., 1998) was used at a concentration of 4 µg/ml. Secondary antibodies of biotinylated goat anti-rabbit and goat anti-human (Vector Laboratories, Burlingame, CA), respectively, were used at a 1:250 dilution in 10% goat serum and incubated at room temperature for 30 minutes. The bound antibody was detected using a Vectastain ABC peroxidase kit (Vector Laboratories) and visualized using 3-3′-diaminobenzidine (DAB). Slides were counterstained with hematoxylin.
GFAP Quantification.
Brightfield images of DAB-labeled GFAP immunohistochemistry on brain slices were obtained with the Axioscan.Z1 slide scanning system (Carl Zeiss, Oberkochen, Germany) using a 20× objective lens and standardized acquisition settings. All images were processed and analyzed using ImageJ software. In each RGB color image, the DAB-GFAP signal was isolated from the hematoxylin nuclear counterstain using the Color Deconvolution plug-in. Images were converted to 8-bit grayscale and thresholded with a standard value prior to quantification. Brain regions of interest (ROIs) were selected using the polygon tool to trace the boundaries of a given ROI using neuroanatomical features as a guide in the raw RGB color images. The ROI selection was then copied to the corresponding thresholded 8-bit image, and the percentage of the total area occupied by GFAP staining in a given brain ROI was measured.
Results
Efficacy of IND24 Was Prion Strain Dependent.
Previous studies on two independent mouse-passaged scrapie strains, RML and ME7, showed a similar survival when dosing was started immediately after inoculation, but differed when dosing was started 60 days after inoculation (Berry et al., 2013). We therefore decided to test a third mouse-passaged scrapie strain, 22L, which was derived from the same scrapie sheep brain pool as RML but had a different passaging history. Treating CAD5 cells infected with RML, ME7, or 22L with different concentrations of IND24 led to strikingly different results. Whereas 2 µM IND24 treatment for 5 days reduced PrPSc in RML-infected cells, it had little effect on the ME7 or 22L strains (Fig. 1A). Following IND24 treatment at 10 µM, the PrPSc signal from ME7-infected cells was reduced, but 22L-infected cells appeared largely unchanged (Fig. 1A).
Fig. 1.

Relationship of IND24 dosing schedule to prion strains and survival. (A) Efficacy of IND24 in cell culture. CAD5 cells infected with RML, ME7, or 22L prions, as indicated, were treated with IND24 at 2 or 10 µM for 5 days, and levels of PK-resistant PrP were determined by immunoblot. PrP was detected using the chimeric Fab D13. RML-infected cells were at passage 14, and ME7- and 22L-infected cells were at passage 10. Molecular weight markers of migrated protein standards represent 20 and 30 kDa. (B) Efficacy of IND24, expressed as the survival index, for mice expressing wild-type levels of PrP inoculated with RML (circles), ME7 (triangles), or 22L (squares) prion strains. IND24 intervention at 210 mg/kg per day was initiated at different time points, which are shown as the relative percentage of the incubation period for each prion strain. Error bars represent S.E.M. (C) Mean incubation periods in wt mice inoculated with RML prions with different IND24 dosing regimens. IND24 dosing shown in gray; no treatment indicated in white. Intermittent dosing is more effective than continuous dosing initiated at 1 dpi; however, continuous dosing prophylactically produces a dramatic increase in survival extension. Error bars represent S.E.M. (D) Efficacy expressed as in (B) but in Tg4053 mice overexpressing PrP inoculated with the RML prion strain.
Time of Intervention with IND24 Impacted Survival Outcome.
When IND24 dosing was initiated at ∼60 days postinoculation (dpi) in wt FVB mice inoculated with RML prions, the survival extension was the same as that achieved when treatment was started the day after inoculation. In contrast, survival of the mice inoculated with the ME7 strain was reduced when treatment with IND24 was introduced at 60 dpi (Berry et al., 2013). To investigate this phenomenon further, we started dosing with IND24 at various points throughout disease progression.
Because prion strains have different incubation periods, we normalized incubation periods of compound-treated mice to vehicle-treated mice to enable a standardized comparison among the experiments and labeled this the “survival index” (Berry et al., 2013). As with the survival index, the time at which treatment was initiated can be expressed as the percentage of time elapsed relative to the incubation period of untreated mice. For example, treatment beginning at 60 dpi of a 118-day incubation period represents an intervention time of 51%. Plotting this value against the survival index provides a visual relationship between these two factors across different prion strains (Fig. 1B).
Despite the limited efficacy in cell culture, dosing 22L-infected mice with IND24 at 210 mg/kg per day starting the day after inoculation still produced a survival index of ∼150. However, starting dosing at ∼50% of the incubation period reduced the survival index to 114 (Fig. 1B; Table 1). The reduced efficacy was analogous to mice inoculated with ME7 prions but differed substantially from that of mice inoculated with RML prions.
TABLE 1.
Efficacy of 210 mg/kg per day of IND24 given to prion-infected wt and Tg4053 mice at various times before and after inoculation
| Inoculum | PrP Expression Level | Dosing Interventiona | Mean Incubation Period ± S.E.M. | n/n0 | Survival Index ± S.E.M. |
|---|---|---|---|---|---|
| dpi | days | ||||
| RML | wt | None | 118 ± 1b | 12/12 | 100c |
| 1 > | 204 ± 5b | 12/12 | 173 ± 4 | ||
| 34 > | 202 ± 2 | 12/12 | 171 ± 2 | ||
| 46 > | 197 ± 1 | 6/6 | 167 ± 2 | ||
| 60 > | 211 ± 1b | 6/6 | 179 ± 1 | ||
| 61 > | 221 ± 1 | 12/12 | 187 ± 2 | ||
| 74 > | 192 ± 14 | 5/5 | 163 ± 12 | ||
| 75 > | 162 ± 3 | 6/6 | 137 ± 3 | ||
| 77 > | 208 ± 13 | 9/9 | 176 ± 11 | ||
| 90 > | 118 ± 6 | 12/12 | 100 ± 5 | ||
| 1–61, 124–187, 243 > | 239 ± 7 | 8/8 | 203 ± 6 | ||
| 61–124 | 163 ± 6 | 7/7 | 138 ± 5 | ||
| −14 > | 452 ± 18 | 7/7 | 383 ± 16 | ||
| ∼4× | None | 51 ± 3b | 8/8 | 100c | |
| 1 > | 112 ± 4b | 9/9 | 220 ± 14 | ||
| 15 > | 122 ± 7 | 8/8 | 239 ± 20 | ||
| 26 > | 156 ± 3 | 8/8 | 306 ± 19 | ||
| 40 > | 64 ± 11 | 5/5 | 125 ± 23 | ||
| ME7 | wt | None | 126 ± 2b | 5/5 | 100c |
| 1 > | 214 ± 4b | 9/9 | 170 ± 4 | ||
| 60 > | 158 ± 5b | 7/7 | 125 ± 5 | ||
| 22L | wt | None | 111 ± 3 | 8/8 | 100c |
| 1 > | 164 ± 1 | 8/8 | 148 ± 4 | ||
| 60 > | 126 ± 2 | 8/8 | 114 ± 4 |
n, number of ill mice; n0, number of inoculated mice.
