

Abstract
Sphingosine 1-phosphate (S1P) levels are significantly higher in blood and lymph than in tissues. This S1P concentration difference is necessary for proper lymphocyte egress from secondary lymphoid tissue and to maintain endothelial barrier integrity. Studies with mice lacking either sphingosine kinase (SphK) type 1 and 2 indicate that these enzymes are the sole biosynthetic source of S1P, but they play different roles in setting S1P blood levels. We have developed a set of drug-like SphK inhibitors, with differing selectivity for the two isoforms of this enzyme. Although all SphK inhibitors tested decrease S1P when applied to cultured U937 cells, only those inhibitors with a bias for SphK2 drove a substantial increase in blood S1P in mice and this rise was detectable within minutes of administration of the inhibitor. Blood S1P also increased in response to SphK2 inhibitors in rats. Mass-labeled S1P was cleared more slowly after intravenous injection into SphK2 inhibitor–treated mice or mice lacking a functional SphK2 gene; thus, the increased accumulation of S1P in the blood appears to result from the decreased clearance of S1P from the blood. Therefore, SphK2 appears to have a function independent of generating S1P in cells. Our results suggest that differential SphK inhibition with a drug might afford a method to manipulate blood S1P levels in either direction while lowering tissue S1P levels.
Introduction
The distribution of the lipid mediator sphingosine 1-phosphate (S1P) in the body is highly compartmentalized. Although virtually all cells are capable of synthesizing S1P, the tissue levels are low compared with blood and lymph, where S1P circulates at single digit micromolar levels. The fraction of whole body S1P that is in the tissue versus circulation is difficult to know with precision. Although S1P is readily measured in blood and lymph, tissue S1P levels are low, which makes its quantification problematic. The S1P concentration differential between the circulatory and tissue compartments is necessary for both proper egress of lymphocytes from secondary lymphoid tissues and the thymus and maintaining endothelial barrier integrity (Rosen et al., 2007; Schwab and Cyster, 2007).
S1P is generated solely by sphingosine kinases (SphKs), of which there are two isoforms (SphK1 and SphK2) (Mizugishi et al., 2005). A variety of studies, summarized by Gräler and colleagues, have helped to develop a picture of bodily distribution of S1P (Sensken et al., 2010). In mice and rats (and presumably other mammals) S1P levels are high in the blood (particularly in erythrocytes), plasma, and lymph, but low in the tissues. The paucity of S1P in tissue is likely to be due to the combined actions of S1P lyase, which cleaves S1P to a long chain aldehyde and phosphoethanolamine; S1P phosphatases, which hydrolyze S1P to yield sphingosine (Sph); and extrusion of S1P into the extracellular environment via SPNS2 and other transporters. Sphingosine levels are the opposite, i.e., higher in cells than in the extracellular environment. This is probably because sphingoid bases, such as sphingosine, are avidly taken up by cells from their environment, unlike S1P.
Mice lacking functional SphK1 alleles have about one-half the circulating S1P levels of wild-type mice (Allende et al., 2004; Kharel et al., 2011). Curiously, ablation of the other SphK gene, Sphk2, in the germ line results in mice with substantially higher blood steady-state S1P levels than corresponding wild-type mice (Mizugishi et al., 2005; Zemann et al., 2006; Sensken et al., 2010; Kharel et al., 2012). Mice with no functional SphK alleles die in utero, with no detectable S1P (Mizugishi et al., 2005). We have previously documented that the results of these genetic manipulations can be recapitulated with small-molecule inhibitors of SphKs in that administration of an SphK1 inhibitor to wild-type mice or rats decreases blood S1P (Kharel et al., 2011; Patwardhan et al., 2015), whereas administration of an SphK2 inhibitor to mice raises blood S1P levels (Kharel et al., 2012).
In this paper, we describe additional investigations of the mechanisms, whereby circulating S1P levels are regulated, in particular, the phenomenon of blood S1P rise after SphK2 inhibition. To this end, we have used additional SphK inhibitors, with improved potency, selectivity, and pharmacokinetics.
Methods and Materials
Sphk1−/− and Sphk2−/− mice were gifts from Dr. Richard Proia (National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases). Male C57BL/6j mice were from the Jackson Laboratories (Bar Harbor, ME), and male Sprague-Dawley rats were from Taconic (Hudson, NY). Deuterated S1P (d7-S1P), S1P, sphingosine, C17-S1P, and C17 sphingosine were purchased from Avanti Polar Lipids (Alabaster, AL). FTY720 and S-phosho-FTY720 were gifts from Dr. Volker Brinkmann (Novartis, Basel, Switzerland).
Synthesis of SLM6041434 and SLC5111312.
The details of the synthesis of SLM6041434 [S-2-(3-[4-(octyloxy)-3-(trifluoromethyl)phenyl]-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide], SLM6081442 [R-2-(3-[4-(octyloxy)-3-(trifluoromethyl)phenyl]-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide] and SLC5111312 [(2S,3S)-3-hydroxy-2-(3-[6-(pentyloxy)naphthalen-2-yl]-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide] will be reported elsewhere (E. A. Morris, M. D. Congdon, Y. Kharel, K. R. Lynch, and W. L. Santos, manuscript in preparation).
