

Abstract
Aldosterone interacts with mineralocorticoid receptor (MR) to stimulate sodium reabsorption in renal tubules and may also affect the vasculature. Caveolin-1 (cav-1), an anchoring protein in plasmalemmal caveolae, binds steroid receptors and also endothelial nitric oxide synthase, thus limiting its translocation and activation. To test for potential MR/cav-1 interaction in the vasculature, we investigated if MR blockade in cav-1–replete or –deficient states would alter vascular function in a mouse model of low nitric oxide (NO)–high angiotensin II (AngII)–induced cardiovascular injury. Wild-type (WT) and cav-1 knockout mice (cav-1−/−) consuming a high salt diet (4% NaCl) received Nω-nitro-l-arginine methyl ester (L-NAME) (0.1–0.2 mg/ml in drinking water at days 1–11) plus AngII (0.7–2.8 mg/kg per day via an osmotic minipump at days 8–11) ± MR antagonist eplerenone (EPL) 100 mg/kg per day in food. In both genotypes, blood pressure increased with L-NAME + AngII. EPL minimally changed blood pressure, although its dose was sufficient to block MR and reverse cardiac expression of the injury markers cluster of differentiation 68 and plasminogen activator inhibitor-1 in L-NAME+AngII treated mice. In aortic rings, phenylephrine and KCl contraction was enhanced with EPL in L-NAME+AngII treated WT mice, but not cav-1−/− mice. AngII-induced contraction was not different, and angiotensin type 1 receptor expression was reduced in L-NAME + AngII treated WT and cav-1−/− mice. In WT mice, acetylcholine-induced relaxation was enhanced with L-NAME + AngII treatment and reversed with EPL. Acetylcholine relaxation in cav-1−/− mice was greater than in WT mice, not modified by L-NAME + AngII or EPL, and blocked by ex vivo L-NAME, 1H-(1,2,4)oxadiazolo(4,3-a)quinoxalin-1-one (ODQ), or endothelium removal, suggesting the role of NO-cGMP. Cardiac endothelial NO synthase was increased in cav-1−/− versus WT mice, further increased with L-NAME + AngII, and not affected by EPL. Vascular relaxation to the NO donor sodium nitroprusside was increased with L-NAME + AngII in WT mice but not in cav-1−/− mice. Plasma aldosterone levels increased and cardiac MR expression decreased in L-NAME + AngII treated WT and cav-1−/− mice and did not change with EPL. Thus, during L-NAME + AngII induced hypertension, MR blockade increases contraction and alters vascular relaxation via NO-cGMP, and these changes are absent in cav-1 deficiency states. The data suggest a cooperative role of MR and cav-1 in regulating vascular contraction and NO-cGMP–mediated relaxation during low NO–high AngII–dependent cardiovascular injury.
Introduction
The renin-angiotensin-aldosterone (ALDO) system (RAAS) is a major regulator of sodium balance, plasma volume, and blood pressure (BP) (Hall, 1986; Guyton, 1991; Ferrario and Strawn, 2006). Likewise, the endothelial nitric oxide (NO) system is an important regulator of vascular function and a key factor in the vascular control of BP (Fleming and Busse, 1999; Murad, 2006). Increased circulating angiotensin II (AngII) and decreased vascular NO levels are associated with several pathologic cardiovascular conditions, such as heart failure, diabetes mellitus, and arteriosclerosis. Mineralocorticoid receptor (MR) blockade has been shown to provide an added benefit to individuals suffering from these conditions, suggesting that excessive ALDO/MR signaling is an important mechanism in the etiology of these diseases. In support of this concept, our group has shown that low NO–high AngII in mice cause myocardial injury and cardiovascular inflammation (Martinez et al., 2002; Oestreicher et al., 2003), and that MR blockade prevents Nω-nitro-l-arginine methyl ester (L-NAME)+AngII–induced cardiovascular injury (Martinez et al., 2002). These studies have suggested that the cardiovascular damage associated with low NO–high AngII states is mediated by ALDO/MR and involves mechanisms independent of changes in BP or AngII levels (Martinez et al., 2002; Oestreicher et al., 2003).
Caveolin-1 (cav-1) is a transmembrane protein in the plasma membrane caveolae of many cell types. In the heart, cav-1 plays a role in regulating systolic and diastolic cardiac function (Wunderlich et al., 2006). In endothelial cells, cav-1 anchors endothelial NO synthase (eNOS), thus limiting its translocation and phosphoactivation (Feron et al., 1998; Segal et al., 1999; Batova et al., 2006) and its capacity to produce NO and activate the NO-cGMP relaxation pathway (Fleming and Busse, 1999; Ignarro, 2002; Murad, 2006). Studies in cav-1 null mice on a normal salt diet have suggested a role for cav-1 in mechanotransduction, vascular remodeling, and cardiovascular function (Drab et al., 2001; Razani et al., 2001; Wunderlich et al., 2006; Yu et al., 2006). Also, we have reported that on a high salt diet, cav-1–deficient mice display altered vascular reactivity and eNOS activity, which can be reversed in part by dietary sodium restriction or MR blockade (Pojoga et al., 2008, 2010a). We have also shown that MR colocalizes with cav-1 in endothelial cells (Coutinho et al., 2014) and cardiac and renal vessels (Pojoga et al., 2010b), suggesting a role for cav-1 as a modulator of ALDO/MR signaling in the vasculature. Importantly, we have shown that in low NO–high AngII states, cav-1–deficient mice develop less myocardial damage compared with their wild-type (WT) counterparts (Pojoga et al., 2010b). However, it is unknown whether cav-1 deficiency in these low NO–high AngII states is also associated with altered vascular MR signaling. In this report, we assess whether cav-1 has a mechanistic role in ALDO/MR-mediated vascular responses in a mouse model of low NO–high AngII-induced end organ damage.
Materials and Methods
Animals.
Male 12-week-old cav-1 knockout (cav-1−/−) and genetically matched WT mice purchased from The Jackson Laboratory (Bar Harbor, ME). The genotypes were confirmed by polymerase chain reaction (PCR) according to the guidelines of the The Jackson Laboratory. Mice were housed in the animal facility in a 12-hour light/dark cycle at 22 ± 1°C ambient temperature and maintained on ad libitum normal Purina rodent chow (0.8% NaCl; Purina, St. Louis, MO) and tap water. After 3 days of acclimatization, mice from each genotype were placed on a high-salt diet (HS) (4% NaCl) for 5 days to achieve sodium balance (Holtzman et al., 1988; Oliverio et al., 2000) and were maintained on the HS diet for an additional 11 days. To induce cardiovascular injury, the mice received L-NAME in drinking water during days 1–11 plus AngII SQ via an osmotic minipump during days 8–11 as previously described (Martinez et al., 2002; Oestreicher et al., 2003; Pojoga et al., 2010b). To induce moderate cardiovascular injury, low-dose (LD) L-NAME (0.1 mg/ml)+AngII (0.7 mg/kg per day) was used. To induce severe cardiovascular injury, high-dose (HD) L-NAME (0.2 mg/ml)+AngII (2.8 mg/kg per day) was used. Some of the L-NAME + AngII treated mice were treated with the MR antagonist eplerenone (EPL) 100 mg/kg per day in food for 11 days. To confirm that the findings are related to MR blockade rather than a specific MR blocker, some of the LD L-NAME + AngII treated mice were treated with the MR antagonist spironolactone 500 mg/kg in drinking water for 11 days, and inclusion of these data did not affect the general direction of the results. All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the guidelines of the Harvard Medical Area Standing Committee on Animals.
BP.
Systolic BP was measured in conscious mice after reaching a sodium balance on days 0 and 13 using tail-cuff plethysmography (BP analyzer, model 179; IITC Life Science, Woodland Hills, CA). Mice were warmed at 30°C and allowed to rest quietly. BP measurements were taken in a quiet room, and the mice were kept calm and handled by the same person. No sedation was used. We have previously demonstrated excellent correlation between systolic BPs assessed simultaneously by tail cuff and telemetry in mice (Guo et al., 2006).