Day treatment started or range; > indicates dosing continued until the end of the experiment.
Data previously reported (Berry et al., 2013).
Vehicle-treated control used to calculate survival index.
FVB mice were inoculated with the RML prion strain, and dosing with IND24 at 210 mg/kg per day was started at 34, 46, ∼60, ∼75, and 90 dpi or intervention times of ∼30–75%. Intervention with IND24 at any point up to ∼50% gave a survival index of ∼175. However, when the dosing intervention was initiated at ∼60%, there was greater variability both among and within experiments, with incubation periods of 162 ± 3, 192 ± 14, and 208 ± 13 dpi from three independent experiments (Table 1). This represents a greater variability than that seen in experiments in which RML-infected mice were treated with vehicle or IND24 from day 1 (Supplemental Fig. 1, A and B; Supplemental Table 1). Starting IND24 dosing at 75% of the incubation period gave no extension in survival compared with the untreated control (Table 1); however, variability was again higher, with some animals showing signs of central nervous system dysfunction soon after starting treatment (90 dpi) and others surviving more than 145 dpi (Supplemental Fig. 1C). We observed a similar phenomenon of mice appearing sick within days of starting treatment of late intervention with Compound B (Lu et al., 2013a).
Intermittent Dosing Extended Survival.
Because initiating IND24 dosing at 60 dpi did not appear to reduce the survival index, we started experiments in which dosing and vehicle were alternated in ∼60-day intervals. Dosing with IND24 from 1 to ∼60 dpi and ∼120 to 180 dpi resulted in a survival index of 203 ± 6, whereas dosing at ∼60–120 dpi resulted in all mice succumbing to disease before dosing could be initiated again at 180 dpi (Fig. 1C; Table 1).
Prolonged Survival after Prophylactic Dosing.
A more dramatic extension in survival was observed when the administration of IND24 was started 14 days before intracerebral inoculation of RML prions. The continuous administration of IND24 produced an extension of the incubation time to over 450 days, corresponding to a survival index of 383 ± 16 (Fig. 1C; Table 1).
PrP Overexpression Altered Survival.
To determine the relationship between survival time and intervention in mice overexpressing MoPrP, Tg4053 mice were inoculated with RML prions, and dosing with IND24 was initiated at 1, 15, 26, or 40 dpi. When dosing was initiated at 1 dpi, the survival index was 220 ± 14 in Tg4053 compared with 173 ± 4 in wt mice (Table 1) (Berry et al., 2013; Lu et al., 2013a). Surprisingly, initiating dosing later in the disease progression appeared to increase the survival index: beginning IND24 at 50% of the incubation period resulted in a survival index of 306 ± 19 (Fig. 1D; Table 1).
IND24 Brain Concentration Depended on Dose and Duration of Exposure.
For compounds with short half-lives, oral dosing led to variable levels in the brain (Lu et al., 2013a). We therefore determined the half-life (t1/2) of IND24 in brain and plasma following a single oral gavage dose at 10 mg/kg. Brain t1/2 was found to be 5.5 hours, and plasma t1/2 was 4.5 hours (Supplemental Fig. 2). Mice feed predominantly at night, but even during the light cycle, the interval between feeding bouts rarely exceeds 3 hours (Goulding et al., 2008), suggesting that under normal conditions, the brain concentration of IND24 is unlikely to fluctuate widely.
To determine the relationship between dose and brain concentration, cohorts of three mice were fed with various amounts of IND24 for 3 days. Mice fed 1 or 5 mg/kg per day had 0.025 and 0.46 µM IND24 in their brains, respectively, whereas doses of 125 mg/kg per day or more resulted in brain concentrations of >10 µM. There was a direct linear correlation between dose and the concentration in the brain after 3 days of feeding (Fig. 2A; R2 = 0.84; P = 0.0013). The brain concentration of IND24 was also determined after longer-term continuous dosing in RML-infected mice. Mice dosed at 210 mg/kg per day for 10–225 days had IND24 brain concentrations of ∼10 µM (Fig. 2B). Dosing at 1 or 5 mg/kg per day for ∼75 days showed a marked escalation of the brain concentrations, suggesting that IND24 accumulated in the brain over time (Fig. 2B).
Fig. 2.
