

Abstract
Cannabidiol (CBD) is a component of cannabis, which does not cause the typical marijuana-type effects, but has a high potential for use in several therapeutic areas. In contrast to Δ9-tetrahydrocannabinol (Δ9-THC), it binds very weakly to the CB1 and CB2 cannabinoid receptors. It has potent activity in both in vitro and in vivo anti-inflammatory assays. Thus, it lowers the formation of tumor necrosis factor (TNF)-α, a proinflammatory cytokine, and was found to be an oral antiarthritic therapeutic in murine collagen-induced arthritis in vivo. However, in acidic media, it can cyclize to the psychoactive Δ9-THC. We report the synthesis of a novel CBD derivative, HU-444, which cannot be converted by acid cyclization into a Δ9-THC–like compound. In vitro HU-444 had anti-inflammatory activity (decrease of reactive oxygen intermediates and inhibition of TNF-α production by macrophages); in vivo it led to suppression of production of TNF-α and amelioration of liver damage as well as lowering of mouse collagen-induced arthritis. HU-444 did not cause Δ9-THC–like effects in mice. We believe that HU-444 represents a potential novel drug for rheumatoid arthritis and other inflammatory diseases.
Introduction
Cannabidiol (CBD), a nonpsychotropic constituent present in most Cannabis sativa varieties, causes a large number of both central and peripheral pharmacological effects (Mechoulam et al., 2002, 2007; Pertwee, 2005; Zhornitsky and Potvin, 2012; Fernández-Ruiz et al., 2013). CBD is a potent antioxidant (Hampson et al., 2000), which may explain—at least in part—its neuroprotective effects in neurodegenerative disorders (Fernández-Ruiz et al., 2013), in amelioration of the progressive degeneration of nigrostriatal dopaminergic neurons occurring in a model of Parkinson's disease (Lastres-Becker et al., 2005), in cerebral ischemia (Braida et al., 2003), in cerebral infarction in mice (Hayakawa et al., 2007), and in hypoxia-ischemia in newborn rats (Pazos et al., 2012). Additional effects of therapeutic relevance, through various mechanisms, are its action in animals on type 1 diabetes (Weiss et al., 2008), on some types of cancer (Massi et al., 2013), on myocardial ischemic reperfusion injury (Durst et al., 2007), on reduction of microglial activation—thus, possibly on the progression of Alzheimer's disease (Martín-Moreno et al., 2011), on brain and liver functions in a fulminant hepatic failure-induced model of hepatic encephalopathy (Avraham et al., 2011), on nausea and emesis (Rock et al., 2012), and others.
The antiepileptic activity of CBD in human patients has been known for nearly 35 years (Cunha et al., 1980), but only recently have marijuana extracts with high levels of CBD been used to suppress pediatric epilepsies (Porter and Jacobson, 2013). In clinical trials CBD has been shown to have antischizophrenic (Leweke et al., 2012) and antianxiety (Bergamaschi et al., 2011) properties.
Of particular relevance to the results presented in this study is the potent anti-inflammatory action of CBD (Mechoulam et al., 2002, 2005; Pertwee, 2005). We have previously shown that, in vitro, CBD suppresses lymphocyte proliferation and blocks zymosan-triggered reactive oxygen burst by peritoneal granulocytes (Malfait et al., 2000). In vivo results reported include blocking lipopolysaccharide (LPS)-induced rise in serum tumor necrosis factor (TNF)-α in mice as well as the progression of arthritis caused by immunization of mice with type II collagen (Malfait et al., 2000).
It is somewhat surprising that, in spite of the very promising pharmacological effects of CBD and its lack of toxicity, it has not been developed as a single drug. CBD is marketed together with Δ9-tetrahydrocannabinol (Δ9-THC; in a 1:1 ratio) by GW Pharmaceuticals (London, UK) as Sativex, sold in Canada and several European countries for spasticity, due to multiple sclerosis (Syed et al., 2014).
We report now the synthesis of a novel CBD derivative, HU-444, using CBD as the starting material, and the evaluation of its anti-inflammatory properties in vitro and in vivo. This novel compound was chosen, as the conversion of the C-1 methyl group into a carboxyl group in the cannabinoid series has been shown to enhance anti-inflammatory activity (Burstein et al., 1992; Sumariwalla et al., 2004) and the reduction of the 8,9-double bond would preclude possible ring cyclization under acidic conditions with one of the phenolic groups leading to psychoactive THC-like compounds.
The results reported now show that HU-444 is a potent anti-inflammatory compound, both in vitro and in vivo.
Materials and Methods
Reagents.
All solvents were purchased from Biolab (Jerusalem, Israel) and J. T. Baker (Deventer, Holland). Chemicals were purchased from Sigma-Aldrich (Rehovot, Israel), Acros, (Geel, Belgium) Holland Moran (Yehud, Israel), Alfa Aesar (Lancashire, UK), Merck (Darmstadt, Germany), J. T. Baker (Center Valley, PA), and Penta (Prague, Czech Republic). They were used without further purification in the reactions, except of dry diethyl ether and CH2Cl2, which were refluxed over sodium and phosphorous pentoxide (P2O5), respectively, and freshly distilled prior to its use.
1H NMR spectra were obtained on a Bruker AMX 300 MHz apparatus using CDCl3 (δ = 7.25 ppm) and TMS (tetramethyl silane) as internal standard for 1H NMR. Thin-layer chromatography was run on silica gel 60F254 plates (Merck). Column chromatography was performed on silica gel 60 A° (Merck). The compounds were localized at 254 nm using a UV lamp.
Syntheses.
The synthesis of HU-444 is presented in Scheme 1. The synthetic steps a, b, and c are published in Kozela et al. (2015). Below we describe in detail the subsequent reaction steps d–i.
Scheme 1.

Syntheses of HU-444 and HU-445. Reagents and Conditions: (a) Pt(IV) oxide/H2, EtOAc, 10psi, rt, 2min; (b) pyridine, acetic anhydride, rt, 12h; (c) SeO2, EtOH, rt, 3h; (d) pyridine; (e) CrO3, CH2Cl2/DMF, rt, 1h; (f) NaClO2; (g) 2-methyl-2-butene; (h) KH2PO4, t-butanol, rt, 1h; (i) NaBH4, EtOH, reflux, 1h.
Syntheses of (-)-8,9-Dihydro-CBD-7-Oic Acid-Diacetate (HU-444) (5) and (-)-8,9-Dihydro-CBD-7-Oic Acid (HU-445) (6)
Cannabidiol.
Crystalline CBD was isolated from hashish following the procedure described by Gaoni and Mechoulam (Gaoni and Mechoulam, 1971). Yield 15%; melting point: 62°C; [α]D20 = −56° (CHCl3).