Plasma ALDO Levels.
Mice were euthanized under deep anesthesia with isoflurane. Blood was collected in purple-top BD Microtainer tubes (Franklin Lakes, NJ) (EDTA), plasma was separated by centrifugation, and ALDO levels were determined in duplicates (200 μl each) using a solid-phase RIA kit (Diagnostic Products Corp., Los Angeles, CA).
Tissue Preparation.
In euthanized mice, the thoracic cavity was opened, and the aorta and heart were rapidly excised. The thoracic aorta was placed in oxygenated Krebs solution, carefully dissected, cleaned of connective tissue under microscopic visualization, and cut into 2-mm-wide rings. Sections of the aorta and heart were placed in liquid nitrogen immediately after collection in preparation for mRNA and protein analysis.
Isometric Contraction and Relaxation.
Aortic rings were suspended between two tungsten wire hooks. One hook was fixed at the bottom of a tissue bath, and the other hook was connected to a Grass force transducer (FT03; Astro-Med Inc., West Warwick, RI). Aortic rings were stretched under 0.5 g of resting tension and allowed to equilibrate in a temperature controlled, water-jacketed tissue bath filled with 50 ml of Krebs solution bubbled with 95% O2 and 5% CO2 at 37°C. The changes in isometric contraction were recorded on a Grass polygraph (Model 7D; Astro-Med).
After tissue equilibration, a control contraction to 96 mM KCl was elicited. Once maximum KCl contraction was reached, the tissue was rinsed with Krebs’ three times, 10 minutes each. Aortic segments were stimulated with increasing concentrations of phenylephrine (Phe) (10−9 to 10−5 M), concentration-contraction curves were constructed, and the maximal Phe contraction was measured. The individual Phe concentration-response curves were further analyzed using a nonlinear regression curve (best-fit sigmoidal dose-response curve; Sigmaplot, San Jose, CA), and the effective concentration that produced half the maximal contraction (ED50) was measured and presented as pED50 (−log M). In other experiments, the tissues were precontracted with Phe (10−5 M), increasing concentrations (10−9 to 0−5 M) of acetylcholine (ACh) were added, and the percentage of relaxation of Phe contraction was measured. Parallel contraction and relaxation experiments were performed in endothelium-intact aortic rings pretreated with the NOS inhibitor L-NAME (3×10−4 M) or the guanylate cyclase inhibitor 1H-(1,2,4)oxadiazolo(4,3-a)quinoxalin-1-one (ODQ) (10−5 M) for 15 minutes. The contraction to Phe and relaxation to ACh were also measured in endothelium-denuded aortic rings prepared by rubbing the vessel interior five times around the tip of a fine forceps. To test the ability of vascular smooth muscle (VSM) to respond to vasodilators, the relaxation of Phe-precontracted aortic rings to the exogenous NO donor sodium nitroprusside (SNP) was measured. Individual ACh and SNP concentration-relaxation curves were further analyzed using a nonlinear regression best-fit sigmoidal dose-response curve, and ACh and SNP ED50 were calculated.
Real-Time PCR.
Total mRNA was extracted from the hearts using the RNeasy mini kit (Qiagen, Germantown, MD). cDNA was synthesized from 1.5 μg RNA with the first-strand cDNA synthesis kit (GE Healthcare, Piscataway, NJ). PCR amplification reactions to detect MR, angiotensin type 1 receptor (AT1R), eNOS, the macrophage and inflammation markers CD68, plasminogen activator inhibitor-1 (PAI-1), and housekeeping 18S ribosomal RNA were performed in duplicate using TaqMan gene expression assays (proprietary primers and probes designed and synthesized by Applied Biosystems, Foster City, CA) and the ABI Prism 7000 sequence detection system (Applied Biosystems). The mRNA expression levels, as normalized to 18S ribosomal RNA levels, were determined using the ΔΔCT method, and the data were presented as a fold increase relative to the measurements in control WT mice.
Western Blot Analysis.
Protein was extracted by homogenizing cardiac tissue with RIPA lysis buffer (Santa Cruz Biotechnology Inc., Dallas, TX). Protein extracts (40 μg) were combined with an equal volume of 2× Laemmli loading buffer, boiled for 5 minutes, and size fractionated by electrophoresis on 7.5% SDS-polyacrylamide gels. Proteins were transferred from the gel to a nitrocellulose membrane by electroblotting. Membranes were incubated in 5% nonfat dried milk in Tris-buffered saline–Tween (USB Corporation, Cleveland, OH) for 1 hour, and then overnight at 4°C with mouse anti-eNOS antibody (1:2500; BD Transduction Laboratories, San Diego, CA). The nitrocellulose membranes were washed 5 × 15 minutes in Tris-buffered saline–Tween, incubated with horseradish peroxidase–conjugated secondary antibody for 1.5 hours, and the reactive proteins were detected using enhanced chemiluminescence (PerkinElmer Life Sciences, Boston, MA) and analyzed with optical densitometry. The blots were subsequently reprobed for β-actin (1:5000; Sigma-Aldrich, St. Louis, MO), and the data were normalized to β-actin to correct for loading. Data were presented as a fold increase relative to the measurements in control WT mice.
Solutions and Drugs.
Krebs solution contained (in mM) 120 NaCl, 5.9 KCl, 25 NaHCO3, 1.2 NaH2PO4, 11.5 dextrose, 2.5 CaCl2, and 1.2 MgCl2, at pH 7.4, and was bubbled with 95% O2 and 5% CO2. High (96 mM) KCl was prepared as Krebs’ with equimolar substitution of NaCl with KCl. Stock solutions of Phe, ACh, SNP, and L-NAME (10−1 M; Sigma-Aldrich) were prepared in distilled water. A stock solution of ODQ (10−1 M) was prepared in dimethylsulfoxide. The final concentration of dimethylsulfoxide in the experimental solution was <0.1%. All other chemicals were of reagent grade or better.
Statistical Analysis.
Data were presented as mean ± S.E.M., with n equaling the number of mice. Data were first analyzed using analysis of variance for the comparison of different groups (cav-1 status versus L-NAME + AngII treatment). When a statistical difference was observed, the data were further analyzed using the Student-Newman-Keuls post hoc test for multiple comparisons and Student’s t test for the comparison of two means. Differences were considered significant if P < 0.05. In all studies, experiments were performed blinded to the animal genotype and treatment. Some of the initial measurements of body weight, BP, CD68, and PAI-1 and AT1R mRNA in control and HD L-NAME + AngII groups were partially included in a previous report (Pojoga et al., 2010b).
Results
Body weight was similar in WT and cav-1−/− mice and did not differ across treatment groups, although there was a tendency for cav-1−/− mice to have lower body weights, especially when receiving L-NAME + AngII (Table 1). In WT and cav-1−/− mice, heart weight and heart/body weight ratio were similar across treatment groups, except for the HD L-NAME + AngII treatment group, as compared with control, which was associated with a significant increase in heart weight in WT mice and heart/body weight ratio in cav-1−/− mice (Table 1).
TABLE 1.