Sphingosine Kinase Inhibition Assay.
The activity of SphK inhibitors was measured using recombinant SphK1 and SphK2 (Kharel et al., 2011). Tests were carried out by mixing cleared cell lysates from baculovirus-infected Sf9 insect cells (0.02–0.03 mg protein) with 200 μl of a reaction mixture consisting of 20 mM Tris-Cl (pH 7.4), 1 mM 2-mercaptoethanol, 1 mM EDTA, 5 mM sodium orthovanadate, 40 mM β-glycerophosphate, 15 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 10 mM MgCl2, 0.5 mM 4-deoxypyridoxine, 10% glycerol, and 0.1 mg/ml each of leupeptin, aprotinin, and soybean trypsin inhibitor. For measuring SphK2 activity, the buffer was supplemented with KCl to 100 mM. Other additions included substrates (D-erythro-sphingosine, 5–10 μM, or FTY720, 1 μM, with or without inhibitors for up to 10 μM) and [γ-32P]ATP (10 μM; specific activity = 8.3 Ci/mmol). After 30 minutes at 37°C, the reaction was stopped by placing the reaction tubes on ice. An aliquot of the reaction mixture was spotted onto 2 × 2 cm squares of P81 phosphocellulose cation exchange paper, which was washed three times with 75 mM orthophosphoric acid, followed by soaking in acetone for 2 minutes. The amount of radionuclide bound to the dried P81 paper was measured by liquid scintillation counting. For determination of KM and Vmax, SphK activity was measured with or without a fixed concentration of the inhibitor, and the effect on KM and Vmax was determined as previously described (Kharel et al., 2011, 2012).
SphK Inhibition in Cultured Cells.
The in \vivo effects of SphK inhibitors were tested on U937 cells grown in RPMI 1640 media supplemented with L-glutamate, 10% fetal bovine serum, and 1% penicillin/streptomycin at 37°C in the presence of 5% CO2. Twenty-four hours before adding the inhibitors, the growth media was replaced with media containing 0.5% fetal bovine serum. Cells were then treated for 2 hours with a SphK inhibitor in the presence or absence of the SphK2 selective substrate FTY720. Finally, levels of S1P and FTY720 phosphate (FTY720-P) in cells were assayed by liquid chromatography mass spectrometry (LC/MS).
Pharmacokinetic Analysis.
Groups of 6–12 week old mice, strain C57BL/6j, were injected i.p. with either 5 or 10 mg/kg SLM6031434, SLM6081442, or SLC5111312 or an equal volume of vehicle (2% solution of hydroxypropyl-β-cyclodextrin; Cargill Cavitron 82004; Cargill Inc., Cedar Rapids, IA). After injection, animals were bled at the specified time points (as soon as possible time points were 1–2 minutes after dosing). Whole blood was processed immediately for LC/MS analysis as described below. The use of mice and rats for these studies was approved by the University of Virginia’s School of Medicine Animal Care and Use Committee.
LC/MS Sample Preparation.
Sample preparation protocols were as described by Shaner et al. (2009), with minor modifications. Cell pellets (2 × 106 to 4 × 106 cells) or whole blood (5 μl) were mixed with 2 ml of methanol/chloroform (3:1) and transferred to a capped glass vial. Suspensions were supplemented with 10 μl of an internal standard solution containing 10 pmol each of C17 S1P or d7-S1P, C17 sphingosine, or deuterated sphingosine and deuterated SLM6031434-d17. Mixtures were placed in a bath sonicator for 10 minutes, incubated at 48°C for 16 hours, cooled to an ambient temperature, mixed with 200 μl of 1 M KOH in methanol for saponification, sonicated, and incubated for 2 hours at 37°C. Following saponification, mixtures were neutralized by the addition of 20 μl of glacial acetic acid, transferred to 2-ml microcentrifuge tubes, and centrifuged at 12,000g for 12 minutes at 4°C. The supernatant fluid was collected in a separate glass vial and evaporated under a stream of nitrogen gas. Immediately prior to LC/MS analysis, the dried material was dissolved in 0.3 ml of methanol and centrifuged at 12,000g for 12 minutes at 4°C. Ten microliters of the resulting supernatant fluid was analyzed by LC/MS.
LC/MS Analysis.
Analyses were performed by LC-ESI MS using a Waters system (Milford, MA) consisting of a triple quadrupole mass spectrometer (Xevo TQ-S) and a solvent pump (Acquity UPLC). A binary solvent gradient with a flow rate of 0.4 ml/min was used to separate sphingolipids and compounds by reverse phase chromatography using an Acquity UPLC CSH C18 column (2.1 × 100 mm, 1.7-μm bead size). Mobile phases consisted of 6:4 acetonitrile/water (mobile phase A) and 9:1 isopropanol/acetonitrile (mobile phase B). Both phases were supplemented with 0.1% formic acid. Chromatographic runs started with 100% A for 1 minute. Solvent B was then increased linearly to 100% B in 7 minutes and held at 100% for 2 minutes. The column was finally re-equilibrated to 100% A for 2 minutes. Natural sphingolipids were detected using multiple reaction monitoring in the positive mode as previously described (Kharel et al., 2012): C17-S1P (366.4 → 250.4); S1P (380.4 → 264.4); dihydroS1P (382.4 → 266.4); deuterated C18S1P-d7 (387.4 → 271.3); C17sphingosine (286.4 → 250.3); sphingosine (300.5 → 264.4); sphinganine (302.5 → 260.0); and deuterated (d7) sphingosine (307.5 → 271.3). Fragmentation of compounds SLM6031434 (as well as its enantiomer SLM6081442), its deuterated form (SLM6031434-d17), and SLC5111312 were analyzed by direct infusion of a 1 μM solution in methanol supplemented with 0.1% formic acid. It was found that the following transitions and cone and collision voltages gave the most intense signals in the positive mode: SLM6031434 and SLM6081442, 454.2→112.1, 66 and 28 V; SLM6031434-d17, 471.4→140.1, 54 and 24 V; and SLC5111312, 410.2→156.1, 54 and 18 V. Retention times for all analytes under our experimental conditions were about 2 minutes. Quantification was carried out by measuring peak areas using commercially available software (Target Lynx, Waters Corp.).