Synthesis of (-)-8,9-Dihydro-7-Oxo-CBD-Diacetate and (-)-8,9-Dihydro-6-Oxo-CBD-Diacetate (Mixture 4).
Dry chromium oxide (353 mg, 3.53 mmol) was added to a stirred solution of dry pyridine (0.951 ml, 11.83 mmol) in dry CH2Cl2:DMF dimethyl formamide (4:1) (4 ml) and stirred at room temperature for 15 minutes under nitrogen. A solution of mixture 3 (378 mg, 0.91 mmol) in dry CH2Cl2:DMF (4:1) (1.5 ml) was added, and the reaction was stirred at room temperature for 1 hour. Ethanol (2 ml) was added, and the reaction was stirred at room temperature for 10 minutes. The mixture was filtrated on silica gel covered with thin layer of Na2SO4, washed several times with ethyl acetate, and evaporated to dryness. The residue was chromatographed on silica gel with 10% ether–petroleum ether to give mixture 4 without separation with a yield of 63%.
Synthesis of (-)-8,9-Dihydro-CBD-7-Oic Acid-Diacetate-(HU-444) (5).
NaClO2 (221 mg, 2.44 mmol) was added in small quantities to a stirred mixture of mixture 4 (235 mg, 0.568 mmol), 2-methyl-2-butene (1.5 ml, 14.22 mmol), and a saturated aqueous solution of KH2PO4 (0.67 ml) in tert-butanol (13.37 ml). The reaction was stirred at room temperature for 5 hours and monitored by thin-layer chromatography. Water was added (60 ml), and the mixture was extracted several times with ethyl acetate. The organic phase was washed with brine, dried over MgSO4, and filtered. Removal of the solvent under reduced pressure afforded a residue that was chromatographed on silica gel with 30% ether–petroleum ether to give compound 5 as a white solid. Yield 13%, Mp: 152°C; [α]D20 = −57° (CHCl3); high-pressure liquid chromatography (HPLC; 60% acetonitrile, 15% water, and 25% methanol): tR = 7.69 minutes, 96%. 1H NMR (CDCl3): δ 6.94 (1H,s,olefin), 6.88 (2H,s,Ar), 3.58 (1H,m,benzyl), 2.51 (3H, m, allyl + benzyl), 2.22 (6H, s. OAc), 1.97 (1H, m), 1.86 (1H, m), 1.54 (5H, br s), 1.32 (5H, m), 0.98 (9H, t, terminal CH3). MS m/z: 502 (silylation), 472, 430, 415, 400. Exact mass calculated for C25H34O6 430.2199, found 430.2198.
Synthesis of (-)-8,9-Dihydro-CBD-7-Oic Acid-(HU-445) (6).
Compound 5 (30 mg, 0.069 mmol) was dissolved in ethanol (8 ml). NaBH4 (3 mg, 0.092 mmol) was added, and the reaction was refluxed for 1 hour. The ethanol was removed under pressure, the residue was diluted with water (10 ml), and the solution was extracted with ether. The combined organic extracts were washed with brine, dried over MgSO4, and filtered. Removal of the solvents under reduced pressure afforded a residue that was chromatographed on silica gel with 30% ether–petroleum ether to give compound 6 as a white solid with a yield of 90%, Mp: 88–90°C; [α]D20 = −56.2° (CHCl3); HPLC (60% acetonitrile, 15% water, and 25% methanol): tR = 8.86 minutes, 95%. 1H NMR (CDCl3): δ 6.94 (1H,s,olefin), 6.88 (2H,s,Ar), 4.12 (2H, br s), 3.59 (1H,m,benzyl), 2.51 (3H, m, allyl + benzyl), 2.22 (3H, s. OAc), 1.97 (1H, m), 1.86 (1H, m), 1.55 (5H, br s), 1.32 (5H, m), 0.98 (9H, t, terminal CH3). MS m/z: 562 (silylation), 489, 416, 346, 343. Exact mass calculated for C21H28O4 346.1987, found 346.1948.
Binding to the Cannabinoid Receptors.
The binding of HU-444 and HU-445 to the cannabinoid receptors CB1 and CB2 was performed, as previously described (Devane et al., 1992a,b; Bayewitch et al., 1996).
Mice.
In Jerusalem, C57BL/6 and Sabra female mice, 8–10 weeks old, were obtained from Harlan Laboratories (Jerusalem, Israel); in London, DBA/1 male mice, 12 weeks old, were obtained from Harlan Laboratories, (Blackthorn, UK). Mice were housed in a room with controlled temperature (22 ± 1°C), humidity (60% ± 1%), and light 12 hours per day. Mice were fed standard animal chow and tap water ad libitum and were kept in a specific pathogen-free facility. The animal research in Jerusalem was conducted in compliance with international laws and approved by the Ethical Committee of the Hebrew University Medical School, whereas in London all animal studies conducted had received prior approval of the local ethical review process committee and were performed under the guidance of the Home Office Animals (Scientific Procedures) Act 1986 (PPL: 70/5446).
Macrophages.
Peritoneal cells were harvested from C57BL/6 female mice 4 days after i.p. injection of 1.5 ml 3% thioglycolate (TG) medium (Difco, Oxford, UK). The cells (TG macrophages) were washed with phosphate-buffered saline; resuspended in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS), sodium pyruvate, glutamine, and antibiotics; and plated (1.2 × 105) in 96-microwell flat-bottom plates (Nunc, Roskilde, Denmark). Following 2- to 3-hour incubation at 37°C, the nonadherent cells were removed by intensive rinsing with phosphate-buffered saline. About 95% of the adherent cells were macrophages.
THC-Like Activity in Mice.
THC and other agonists of the CB1 cannabinoid receptor cause a typical tetrad of pharmacological effects in Sabra mice—namely, ring immobility (catalepsy), which measures the percentage of time, over 4 minutes; mice remain immobile on a ring (5.5 cm diameter), the open field test that measures locomotor activity, hypothermia, and hot plate latency (antinociception) (Martin et al., 1991; Fride and Mechoulam, 1993). Sabra mice were administered HU-444 i.p. and assayed in the above tetrad.
Macrophage Cell Lines.
RAW 264.7 cells, a monocytic–macrophage cell line, derived from BALB/c mice, were obtained from American Type Culture Collection (Rockville, MD). The cells were cultured in DMEM supplemented with 10% FCS and sodium pyruvate, glutamine, and antibiotics. For activation, the cells (105 cells/microwell) were incubated with 1 μg/ml LPS (Escherichia coli; Sigma-Aldrich, Jerusalem, Israel) for 24 hours.
Treatment of Macrophages with HU-444.