Body and heart weight, Phe and KCl contraction, and ACh and SNP relaxation in aortic rings of control and LD or HD L-NAME + AngII ± EPL–treated WT and cav-1−/− mice
Data represent mean ± S.E.M.; n = 4–13
| WT |
cav-1−/− |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Control | L-NAME + AngII LD | L-NAME + AngII HD | L-NAME + AngII LD + EPL | L-NAME + AngII HD + EPL | Control | L-NAME + AngII LD | L-NAME + AngII HD | L-NAME + AngII LD + EPL | L-NAME + AngII HD + EPL | |
| Body weight (g) | 25.9 ± 0.9 | 26.3 ± 1.4 | 28.4 ± 1.1 | 25.6 ± 1.8 | 26.7 ± 2.0 | 23.5 ± 0.09 | 22.3 ± 0.9# | 21.8 ± 0.7# | 21.2 ± 0.2# | 22.8 ± 1.3 |
| Heart weight (mg) | 154.4 ± 8.3 | 175.5 ± 28.4 | 182.4 ± 5.6* | 158.5 ± 17.0 | 186.0 ± 15.2 | 150.6 ± 7.2 | 158.8 ± 11.6 | 169.0 ± 9.0 | 168.3 ± 19.2 | 198.9 ± 18.8 |
| Heart/body (mg/g) | 6.0 ± 0.2 | 6.6 ± 0.9 | 6.5 ± 0.2 | 6.1 ± 0.3 | 7.4 ± 1.1 | 6.5 ± 0.3 | 7.1 ± 0.5 | 7.9 ± 0.4*,# | 7.9 ± 0.9* | 8.9 ± 0.9 |
| Phe maximum (10−5 M) | ||||||||||
| Contraction (g) | 0.42 ± 0.09 | 0.50 ± 0.10 | 0.37 ± 0.08 | 0.90 ± 0.28* | 0.88 ± 0.12*,§ | 0.35 ± 0.12 | 0.50 ± 0.18 | 0.28 ± 0.06 | 0.37 ± 0.15 | 0.35 ± 0.07# |
| +L-NAME (3 × 10−4 M) | 0.94 ± 0.20† | 0.74 ± 0.24 | 0.36 ± 0.06 | 1.07 ± 0.13 | 1.10 ± 0.34 | 0.78 ± 0.18 | 1.27 ± 0.08† | 0.65 ± 0.10† | 0.68 ± 0.30 | 0.41 ± 0.14 |
| +ODQ (10−5 M) | 0.79 ± 0.25 | 0.75 ± 0.15 | 0.42 ± 0.03 | 1.39 ± 0.42 | 0.86 ± 0.30 | 0.63 ± 0.30 | 0.97 ± 0.17 | 0.40 ± 0.05 | 0.48 ± 0.32 | 0.71 ± 0.13† |
| −Endo | 0.73 ± 0.07 | 0.50 ± 0.34 | N.D. | 0.89 ± 0.21 | N.D. | 0.45 ± 0.14 | 0.88 ± 0.13 | N.D. | 0.52 ± 0.14 | N.D. |
| Phe pED50 (−log M) | 7.19 ± 0.13 | 7.37 ± 0.23 | 7.09 ± 0.21 | 7.75 ± 0.85 | 6.85 ± 0.12 | 6.91 ± 0.12 | 7.26 ± 0.23 | 6.90 ± 0.10 | 7.12 ± 0.32 | 6.88 ± 0.15 |
| +L-NAME | 7.37 ± 0.12 | 6.96 ± 0.29 | 7.22 ± 0.77 | 7.28 ± 0.50 | 6.47 ± 0.44 | 6.72 ± 0.20# | 8.12 ± 0.36* | 6.08 ± 0.43# | 6.26 ± 0.38 | 6.43 ± 0.22 |
| +ODQ | 7.27 ± 0.17 | 7.07 ± 0.32 | 6.05 ± 0.01*,† | 7.63 ± 1.08 | 6.38 ± 0.69 | 6.62 ± 0.24 | 7.04 ± 0.64 | 6.26 ± 0.38 | 8.22 ± 0.52* | 6.45 ± 0.31 |
| −Endo | 7.13 ± 0.10 | 7.64 ± 0.40 | N.D. | 7.93 ± 1.18 | N.D. | 6.92 ± 0.07 | 7.71 ± 0.79 | N.D. | 7.02 ± 0.34 | N.D. |
| KCl contraction (g) | 0.30 ± 0.06 | 0.39 ± 0.08 | 0.33 ± 0.07 | 0.66 ± 0.19 | 0.63 ± 0.10* | 0.28 ± 0.10 | 0.45 ± 0.17 | 0.33 ± 0.07 | 0.32 ± 0.13 | 0.40 ± 0.04 |
| Phe maximum contraction | ||||||||||
| (% 96 mM KCl) | 137.1 ± 11.4 | 129.4 ± 11.1 | 117.4 ± 10.2 | 153.3 ± 20.3 | 141.8 ± 8.9 | 125.6 ± 10.9 | 132.2 ± 15.2 | 101.9 ± 20.2 | 124.7 ± 19.8 | 90.1 ± 10.5*,# |
| +L-NAME | 196.8 ± 22.4† | 141.7 ± 17.3 | 121.4 ± 30.2 | 160.1 ± 2.5 | 124.7 ± 3.2 | 179.4 ± 42.4 | 176.7 ± 6.7 | 180.5 ± 11.3 | 130.3 ± 16.0 | 98.2 ± 25.6 |
| +ODQ | 186.3 ± 17.1† | 164.0 ± 13.0 | 108.4 ± 19.3* | 139.2 ± 46.7 | 133.9 ± 2.5 | 115.1 ± 27.5 | 94.1 ± 9.5# | 176.6 ± 9.9 | 145.0 ± 42.0 | 124.5 ± 24.3 |
| −Endo | 115.4 ± 7.6 | 100.0 ± 16.7 | N.D. | 150.4 ± 33.2 | N.D. | 158.2 ± 10.1# | 225.6 ± 66.5 | N.D. | 202.8 ± 44.0 | N.D. |
| ACh (10−5 M) % relaxation | 22.3 ± 4.3 | 19.6 ± 3.8 | 51.8 ± 9.6* | 26.2 ± 10.8 | 35.6 ± 13.1 | 68.3 ± 6.9 | 70.7 ± 9.1 | 77.1 ± 7.2 | 69.4 ± 10.6 | 72.7 ± 10.9 |
| pED50 (−log M) | 7.00 ± 0.35 | 6.50 ± 0.21 | 7.53 ± 0.35 | 6.29 ± 0.35 | 6.96 ± 0.13 | 6.95 ± 0.18 | 6.60 ± 0.20 | 6.88 ± 0.23 | 6.66 ± 0.29 | 6.97 ± 0.27 |
| SNP (10−8 M) % relaxation | 45.6 ± 7.1 | 47.5 ± 5.9 | 78.1 ± 11.4* | 73.3 ± 13.1 | 67.6 ± 18.3 | 72.61 ± 7.91 | 67.2 ± 6.8 | 66.5 ± 16.4 | 85.4 ± 9.1 | 51.8 ± 21.5 |
| pED50 (−log M) | 7.94 ± 0.13 | 7.88 ± 0.23 | 8.63 ± 0.38 | 8.38 ± 0.14 | 8.41 ± 0.38 | 8.32 ± 0.20 | 8.38 ± 0.14 | 8.26 ± 0.26 | 8.74 ± 0.15 | 8.11 ± 0.40 |
N.D., not determined because of insufficient mouse tissue.
Significantly different (P < 0.05) from respective control WT or cav-1−/− mice; §P < 0.05, EPL-treated versus without EPL treatment; #P < 0.05, cav-1−/− versus WT; †P < 0.05, L-NAME or ODQ-treated or endothelium-denuded arteries versus control nontreated arteries.
Effect of L-NAME + AngII on BP.
In WT mice, there was a dose-response relationship between L-NAME + AngII treatment and BP, with a minimal BP reduction when EPL was added to the HD L-NAME + AngII (Fig. 1A). In contrast, in cav-1−/− mice, two notable differences were observed when compared with WT mice. First, consistent with the reported increase in eNOS activity and NO levels in cav-1−/− mice (Razani et al., 2001; Wunderlich et al., 2006; Pojoga et al., 2008, 2010a), BP significantly increased even with only a low dose of L-NAME (Fig. 1B). Second, the BP responses to all treatments were generally greater in cav-1−/− versus WT mice. However, similar to WT mice, EPL also had little effect on reducing the elevated BP in cav-1−/− mice (Fig. 1B).
Fig. 1.