Results
Inhibitor Development.
The new SphK2 inhibitors featured in this report, SLM6031434, its enantiomer (SLM6081442), and SLC5111312 were discovered in a program to develop SphK inhibitors using SLR080811 (Kharel et al., 2012) as a lead compound. This scaffold, which yields inhibitors that are competitive with sphingosine, features a 2-substituted pyrrolidyl guanidine “warhead.” We have postulated, based on molecular modeling studies of earlier SphK inhibitors with carboximidamide groups, that the guanidine moiety chelates the gamma phosphate of ATP when bound to the SphK active site (Kennedy et al., 2011). The inhibitory constants for SLR080811, SLM6031434, SLM6081442, and SLC5111312 are presented in Table 1.
TABLE 1.
Potency of SphK inhibitors assessed in vitro with recombinant mouse and rat SphK1 and SphK2
Inhibitory constants for recombinant enzymes were obtained by kinetic analysis of S1P production using a variable concentration of sphingosine and a fixed concentration of ATP in the presence and absence of inhibitors as described previously (Kharel et al., 2012). Values corresponding to KI are expressed in micromolar units.
| Inhibitor | mSphK1 | mSphK2 | rSphK1 | rSphK2 |
|---|---|---|---|---|
| SLR080811 | 15 | 1.3 | N.D. | 1.4 |
| SLM6031434 | >20 | 0.4 | 22 | 0.5 |
| SLM6081442 | >20 | 19 | N.D. | ND |
| SLC5111312 | 20 | 1 | 0.8 | 1.1 |
mSphK1, mouse sphingosine kinase 1; mSphK2, mouse sphingosine kinase 2; N.D., not determined; rSphK1, rat sphingosine kinase 1; rSphK2, rat sphingosine kinase 2.
The lead compound, SLR080811, exhibits a KI of 1 μM and is a moderately selective (10-fold) SphK2 inhibitor (see Table 1), with a half-life in mice of about 4 hours (Kharel et al., 2012). Introduction of a 3′-trifluoromethyl group on the phenyl ring and an ether linkage to the 4′-octyl group resulted in SLM6031434, which is more potent and more SphK2 selective than the lead compound (refer to Fig. 1 for the structures). Not surprisingly, the R-enantiomer of SLM6031434, SLM6081442, was considerably less potent. Installation of a 3-OH on the pyrrolidine ring and replacement of the 4-octylphenyl group with a 6-pentoxylnaphthyl group resulted in SLC5111312. This compound was selective for mouse SphK2 as compared with mouse SphK1, with a 20-fold difference in potency (Table 1). In contrast, at the rat enzymes, SLC5111312 is a nonselective inhibitor. We and others have observed a similar differential in potency between mouse and human SphK1 over a variety of chemical scaffolds (Schnute et al., 2012; Rex et al., 2013; Zhang et al., 2014; Patwardhan et al., 2015). Importantly, SLC5111312 provides a tool that is simultaneously a SphK2 selective (mouse) and dual SphK (rat and human) inhibitor.
Fig. 1.

Chemical structures of the pyrrolidyl guanidine SphK inhibitors used in these studies.
Characterization of SphK Inhibitors in Cultured Cells.
To ascertain whether SLM6031434, SLM6081442, and SLC5111312 are active on intact cells, we used a human monocytic leukemia cell line, U937, which has robust SphK1 and SphK2 activities. Incubation of U937 cells for 2 hours with our inhibitors resulted in a concentration-dependent decrease in cell-associated S1P, with a concomitant increase in sphingosine (Fig. 2). As expected, the R-enantiomer (SLM6081442) was less potent than the S-enantiomer SLM6031434, and the dual inhibitor SLC5111312 lowered S1P levels further than the SphK2 selective inhibitor. To confirm that the inhibitors were active at SphK2, we carried out the experiments in the presence of the SphK2-selective substrate FTY720 and quantified FTY720 and FTY720-P in cell extracts. Both the SphK2 selective and SphK1/2 dual inhibitors blocked the formation of FTY720-P (Fig. 3).
Fig. 2.