HU-444 was first dissolved in absolute ethanol (1 mg/50–100 μl ethanol), and the solutions were further diluted with DMEM. Various nontoxic concentrations were added to the macrophages, followed by addition of 1 μg/ml LPS dissolved in saline for activation. The macrophages were then cultivated in a humid atmosphere with 5% CO2 for 24 hours. The supernatant fluids were harvested and kept at −20°C until assayed for TNF-α.
Reactive Oxygen Species Production by RAW 264.7 Macrophages.
RAW 264.7 cells were removed using a scraper, washed, and resuspended in Hanks’ balanced salt solution (without phenol red). For measurement of chemiluminescence, 0.5 ml cell suspension (5 × 105 cells) was added to each luminometer tube, together with various doses of HU-444 (dissolved in ethanol and diluted with Hanks’ salt solution). Then, 10 μL luminol (Sigma-Aldrich) and 30 μL containing 40 μg zymosan (Sigma-Aldrich) were added to the tubes; the chemiluminescence was measured immediately in a Luminometer (Biolumate LB 95; Berhold, Wilbad, Germany).
Evaluation of TNF-α Levels Produced In Vitro and In Vivo.
Peritoneal thioglycolate macrophages were cultured (1.2 × 105/microwell) for 2–3 hours, rinsed in phosphate-buffered saline, and further incubated in DMEM with 5% FCS, in the presence of varying concentrations of HU-444. LPS was used as a stimulus at 1 μg/ml. The viability of the cells at the end of the experiment was >90%. The supernatants were collected and kept at −20°C until assayed.
To determine TNF-α levels in mice sera, C57BL/6 female mice (8–10 weeks old; 24 mice/experiment) were injected with 5 mg/kg LPS (i.p.) and then with various doses of HU-444. Ninety minutes later, the mice were bled; the sera were kept at −20°C until assayed (Malfait et al., 2000). TNF-α levels both in sera and the culture supernatants were determined by enzyme-linked immunosorbent assay (ELISA; R&D Systems, San Diego, CA), according to the manufacturer's procedure.
Nitric Oxide Detection Assay.
Nitric oxide (NO) generation was determined by measuring the nitrite accumulated in the supernatants (100 μL) of LPS (1 µg/ml)- and HU-444–treated RAW macrophages as follows. An equal volume (100 μL) of Griess reagent (1% sulfanilamide, 0.1% naphthalene diamine HCl, 2% H3PO4) was added to each supernatant. Following 10 minutes of incubation at room temperature, the color production was measured at 550 nm with an ELISA reader. The concentration of nitrate was calculated according to a standard curve.
Liver Injury.
C57BL/6 male mice, 8–10 weeks old, total of 48 mice (in three experiments), were injected i.v. with 20 mg/kg concanavalin A (Con A), dissolved in pathogen-free saline (Tiegs et al., 1992). Immediately thereafter, the mice were injected i.p. with various doses of HU-444. Twenty hours later, the mice were bled through the orbital plexus (for collection of serum for liver enzymes and TNF-α assays) and sacrificed. The livers were removed, fixed with buffer formalin, and stained with H&E, for microscope evaluation. TNF-α levels in the sera of Con A–treated mice were determined by ELISA (R&D Systems).
Determination of Alanine Aminotransferase and Aspartate Aminotransferase Levels.
The levels of two aminotransferases, alanine aminotransferase (ALT) and aspartate aminotransferase (AST), were assayed in the sera of Con A–treated mice, with or without HU-444 treatment, by ALT and AST strips, respectively (Refloram-Mannheim, Mannheim, Germany), and quantitated by an automated analyzer (Reflotran Plus; Roche, Basel, Switzerland).
Histologic Analysis.
The livers of Con A–injected mice, with or without HU-444 treatment, were fixed in 10% buffer formalin. Paraffin sections of 4–5 µm thickness were stained with H&E.
For microscopic evaluation, sections were examined (40×) blindly and scored 0–5, as follows: 0, normal liver morphology; 1, minimal changes; 2, mild, few lesions; 3, moderate number of lesions; 4, marked changes in the liver; and 5, severe liver damage.
Collagen-Induced Arthritis Study: Induction Monitoring and Treatment with HU-444.
Collagen-induced arthritis (CIA) was induced in genetically susceptible DBA/1 mice (H-2q; Harlan Laboratories, UK) by subcutaneous immunization, at 16 weeks with purified type II chicken collagen emulsified with Freund’s complete adjuvant (100 µl 2 mg/ml, divided over two sites in the lower dorsal region). Mice were purchased at 12 weeks of age and acclimatized in the animal house over a 20-hour light/dark cycle. Mice were housed in individual ventilated cages, and the ambient temperature was maintained at 21°C. Mice were fed with standard laboratory chow and tap water ad libitum. Onset was characterized by visible clinical signs of arthritis, which were a varying degree of redness and swelling of any of the paws. Mice were treated with HU-444 either i.p. (graded doses of 2.5, 5.0, and 10 mg/kg) or orally (15 mg/kg), daily for 10 days. For the purpose of administering various doses of HU-444 to mice, the average weight of a mouse was considered as 25 g and nine arthritic mice were recruited in all study groups. All treatments were carried out from day 1, which is considered as the day when the first visible clinical signs of arthritis developed in a mouse. Vehicle (ethanol: cremophor-EL: saline; 1:1:18 v/v/v for i.p. or 1:1:8 v/v/v for oral administrations, respectively) was administered to a group of arthritic mice used as controls. Vehicle on its own and HU-444 in vehicle were administered in experiments at either 0.1 (oral gavage) or 0.2 (i.p.) ml. All paws were graded daily using previously well-validated clinical score readouts (Sumariwalla et al., 2004). Hind paw thickness was measured (mm) using a caliper device. All treatment studies had the prior approval from a local ethical review process committee and were conducted in accordance with the Home Office–approved project license PPL: 70/5446.
Histology of Arthritic Feet.
Mice were euthanized at the end of treatments and hind paws fixed in buffered formalin, decalcified in 10% EDTA for 3–4 weeks, embedded in paraffin wax, sectioned to 5 µm thicknesses, and stained with H&E. All joints were scored blindly for joint architectural changes by a well-validated scoring system (Sumariwalla et al., 2003). In brief, score 0 represented normal joints; 1, mild, change involving synovial hyperplasia, chondrocyte denucleation, and a few focal bone and cartilage erosions; 2, moderate, change involving extensive synovial hyperplasia and pannus front accompanying large areas of bone and cartilage erosions; and 3, severe, total loss of joint architecture. For the purpose of scoring, all joints (distal, proximal phalangeal, first metatarsal, medial cuneiform, and tibia-tarsus) present within the stained section were scored. Histologic evaluation was done on all mice paw sections from all groups. Percentage of joints involved as normal, mild, moderate, and severe was then calculated for each group. Data were presented as percentage joints scored and classified as either protected (normal to mild arthritic changes) or damaged (moderate to severe arthritic changes). Images were captured using Olympus BX51 optical microscope and DP Controller and Manager Software (Olympus, version 3.3.1.292/222; Center Valley, PA), at original magnification, 100×. For publication purposes, only the proximal phalangeal joint images were depicted for all treatment groups.