Systolic BP in control and LD or HD L-NAME + AngII ± EPL treated WT (A) and cav-1−/− mice (B). Data represent mean ± S.E.M. (n = 4–8). *Significantly different (P < 0.05) from respective control WT or cav-1−/− mice; §P < 0.05, HD versus respective LD; #P < 0.05, cav-1−/− versus WT mice.
Effects of L-NAME + AngII on RAAS.
Because the effects of L-NAME + AngII treatment on BP could partly involve the RAAS, we assessed the RAAS in WT and cav-1−/− mice. LD L-NAME + AngII had a minimal effect on plasma ALDO levels in WT mice and a relatively greater effect in cav-1−/− mice (Fig. 2A). The EPL dose used was effective in blocking the MR, as reflected in the increase in ALDO levels. Importantly, the increase in ALDO levels with EPL was greater in cav-1−/− mice than in WT mice, suggesting a sufficient blockade of MR with the EPL dose used (Fig. 2A). Real-time PCR analysis revealed that under control conditions, cardiac AT1R mRNA, but not MR mRNA levels, were greater in cav-1−/− mice than in WT mice (Fig. 2, B and C). However, both MR and AT1R were appropriately downregulated in the L-NAME + AngII treated groups secondary to the increased levels of their agonists ALDO and AngII, respectively (Fig. 2, B and C). The decreases in MR and AT1R with L-NAME + AngII could still be observed with EPL treatment in both genotypes (Fig. 2, B and C), although there was a small but significant increase in AT1R mRNA with EPL in the L-NAME + AngII treated cav-1−/− mice (Fig. 2C).
Fig. 2.

Plasma ALDO (A), MR mRNA (B), and AT1R mRNA (C) levels in cardiac tissue of control and LD or HD L-NAME + AngII ± EPL treated WT and cav-1−/− mice. The effects of HD L-NAME + AngII on ALDO levels are not shown as they were published in a previous report (Pojoga et al., 2010b). Data represent mean ± S.E.M. (n = 4–8). *Significantly different (P < 0.05) from respective control WT or cav-1−/− mice; †P < 0.05, EPL-treated versus without EPL treatment; #P < 0.05, cav-1−/− versus WT mice.
Effect of EPL on L-NAME + AngII–Induced Cardiac Injury.
It could be argued that the lack of effects of EPL on reversing the L-NAME + AngII–induced increases in BP is due to an inadequate dose of EPL. We have previously documented, both with histologic and molecular biology tools, that this dose of EPL can reduce the cardiac injury observed with L-NAME + AngII administration (Rocha et al., 2000; Martinez et al., 2002; Oestreicher et al., 2003). In this study, we assessed cardiovascular injury by measuring cardiac mRNA levels for the macrophage marker CD68 and the inflammation marker PAI-1. In WT mice, L-NAME + AngII treatment, as compared with control, increased cardiac mRNA levels for CD68 (Fig. 3A) and PAI-1 (Fig. 3B). The L-NAME + AngII–induced increases in CD68 and PAI-1 expression were blunted when WT mice received EPL. In cav-1−/− mice, L-NAME + AngII treatment also increased CD68 mRNA levels, and this increase was blunted with EPL (Fig. 3A). In contrast to the results in WT mice, L-NAME + AngII treatment did not increase PAI-1 mRNA levels in cav-1−/− mice. Further, the addition of EPL to L-NAME + AngII treatment led to an increase in PAI-1 rather than a decrease in PAI-1, as observed in WT mice (Fig. 3).
Fig. 3.

Cardiac mRNA expression of the inflammation markers CD68 (A) and PAI-1 (B) in control and LD or HD L-NAME + AngII ± EPL treated WT and cav-1−/− mice. Data represent mean ± S.E.M. (n = 4–8). *Significantly different (P < 0.05) from respective control WT or cav-1−/− mice; †P < 0.05, EPL-treated versus without EPL treatment; #P < 0.05, cav-1−/− versus WT mice.
EPL Enhances Vascular Contraction Mechanisms in WT but Not Cav-1−/−.
To assess if the observed changes in BP are related to changes in vascular contraction mechanisms, aortic rings were isolated from control and L-NAME + AngII±EPL treated WT and cav1−/− mice and their vasoreactivity was measured. Phe contraction was reduced in the aorta of cav-1−/− mice as compared with WT mice (Fig. 4). L-NAME + AngII treatment, as compared with control, did not significantly influence Phe contraction in either WT or cav1−/− mice. However, the vascular response to Phe during combined L-NAME + AngII + EPL treatment differed by genotype and was enhanced in L-NAME + AngII + EPL treated versus control in WT mice (Fig. 4A) but not in cav-1−/− mice (Fig. 4B). When Phe contraction was presented as the percentage of the maximum and Phe ED50 was calculated, Phe was equally potent in the aorta of L-NAME + AngII±EPL treated mice versus control WT (Fig. 4C) or cav-1−/− mice (Fig. 4D; Table 1).
Fig. 4.
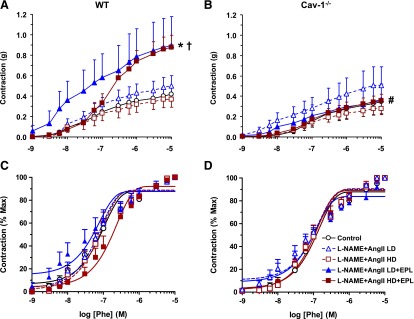
Phe-induced contraction in aortic rings of control and LD or HD L-NAME + AngII ± EPL treated WT (A and C) and cav-1−/− mice (B and D). Aortic rings were stimulated with increasing concentrations of Phe, and the contractile response was measured and presented in grams (A and B) or as a percentage of maximum Phe contraction (C and D). Data represent mean ± S.E.M. (n = 4–8). *Significantly different (P < 0.05) from respective control WT mice; †P < 0.05, EPL-treated versus without EPL treatment; #P < 0.05, cav-1−/− versus WT mice.
We tested the vascular response to other vasoconstrictor agonists. AngII (10−6 M) caused a small contractile response in aortic rings that was not significantly different among control and L-NAME + AngII±EPL treated WT (Fig. 5A) and cav-1−/− mice (Fig. 5B).
Fig. 5.
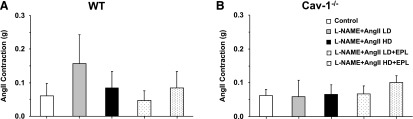
AngII-induced contraction in aortic rings of control and LD or HD L-NAME + AngII ± EPL treated WT (A) and cav-1−/− mice (B). Aortic rings were stimulated with AngII 10−6 M, and the contractile response was measured in grams. Data represent mean ± S.E.M. (n = 4–8).
High KCl (96 mM), which causes membrane depolarization and stimulates Ca2+ entry into the VSM (Khalil and van Breemen, 1990; Murphy and Khalil, 1999), caused aortic contraction that did not change with L-NAME + AngII treatment, but was enhanced in L-NAME + AngII + EPL treated mice compared with control WT mice (Fig. 6A). KCl contraction was not different between cav-1−/− and WT mice or between L-NAME + AngII ± EPL treated and control cav-1−/− mice (Fig. 6B). We also assessed possible changes in other mechanisms of VSM contraction that could enhance the myofilament force sensitivity to Ca2+ and found that Phe contraction as the percentage of the Ca2+-dependent KCl contraction was not different in control and L-NAME + AngII ± EPL treated WT (Fig. 6C) or cav-1−/− mice (Fig. 6D) .
Fig. 6.