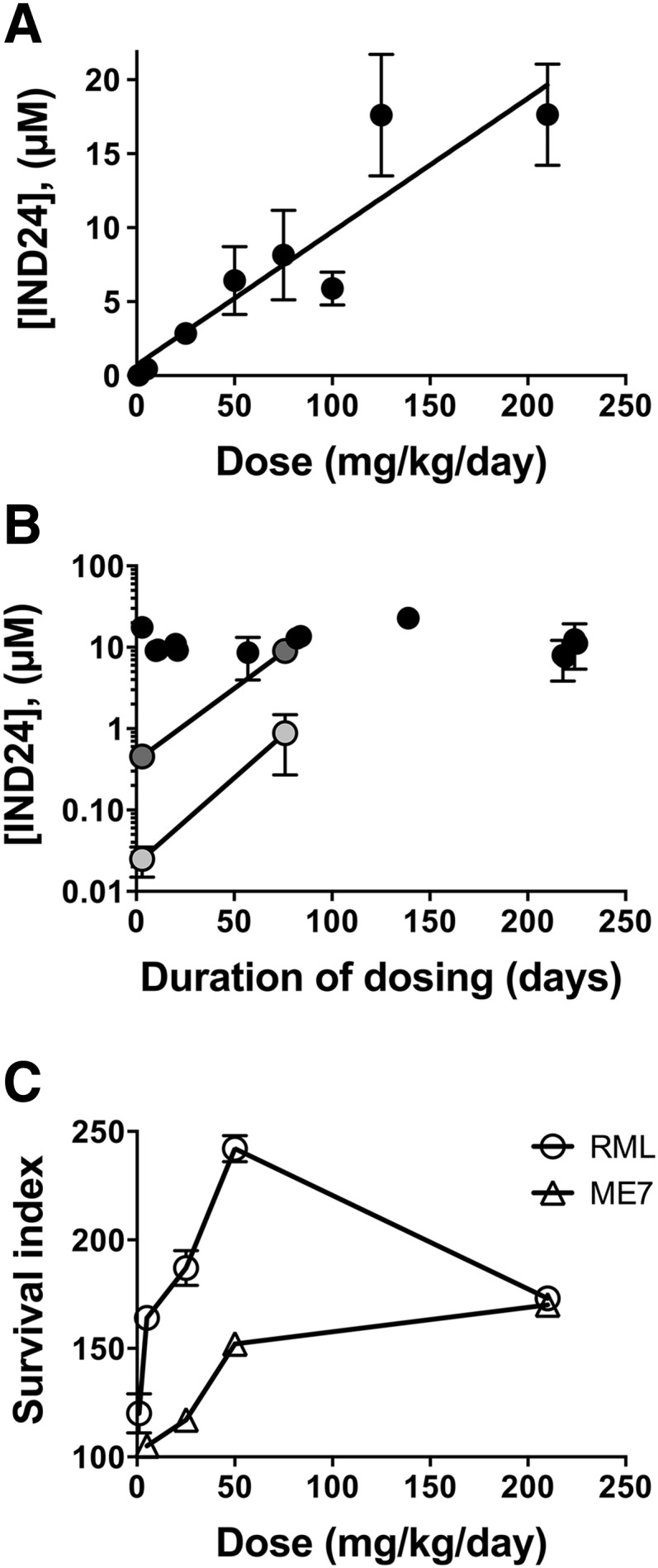
Relationship of IND24 dose to brain concentration and survival. (A) Brain concentration of IND24 was determined from groups of three mice fed a complete liquid diet containing various amounts of IND24 for 3 days; each point represents up to 23 independent replicates. A strong direct correlation (R2 = 0.84) was observed between the amount administered and that reaching the brain. Error bars represent S.D. (B) Brain levels of IND24 with long-term dosing at 210 mg/kg per day (black), 5 mg/kg per day (gray), and 1 mg/kg per day (light gray). Marked dose accumulation was observed at 1 and 5 mg/kg per day. Error bars represent S.D. (C) The relationship between dose and survival was not linear for wt mice inoculated with RML (circles) or ME7 (triangles) prions. Error bars represent S.E.M.
IND24 Dose and Survival Times.
To determine whether the dose correlated with an extension in survival, groups of wt mice were inoculated with RML prions and administered IND24 at 1, 5, 25, 50, or 210 mg/kg per day in a complete liquid diet starting the day after inoculation. The lowest dose of 1 mg/kg per day IND24 gave a survival index of 120 or a 20% extension in survival over uninoculated controls (Fig. 2C; Table 2). A dose of 5 mg/kg per day IND24 resulted in a survival index of 164 ± 2 compared with 110 ± 4 at the same dose administered beginning at 60 dpi. The survival index increased steadily from the 50 mg/kg per day dose, which resulted in a survival index of ∼240 (Table 2). At 210 mg/kg per day, the survival index was ∼170 (Supplemental Table 1; Table 1).
TABLE 2.
Efficacy of IND24 given at increasing doses to prion-infected mice
| Inoculum | PrP Expression Level | Dose | Dosing Interventiona | Mean Incubation Period ± S.E.M. | n/n0 | Survival Index ± S.E.M. |
|---|---|---|---|---|---|---|
| mg/kg per day | dpi | days | ||||
| RML | wt | None | None | 118 ± 1b | 12/12 | 100c |
| 1 | 1 > | 142 ± 10 | 3/3 | 120 ± 9 | ||
| 5 | 1 > | 193 ± 2 | 3/3 | 164 ± 2 | ||
| 5 | 60 > | 130 ± 5 | 9/9 | 110 ± 4 | ||
| 25 | 1 > | 221 ± 9 | 5/5 | 187 ± 8 | ||
| 50 | 1 > | 286 ± 7 | 8/8 | 242 ± 6 | ||
| ME7 | wt | None | None | 126 ± 2b | 5/5 | 100c |
| 5 | 1 > | 132 ± 3 | 6/6 | 105 ± 3 | ||
| 25 | 1 > | 148 ± 4 | 7/7 | 117 ± 4 | ||
| 50 | 1 > | 192 ± 4 | 6/6 | 152 ± 4 |
n, number of ill mice; n0, number of inoculated mice.
Day treatment started; > indicates dosing continued until the end of the experiment.
Data previously reported (Berry et al., 2013).
Vehicle-treated control used to calculate survival index.
We next examined different doses of IND24 on wt mice inoculated with the ME7 strain. Although dosing at 210 mg/kg per day gave a similar survival index to RML-infected mice, administering IND24 to ME7-infected mice at lower doses was less efficacious than in RML-infected mice (Fig. 2C; Table 2).
New 2-AMTs Exhibited Good Brain Penetration.
To extend our studies of 2-AMTs, we determined the pharmacokinetics for compounds from three structural subgroups (Fig. 3). IND114338 (1) is a close analog of IND24, differing only by replacing the terminal benzene ring with a 3-pyridyl group (Li et al., 2013) (compound #15). Hybrids of the best AMT A/B-ring chemotypes were combined with phenyloxazole C-ring (IND124–IND126; 2–4). IND124 is the 2-AMT analog of the biaryl hydrazone Compound B (Kawasaki et al., 2007). We also tested the most potent compound from a series of 2-AMTs with a modified amide C-ring, IND126461 (5) (Li et al., 2013) (compound #47).
Fig. 3.