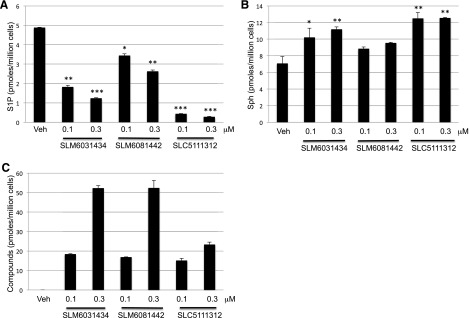
Effect of SphK2 inhibitors on sphingolipid levels in cultured U937 cells. Cells were treated for 2 hours with two different concentrations of inhibitors and then harvested and lysed, and the amounts of sphingolipids were measured by LC/MS as described in the Materials and Methods section. The amount of sphingolipids and inhibitors in the cells are expressed in pmoles per million cells: (A) S1P; (B) sphingosine; and (C) inhibitors. Data correspond to the mean ± S.D. of three independent experiments. The level of significance is indicated for each experiment (*P < 0.05; **P < 0.01; and ***P < 0.001) using one-way analysis of variance with the Bonferroni multiple comparison test compared with control.
Fig. 3.

Blockade of FTY720 phosphorylation by SphK2 inhibitors in cultured U937 cells. U937 cells were exposed to 1 μM of FTY720 and different concentrations of SLM6031434, SLM6081442, or SLC5111312, as indicated, for 2 hours. Cells were harvested by centrifugation and processed for LC/MS. Accumulation of FTY720-P was measured by LC/MS as described in the Materials and Methods section. Data correspond to the mean ± S.D. of three independent experiments. The level of significance is indicated for each experiment (**P < 0.01 and ***P < 0.001) using one-way analysis of variance with the Bonferroni multiple comparison test.
Characterization of SphK Inhibitors in Mice.
The rapidity with which blood S1P levels change in mice (see below) provides a useful pharmacodynamic marker for assessing target engagement; consequently, the SphK1- and SphK2-null mice provide a platform for assessing the selectivity of SphK inhibitors. As documented in Fig. 4, administration of the SphK2 selective inhibitors SLM6031434 or SLC5111312 to mice lacking functional SphK1 alleles (Sphk1−/−) resulted in a decrease in blood S1P. However, these inhibitors did not affect the concentration of blood S1P in SphK2-null (Sphk2−/−) mice. These results are predictable for SphK2-selective inhibitors and confirm the results that we reported previously with our lead compound SLR080811 (Kharel et al., 2012).
Fig. 4.

Blood S1P levels in Sphk1−/− (A) and Sphk2−/− (B) mice treated with SphK2 inhibitors. Cohorts of male mice (congenic on a C57BL/6j background) of the specified genotypes were injected intravenously with 5 mg/kg SLM6031434 or 10 mg/kg SLC5111312. Blood was drawn after 2 hours and analyzed for S1P levels by LC/MS as described in the Materials and Methods section. The level of significance is indicated for each experiment (**P < 0.01) using one-way analysis of variance with the Bonferroni multiple comparison test.
In contrast, the response of wild-type mice to the SphK2 inhibitors, i.e., mice that have both SphK1 and SphK2 alleles, was very different. In these mice, blood S1P increased in response to SphK2 inhibitors (Fig. 5A), which is consistent with previous observations with our lead SphK2 inhibitor (Kharel et al., 2012). Administration of the same dose of the R-enantiomer of SLM6031434 (SLM6081442) did not change blood S1P levels (Fig. 5C). The results with null mice and the less active enantiomer strongly suggest that the increase in blood S1P that we observe is due to SphK2 inhibition rather than to some off-target effect.
Fig. 5.
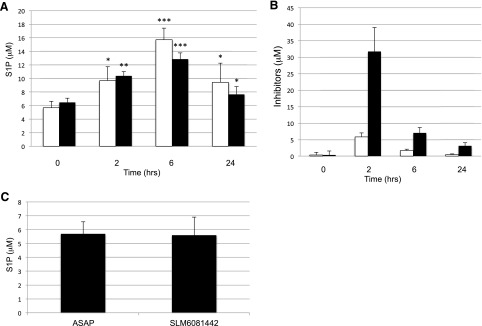
Blood S1P levels in wild-type mice injected with SphK inhibitors. C57BL/6j male mice (n = 3–4 per group) were injected (i.p.) with 5 mg/kg of SLM6031434, 10 mg/kg of SLM6081442, or 10 mg/kg SLC5111312. After the indicated periods, blood was drawn and S1P and inhibitors were extracted and measured by LC/MS. Levels of S1P (A) and inhibitors (B) in mice treated with SLM6031434 (open bars) and SLC5111312 (filled bars) are shown. (C) S1P levels in mice treated with (10 mg/kg i.p.) SLM6081442 for 6 hours. Data are presented as the mean ± S.D. of three independent experiments. The level of significance is indicated for each experiment (*P < 0.05; **P < 0.01; and ***P < 0.001) using one-way analysis of variance and the Bonferroni multiple comparison test.
Determination of the Balance Point between Raising and Lowering Blood S1P.