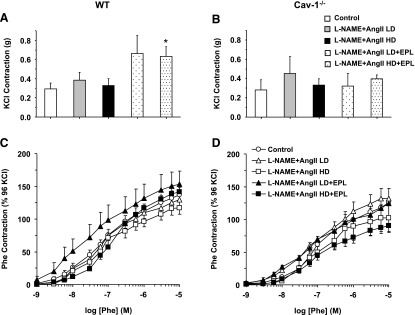
KCl-induced contraction and Phe-induced contraction as the percentage of KCl in aortic rings of control and LD or HD L-NAME + AngII ± EPL treated WT (A and C) and cav-1−/− mice (B and D). Aortic rings were stimulated with high 96 mM KCl depolarizing solution, and the contractile response was measured in grams (A and B). The contractile response to increasing concentrations of Phe was also measured and presented as the percentage of KCl contraction (C and D). Data represent mean ± S.E.M. (n = 4–8). *Significantly different (P < 0.05) from respective control WT mice.
Effects of Modulators of Endothelium-Derived NO-cGMP Pathway on Vascular Contraction.
To test for the role of endothelium-derived factors in the observed differences in vascular contraction during chronic treatment with L-NAME + AngII ± EPL, vascular contraction was measured in vessels treated ex vivo with the NO synthase (NOS) blocker L-NAME or guanylate cyclase inhibitor ODQ and in endothelium-denuded vessels. Analysis of vascular contraction in grams showed that ex vivo L-NAME significantly enhanced Phe contraction in control WT (Fig. 7A), but this enhancement was less apparent in L-NAME + AngII ± EPL–treated WT (Fig. 7, C, E, G, and I; Table 1). Ex vivo ODQ and endothelium removal insignificantly enhanced Phe contraction in control WT mice (Fig. 7A) and showed fewer effects in L-NAME + AngII ± EPL–treated WT mice (Fig. 7, C, E, G, and I). Ex vivo L-NAME enhanced Phe contraction significantly in cav-1−/− mice treated with L-NAME + AngII (Fig. 7, D and F), but had fewer effects in control (Fig. 7B) and L-NAME + AngII + EPL treated cav-1−/− mice (Fig. 7, H and J; Table 1). Ex vivo ODQ enhanced Phe contraction mainly in HD L-NAME + AngII + EPL cav-1−/− mice (Fig. 7J), and both ex vivo ODQ and endothelium removal showed fewer effects in control (Fig. 7B), L-NAME + AngII treated (Fig. 7, D and F), and LD L-NAME + AngII + EPL treated cav-1−/− mice (Fig. 7H; Table 1).
Fig. 7.

Effect of ex vivo blockade of NO-cGMP or endothelium removal on Phe-induced contraction in aortic rings of control and LD or HD L-NAME + AngII ± EPL treated WT (A, C, E, G, and I) and cav-1−/− mice (B, D, F, H, and J). Aortic rings were either kept endothelium-intact (open circles), pretreated ex vivo with the NOS inhibitor L-NAME (3 × 10−4 M) (closed circles) or the guanylate cyclase inhibitor ODQ (10−5 M) (open triangles) for 15 minutes, or endothelium denuded (closed triangles). The vessels were stimulated with increasing concentrations of Phe, and the contractile response was presented in grams. Data represent mean ± S.E.M. (n = 9–17). *P < 0.05, L-NAME–treated versus control nontreated arteries; #P < 0.05, ODQ-treated versus control nontreated arteries.
When Phe contraction was presented as the percentage of the maximum and Phe ED50 was calculated, ex vivo L-NAME, ODQ, or endothelium removal did not significantly enhance the sensitivity to Phe in any of the treatment groups or genotypes (Fig. 8; Table 1). However, ex vivo ODQ appeared to cause a decrease in Phe potency in aortic rings of HD L-NAME + AngII treated WT mice (Fig. 8E).
Fig. 8.

Effect of ex vivo blockade of NO-cGMP or endothelium removal on vascular sensitivity to Phe in aortic rings of control and LD or HD L-NAME + AngII ± EPL treated WT (A, C, E, G, and I) and cav-1−/− mice (B, D, F, H, and J). Aortic rings were either kept endothelium-intact (open circles), pretreated ex vivo with the NOS inhibitor L-NAME (3 × 10−4 M) (closed circles) or the guanylate cyclase inhibitor ODQ (10−5 M) (open triangles) for 15 minutes, or endothelium denuded (closed triangles). The vessels were stimulated with increasing concentrations of Phe, and the contractile response was presented as the percentage of maximum Phe contraction. Data represent mean ± S.E.M. (n = 9–17). #P < 0.05, ODQ-treated versus control nontreated arteries.
When Phe contraction was presented as the percentage of KCl contraction, ex vivo L-NAME or ODQ significantly enhanced the Phe contractile response in control mice (Fig. 9A) but not L-NAME + AngII ± EPL treated WT mice (Fig. 9, C, E, G, and I). Endothelium removal did not enhance Phe contraction as the percentage of KCl in any of the WT groups (Fig. 9, A, C, E, G, and I). There was no significant effect of ex vivo L-NAME, ex vivo ODQ, or endothelium removal in any of the cav-1−/− groups. However, examination of Fig. 9 reveals tendencies for enhanced Phe contraction as the percentage of KCl in ex vivo L-NAME treated aortic rings from control and L-NAME + AngII treated cav-1−/− mice (Fig. 9, B, D, and F), in ex vivo ODQ-treated aortic rings from HD L-NAME + AngII treated mice, and in endothelium-denuded aortic rings from control and LD L-NAME + AngII ± EPL treated cav-1−/− mice (Fig. 9, B, D, and H; Table 1).
Fig. 9.

Effect of ex vivo blockade of NO-cGMP or endothelium removal on Phe-induced contractile response relative to KCl contraction in aortic rings of control and LD or HD L-NAME + AngII ± EPL treated WT (A, C, E, G, and I) and cav-1−/− mice (B, D, F, H, and J). Aortic rings were either kept endothelium-intact (open circles), pretreated ex vivo with the NOS inhibitor L-NAME (3 × 10−4 M) (closed circles) or the guanylate cyclase inhibitor ODQ (10−5 M) (open triangles) for 15 minutes, or endothelium denuded (closed triangles). After measuring contraction to KCl (96 mM), the vessels were stimulated with increasing concentrations of Phe, and the contractile response was presented as the percentage of KCl contraction. Data represent mean ± S.E.M. (n = 9–17). *P < 0.05, L-NAME-treated versus control nontreated arteries; #P < 0.05, ODQ-treated versus control nontreated arteries.
EPL Reverses Enhanced NO-cGMP Relaxation in L-NAME + AngII Treated WT but Not Cav-1−/−.
We also assessed if the changes in BP are related to the changes in vascular relaxation mechanisms. ACh-induced vascular relaxation did not significantly change with LD L-NAME + AngII ± EPL, but was enhanced in HD L-NAME + AngII treated versus control WT mice, and this enhancement was prevented by the addition of EPL to HD L-NAME + AngII treatment (Fig. 10A). ACh-induced relaxation was enhanced in control cav-1−/− mice (Fig. 10B) as compared with WT mice (Fig. 10A). However, in contrast to what was observed in WT mice, L-NAME + AngII treatment with or without EPL did not affect ACh-induced relaxation in cav-1−/− mice (Fig. 10B). Real-time PCR revealed that cardiac eNOS mRNA levels were similar in cav-1−/− mice versus WT mice. In both genotypes, cardiac eNOS mRNA levels were reduced with L-NAME + AngII ± EPL treatment as compared with control groups (Fig. 10C). Western blots revealed that cardiac total eNOS protein was higher in cav-1−/− mice as compared with WT mice under control and L-NAME + AngII treatment. Further, and in contrast to the decrease in eNOS mRNA levels with L-NAME + AngII ± EPL, cardiac eNOS protein was higher in L-NAME + AngII ± EPL treated mice as compared with control WT and cav-1−/− mice (Fig. 10D).
Fig. 10.