Structures of novel 2-AMTs from three structural subgroups. IND24 analog IND114338 (1); compounds with a phenyloxazole C-ring, including the 2-AMT analog of Compound B, IND124 (2), and hybrids with other A/B-ring chemotypes, IND125 (3) and IND126 (4); and a compound with a modified C-ring, IND126461 (5). Synthesis and initial PK parameters of (1) and (5) were reported previously (Li et al., 2013).
Using PEG400 as the excipient, we previously reported that chronic dosing in male mice led to mortality after ∼3–4 months, even when the excipient concentration was reduced 10-fold (Lu et al., 2013a) (Supplemental Table 2) and therefore sought an alternative excipient. Brain concentration for IND114338 after 3-day dosing (C3day) at 210 mg/kg per day in a complete liquid diet using 0.5% carboxy methyl cellulose (CMC) as the excipient was 2.9 µM, which is more than 40 times the cellular EC50 (Table 3). IND124, IND125, and IND126 all had good brain exposure after oral gavage, yielding micromolar Cmax values; however, the C3day to EC50 ratios for IND124, IND125, and IND126 varied greatly at 0.04, 860, and 0.2, respectively. Dosing IND126461 at 200 mg/kg per day for 3 days yielded a brain concentration of 0.1 µM, which is 1.4 times the cellular EC50.
TABLE 3.
Pharmacokinetic parameters of five novel 2-AMTs
| Compound | EC50 | Tissue | Oral Gavage, 10 mg/kg | 3-Day Feeding | ||||
|---|---|---|---|---|---|---|---|---|
| Cmax | tmax | AUClast | C3day | Dose | Excipient | |||
| µM | µM | hours | µM⋅h | µM | ||||
| IND114338 | 0.068a | Brain | 2.5a | 2 | 10.9a | 0.15 | 25 | 0.5% CMC |
| 2.9 | 210 | 0.5% CMC | ||||||
| 13.3 | 200 | 0.125% PEG400 | ||||||
| Plasma | 4.1 | 2 | 15.6a | 0.24 | 25 | 0.5% CMC | ||
| 3.1 | 210 | 0.5% CMC | ||||||
| 4.7 | 200 | 0.125% PEG400 | ||||||
| IND124 | 0.124 | Brain | 2.8 | 0.25 | 3.8 | 0.005 | 200 | 0.125% PEG400 |
| Plasma | 1.2 | 0.25 | 1.4 | 0.003 | 200 | 0.125% PEG400 | ||
| IND125 | 0.057 | Brain | 1.1 | 4.0 | 4.3 | 49 | 200 | 0.125% PEG400 |
| Plasma | 0.8 | 4.0 | 2.9 | 6.0 | 200 | 0.125% PEG400 | ||
| IND126 | 0.228 | Brain | 3.6 | 4.0 | 4.3 | 0.04 | 200 | 0.125% PEG400 |
| Plasma | 0.3 | 4.0 | 0.5 | 0.02 | 200 | 0.125% PEG400 | ||
| IND126461 | 0.070a | Brain | 0.1a | 0.5 | 0.2a | 0.10 | 200 | 0.125% PEG400 |
| Plasma | 0.1 | 4 | 0.4a | 0.20 | 200 | 0.125% PEG400 | ||
Data previously reported (Li et al., 2013).
The relationship between cellular EC50 and the required concentrations in the brain needed to achieve efficacy in animal models of prion disease is not well defined. Because IND24 resulted in a substantial extension of survival when administered at 5 mg/kg per day, where the C3day to EC50 ratio was 0.3, we selected IND114338, IND125, IND126, and IND126461 for efficacy studies. Because the ratio for IND124 was so low, suggesting that transit through the blood-brain barrier was likely to be poor, we did not determine the efficacy of this compound.
Novel AMT Analogs Extended Survival Up to 3-Fold.
IND114338 was dosed in 0.5% CMC at either 25 or 200 mg/kg per day in wt FVB mice inoculated with RML prions. Although vehicle containing 0.5% CMC was not toxic to male or female mice when administered for 220 days in uninoculated mice, the incubation periods in male prion-infected mice were ∼20% shorter than those in female mice (Supplemental Table 2). Small sex differences previously reported for prion-infected mice have often been contradictory and were generally less than 10% of the incubation period (Akhtar et al., 2011). For comparative purposes, we therefore only used data on female mice.
The survival extension in mice treated with IND114338 was greater with an increasing dose, with survival indices of 122 ± 11 and 197 ± 17 following treatment with 25 and 200 mg/kg per day, respectively (Table 4). Due to the problems encountered with CMC, subsequent studies were performed with 0.125% PEG400 as the excipient. IND125 and IND126 were dosed at 200 mg/kg per day in Tg4053 mice inoculated with RML prions. Although IND126 only marginally extended survival, with a survival index of 120 ± 9, treatment with IND125 tripled the lifespan of RML-infected mice (Table 4). RML-inoculated Tg4053 mice dosed with IND126461 at 200 mg/kg per day exhibited a survival index of 133 ± 10 (Table 4).
TABLE 4.
Efficacy of four novel 2-AMTs in scrapie and CJD prion-infected mice
| Mouse Line (PrP Sequence) | PrP Expression Level | Inoculum | Treatment | Dose | Excipient | Mean Incubation Period ± S.E.M. | n/n0 | Survival Index ± S.E.M. |
|---|---|---|---|---|---|---|---|---|
| mg/kg per day | days | |||||||
| FVB (mouse) | wt | RML | None | — | 0.5% CMC | 115 ± 2 | 4/4 | 100b |
| IND114338 | 25 | 0.5% CMC | 140 ± 12 | 4/4 | 122 ± 11 | |||
| 200 | 0.5% CMC | 227 ± 19 | 4/4 | 197 ± 17 | ||||
| None | — | 1.25% PEG400 | 118 ± 1a | 12/12 | 100b | |||
| IND24 | 210 | 1.25% PEG400 | 204 ± 5a | 12/12 | 173 ± 4 | |||
| IND125 | 200 | 0.125% PEG400 | 188 ± 4 | 8/8 | 159 ± 4 | |||
| Tg4053 (mouse) | ∼4× | RML | None | — | 0.125% PEG400 | 51 ± 3a | 8/8 | 100b |
| IND24 | 210 | 0.125% PEG400 | 112 ± 4a | 9/9 | 220 ± 14 | |||
| IND125 | 200 | 0.125% PEG400 | 154 ± 5 | 7/7 | 302 ± 20 | |||
| IND126 | 200 | 0.125% PEG400 | 61 ± 3 | 6/6 | 120 ± 9 | |||
| IND126461 | 200 | 0.125% PEG400 | 68 ± 3 | 6/6 | 133 ± 10 | |||
| Anle138b | 200 | 0.125% PEG400 | 125 ± 4 | 5/5 | 245 ± 16 | |||
| Tg1014 (chimeric human/ mouse) | ∼4× | sCJD(MM1) Preparation A | None | — | 0.125% PEG400 | 78 ± 1a | 7/7 | 100b |
| IND114338 | 200 | 0.125% PEG400 | 76 ± 2 | 8/8 | 97 ± 3 | |||
| IND125 | 200 | 0.125% PEG400 | 78 ± 3 | 7/7 | 100 ± 4 | |||
| IND126461 | 200 | 0.125% PEG400 | 78 ± 3 | 7/7 | 100 ± 4 | |||
| Anle138b | 200 | 0.125% PEG400 | 76 ± 1 | 6/6 | 97 ± 2 | |||
| sCJD(MM1) Preparation B | None | — | 0.125% PEG400 | 91 ± 1 | 8/8 | 100b | ||
| IND126 | 200 | 0.125% PEG400 | 86 ± 1 | 8/8 | 95 ± 2 |
n, number of ill mice; n0, number of inoculated mice.