Experimental Design and Statistics.
Experiments, both in vitro and in vivo (4–5 mice/group), were repeated three times, and the results were expressed as the mean ± S.E.M. of triplicate values. Evaluation of statistical significance was done by using analysis of variance. Joint histology data were evaluated by Fischer’s exact test. The P ≤ 0.05 values were considered significant.
Results
Chemistry.
CBD, which was extracted from hashish (Gaoni and Mechoulam, 1971), was hydrogenated with Pt (IV) oxide in ethyl acetate to yield 8,9-dihydro-CBD 1, which was then converted into the diacetate 2 by pyridine and acetic anhydride. Compound 2 was oxidized with selenium IV oxide in ethanol to give a mixture of the allyllic alcohols 3. The allylic hydroxylation reaction by selenium IV oxide has been widely investigated (Sharpless and Lauer, 1972; Lander et al., 1976; Stephenson and Speth, 1979). Apparently, it involves a (2,3)sigmatropic migration reaction of intermediate allylselenilic acids, leading mostly to allylic hydroxylation products. It has been widely used for the oxidation of cyclohexenyl systems. As mixture 3 was difficult to separate, it was directly oxidized with chromium VI oxide in the presence of pyridine, to give a mixture 4 of the expected aldehyde and ketone. The chromium (VI) oxide–pyridine complex is used as an oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones (Holum, 1961; Ratcliffe and Rodehorst, 1970) at room temperature. On further oxidation of the mixture 4 with sodium chlorite (Pellegata et al., 1986; Burstein et al., 1992), we obtained the acid diacetate HU-444 (compound 5) from the aldehyde, which was easy to separate from the unchanged ketone. The overall yield from CBD was 13% (96% purity by HPLC). The conversion of the diacetate to the free diphenol was carried out by sodium borohydride in ethanol to give HU-445 (compound 6) in 90% yield (95% purity by HPLC). All compounds were purified on silica gel chromatography (Scheme 1).
While the overall yield of HU-444 is relatively low, in view of the ready availability of CBD, its synthesis represents a practical route.
Binding to the Cannabinoid Receptors.
Neither 5 (HU-444) nor 6 (HU-445) was found to bind to either the CB1 or the CB2 receptor (Ki above 10 µM).
Pharmacology.
Compound 5 (HU-444) was first investigated for its possible THC-like psychoactivity in the tetrad assay, which measures cannabinoid-induced hypokinesia, hypothermia, and antinociception in a tail flick or hot plate test in mice (Martin et al., 1991; Fride and Mechoulam, 1993; Mechoulam et al., 2014). The mouse tetrad serves as a useful in vivo screen for psychotropic cannabinoids, which, in contrast to many other types of drugs, displays potency in all four of these bioassays. As no psychoactivity was noted (data not presented), we evaluated HU-444 in a wide range of assays relevant to inflammation.
The in vitro anti-inflammatory assays were mostly done with macrophages, whereas for the in vivo studies we used an animal model for both autoimmune hepatitis and rheumatoid arthritis (RA). The results showed that HU-444 is a potent anti-inflammatory compound, both in vitro and in vivo.
Suppression of Reactive Oxygen Species Production by HU-444.
To study the effect of HU-444 on the ability of macrophages to produce ROS, RAW 264.7 cells were stimulated with zymosan together with various doses of HU-444. A 39% and 62% decrease of reactive oxygen species (ROS) generation was observed in the presence of 20 and 40 μg/ml HU-444, respectively (Fig. 1A).
Fig. 1.

Effect of HU-444 on in vitro macrophage function. (A) Generation of ROS (assayed by chemiluminescence) (Ben-Shabat et al., 2006) by RAW 264.7 mouse macrophage cell line, following stimulation with zymosan, in the absence or presence of HU-444. Control chemiluminescence was 1370 cpm. *Statistically significant difference with a P value ≤0.05. (B) Generation of NO (assayed by Griess reagent) (Ben-Shabat et al., 2006) by RAW 264.7 cells, following stimulation by 1 μg/ml LPS for 24 hours in the presence of HU-444. The level of NO2/NO3 in the control supernatants was 42.5 nmol/ml. *Statistically significant difference with a P value ≤0.05.
Suppression of NO Production by HU-444.
To study the effect of HU-444 on macrophage production of NO, RAW 264.7 cells were incubated for 24 hours with LPS (1 μg/ml) and various doses (5–60 µg/ml) of HU-444. NO production was suppressed by 51% and 64% following incubation of the cells with 40 and 60 µg/ml HU-444, respectively (Fig. 1B).
Inhibition of TNF-α Production in Macrophages and Mice.
TNF-α production by TG macrophages was determined following their incubation with LPS (1 µg/ml) and various doses of HU-444. Inhibition of up to 69% was noted upon cell incubation with 40 µg/ml HU-444 for 24 hours (Fig. 2A). HU-444 also reduced the TNF-α serum levels by 34–45% in C57BL/6 mice after i.p. injection of LPS (100 μg/mouse). Already, at 2.5 mg/kg HU-444, a strong inhibition in TNF-α serum levels of 34% was observed, with only a slightly higher inhibition (45%) when increasing the dose to 10 mg//kg, suggesting that a plateau effect has been reached (Fig. 2B).
Fig. 2.

Effect of HU-444 on TNF-α production in vitro and in vivo. (A) TNF-α (determined by ELISA) production by TG macrophages following incubation with 1 μg/ml LPS in presence of HU-444 for 24 hours. Control macrophages (105) produced 59 ± 3 pg/ml TNF-α during the 24 hours. *Statistically significant difference with a P value ≤0.05. (B) TNF-α in mice sera 90 minutes after injections of LPS and HU-444. TNF titer in the control LPS sera was 732 ± 60 pg/ml. *Statistically significant difference with a P value ≤0.05.
Effect of HU-444 on Con A–Induced Liver Damage.
Intravenous administration of Con A to mice causes a CD4+ T cell–driven, TNFR1-dependent acute hepatitis, leading to pathologic damage of the liver. This is accompanied by elevation of liver enzymes, interleukin-2, and inflammatory cytokines (Kusters et al., 1997; Ohta and Sitkovsky, 2001). When Con A–treated mice were injected i.p. with 5 mg/kg HU-444, a marked reduction in the ALT and AST aminotransferase serum levels was observed (Fig. 3). ALT was reduced by 87% (Fig. 3A), and AST by 85% (Fig. 3B). We observed a bell-shaped dose response with both enzymes, with an optimal dose of 5 mg/kg. A bell-shaped dose response is often observed with CBD drugs (Jamontt et al., 2010). The TNF-α serum levels in Con A–treated C57BL/6 mice were also reduced after treatment with HU-444. Inhibition of 53% in TNF-α titers was scored following injection of 5 mg/kg HU-444 (Fig. 4).