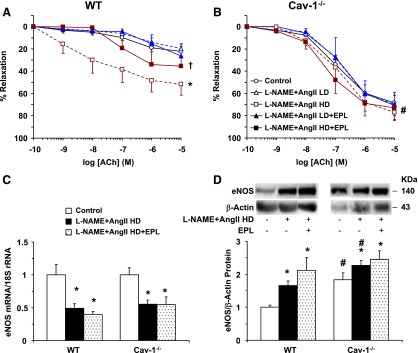
ACh-induced relaxation in aortic rings (A and B), eNOS mRNA (C), and total eNOS protein (D) in the heart of control and LD or HD L-NAME + AngII ± EPL treated WT and cav-1−/− mice. Aortic rings were precontracted with Phe (10−5 M), increasing concentrations of ACh were added, and the percentage of relaxation of the Phe contraction was measured. Cardiac tissue homogenates were also prepared for measurement of eNOS mRNA expression using real-time PCR and eNOS protein using Western blots. Data represent mean ± S.E.M. (n = 5–8). *Significantly different (P < 0.05) from respective control WT or cav-1−/− mice; †P < 0.05, EPL-treated versus without EPL treatment; #P < 0.05, cav-1−/− versus WT mice.
Endothelium removal and ex vivo treatment of aortic rings with the NOS inhibitor L-NAME or guanylate cyclase inhibitor ODQ significantly inhibited ACh-induced relaxation in vessels from control or L-NAME + AngII ± EPL treated WT and cav-1−/− mice (Fig. 11).
Fig. 11.

Effect of endothelium removal and ex vivo blockade of the NO-cGMP pathway on ACh-induced relaxation in aortic rings of control and LD or HD L-NAME + AngII ± EPL treated WT (A, C, E, G, and I) and cav-1−/− mice (B, D, F, H, and J). Aortic rings were either kept endothelium intact (open circles), pretreated ex vivo with the NOS inhibitor L-NAME (3 × 10−4 M) (closed circles) or the guanylate cyclase inhibitor ODQ (10−5 M) (open triangles) for 15 minutes, or endothelium denuded (closed triangles). The tissues were precontracted with Phe (10−5 M), increasing concentrations of ACh were added, and the percentage of relaxation of the Phe contraction was measured. Data represent mean ± S.E.M. (n = 4–8). * P < 0.05, L-NAME- or ODQ-treated or endothelium-denuded versus intact nontreated vessels.
The exogenous NO donor SNP caused a concentration-dependent relaxation that was enhanced in HD L-NAME + AngII versus control WT mice, and addition of EPL to HD L-NAME + AngII treatment reversed this enhancement, resulting in relaxation that did not differ significantly from control WT mice (Fig. 12A). In comparison, SNP-induced relaxation was not significantly different in L-NAME + AngII ± EPL treated versus control cav-1−/− mice (Fig. 12B).
Fig. 12.

SNP-induced relaxation in aortic rings of control and LD or HD L-NAME + AngII ± EPL treated WT (A) and cav-1−/− (B) mice. Aortic rings were precontracted with Phe (10−5 M), increasing concentrations of SNP were added, and the percentage of relaxation of the Phe contraction was measured. Data represent mean ± S.E.M. (n = 4–8). *Significantly different (P < 0.05) from respective control WT mice.
Discussion
The main findings of this study are 1) BP is increased in WT and cav-1−/− mice treated with L-NAME + AngII; 2) EPL does not reverse the L-NAME + AngII–induced increases in BP, although its dose is sufficient to reverse the L-NAME + AngII–induced cardiac expression of the inflammation marker CD68; 3) Phe- and KCl-induced vascular contraction are enhanced by EPL in L-NAME + AngII–treated WT mice, but not in cav-1−/− mice; 4) ACh- and SNP-induced relaxation is enhanced by L-NAME + AngII, and EPL reverses these effects in WT mice but not cav-1−/− mice; and 5) in both WT and cav-1−/− mice, L-NAME + AngII treatment is associated with increased eNOS protein and plasma ALDO levels and decreased cardiac eNOS, AT1R, and MR mRNA expression, and EPL minimally changes these levels.
L-NAME + AngII Increase BP in WT and Cav-1−/− Mice.
BP is tightly regulated by neural, renal, hormonal, and vascular control mechanisms that prevent any sudden or long-term variations during various environmental changes (Cain and Khalil, 2002; Ponnuchamy and Khalil, 2009). For example, HS diet alone may not significantly change BP, vascular reactivity (Giardina et al., 2001; Smith et al., 2003), or cardiac tissue integrity (Rocha et al., 2000; Martinez et al., 2002; Oestreicher et al., 2003; Turchin et al., 2006). In the presence of an intact renal feedback mechanism, any HS-induced increases in plasma volume cause feedback inhibition of RAAS, leading to decreased salt and water reabsorption (Hall, 1986; Guyton, 1991). Also, in the presence of an intact vascular endothelium, any HS-induced vasoconstriction is normally counterbalanced by compensatory NO production and vascular relaxation (Giardina et al., 2001; Smith et al., 2003). However, in the presence of aberrant renal or cardiovascular mechanisms, an HS diet could lead to increased BP and cardiovascular injury. For instance, in rats chronically treated with L-NAME, an HS diet causes increases in BP and vasoconstriction (Giardina et al., 2001). Also, in mice chronically treated with L-NAME + AngII, an HS diet is associated with cardiovascular injury and inflammation (Martinez et al., 2002; Oestreicher et al., 2003). Consistent with these reports, the present data demonstrate that in WT mice consuming an HS diet, chronic L-NAME + AngII increased BP. BP was insignificantly greater in cav-1−/− mice than in WT mice, an observation that we previously attributed to excessive NO production and increased formation of peroxinitrites and oxidative stress (Pojoga et al., 2008, 2010a). BP also showed greater sensitivity to L-NAME in cav-1−/− mice than in WT mice, supporting differential regulation of BP in cav-1−/− mice versus WT mice. Additionally, the body weight was different in L-NAME + AngII–treated cav-1−/− mice versus WT mice, suggesting differences in volume regulation. L-NAME functions by blocking NOS, whereas AngII activates vascular AT1R to increase vasoconstriction (Touyz and Schiffrin, 2000) and increases salt and water reabsorption either directly via renal tubular AT1R or indirectly by stimulating ALDO secretion, which in turn activates MR in the renal collecting ducts and further increases plasma volume (Hall, 1986; Meneton et al., 2005; Khalil, 2006; Adler and Williams, 2007). We reasoned that if the pressor effects of L-NAME + AngII involve ALDO/MR-mediated renal mechanisms, then an adequate dose of the MR antagonist EPL should be sufficient to prevent the effects of ALDO on renal MR and in turn decrease sodium absorption, plasma volume, and BP.
MR Antagonism Reverses L-NAME + AngII–Induced Cardiac Injury but Not BP.
To confirm the use of an adequate dose of EPL, we made use of our previous observations that ALDO/MR could mediate L-NAME + AngII–induced renal arteriopathy and myocardial necrosis (Rocha et al., 2000; Oestreicher et al., 2003; Pojoga et al., 2010b) and that EPL treatment prevents the cardiac and renal damage in the L-NAME + AngII model of cardiovascular injury (Rocha et al., 2000; Martinez et al., 2002; Oestreicher et al., 2003). Consistent with our previous report (Pojoga et al., 2010b), L-NAME + AngII increased cardiac expression of the inflammation markers CD68 and PAI-1 in WT mice and to a lesser extent in cav-1−/− mice, and EPL reversed the increases in CD68 in both genotypes and PAI-1 in WT mice. Thus, although L-NAME + AngII treatment could have different systemic effects on the cardiac and renal systems, our use of an adequate dose of EPL should rule out the contribution of MR-mediated heart or kidney damage. Although we used EPL at doses that effectively prevent MR-mediated cardiac and renal injury, it did not decrease BP in L-NAME + AngII–treated WT or cav-1−/− mice, suggesting the involvement of factors other than the renal MR or MR-mediated cardiac injury. We are not aware of any data to support the possibility that EPL has other non-MR off-target effects, and thus propose that the lack of effect of EPL on BP in the L-NAME + AngII–treated mouse may involve the presence of opposing mechanisms, whereby EPL blockade of renal MR-mediated salt and water reabsorption is counterbalanced by concomitant blockade of a vascular MR-mediated compensatory mechanism.