The observations presented in this paper with our more selective SphK2 inhibitors SLM6031434 and SLC5111312 corroborate our previous observation with a less selective inhibitor, SLR080811 (Kharel et al., 2012), and further demonstrate that SphK2 inhibition in vivo leads to an increase of blood S1P. On the other hand, as we previously demonstrated (Kharel et al., 2011), SphK1 inhibition produces a reduction of blood S1P. These data led us to ask whether a dual SphK1/SphK2 inhibitor would increase or decrease blood S1P. We were unable to perform this experiment in mice because of the loss of potency at mouse SphK1; therefore, we performed this experiment using rats. Similar to mice, SphK2 and SphK1 inhibition in this species produces an elevation and reduction of blood S1P levels, respectively, as demonstrated in Fig. 6A and a previous report (Patwardhan et al., 2015). We dosed rats with SLC5111312, a dual SphK1/SphK2 inhibitor in this species, and observed that blood S1P was lowered to about 40% of the initial level (Fig. 6B). In total, these data suggest that the balance point between blood S1P rising and falling in response to SphK inhibition lies somewhere between dual and 10-fold SphK2 selective inhibition.
Fig. 6.

Blood S1P levels in rats treated with SphK inhibitors. Sprague-Dawley male rats (8–10 weeks; n = 4) were injected with 10 mg/kg of SLR080811 (A) or SLC5111312 (B) for the indicated period. Blood was drawn, and S1P was extracted and measured by LC/MS as described in the Materials and Methods section. Data correspond to the mean ± S.D. of three independent experiments. The level of significance is indicated for each experiment (*P < 0.05 and **P < 0.01) using one-way analysis of variance and the Bonferroni multiple comparison test.
Kinetics of Blood S1P in Response to SphK2 Inhibition.
We next measured the time course of the response to SphK2 inhibition in mice. To this end, we injected SLM6031434 or vehicle i.v., drew blood over the course of an hour, and quantified S1P in blood extracts. As depicted in Fig. 7, we detected an increase in blood S1P levels as early as 5 minutes after inhibitor injection and this differential widened at 20 and 50 minutes. To determine the effect of SphK2 inhibition on the clearance of exogenous S1P from the blood, we injected vehicle or the SphK2 inhibitor SLM6031434 i.p., and after 4 hours, we administered C17-S1P (nearly all S1P in mammals is C18) by i.v. injection. We drew blood at 5, 20, and 50 minutes thereafter and quantified S1P in blood extracts. In addition, we performed the analogous experiment using C17-S1P and vehicle-injected Sphk2−/− mice. As documented in Fig. 8 and as reported by others (Peest et al., 2008; Venkataraman et al., 2008), exogenous S1P introduced by the i.v. route is rapidly removed from the blood. In the absence of SphK2 (Sphk2−/− mice) or the presence of the SphK2 inhibitor, the clearance of S1P from the blood was markedly slowed. These data suggest that changes in blood S1P in response to diminished SphK2 activity are at least in part due to changes in the rate of clearance of S1P from the blood.
Fig. 7.

Time course of blood S1P levels in wild-type mice injected intravenously with a SphK2 inhibitor. C57BL/6 male mice (6–8 weeks; n = 3–4) were injected i.v. with vehicle (square) or 2 mg/ml SLM6031434 (diamond). Blood S1P levels at various times (2, 5, 20, and 50 minutes) were measured by LC/MS. Data correspond to the mean ± S.D. of two independent experiments. The level of significance is indicated for each experiment (*P < 0.05 and **P < 0.01) using one-way analysis of variance and the Bonferroni multiple comparison test.
Fig. 8.

Decay rate of C17-S1P intravenously injected to wild-type mice treated with a SphK2 inhibitor. C57BL/6 male mice (6–8 weeks; n = 3–4) were treated with vehicle or SLM6031434 (5 mg/kg i.p.) for 4 hours. C17-S1P dissolved in mouse plasma was injected i.v. in an amount targeting an estimated initial concentration of 2 μM (assuming blood volume of the mice is 1.75 ml). Blood samples were drawn at 2, 5, 20, and 50 minutes post–C17- S1P injection, and C17-S1P was quantified by LC/MS. Data correspond to the mean ± S.D. of two independent experiments. The level of significance is indicated for each experiment (**P < 0.01) using one-way analysis of variance and the Bonferroni multiple comparison test.
Stability of S1P in Rat Blood Ex Vivo.
A potential mechanism for the clearance of blood S1P is its dephosphorylation, followed by the uptake of the resulting sphingosine by tissues. We reasoned that S1P, like the analogous lysophospholipid mediator lysophosphatidic acid, might be unstable in blood ex vivo because of the presence of the ubiquitous lipid exo-phosphatases LPP1 and LPP3. However, in contrast to lysophosphatidic acid (Tomsig et al., 2009), endogenous S1P or exogenous d7-S1P levels in blood ex vivo were unchanged over 24 hours (Fig. 9). Moreover, the stability of S1P was not due to a balanced degradation and synthesis because SphK inhibitors had no effect on S1P under these conditions (data not shown).
Fig. 9.

Time course of S1P levels in rat blood ex vivo. Blood was drawn from Sprague-Dawley male rats (8–10 weeks; n = 3) and incubated with vehicle (open bar), 1 μM SLM6031434 (filled bar), or 1 μM d7-S1P (gray bar) for the indicated time periods. Data are presented as the mean ± S.D. of three independent experiments.