Fig. 3.

Levels of liver aminotransferases (ALT, AST) in sera of Con A–treated mice following injection of HU-444. (A) ALT, alanine aminotransferase. *Statistically significant difference with a P value ≤0.05. (B) AST, aspartate aminotransferase. *Statistically significant difference with a P value ≤0.05.
Fig. 4.

TNF-α level in serum of mice 20 hours after i.v. injection of Con A and i.p. injection of HU-444. *Statistically significant difference with a P value ≤0.05.
Prevention of Con A–Induced Liver Damage by HU-444.
To study the effect of HU-444 on Con A–induced liver damage, histopathological evaluations of the livers were performed (Fig. 5; Table 1). Con A treatment caused marked liver damage with necrosis and mononuclear cell infiltration (Fig. 5A). Administration of HU-444 attenuated significantly liver damage and reduced mononuclear infiltration, leading to almost normal histology (Fig. 5, C and D). An optimally preserved normal liver histology was observed after treatment with 5 mg/kg HU-444 (Fig. 5C).
Fig. 5.

Histopathology of liver, 20 hours after Con A injections and HU-444 injections. Staining with H&E. (A) Con A–injected mice. Severe damage can be seen; areas of necrosis and infiltration of inflammatory cells. (B) Injection of 2.5 mg/kg HU-444, concomitant with Con A. Normal liver histology was largely preserved, although some infiltration of inflammatory cells was seen. (C) Injection of 5 mg/kg HU-444, concomitant with Con A. Near-normal liver structure seen microscopically. (D) Injection of 10 mg/kg HU-444, concomitant with Con A. Although without area of necrosis, changes of normal liver cell morphology can be seen microscopically.
TABLE 1.
Concomitant treatment of mice with HU-444 confers protection against Con A–induced liver damage
| Treatment (HU-444 mg/kg, i.p.) | Pathologic Score |
|---|---|
| Con A | 3–4 |
| Con A + 2.5 | 3 |
| Con A + 5.0 | 1.5 |
| Con A + 10.0 | 2.5 |
Treatment with HU-444 Provides Beneficial Outcome in an Established Mouse Model of Arthritis.
We explored the potential anti-inflammatory and disease-modulating properties of HU-444 using our mouse CIA model (Malfait et al., 2000; Sumariwalla et al., 2004). Administration of HU-444 systemically by i.p. or oral route ameliorated clinical signs of arthritis (Fig. 6). Treatments commenced from day 1 of arthritis, which is considered as the first day of the appearance of visible clinical signs of arthritis (redness/swelling) in any of the paws. The average day of onset of disease is about day 14–28 postimmunization. This period can, however, be variable between mice and experiments. As seen from Fig. 6, an inverse proportion dose curve was established with the highest dose of 10 mg/kg, daily being clinically nonbeneficial, whereas the two lower doses of 2.5 and 5.0 mg/kg showed beneficial clinical efficacy. Doses below 2.5 mg/kg were inefficient, indicating for the existence of an inverse bell-shaped curve, as is seen with CBD (Malfait et al., 2000) and a synthetic CBD derivative (Sumariwalla et al., 2004).
Fig. 6.
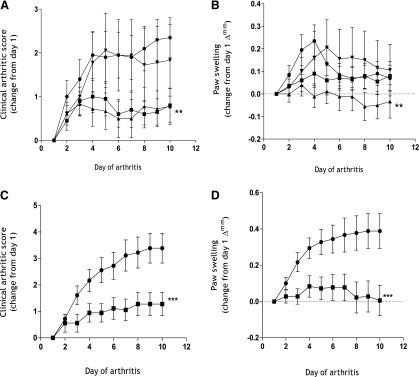
Amelioration of clinical signs of arthritis in a mouse CIA model following treatment with HU-444. (A) Clinical arthritic score (B), paw swelling (∆mm) as a change from day 1 of arthritis over a 10-day period following daily i.p. treatment with vehicle (●), and HU-444 at 2.5 (▪), 5.0 (▴), and 10 (▾) mg/kg doses. (C) Clinical arthritic score, (D) paw swelling (∆mm) as a change from day 1 of arthritis over a 10-day period following daily oral treatment with vehicle (●) and HU-444 at 15 mg/kg (▪). All treatments were initiated at day 1 of visible clinical signs of arthritis and lasted 10 days. Each symbol represents the mean ± S.E.M. of nine mice. Statistically significant difference **P < 0.05 (one-way analysis of variance with Tukey’s posttest), ***P < 0.0001 (two-way analysis of variance with Bonferroni’s posttest).
Clinical signs of arthritis were recorded daily as a composite clinical score of the four limbs, which was significantly reduced at 2.5 and 5.0 mg/kg over the entire 10-day period of HU-444 administration (Fig. 6A). In addition, hind paw thickness (mm) readings recorded as paw-swelling changes from day 1 of arthritis (∆mm) showed significant reduction with 5 mg/kg, but not with 2.5 or 10 mg/kg (Fig. 6B). Only a modest reduction in paw swelling was recorded upon i.p. injection of vehicle from days 5 to 10 of CIA. More importantly, daily oral administration of HU-444 at 15 mg/kg to arthritic mice for 10 days significantly ameliorated the clinical score and paw-swelling profiles (Fig. 6, C and D).
HU-444 also provided clinical benefits in terms of conferring protection to joints and prevention of damage caused by pathologic arthritic alterations (Fig. 7). H&E staining of joint sections of mice treated i.p. with HU-444 (2.5 mg/kg) showed a significant reduction (36.7%) in damaged joints, with a concomitant increase (18.8%) in protected joints, when compared with a vehicle-treated group (Fig. 7A). Similarly, a 45.5% reduction in damaged joints and 48.9% increase in protected joints was seen upon oral treatment of arthritic mice with HU-444 (15 mg/kg), when compared with the vehicle-treated group (Fig. 7B). Image analysis of representative proximal phalanx joints from both i.p. and oral-treated mice showed preserved joint architecture when compared with vehicle-treated mice (Fig. 7, C and D). A nonarthritic joint from an immunized mouse, as well as an arthritic joint from an untreated CIA mouse, are shown alongside for comparison (Fig. 7E). However, despite the significant joint protection seen upon treatment with HU-444, some residual synovial hypertrophy remained.