MR Antagonism Enhances Vascular Contraction in L-NAME + AngII–Treated WT Mice but Not Cav-1−/− Mice.
We have recently reported the presence of basal MR-mediated vascular activity during an HS diet, and that this activity is modulated in cav-1 null mice (Pojoga et al., 2010a). In the present study, we tested if any MR-mediated vascular activity would change during low NO–high AngII–induced cardiovascular injury. The observed enhancement of Phe-induced contraction with EPL in L-NAME + AngII–treated WT supports the presence of MR-mediated vasodilator activity. This MR-mediated vascular activity appears to involve cav-1 because EPL did not change Phe contraction in L-NAME + AngII–treated cav-1−/− mice. The EPL-induced enhancement of Phe contraction in WT mice is unlikely due to increased sensitivity of α-adrenergic receptors because Phe was equally potent in L-NAME + AngII–treated mice with or without EPL. To test if EPL-induced enhancement of vascular contraction in WT mice was specific to a particular agonist/receptor, we examined the response to AngII. However, AngII contraction was very small and was not different in control or L-NAME + AngII ± EPL–treated WT or cav-1−/− mice. The small AngII contractile response is likely due to tachyphylaxis and downregulation of AT1R during chronic exposure to AngII. This is supported by the observation that cardiac AT1R expression was reduced in L-NAME + AngII–treated versus control mice of both genotypes. Interestingly, AT1R expression was greater in cav-1−/− mice than in WT mice, which may be related to the report that cav-1 is necessary for AT1R trafficking and signaling (Ushio-Fukai and Alexander, 2006). Thus, the overexpression of AT1R in cav-1–deficient mice may represent a feedback rescue mechanism to compensate for the loss of cav-1. On the other hand, vascular contraction to high KCl, which mainly stimulates receptor-independent Ca2+ entry into VSM (Khalil and van Breemen, 1990; Murphy and Khalil, 1999), was enhanced with EPL in L-NAME + AngII–treated WT mice, supporting that L-NAME + AngII may activate a common MR-mediated vascular relaxation mechanism. KCl-induced contraction was not enhanced with EPL in L-NAME + AngII–treated cav-1−/− mice, supporting that the common MR-mediated vascular relaxation mechanism requires an interaction between MR and cav-1.
Evidence for L-NAME + AngII–Induced MR-Mediated Enhanced Vascular Relaxation in WT.
To further examine the potential MR-mediated vascular mechanisms, we tested the effects of ACh, a known stimulator of endothelium-derived vasodilator substances, such as NO. Under basal conditions, eNOS is bound to cav-1 in endothelial cell caveolae. ACh-induced increase in endothelial cell Ca2+ promotes the release of Ca2+-dependent eNOS from cav-1 to the cytosol, where it is phosphorylated and fully activated to produce NO (Feron et al., 1996; Segal et al., 1999; Minshall et al., 2003; Batova et al., 2006). NO diffuses from endothelial cells into VSM, where it activates guanylate cyclase to produce cGMP, which causes vascular relaxation by inhibiting Ca2+ entry, stimulating Ca2+ extrusion, and decreasing Ca2+ sensitivity of the contractile proteins (Fleming and Busse, 1999; Ignarro, 2002; Murad, 2006). The observed enhanced ACh relaxation in L-NAME + AngII–treated WT mice is likely due to activation of the MR-mediated NO-cGMP vascular relaxation pathway because 1) it was reversed with EPL; 2) it was inhibited by endothelium removal or ex vivo treatment with the NOS blocker L-NAME or guanylate cyclase inhibitor ODQ; 3) it was associated with increased cardiac eNOS protein; and 4) vascular relaxation to the NO donor and guanylate cyclase activator SNP was enhanced in L-NAME + AngII–treated WT, and this enhancement was reversed with EPL.
Loss of L-NAME + AngII–Induced MR-Mediated Vascular Relaxation in Cav-1−/− Mice.
Studies in cav-1−/− mice have suggested a role of cav-1 in vascular mechanotransduction, function, and remodeling (Drab et al., 2001; Razani et al., 2001; Wunderlich et al., 2006; Yu et al., 2006). Consistent with our previous report (Pojoga et al., 2008), ACh-induced aortic relaxation was greater in cav-1−/− mice than in WT mice, supporting an increase in endothelium-derived relaxing factor(s) in cav-1 deficiency states. This is also supported by the report that pharmacological disruption of caveolae and cav-1 using methyl-β-cyclodextrin in isolated aorta from AngII-infused apolipoprotein E knockout mice caused enhancement of the ACh-induced concentration-relaxation curve when compared with vehicle control (Seto et al., 2013). Also, the present Western blots revealed an increase in cardiac eNOS protein in cav-1−/− versus WT mice. Since cav-1−/− mice have intact cav-3 expression in cardiac myocytes, the increased cardiac eNOS may reflect changes in the heart vessels rather than in the myocytes. In contrast with WT, ACh-induced relaxation was not affected by L-NAME + AngII ± EPL in cav-1−/− mice, supporting the loss of MR-mediated vascular relaxation mechanisms likely because the artery may not be able to relax any further because eNOS and the NO-cGMP pathway are maximally activated. Similarly, the enhancement of SNP-induced relaxation in L-NAME + AngII–treated WT mice and its reversal with EPL were not observed in cav-1−/− mice, supporting a role for cav-1 in MR-mediated vascular relaxation via NO-cGMP.
Mechanisms of L-NAME + AngII–Induced Enhanced Vascular MR Activity in WT Mice but Not Cav-1−/− Mice.
The changes in vascular MR activity during L-NAME + AngII treatment could be related to several factors, including ALDO levels, MR expression, and/or MR activity. AngII activates AT1R in the adrenal gland to increase ALDO production. However, an increase in ALDO production may not be the sole mechanism for the vascular changes observed in WT mice but not cav-1−/− mice, as plasma ALDO levels were increased in WT mice and further increased in cav-1−/− mice, particularly during treatment with L-NAME + AngII and EPL. Also, vascular MR can be activated not only by ALDO, but also by other steroid and nonsteroid compounds (Funder, 2006, 2007, 2009; Di Zhang et al., 2008), including direct activation by AngII (Strawn, 2005). Another possible explanation for the changes in vascular MR activity during L-NAME + AngII treatment may involve changes in MR expression. MR has been identified in cardiac tissues, and ALDO/MR interaction may affect cardiac function during an HS diet (Rocha et al., 2000; Martinez et al., 2002; Joffe and Adler, 2005; Turchin et al., 2006; Garg et al., 2015). Also, vascular MR has been implicated in vasospasm, vascular hypertrophy (Funder, 2006, 2007, 2009; Schiffrin, 2006), inflammation, and atherosclerosis (Strawn, 2005; Ferrario and Strawn, 2006). However, the present data showed reduced cardiac MR expression during L-NAME + AngII treatment in both WT and cav-1−/− mice, and EPL did not cause any further changes in MR expression. Alternatively, the vascular effects of L-NAME + AngII and their reversal with EPL in WT mice, but not cav-1−/− mice, could be related to differences in the sensitivity of vascular MR and MR–cav-1–NO coupling mechanisms. Another intriguing possibility is that ALDO may act independently of MR via G protein–coupled receptor 30 to induce rapid vascular effects, a setting in which classic MR blockers may act as partial antagonists (Gros et al., 2011). These ALDO/G protein–coupled receptor 30–mediated endothelium-dependent vasodilator effects could counteract any ALDO/MR-mediated vasoconstrictor effects on VSM (Gros et al., 2013) and hence determine the net effect of ALDO on vascular reactivity.
Potential Mechanisms Linking Vascular MR, Cav-1, and NO-cGMP Pathway.