Discussion
A key feature of S1P biology in rodents, and perhaps all vertebrates, is the compartmentalization of this signaling molecule. Most S1P appears to be in the blood and lymph, probably because intracellular S1P is either degraded by S1P lyase or extruded by transporters, such as SPNS2. Most importantly, the asymmetric distribution of S1P serves as a signaling mechanism. Indeed, the best understood physiologic functions of S1P, e.g., control of lymphocyte egress from lymphoid tissues and maintenance of endothelial barrier integrity, are dependent on the presence of a steep gradient of S1P between blood/lymph and tissue. Therefore, understanding the mechanisms that control the compartmentalization of S1P is important.
At the onset of our study, we focused on SphK2 because several groups of investigators, using two independently derived lines of SphK2-null mice, had reported substantially higher (2- to 4-fold) circulating S1P levels (blood and/or plasma) as compared with wild-type control mice (Mizugishi et al., 2005; Zemann et al.,2006; Sensken et al., 2010; Kharel et al., 2012). In contrast, mice with germ line ablation of the Sgpp1 gene, which encodes the degrading enzyme S1P phosphatase 1 (Allende et al., 2013) and mice with forced over-expression SphK1 via an Sphk1 transgene driven by a broad spectrum promoter (Takuwa et al., 2010) do not have differences in circulating S1P (plasma) as compared with wild-type control mice. However, mice lacking the S1P-degrading enzyme S1P lyase (via germ line ablation of Sgpl1) have serum S1P levels that are 4-fold higher than wild-type littermates (Bektas et al., 2010).
We have previously demonstrated that administration of our lead SphK2 inhibitor, SLR080811, recapitulates the blood S1P levels observed in SphK2 null mice in that it raises blood S1P levels substantially. In the present study, we found that more selective SphK2 inhibitors (SLM6031434 and SLC5111312) also raise blood S1P levels when administered to mice, further suggesting that SphK2 is the site of action of these inhibitory molecules. In addition, we extended this observation to rats. Although the response to SphK2-biased inhibitors in vivo, an increase in the enzyme’s product in response to inhibition of its catalytic activity, is counterintuitive, the response of cultured cells to the SphK2 inhibitors is not. That is, S1P levels in cultured cells decrease in response to SphK1 or SphK2 inhibitors, and the effect is more pronounced when both SphKs are blocked. Straightforward explanations for a rise in circulating S1P in response to inhibition of SphK2 include the protein having some heretofore undetected S1P-degrading capacity (for example, lyase or phosphatase activity) or an off-target effect (for example, blockade of S1P lyase). However, these scenarios would lead to increases in S1P in cultured cells treated with SphK2 inhibitors as well as in vivo, but we failed to observe such an effect in U937 cells; indeed, we observed the opposite, expected effect, which makes such explanations unlikely.
The steady-state level of blood S1P is necessarily due to a balanced appearance (i.e., synthesis) and disappearance (clearance). The increase in blood S1P in response to the SphK2-biased inhibitors requires functional SphK1, which is logical since SphK1 and SphK2 are the only biosynthetic routes to S1P (Mizugishi et al., 2005). The decrease in blood S1P in response to an unbiased (dual) inhibitor reflects the inhibition of SphK1 as well as SphK2. Presumably, dose escalation of SphK2-biased inhibitors, such as SLR080811, would decrease blood S1P once sufficient SphK1 inhibition is achieved. Conversely, a highly selective (“pure”) SphK2 inhibitor would not diminish blood S1P, regardless of the dose. Testing these predictions awaits the development of more potent and selective SphK inhibitors than are currently available. The relationship between blood and tissue S1P levels that we envision in response to SphK inhibitors, with a range of selectivity, is illustrated in Fig. 10.
Fig. 10.

Illustration of the effect of SphK inhibitors, with varying selectivity for SphK1 and SphK2, on blood and tissue S1P levels. Intracellular S1P levels decrease in response to SphK inhibitors across a spectrum of selectivity, but the magnitude of decrease after application of a biased inhibitor would depend on the relative activity of SphK1 versus SphK2. In our model, blood S1P levels are lowest with a pure SphK1 inhibitor and increase as SphK2 inhibition is added. When SphK2 inhibition is prominent (i.e., a SphK2-biased inhibitor), the effect on the decreased clearance from blood becomes dominant, as evidenced by an increase in blood S1P. Note that dose escalation of a SphK2-biased inhibitor would, in theory, shift the inflection point of the blood S1P curve rightward as the degree of SphK1 inhibition increases at higher doses.
It is conceivable that SphK2 inhibition could activate an existing SphK1 or increase the number of SphK1 molecules and thereby increase synthesis of S1P. However, two observations argue against this scenario as the sole mechanism for raising blood S1P. First, as mentioned above, forced expression of SphK1 via a transgene does increase tissue S1P but does not increase plasma S1P levels (Takuwa et al., 2010). Second, the rise in blood S1P is detectable 5 minutes after inhibitor administration, which argues against a response at the genomic level. According to our experiments, the actual mechanism seems to be related to the role of SphK2 in S1P clearance from the blood, as demonstrated by the reduced clearance of exogenous C17-S1P in SphK2-null mice and wild-type mice treated with the SphK2-selective inhibitor SLM6031434. Our results are consistent with those of Sensken et al., who reported that C17-S1P loaded into erythrocytes is cleared less efficiently by Sphk2−/− mice than by wild-type mice (Sensken et al., 2010). Regarding the phenomenon of rapid S1P clearance, our results (see Fig. 9) are consistent with previous observations on the rapid disappearance of C17-S1P injected i.v. into mice (Peest et al., 2008; Venkataraman et al., 2008; Salous et al., 2013). Further, we have reported previously that i.v. administration of a SphK1 inhibitor rapidly depletes endogenous blood S1P in mice (Kharel et al., 2011), and we have extended this observation to rats using a later generation SphK1 inhibitor (Patwardhan et al., 2015). Thus, the available data consistently indicate that blood S1P is replaced rapidly, at least in the species studied.