Fig. 7.

Treatment of established CIA with HU-444 protects against pathologic joint damage. Percentage of joints scored as either protected (□) or damaged (▨), following (A) i.p. treatment at different doses of HU-444 (as indicated in figure) or (B) oral treatment with HU-444 at 15 mg/kg. Statistically significant difference *P = 0.0332, ***P = 0.0006 by Fisher’s exact test. Representative H&E-stained images (captured at 100×) of proximal phalanx joints from (C) intraperitoneal, (D) oral treatment with vehicle or HU-444, as indicated, (E) nonarthritic and arthritic joint, as indicated from immunized CIA mice from an unrelated experiment are shown for comparison. Figure annotations refer to C (cartilage), B (bone), E (erosions), P (pannus), SY (synovium), and JS (joint space). Scale bars represent 200 µm optical distances.
Discussion
In the present study, we report the synthesis of a novel resorcinol derivative HU-444 (compound 5) that exhibits anti-inflammatory effects both in vitro and in vivo using mouse models of RA and inflammation-induced liver damage.
RA is an autoimmune, chronic, debilitating musculoskeletal disease (Feldmann et al., 1995). Increasingly, RA is known to be associated with cardiovascular inflammatory diseases leading to enhanced mortality in patients (Full et al., 2009). Key cellular and molecular components associated with the onset and ongoing inflammatory processes in RA have been identified, although yet the specific antigen/environmental agent triggering the onset of the autoimmune pathways remains uncharacterized (Feldmann et al., 2010).
We have previously observed that CBD and its synthetic derivate HU-320 have potent anti-inflammatory effects and ameliorate the clinical signs of RA in a mouse model (Malfait et al., 2000; Sumariwalla et al., 2004). The anti-inflammatory disease-remitting activity of CBD and HU-320 led us to explore the potential of HU-444, a new, chemically related cannabinoid compound.
We decided to synthesize HU-444 to get a compound with enhanced anti-inflammatory activity that cannot be converted under acidic conditions to a psychoactive THC-like molecule. The synthesis uses CBD as the starting material. CBD is formed in the plant (or by heating) from its precursor cannabidiolic acid (CBD acid). CBD acid is present in most Cannabis sativa varieties, including hemp, which is used for industrial purposes. In view of the growing interest in CBD (Porter and Jacobson, 2013), several C. sativa varieties have been developed that contain up to 20% CBD (or CBD acid). Hence, CBD is potentially an inexpensive natural product. The synthetic pathway from CBD to HU-444 is short, but the yield is relatively low. In view of the simplicity of the reactions used, the yields can presumably be increased, if needed.
Whereas in the past marijuana and hashish, the most widely used preparations of cannabis, contained CBD and the psychoactive ∆9-tetrahydrocannabinol (THC) in almost equal amounts (about 2–5%), many marijuana varieties today contain high levels of THC (15–20%), presumably due to mostly illegal commercial interests. These varieties contain almost no CBD, a constituent that does not cause the typical marijuana ''high'' and is hence apparently of minor interest to the illicit growers.
The in vitro assays used in the present study are well-established and widely used in anti-inflammatory research. They parallel to a large extent the assays previously used by us for the examination of the anti-inflammatory properties of CBD (Malfait et al., 2000) and HU-320 (Sumariwalla et al., 2004). We observed that HU-444 is a potent anti-inflammatory drug both in vitro and in vivo. As expected, HU-444 did not cause any of the typical THC-like pharmacological effects in mice.
We show that HU-444 suppresses in vitro the generation of ROS and NO by RAW mouse monocytes induced by zymosan and inhibits TNF-α production by TG macrophages induced by LPS. In vivo production of TNF-α elicited by LPS is also prevented by HU-444. These findings clearly show an immunosuppressive function of HU-444 on macrophage function. The most striking effect following administration of HU-444 was the amelioration of the Con A–mediated liver damage (Fig. 5). Administration of 5 mg/kg HU-444 restored to a considerable extent the normal histology of the liver. This is reflected in the reduced serum levels of the liver enzymes ALT and AST following treatment with HU-444. As Con A–induced hepatitis is considered to mimic autoimmune hepatitis in humans (Krawitt, 2006), the marked amelioration of hepatitis by HU-444 seen in our study suggests a promising curative therapeutic role for this cannabinoid compound in human autoimmune hepatitis.
In light of the encouraging anti-inflammatory properties of HU-444, we decided to explore the therapeutic potential of HU-444 in an animal model of arthritis. Mouse CIA remains a well-characterized and widely used model of RA (Williams, 1998, 2004). The CIA model is routinely used to evaluate the possible therapeutic potential of novel anti-inflammatory/disease-ameliorating compounds for use in RA (Williams, 2007). Indeed, anti–TNF-α, interleukin-Ra, and other antibody-based biologics targeting key cellular and molecular targets had proven their merits originally in this model and then in clinical settings of RA (Taylor et al., 2001). However, most of these treatments are delivered by i.v. administration and are beyond the reach of many patients due to their cost factor. Thus, the search for easily prepared, small mol. wt. compounds, which can be delivered per os, continues. A hitherto mostly untapped wealth of novel compounds is natural products from plants, known to possess anti-inflammatory properties. Indeed, C. sativa preparations and pure cannabinoids have been used to relieve symptoms of pain associated with several clinical disease conditions and ailments (Marmor, 1998; Zurier, 2003). We show in this work that our semisynthetic compound HU-444 based on the naturally occurring CBD has profound anti-inflammatory effects that relieves paw swelling and arthritis symptoms in CIA, and may thus be a potent oral drug in the therapy of human RA.
Conclusion.
Our results on the anti-inflammatory potential of HU-444 indicate that it lowers the levels of a variety of inflammatory mediators, such as TNF-α, ROS, and NO in vitro, and ameliorates arthritis in a mouse model at both the macroscopic and pathologic levels, when administered either i.p. or orally.
We believe that HU-444, a low mol. wt. compound, has the potential to be developed as a novel drug for use in inflammatory conditions, particularly in RA.
Acknowledgments
The authors thank all staff members of the Biological Services Unit at the Kennedy Institute of Rheumatology (KIR) in London for help provided in the care and maintenance of laboratory mice. Special thanks to David Essex for assistance with histology of arthritic paws. Arthritis Research UK provided a core grant facility to KIR, which helped facilitate part of this work.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CBD
cannabidiol
- CIA
collagen-induced arthritis
- Con A
concanavalin A
- DMEM
Dulbecco’s modified Eagle medium
- ELISA
enzyme-linked immunosorbent assay
- FCS
fetal calf serum
- HPLC
high-pressure liquid chromatography
- LPS
lipopolysaccharide
- NO
nitric oxide
- RA
rheumatoid arthritis
- ROS
reactive oxygen species
- TG
thioglycolate
- THC
tetrahydrocannabinol
- TNF
tumor necrosis factor
Authorship Contributions
Participated in research design: Haj, Sumariwalla, Mechoulam, Feldmann, Gallily.