Data previously reported (Berry et al., 2013).
Vehicle-treated control used to calculate survival index.
We previously reported a significantly reduced monoglycosylated/diglycosylated glycoform ratio of protease-resistant PrP upon treatment with IND24 (Berry et al., 2013). Treatment of wt mice with IND114338 led to a small but significant increase in monoglycosylated/diglycosylated PrPSc (Supplemental Fig. 3B). Tg4053 mice treated with IND125, IND126, and IND126256 did not have significantly different monoglycosylated/diglycosylated protease-resistant PrP ratios compared with vehicle-treated mice (Supplemental Fig. 3, D–F). However, in contrast to IND24 treatment of mice expressing wt levels of PrP (Berry et al., 2013) (Supplemental Fig. 3A), RML-infected Tg4053 mice treated with IND24 did not show a significantly different monoglycosylated/diglycosylated PrPSc ratio (Supplemental Fig. 3C).
2-AMTs Altered Patterns of PrPSc Deposition and Neuropathological Lesions.
IND24 treatment of Tg4053 mice resulted in a markedly different distribution of PrPSc from vehicle-treated mice (Fig. 4, B and C). In RML-infected Tg4053 mice treated with vehicle alone, PrPSc staining was intense throughout the neocortex, hippocampus, thalamus, and dorsal midbrain. Following IND24 treatment, PrPSc was less intense in the neocortex, and a lower degree of staining was observed throughout the thalamus, midbrain, and cerebellum. In all cases, PrP deposition was accompanied by astrocytic gliosis and vacuolation (Fig. 5, A and B); however, IND24 treatment led to an ∼3-fold reduction in GFAP immunoreactivity compared with vehicle-treated controls(Fig. 6, A and B).
Fig. 4.

Neuropathological characterization of brains from Tg4053 mice treated with 2-AMTs. (A) Schematic sagittal view (top) of a mouse brain, with the location of the four coronal sections (bottom) indicated by dotted lines. BF, basal forebrain; Cd, caudate nucleus; Hp, hippocampus; Hth, hypothalamus; Mb, midbrain; Nc, neocortex; OB, olfactory bulb; Th, thalamus. Relative PrPSc intensities shown as high (red), medium (orange), and low (green) in four coronal sections following treatment with (B) vehicle (n = 5), (C) IND24 (n = 7), (D) IND125 (n = 6), (E) IND126 (n = 7), and (F) IND126461 (n = 6). In each of the treatments with IND126 and IND126461, three mice had substantially less PrPSc staining than the rest and were not included in the average distribution calculations.
Fig. 5.

Representative immunohistochemical staining in the thalamus and pons for PrP and GFAP, together with hematoxylin and eosin staining, following treatment with (A) vehicle, (B) IND24, (C) IND125, (D) IND126, and (E) IND126461. The level of GFAP staining and vacuolation are closely correlated with PrPSc deposition. Most notably, in the thalamus of IND125-treated mice, PrPSc and GFAP staining were minimal, and there was no vacuolation. The scale bar in the bottom right panel represents 50 µm and applies to all of the micrographs. PrP was stained with HuM Fab R2 antibody, and GFAP was stained with the anti-GFAP rabbit polyclonal antibody Z0334.
Fig. 6.

Quantification of GFAP staining in the brains of 2-AMT–treated Tg4053 mice. GFAP immunohistochemistry in representative coronal sections shown for the neocortex (Nc), hippocampus (Hp), and thalamus (Th) (upper panels) and cerebellum (Cb) and pons (Po) (middle panels) as well as quantification of GFAP staining in mice treated with (A) vehicle (n = 5, Nc/Hp/Th; n = 6, Po/Cb); (B) IND24 (n = 9, Nc/Hp/Th; n = 8, Po/Cb); (C) IND125 (n = 4, Nc/Hp/Th; n = 7, Po/Cb); (D) IND126 (n = 7, all regions); and (E) IND126461 (n = 6, Nc/Hp/Th; n = 3, Po/Cb). Error bars represent S.D. Note that graph scales in (B–E) are magnified approximately 3-fold compared with (A).
Other 2-AMT treatments also resulted in distinct patterns of PrPSc distribution (Fig. 4, D–F). Remarkably, in IND125-treated mice, which survived 3-fold longer than controls, PrPSc deposition was essentially absent throughout the cortex, hippocampus, and thalamus (Fig. 4D), which was accompanied by minimal astrocytic gliosis and no noticeable vacuolation (Fig. 5C). However, IND125-treated mice displayed robust PrPSc staining in the brainstem (Fig. 4D; Fig. 5C). Quantification of GFAP immunohistochemistry in IND125-treated mice showed heavier staining in the pons (Fig. 6C). Although treatment with IND126 and IND126461 led to a modest extension in survival, both had a dramatically lower PrPSc deposition than vehicle-treated controls throughout much of the brain, but robust staining in the caudate nucleus/striatum (Fig. 4, E and F) and moderate levels of astrocytic gliosis and vacuolation (Fig. 5, D and E; Fig. 6, D and E).