Taken together, the results of our present study and the aforementioned previous studies suggest that blood S1P turns over rapidly and that SphK2, in addition to its role in synthesizing S1P in cells, has a role in the clearance of S1P from the blood. The mechanism whereby S1P is cleared from the blood is not fully understood, and the role of SphK2 in this process is unknown. The clearance of S1P from the circulation has been investigated by Morris et al. (Salous et al., 2013), who reported that exogenous (i.e., i.v. injected) lysophospholipids, including C17-S1P, disappeared rapidly from mouse circulation. Further, the liver appeared to be the primary site of clearance of these exogenous lipids in those studies. Gräler et al. have proposed a facile explanation for the role of SphK2 in the removal of S1P from the blood (Mizugishi et al., 2005). These authors posited that S1P is dephosphorylated to form Sph by the ubiquitous lipid exo-phosphatase LPP1 at the cell surface and that SphK2 at the cell membrane traps Sph in its phosphorylated form, i.e., S1P. In our view, this explanation is not supported by the available data. As Peest et al. have reported and we have now confirmed (see Fig. 9), S1P is very stable in blood ex vivo. This is in contrast to the related lysophospholipid mediator lysophosphatidic acid, which is rapidly degraded in blood ex vivo, and this degradation is markedly slower in blood lacking LPP1 (Tomsig et al., 2009). Further, it is SphK1, and not SphK2, that is known to be plasma membrane associated (Pitson et al., 2003); thus, it is unclear why SphK1 could not serve to trap imported Sph in the cell in SphK2 inhibitor–treated mice or SphK2-null mice. However, we currently have no cogent explanation of the mechanism whereby SphK2 functions to remove S1P from the blood.
The effect of the SphK2 inhibitors suggests that the catalytic activity of SphK2 is required for its role in clearing S1P from the blood. However, it remains a formal possibility that inhibition of SphK2 catalysis and blockade of S1P clearance by our inhibitors are unrelated. That is, binding of, for example, SLM6031434 could stabilize a conformation that interferes with a function of SphK2 and is independent of catalysis. A mouse, wherein SphK2 has been rendered catalytically inert via a point mutation in the Sphk2 gene, would be instructive regarding the question of whether SphK2 has functions independent of catalysis.
In sum, the available data indicate that SphK2 plays a pivotal role in removing S1P from the blood and that this removal is rapid. Further, mixed SphK inhibitors with a SphK2 bias (≥10-fold) raise circulating S1P levels. The effect of SphK2 inhibitors suggests that the catalytic activity of SphK2 is required for the rise of blood S1P and that changes of S1P in the blood occur very quickly after an inhibitor is administered. The rapidity whereby S1P is cleared from circulation suggests SphK2 inhibition is due to changes in clearance rather than synthesis, and this supposition is supported by our present results. The limitations of our investigation include that measurements were made after a single dose of the inhibitor, the inhibitors tested consisted of a single scaffold (albeit with multiple chemical species), and finally, that our in vivo studies were limited to rodents. However, if this pharmacological effect is reproduced in primates and the response is sustained with chronic dosing regimens, therapeutic intervention with a SphK2-biased inhibitor could then be used to raise blood S1P while decreasing tissue S1P and thereby increasing the blood/tissue S1P gradient in patients. Such a maneuver might be useful in conditions involving ischemia reperfusion injury, wherein endothelial barrier function is compromised. Such pathologies include occlusive stroke, myocardial infarction, and acute kidney injury.
Acknowledgments
The authors thank Richard Proia (National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD) for the gifts of Sphk1−/− and Sphk2−/− mice, Volker Brinkmann (Novartis, Basel, Switzerland) for the gifts of FTY720 and FTY720-P, and Sean Lynch for the LC purification of the SphK inhibitors. The authors also thank Sayeh Agah (Pharmacology, University of Virginia) and Andrew Bolt (SphynKx Therapeutics) for their helpful suggestions during the preparation of this manuscript. Andrew Bolt also supplied an early version of the illustration that is Fig. 10.
Abbreviations
- d7-S1P
deuterated S1P
- FTY720-P
FTY720-P
- LC/MS
liquid chromatography mass spectrometry
- S1P
sphingosine 1-phosphate
- Sph
sphingosine
- SphK
sphingosine kinase
Authorship Contributions
Participated in research design: Kharel, Lynch.
Conducted experiments: Kharel, Lynch.
Contributed new reagents or analytic tools: Morris, Congdon, Thorpe, Tomsig, Santos.
Performed data analysis: Kharel, Lynch.