Conducted experiments: Haj, Sumariwalla, Hanuš, Kogan, Yektin, Gallily.
Performed data analysis: Haj, Sumariwalla, Mechoulam, Feldmann, Gallily.
Wrote or contributed to the writing of manuscript: Haj, Sumariwalla, Mechoulam, Gallily.
Footnotes
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grant DA 978 to R.M.]; Kessler Foundation of Boston [to R.M.]; and Israel Ministry of Science [to C.G.H.].
References
- Avraham Y, Grigoriadis N, Poutahidis T, Vorobiev L, Magen I, Ilan Y, Mechoulam R, Berry E. (2011) Cannabidiol improves brain and liver function in a fulminant hepatic failure-induced model of hepatic encephalopathy in mice. Br J Pharmacol 162:1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayewitch M, Rhee MH, Avidor-Reiss T, Breuer A, Mechoulam R, Vogel Z. (1996) (-)-Delta9-tetrahydrocannabinol antagonizes the peripheral cannabinoid receptor-mediated inhibition of adenylyl cyclase. J Biol Chem 271:9902–9905. [DOI] [PubMed] [Google Scholar]
- Ben-Shabat S, Hanuš LO, Katzavian G, Gallily R. (2006) New cannabidiol derivatives: synthesis, binding to cannabinoid receptor, and evaluation of their antiinflammatory activity. J Med Chem 49:1113–1117. [DOI] [PubMed] [Google Scholar]
- Bergamaschi MM, Queiroz RH, Chagas MH, de Oliveira DC, De Martinis BS, Kapczinski F, Quevedo J, Roesler R, Schröder N, Nardi AE, et al. (2011) Cannabidiol reduces the anxiety induced by simulated public speaking in treatment-naive social phobia patients. Neuropsychopharmacology 36:1219–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braida D, Pegorini S, Arcidiacono MV, Consalez GG, Croci L, Sala M. (2003) Post-ischemic treatment with cannabidiol prevents electroencephalographic flattening, hyperlocomotion and neuronal injury in gerbils. Neurosci Lett 346:61–64. [DOI] [PubMed] [Google Scholar]
- Burstein SH, Audette CA, Breuer A, Devane WA, Colodner S, Doyle SA, Mechoulam R. (1992) Synthetic nonpsychotropic cannabinoids with potent antiinflammatory, analgesic, and leukocyte antiadhesion activities. J Med Chem 35:3135–3141. [DOI] [PubMed] [Google Scholar]
- Cunha JM, Carlini EA, Pereira AE, Ramos OL, Pimentel C, Gagliardi R, Sanvito WL, Lander N, Mechoulam R. (1980) Chronic administration of cannabidiol to healthy volunteers and epileptic patients. Pharmacology 21:175–185. [DOI] [PubMed] [Google Scholar]
- Devane WA, Breuer A, Sheskin T, Järbe TUC, Eisen MS, Mechoulam R. (1992a) A novel probe for the cannabinoid receptor. J Med Chem 35:2065–2069. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanuš L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. (1992b) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949. [DOI] [PubMed] [Google Scholar]
- Durst R, Danenberg H, Gallily R, Mechoulam R, Meir K, Grad E, Beeri R, Pugatsch T, Tarsish E, Lotan C. (2007) Cannabidiol, a nonpsychoactive Cannabis constituent, protects against myocardial ischemic reperfusion injury. Am J Physiol Heart Circ Physiol 293:H3602–H3607. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Brennan FM, Elliott MJ, Williams RO, Maini RN. (1995) TNF alpha is an effective therapeutic target for rheumatoid arthritis. Ann N Y Acad Sci 766:272–278. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Williams RO, Paleolog E. (2010) What have we learnt from targeted anti-TNF therapy? Ann Rheum Dis 69 (Suppl 1):i97–i99. [DOI] [PubMed] [Google Scholar]
- Fernández-Ruiz J, Sagredo O, Pazos MR, García C, Pertwee R, Mechoulam R, Martínez-Orgado J. (2013) Cannabidiol for neurodegenerative disorders: important new clinical applications for this phytocannabinoid? Br J Clin Pharmacol 75:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fride E, Mechoulam R. (1993) Pharmacological activity of the cannabinoid receptor agonist, anandamide, a brain constituent. Eur J Pharmacol 231:313–314. [DOI] [PubMed] [Google Scholar]
- Full LE, Ruisanchez C, Monaco C. (2009) The inextricable link between atherosclerosis and prototypical inflammatory diseases rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res Ther 11:217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaoni Y, Mechoulam R. (1971 The isolation and structure of delta-1-tetrahydrocannabinol and other neutral cannabinoids from hashish. J Am Chem Soc 93:217–224. [DOI] [PubMed] [Google Scholar]
- Hampson AJ, Grimaldi M, Lolic M, Wink D, Rosenthal R, Axelrod J. (2000) Neuroprotective antioxidants from marijuana. Ann N Y Acad Sci 899:274–282. [PubMed] [Google Scholar]
- Hayakawa K, Mishima K, Nozako M, Hazekawa M, Irie K, Fujioka M, Orito K, Abe K, Hasebe N, Egashira N, et al. (2007) Delayed treatment with cannabidiol has a cerebroprotective action via a cannabinoid receptor-independent myeloperoxidase-inhibiting mechanism. J Neurochem 102:1488–1496. [DOI] [PubMed] [Google Scholar]
- Holum JR. (1961) Study of the chromium (VI) oxide-pyridine complex. J Org Chem 26:4814–4816. [Google Scholar]
- Jamontt JM, Molleman A, Pertwee RG, Parsons ME. (2010) The effects of Δ-tetrahydrocannabinol and cannabidiol alone and in combination on damage, inflammation and in vitro motility disturbances in rat colitis. Br J Pharmacol 160:712–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozela E, Haj C, Hanuš L, Chourasia M, Shurki A, Juknat A, Kaushansky N, Mechoulam R, Vogel Z. (2015) HU-446 and HU-465, derivatives of the non-psychoactive cannabinoid cannabidiol, decrease the activation of encephalitogenic T cells. Chem Biol Drug Des, in press. [DOI] [PubMed] [Google Scholar]
- Krawitt EL. (2006) Autoimmune hepatitis. N Engl J Med 354:54–66. [DOI] [PubMed] [Google Scholar]
- Küsters S, Tiegs G, Alexopoulou L, Pasparakis M, Douni E, Künstle G, Bluethmann H, Wendel A, Pfizenmaier K, Kollias G, et al. (1997) In vivo evidence for a functional role of both tumor necrosis factor (TNF) receptors and transmembrane TNF in experimental hepatitis. Eur J Immunol 27:2870–2875. [DOI] [PubMed] [Google Scholar]
- Lander N, Ben-Zvi Z, Mechoulam R, Martin B, Nordqvist M, Agurell S. (1976) Total synthesis of cannabidiol and ∆1-THC metabolites. J Chem Soc Perkin Trans 1:8–16. [PubMed] [Google Scholar]