2-AMTs Were Not Efficacious for CJD Prions.
Compounds efficacious against mouse-passaged sheep scrapie prions were unable to extend the lives of Tg mice inoculated with CJD prions in human brain homogenates (Berry et al., 2013; Lu et al., 2013a). We inoculated Tg1014 mice expressing a chimeric Hu/Mo PrP, with either of the two brain homogenates of PrP prions from a single sCJD(MM1) case, and treated them with one of four different 2-AMTs (Table 4). Each 2-AMT was administered at a dose of 200 mg/kg per day, and each failed to extend survival.
Another compound, termed Anle138b, was recently reported to extend survival in mice infected with RML prions and to be effective against human CJD prions in the protein cyclic misfolding amplification assay (Wagner et al., 2013). Since our dosing strategy differed from that previously used for Anle138b, we first attempted to repeat the result in RML-infected mice. Tg4053 mice inoculated with RML prions were continuously dosed with Anle138b, which resulted in a doubling of the incubation time and a survival index of 245 ± 16 (Table 4). However, Anle138b did not extend survival in Tg1014 mice inoculated with sCJD(MM1) prions (Table 4).
Discussion
As evidenced by the recent high-profile failures of phase 3 clinical trials in AD, developing effective therapeutics for NDs remains a major challenge. With growing understanding of the commonalities among the NDs, where the pathogenic proteins undergo a conformational change that becomes self-propagating, the prototypical PrP prions offer a robust platform on which to develop therapeutics.
The ability to interrogate multiple strains of PrP prions in cellular and animal models may offer an important advantage since recent findings argue that multiple prion strains feature in the pathogenesis of AD (Paravastu et al., 2009; Lu et al., 2013b; Watts et al., 2014) as well as the synucleinopathies (Guo et al., 2013; Watts et al., 2013) and tauopathies (Clavaguera et al., 2013; Sanders et al., 2014).
Scrapie Prion Strains and IND24 Efficacy.
The three PrP prion strains, RML, ME7, and 22L, exhibited similar incubation periods in the vehicle-treated mice; however, they had different responses to IND24 treatment both in vivo and in vitro. IND24 showed little efficacy against 22L prions in cell culture (Fig. 1A), but extended the survival of 22L-infected wt mice by ∼50% when administered beginning at 1 dpi (Table 1). Starting dosing the day after inoculation, IND24 extended the survival of ME7- and RML-infected wt mice by ∼70%. When IND24 was administered beginning at ∼50% of the incubation period, the survival index remained at ∼179 for RML-infected mice, but decreased to 125 and 114 for ME7- and 22L-infected mice, respectively. Similar patterns were observed in cultured CAD5 cells: 2 μM IND24 inhibited replication of RML prions but was ineffective if the cells were infected with either ME7 or 22L prions (Fig. 1A). Increasing the IND24 concentration to 10 μM inhibited ME7 prion formation but did not alter 22L prion multiplication. Clearly, IND24 would not have been discovered if CAD5 cells infected with 22L prions had been used to evaluate this analog. The number of potent analogs rejected for follow-up because they did not show efficacy in the primary screen remains to be established.
Treatment of RML-infected mice with IND24 starting at ∼60% of the incubation period appears to be on the cusp of efficacious intervention, where small changes in disease progression can have a big impact in outcome. Indeed, this may account for much of the variability among the experiments. The failure of treatment when intervention occurred after 75% of the incubation period is consistent with the “window of reversibility” for rescue before too many neurons have died or become irreversibly dysfunctional (Moreno et al., 2012).
PrPC Expression and IND24 Efficacy.
The survival ratios for IND24-treated Tg4053 mice were uniformly higher than those found in wt mice, as we observed in previous prion efficacy studies (Lu et al., 2013a). This observation suggests that PrP overexpression does not simply accelerate prion replication and the attendant neurodegeneration. Collecting the full ensemble of variations in IND24 efficacy requires many more determinations, with various IND24 doses in mice expressing different levels of PrPC, infected with an array of PrP prion strains, and collected at multiple time points.
IND24-Resistant Prions and Drug Holidays.
Because drug-resistant prions were found in mice treated with IND24 (Berry et al., 2013), we asked if drug holidays might extend survival. When dosing at 210 mg/kg per day was alternated 60 days on and 60 days off, mice appeared to live slightly longer than with continuous dosing, whereas with the opposite cycle of off-on-off, all of the mice died before the second dosing section was started. However, the most dramatic extension in survival was when mice were dosed for 14 days before they were inoculated. Prophylactic dosing led to an almost 4-fold extension in survival (Fig. 1C). No overt toxicity was observed after dosing IND24 at 210 mg/kg per day for more than a year, and dosing as low as 5 mg/kg per day extended survival by more than 60%; therefore, a large margin of safety is suggested.
Prophylactic dosing is relevant to the ∼15% of CJD cases that are inherited. Although carriers have the mutation from embryogenesis, disease rarely manifests before the fourth decade of life, suggesting the need for a second event to initiate self-sustaining prion propagation and providing a large therapeutic window for prophylactic treatment.
Although the dose showed a good correlation to brain concentration when IND24 was given in food for 3 days, we observed evidence of drug accumulation in long-term studies. Brain concentrations of >10 µM were achieved within 3 days when IND24 was dosed at 210 mg/kg per day and stayed at this level with continuous dosing (Fig. 2). However, low doses of IND24 (≤5 mg/kg per day) gave low brain levels after 3 days, but these levels increased more than 20-fold after ∼75 days of continuous dosing. When initiated at 1 dpi, doses of 5 mg/kg per day led to almost as great a survival extension as doses of 210 mg/kg per day. However, dosing at 210 mg/kg per day could be delayed to 60 dpi without affecting the overall survival, but doses of 5 mg/kg per day showed a lower survival index when treatment was delayed.
A greater survival extension was observed at 50 mg/kg per day compared with 210 mg/kg per day (Fig. 2C). This finding could not be attributed simply to toxicity at the higher dose since prophylactic dosing at 210 mg/kg per day continued for more than a year (Fig. 1C). However, we cannot exclude the possibility that toxicity may be different at different stages of the disease process. One possibility is that higher levels of IND24 lead to the more rapid generation of an IND24-resistant prion strain, which was ultimately detrimental to survival. This explanation would be consistent with greater survival using intermittent dosing, i.e., drug holidays.