Wrote or contributed to the writing of the manuscript: Kharel, Tomsiq, Lynch.
Footnotes
This study was supported by grants from the National Institutes of Health [Grants R01 GM067958, R01 GM104366, and R43 GM106495]. W.L.S. and K.R.L. are among the cofounders of SphynKx Therapeutics LLC.
References
- Allende ML, Sasaki T, Kawai H, Olivera A, Mi Y, van Echten-Deckert G, Hajdu R, Rosenbach M, Keohane CA, Mandala S, et al. (2004) Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem 279:52487–52492. [DOI] [PubMed] [Google Scholar]
- Allende ML, Sipe LM, Tuymetova G, Wilson-Henjum KL, Chen W, Proia RL. (2013) Sphingosine-1-phosphate phosphatase 1 regulates keratinocyte differentiation and epidermal homeostasis. J Biol Chem 288:18381–18391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bektas M, Allende ML, Lee BG, Chen W, Amar MJ, Remaley AT, Saba JD, Proia RL. (2010) Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J Biol Chem 285:10880–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy AJ, Mathews TP, Kharel Y, Field SD, Moyer ML, East JE, Houck JD, Lynch KR, Macdonald TL. (2011) Development of amidine-based sphingosine kinase 1 nanomolar inhibitors and reduction of sphingosine 1-phosphate in human leukemia cells. J Med Chem 54:3524–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharel Y, Mathews TP, Gellett AM, Tomsig JL, Kennedy PC, Moyer ML, Macdonald TL, Lynch KR. (2011) Sphingosine kinase type 1 inhibition reveals rapid turnover of circulating sphingosine 1-phosphate. Biochem J 440:345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharel Y, Raje M, Gao M, Gellett AM, Tomsig JL, Lynch KR, Santos WL. (2012) Sphingosine kinase type 2 inhibition elevates circulating sphingosine 1-phosphate. Biochem J 447:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. (2005) Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol 25:11113–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan NN, Morris EA, Kharel Y, Raje MR, Gao M, Tomsig JL, Lynch KR, Santos WL. (2015) Structure-activity relationship studies and in vivo activity of guanidine-based sphingosine kinase inhibitors: discovery of SphK1- and SphK2-selective inhibitors. J Med Chem 58:1879–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peest U, Sensken SC, Andréani P, Hänel P, Van Veldhoven PP, Gräler MH. (2008) S1P-lyase independent clearance of extracellular sphingosine 1-phosphate after dephosphorylation and cellular uptake. J Cell Biochem 104:756–772. [DOI] [PubMed] [Google Scholar]
- Pitson SM, Moretti PA, Zebol JR, Lynn HE, Xia P, Vadas MA, Wattenberg BW. (2003) Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J 22:5491–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex K, Jeffries S, Brown ML, Carlson T, Coxon A, Fajardo F, Frank B, Gustin D, Kamb A, Kassner PD, et al. (2013) Sphingosine kinase activity is not required for tumor cell viability. PLoS One 8:e68328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H, Sanna MG, Cahalan SM, Gonzalez-Cabrera PJ. (2007) Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends Immunol 28:102–107. [DOI] [PubMed] [Google Scholar]
- Salous AK, Panchatcharam M, Sunkara M, Mueller P, Dong A, Wang Y, Graf GA, Smyth SS, Morris AJ. (2013) Mechanism of rapid elimination of lysophosphatidic acid and related lipids from the circulation of mice. J Lipid Res 54:2775–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnute ME, McReynolds MD, Kasten T, Yates M, Jerome G, Rains JW, Hall T, Chrencik J, Kraus M, Cronin CN, et al. (2012) Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem J 444:79–88. [DOI] [PubMed] [Google Scholar]
- Schwab SR, Cyster JG. (2007) Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol 8:1295–1301. [DOI] [PubMed] [Google Scholar]
- Sensken SC, Bode C, Nagarajan M, Peest U, Pabst O, Gräler MH. (2010) Redistribution of sphingosine 1-phosphate by sphingosine kinase 2 contributes to lymphopenia. J Immunol 184:4133–4142. [DOI] [PubMed] [Google Scholar]
- Shaner RL, Allegood JC, Park H, Wang E, Kelly S, Haynes CA, Sullards MC, Merrill AH., Jr (2009) Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J Lipid Res 50:1692–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuwa N, Ohkura S, Takashima S, Ohtani K, Okamoto Y, Tanaka T, Hirano K, Usui S, Wang F, Du W, et al. (2010) S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovasc Res 85:484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomsig JL, Snyder AH, Berdyshev EV, Skobeleva A, Mataya C, Natarajan V, Brindley DN, Lynch KR. (2009) Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem J 419:611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman K, Lee YM, Michaud J, Thangada S, Ai Y, Bonkovsky HL, Parikh NS, Habrukowich C, Hla T. (2008) Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res 102:669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemann B, Kinzel B, Müller M, Reuschel R, Mechtcheriakova D, Urtz N, Bornancin F, Baumruker T, Billich A. (2006) Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood 107:1454–1458. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Berka V, Song A, Sun K, Wang W, Zhang W, Ning C, Li C, Zhang Q, Bogdanov M, et al. (2014) Elevated sphingosine-1-phosphate promotes sickling and sickle cell disease progression. J Clin Invest 124:2750–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]