- Lastres-Becker I, Molina-Holgado F, Ramos JA, Mechoulam R, Fernández-Ruiz J. (2005) Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: relevance to Parkinson’s disease. Neurobiol Dis 19:96–107. [DOI] [PubMed] [Google Scholar]
- Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C, Klosterkötter J, Hellmich M, Koethe D. (2012) Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry 2:e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfait AM, Gallily R, Sumariwalla PF, Malik AS, Andreakos E, Mechoulam R, Feldmann M. (2000) The nonpsychoactive cannabis constituent cannabidiol is an oral anti-arthritic therapeutic in murine collagen-induced arthritis. Proc Natl Acad Sci USA 97:9561–9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor JB. (1998) Medical marijuana. West J Med 168:540–543. [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Compton DR, Thomas BF, Prescott WR, Little PJ, Razdan RK, Johnson MR, Melvin LS, Mechoulam R, Ward SJ. (1991) Behavioral, biochemical, and molecular modeling evaluations of cannabinoid analogs. Pharmacol Biochem Behav 40:471–478. [DOI] [PubMed] [Google Scholar]
- Martín-Moreno AM, Reigada D, Ramírez BG, Mechoulam R, Innamorato N, Cuadrado A, de Ceballos ML. (2011) Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: relevance to Alzheimer’s disease. Mol Pharmacol 79:964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi P, Solinas M, Cinquina V, Parolaro D. (2013) Cannabidiol as potential anticancer drug. Br J Clin Pharmacol 75:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Hanuš LO, Pertwee R, Howlett AC. (2014) Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat Rev Neurosci 15:757–764. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Parker LA, Gallily R. (2002) Cannabidiol: an overview of some pharmacological aspects. J Clin Pharmacol 42:11S–19S. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Peters M, Murillo-Rodriguez E, Hanuš LO. (2007) Cannabidiol: recent advances. Chem Biodivers 4:1678–1692. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Sumariwalla PF, Feldmann M, Gallily R. (2005) Cannabinoids in models of chronic inflammatory conditions. Phytochem Rev 4:11–18. [Google Scholar]
- Ohta A, Sitkovsky M. (2001) Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414:916–920. [DOI] [PubMed] [Google Scholar]
- Pazos MR, Cinquina V, Gómez A, Layunta R, Santos M, Fernández-Ruiz J, Martínez-Orgado J. (2012) Cannabidiol administration after hypoxia-ischemia to newborn rats reduces long-term brain injury and restores neurobehavioral function. Neuropharmacology 63:776–783. [DOI] [PubMed] [Google Scholar]
- Pellegata R, Ventura P, Villa M, Palmisano G, Lesma G. (1986) An improved procedure for the synthesis of oleuropeic acid. Synth Commun 15:165–170. [Google Scholar]
- Pertwee RG. (2005) Pharmacological actions of cannabinoids. Handbook Exp Pharmacol 168:1–51. [DOI] [PubMed] [Google Scholar]
- Porter BE, Jacobson C. (2013) Report of a parent survey of cannabidiol-enriched cannabis use in pediatric treatment-resistant epilepsy. Epilepsy Behav 29:574–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe R, Rodehorst R. (1970) Improved procedure for oxidations with the chromium trioxide-pyridine complex. J Org Chem 35:4000–4002. [Google Scholar]
- Rock EM, Bolognini D, Limebeer CL, Cascio MG, Anavi-Goffer S, Fletcher PJ, Mechoulam R, Pertwee RG, Parker LA. (2012) Cannabidiol, a non-psychotropic component of cannabis, attenuates vomiting and nausea-like behaviour via indirect agonism of 5-HT(1A) somatodendritic autoreceptors in the dorsal raphe nucleus. Br J Pharmacol 165:2620–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless KB, Lauer RF. (1972) Selenium dioxide oxidation of olefins: evidence for the intermediacy of allylselenic acids. J Am Chem Soc 94:7154–7155. [Google Scholar]
- Stephenson LM, Speth DR. (1979) Mechanism of allylic hydroxylation by selenium dioxide. J Org Chem 44:4683–4689. [Google Scholar]
- Sumariwalla PF, Cao Y, Wu HL, Feldmann M, Paleolog EM. (2003) The angiogenesis inhibitor protease-activated kringles 1-5 reduces the severity of murine collagen-induced arthritis. Arthritis Res Ther 5:R32–R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumariwalla PF, Gallily R, Tchilibon S, Fride E, Mechoulam R, Feldmann M. (2004) A novel synthetic, nonpsychoactive cannabinoid acid (HU-320) with antiinflammatory properties in murine collagen-induced arthritis. Arthritis Rheum 50:985–998. [DOI] [PubMed] [Google Scholar]
- Syed YY, McKeage K, Scott LJ. (2014) Delta-9-tetrahydrocannabinol/cannabidiol (Sativex®): a review of its use in patients with moderate to severe spasticity due to multiple sclerosis. Drugs 74:563–578. [DOI] [PubMed] [Google Scholar]
- Taylor PC, Williams RO, Maini RN. (2001) Immunotherapy for rheumatoid arthritis. Curr Opin Immunol 13:611–616. [DOI] [PubMed] [Google Scholar]
- Tiegs G, Hentschel J, Wendel A. (1992) A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest 90:196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss L, Zeira M, Reich S, Slavin S, Raz I, Mechoulam R, Gallily R. (2008) Cannabidiol arrests onset of autoimmune diabetes in NOD mice. Neuropharmacology 54:244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RO. (1998) Rodent models of arthritis: relevance for human disease. Clin Exp Immunol 114:330–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RO. (2004) Collagen-induced arthritis as a model for rheumatoid arthritis. Methods Mol Med 98:207–216. [DOI] [PubMed] [Google Scholar]
- Williams RO. (2007) Collagen-induced arthritis in mice. Methods Mol Med 136:191–199. [DOI] [PubMed] [Google Scholar]
- Zhornitsky S, Potvin S. (2012) Cannabidiol in humans: the quest for therapeutic targets. Pharmaceuticals (Basel) 5:529–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurier RB. (2003) Prospects for cannabinoids as anti-inflammatory agents. J Cell Biochem 88:462–466. [DOI] [PubMed] [Google Scholar]