Five Novel 2-AMTs.
To expand the efficacy of 2-AMT drugs to treat people suffering from CJD, we synthesized many analogs and first tested them for their potency by titrations in ScN2a cells infected with RML prions (Li et al., 2013; Silber et al., 2013). Next, we measured the brain concentration after 3 days of administering the 2-AMTs, and from this study, selected compounds for long-term dosing in efficacy studies. The ratio of brain concentration to EC50 is a commonly used metric to predict efficacy. However, evidence of dose accumulation makes such a metric difficult to interpret. Low doses of IND24, with a C3day to EC50 ratio of 0.3, were efficacious, whereas IND126261 and IND126, which had C3day to EC50 ratios of 1.0 and 0.2, respectively, produced little extension in survival.
When IND114338 was given to wt mice, it was slightly more effective than IND24 at extending survival (Table 4). In contrast, IND125 was less effective than IND24 when administered to wt FVB mice, but was the most efficacious of all the compounds when given to Tg4053-overexpressing MoPrP. When IND125 was administered orally at 200 mg/kg per day, it tripled survival over vehicle-treated Tg4053 mice infected with RML prions (Table 4). Moreover, neuropathological analysis of these mice showed that treatment prevented PrPSc deposition throughout most of the cerebrum. The major site of PrPSc deposition was in the brainstem, which probably accounts for the central nervous system dysfunction that limits survival (Figs. 4 and 5). PrPSc staining in the striatum of mice treated with IND126 and IND126461 may suggest neurodegeneration, which could account for the progressive motor phenotype observed in the mice, leading them to be euthanized.
Prion Strain Changes upon Treatment.
We previously used the ratio of monoglycosylated to diglycosylated PrPSc glycoforms as a predictive tool for determining strain change upon treatment. Of the six statistical comparisons presented here, only the two in wt mice (IND24 and IND114338) were significantly different from controls, where the IND24 finding represents an independent repeat of a previously published result. None of the glycoform ratios in Tg4053 mice were different from controls, including IND24 treatment.
Quest for CJD Therapeutics.
Since the first transmission of CJD brain homogenates to apes and monkeys (Gibbs et al., 1968), there have been attempts to propagate CJD prions in cultured cell lines. Although none have been successful to date, it has been possible to replicate sheep, deer, and artificial prions in murine and lagomorph cell lines (Race et al., 1987; Butler et al., 1988; Vilette et al., 2001; Bian et al., 2010). As described previously, Tg1014 mice expressing a chimeric Hu/MoPrP transgene support replication of both human and murine prions (Giles et al., 2010; Berry et al., 2013). To determine if IND24 was efficacious for CJD prions, we gave the drug to Tg1014 mice inoculated with either RML or sCJD(MM1) prions. The incubation times were doubled in Tg1014 mice given IND24 and inoculated with RML prions, but they were unchanged by the drug in mice injected with sCJD(MM1) prions (Berry et al., 2013).
To determine if any of the new 2-AMTs were effective against CJD prions, we administered four different novel 2-AMTs to Tg1014 mice inoculated with sCJD(MM1) homogenates (Table 4). Disappointingly, none of the four new 2-AMTs extended the lives of the Tg1014 mice inoculated with CJD prions. The failure of Anle138b to extend survival in CJD-infected mice suggests that in vitro methods, such as protein cyclic misfolding amplification, may not be predictive of in vivo results. From our findings reported here and previously (Berry et al., 2013), it seems likely that developing a cell culture system that can propagate CJD prions will be critical for discovering drugs that will be efficacious in extending the lives of mice expressing HuPrP and/or chimeric Hu/MoPrP. Once that is achieved, advancing compounds into people dying of CJD would seem warranted.
Supplementary Material
Abbreviations
- ACN
acetonitrile
- AD
Alzheimer’s disease
- 2-AMT
2-aminothiazole
- CJD
Creutzfeldt-Jakob disease
- CMC
carboxy methyl cellulose
- DAB
diaminobenzidine
- dpi
days postinoculation
- Hu
human
- HuM
human-mouse chimeric
- Mo
mouse
- ND
neurodegenerative disease
- PD
Parkinson’s disease
- PEG400
polyethylene glycol 400
- PK
proteinase K
- PrP
prion protein
- PrPC
α-helical cellular isoform of prion protein
- PrPSc
self-propagating, β-sheet-rich scrapie isoform of prion protein
- ROI
region of interest
- sCJD(MM1)
sporadic CJD(MM1)
- wt
wild type
Authorship Contributions
Participated in research design: Giles, Silber, Prusiner.
Conducted experiments: Berry, Oehler, Elepano, Bhardwaj, Patel.
Contributed new reagents or analytic tools: Gallardo-Godoy, Bryant, Renslo.
Performed data analysis: Giles, Berry, Condello, Hawley, Guan, DeArmond.
Wrote or contributed to the writing of the manuscript: Giles, Berry, Prusiner.
Footnotes
This work was supported by the National Institutes of Health National Institute on Aging [Grants AI064709, AG002132, AG010770, AG021601, AG031220, and NS080630] as well as by gifts from the Sherman Fairchild Foundation, Schott Foundation for Public Education, Rainwater Charitable Foundation, Mary Jane Brinton Fund, Dana Foundation (A121428), and G. Harold and Leila Y. Mathers Charitable Foundation.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Akhtar S, Wenborn A, Brandner S, Collinge J, Lloyd SE. (2011) Sex effects in mouse prion disease incubation time. PLoS One 6:e28741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DB, Lu D, Geva M, Watts JC, Bhardwaj S, Oehler A, Renslo AR, DeArmond SJ, Prusiner SB, Giles K. (2013) Drug resistance confounding prion therapeutics. Proc Natl Acad Sci USA 110:E4160–E4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen RA, Marsh RF. (1994) Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68:7859–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian J, Napier D, Khaychuck V, Angers R, Graham C, Telling G. (2010) Cell-based quantification of chronic wasting disease prions. J Virol 84:8322–8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler DA, Scott MRD, Bockman JM, Borchelt DR, Taraboulos A, Hsiao KK, Kingsbury DT, Prusiner SB. (1988) Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J Virol 62:1558–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GA, Ebeling C, Yang S-L, Telling G, Torchia M, Groth D, Westaway D, DeArmond SJ, Prusiner SB. (1994) Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc Natl Acad Sci USA 91:5690–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GA, Kingsbury DT, Goodman PA, Coleman S, Marshall ST, DeArmond S, Westaway D, Prusiner SB. (1986) Linkage of prion protein and scrapie incubation time genes. Cell 46:503–511. [DOI] [PubMed] [Google Scholar]
- Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, et al. (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 110:9535–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, Clarke AR. (2007) A general model of prion strains and their pathogenicity. Science 318:930–936. [DOI] [PubMed] [Google Scholar]
- Dickinson AG. (1976) Scrapie in sheep and goats, in Slow Virus Diseases of Animals and Man (Kimberlin RH. ed) pp 209–241, North-Holland Publishing Company, Amsterdam. [Google Scholar]
- Frost B, Ollesch J, Wille H, Diamond MI. (2009) Conformational diversity of wild-type tau fibrils specified by templated conformation change. J Biol Chem 284:3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo-Godoy A, Gever J, Fife KL, Silber BM, Prusiner SB, Renslo AR. (2011) 2-Aminothiazoles as therapeutic leads for prion diseases. J Med Chem 54:1010–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, May BCH, Renslo AR, Prusiner SB. (2010) Discovery of 2-aminothiazoles as potent antiprion compounds. J Virol 84:3408–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs CJ, Jr, Gajdusek DC, Asher DM, Alpers MP, Beck E, Daniel PM, Matthews WB. (1968) Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 161:388–389. [DOI] [PubMed] [Google Scholar]
- Giles K, Glidden DV, Patel S, Korth C, Groth D, Lemus A, DeArmond SJ, Prusiner SB. (2010) Human prion strain selection in transgenic mice. Ann Neurol 68:151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding EH, Schenk AK, Juneja P, MacKay AW, Wade JM, Tecott LH. (2008) A robust automated system elucidates mouse home cage behavioral structure. Proc Natl Acad Sci USA 105:20575–20582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, et al. (2013) Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Kawagoe K, Chen CJ, Teruya K, Sakasegawa Y, Doh-ura K. (2007) Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J Virol 81:12889–12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Silber BM, Rao S, Gever JR, Bryant C, Gallardo-Godoy A, Dolghih E, Widjaja K, Elepano M, Jacobson MP, et al. (2013) 2-Aminothiazoles with improved pharmacotherapeutic properties for treatment of prion disease. ChemMedChem 8:847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Giles K, Li Z, Rao S, Dolghih E, Gever JR, Geva M, Elepano ML, Oehler A, Bryant C, et al. (2013a) Biaryl amides and hydrazones as therapeutics for prion disease in transgenic mice. J Pharmacol Exp Ther 347:325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. (2013b) Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 154:1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. (2007) Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci USA 104:20908–20913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, et al. (2012) Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485:507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelschlegel AM, Fallahi M, Ortiz-Umpierre S, Weissmann C. (2012) The extended cell panel assay characterizes the relationship of prion strains RML, 79A, and 139A and reveals conversion of 139A to 79A-like prions in cell culture. J Virol 86:5297–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. (2009) Seeded growth of beta-amyloid fibrils from Alzheimer’s brain-derived fibrils produces a distinct fibril structure. Proc Natl Acad Sci USA 106:7443–7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. (2014) Madness and Memory, Yale University Press, New Haven, CT. [Google Scholar]
- Race RE, Fadness LH, Chesebro B. (1987) Characterization of scrapie infection in mouse neuroblastoma cells. J Gen Virol 68:1391–1399. [DOI] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, et al. (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82:1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber BM, Rao S, Fife KL, Gallardo-Godoy A, Renslo AR, Dalvie DK, Giles K, Freyman Y, Elepano M, Gever JR, et al. (2013) Pharmacokinetics and metabolism of 2-aminothiazoles with antiprion activity in mice. Pharm Res 30:932–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöhr J, Condello C, Watts JC, Bloch L, Oehler A, Nick M, DeArmond SJ, Giles K, DeGrado WF, Prusiner SB. (2014) Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc Natl Acad Sci USA 111:10329–10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöhr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, Prusiner SB, Giles K. (2012) Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc Natl Acad Sci USA 109:11025–11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamgüney G, Giles K, Glidden DV, Lessard P, Wille H, Tremblay P, Groth DF, Yehiely F, Korth C, Moore RC, et al. (2008) Genes contributing to prion pathogenesis. J Gen Virol 89:1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. (1996) Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274:2079–2082. [DOI] [PubMed] [Google Scholar]
- Telling GC, Scott M, Hsiao KK, Foster D, Yang S-L, Torchia M, Sidle KCL, Collinge J, DeArmond SJ, Prusiner SB. (1994) Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci USA 91:9936–9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilette D, Andreoletti O, Archer F, Madelaine MF, Vilotte JL, Lehmann S, Laude H. (2001) Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc Natl Acad Sci USA 98:4055–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F, Prix C, Pan-Montojo F, Bertsch U, Mitteregger-Kretzschmar G, et al. (2013) Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol 125:795–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Condello C, Stöhr J, Oehler A, Lee J, DeArmond SJ, Lannfelt L, Ingelsson M, Giles K, Prusiner SB. (2014) Serial propagation of distinct strains of Aβ prions from Alzheimer’s disease patients. Proc Natl Acad Sci USA 111:10323–10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, DeArmond SJ, Prusiner SB. (2013) Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci USA 110:19555–19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson RA, Peretz D, Pinilla C, Ball H, Bastidas RB, Rozenshteyn R, Houghten RA, Prusiner SB, Burton DR. (1998) Mapping the prion protein using recombinant antibodies. J Virol 72:9413–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Huo M, Zhou J, Xie S. (2010) PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed 99:306–314. [DOI] [PubMed] [Google Scholar]
- Zhu L, Ramboz S, Hewitt D, Boring L, Grass DS, Purchio AF. (2004) Non-invasive imaging of GFAP expression after neuronal damage in mice. Neurosci Lett 367:210–212. [DOI] [PubMed] [Google Scholar]
- Zlotnik I, Rennie JC. (1963) Further observations on the experimental transmission of scrapie from sheep and goats to laboratory mice. J Comp Pathol 73:150–162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.