The molecular interaction among vascular MR, cav-1, and the NO-cGMP pathway is unclear but could involve a vascular MR-mediated increase in eNOS protein to counterbalance the renal MR-mediated increase in sodium and water retention and plasma volume (Fig. 13). MR may also function via a plasma membrane channel or pump. For instance, MR is coupled to the nongenomic activation of the sodium channel ENaC in the renal collecting tubules (Zhou and Bubien, 2001; Lee et al., 2008; McEneaney et al., 2008), and ALDO may stimulate the surface expression and activity of the Na+-H+ exchanger (NHE3) in the proximal tubules’ epithelial cells (Drumm et al., 2006). Also, ENaC, NHE3, and the Na+-Ca2+ exchanger have been identified in VSM and endothelial cells (Golestaneh et al., 2001; Jernigan and Drummond, 2006; Ponnuchamy and Khalil, 2009), and MR may regulate the ion-gated sodium channel in endothelial cells through an effect on the cytoskeleton (Golestaneh et al., 2001). It is possible that during an HS diet, high AngII and consequent activation of MR may promote Na+ entry into endothelial cells, and in the presence of a maximally activated Na+-K+ pump, the cellular Na+ load could be extruded via a reverse-mode Na+-Ca2+ exchanger, leading to increased cellular Ca2+ and increased activity of the Ca2+-dependent eNOS.
Fig. 13.
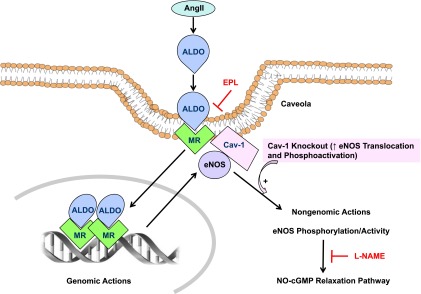
Schematic diagram of the potential interaction between vascular MR and cav-1 during low NO–high AngII–induced cardiovascular injury. Increased circulating AngII is known to increase salt and water retention either directly via renal tubular AT1R or indirectly by stimulating ALDO secretion. AngII also stimulates vascular AT1R to induce vasoconstriction, whereas low NO (by L-NAME) causes further vasoconstriction and vascular injury. In cav-1 replete states (WT), cav-1 anchors eNOS, thus limiting its translocation and phosphoactivation. MR colocalizes with cav-1 in endothelial cells, and L-NAME + AngII–induced cardiovascular injury may trigger compensatory MR-mediated genomic changes in the amount of eNOS mRNA/protein and nongenomic increases in eNOS phosphorylation/activity and thereby the NO-cGMP vascular relaxation pathway. Thus, during L-NAME + AngII treatment in cav-1 replete states, MR blockade with EPL would prevent the compensatory MR-mediated NO-cGMP relaxation pathway and consequently increase vascular contraction. In cav-1 deficiency states (knockout), eNOS is maximally activated, leading to loss of L-NAME + AngII–induced compensatory MR-mediated vasodilator effects, and, in turn, a lack of vascular effects of EPL.
Limitations.
Other considerations or limitations are 1) EPL treatment was associated with increased cardiac PAI-1 in L-NAME + AngII–treated cav-1−/− mice, suggesting additional effects of EPL on cardiovascular inflammation. 2) Treatment with L-NAME + AngII was associated with decreased eNOS mRNA expression, but could also decrease eNOS protein degradation and, as a result, lead to a net increase in eNOS protein levels. 3) Assuming that KCl mainly stimulates Ca2+ entry into VSM, then the lack of difference in Phe contraction as the percentage of KCl in L-NAME + AngII ± EPL–treated versus nontreated vessels suggests no change in the Ca2+ sensitization mechanisms of VSM contraction, such as protein kinase C and Rho kinase. Interestingly, some enhancement of Phe contraction could be observed in L-NAME + AngII–treated mice upon blockade of NOS, guanylate cyclase, or endothelium removal. These observations support the contention that L-NAME + AngII is associated with increased NO, which could inhibit the Ca2+ sensitization pathways, and only when the NO-cGMP pathway is inhibited, the Ca2+ sensitization pathways are manifested. 4) The present study was performed on the aorta, and although working with small vessels from small animals, such as the mouse, could pose a challenge, the interaction of MR, cav-1, and NOS should be further examined in small resistance vessels.
Conclusions and Perspectives.
The present results are consistent with the contention that during sodium overload and low NO–high AngII–induced hypertension and cardiovascularinjury, feedback renal mechanisms to decrease decrease MR-mediated sodium reabsorption and increases in plasma volume and BP could be reinforced by MR-mediated vascular relaxation to decrease vasoconstriction. The MR-mediated vascular relaxation pathway involves enhanced eNOS activity, endothelium-dependent NO-cGMP, and VSM responsiveness to cGMP. Cav-1 has been suggested as a mediator of the effects of steroid hormones on their receptors (Li et al., 2001, 2006) and an important regulator of eNOS expression and activity (Drab et al., 2001; Razani et al., 2001; Wunderlich et al., 2006; Yu et al., 2006), particularly during an HS diet (Pojoga et al., 2008). The changes in vascular function in L-NAME + AngII–treated WT mice with EPL and their absence in cav-1 null mice provides evidence for a potential interaction between vascular MR, cav-1, and eNOS in regulating vascular function during an HS diet and low NO–high AngII–dependent cardiovascular injury (Fig. 13). The differential effects of EPL on the MR-mediated vascular relaxation mechanism in WT versus cav-1−/− mice also raise a note of caution regarding the use of MR antagonists in clinical settings. Cav-1−/− mice show insulin resistance (Cohen et al., 2003; Pojoga et al., 2011; Asterholm et al., 2012), and forearm blood flow, a measure of endothelial cell function, deteriorates with spironolactone treatment in diabetic subjects (Davies et al., 2004). Also, our studies in humans have shown that cav-1 gene variants are associated with insulin resistance in hypertensive individuals (Pojoga et al., 2011). The present findings suggest that hypertensive individuals with minor allele carriers of cav-1 gene variants may have blunted compensatory MR-mediated vascular relaxation mechanisms, and the use of MR antagonists for the management of hypertension and cardiovascular injury should be carefully evaluated in these individuals.
Acknowledgments
The authors acknowledge the technical assistance of Paul Loutraris at Brigham and Women’s Hospital.
Abbreviations
- ACh
acetylcholine
- ALDO
aldosterone
- AngII
angiotensin II
- AT1R
angiotensin type 1 receptor
- BP
blood pressure
- cav-1
caveolin-1
- cav-1−/−
cav-1 knockout
- eNOS
endothelial nitric oxide synthase
- EPL
eplerenone
- HD
high dose
- HS
high salt
- LD
low dose
- L-NAME
Nω-nitro-l-arginine methyl ester
- MR
mineralocorticoid receptor
- NO
nitric oxide
- NOS
nitric oxide synthase
- ODQ
1H-(1,2,4)oxadiazolo(4,3-a)quinoxalin-1-one
- PAI-1
plasminogen activator inhibitor-1
- PCR
polymerase chain reaction
- Phe
phenylephrine
- RAAS
renin-angiotensin-aldosterone system
- SNP
sodium nitroprusside
- VSM
vascular smooth muscle
- WT
wild type
Authorship Contributions
Participated in research design: Pojoga, Williams, Khalil.
Conducted experiments: Pojoga, Yao, Reslan, Khalil.
Performed data analysis: Pojoga, Yao, Opsasnick, Siddiqui, Reslan, Khalil.
Wrote or contributed to the writing of the manuscript: Pojoga, Adler, Williams, Khalil.
Footnotes
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grants HL104032 (to L.H.P.), K24-HL103845 (to G.K.A.), HL114765 and T32-HL007609 (to G.H.W.), and HL65998, HL98724, and HL111775 (to R.A.K.)]; the National Institutes of Health Eunice Kennedy Shriver National Institute of Child Health and Human Development [Grant HD60702 (to R.A.K.)]; and the American Heart Association [Grants 0735609T and 14GRNT2050000 (to L.H.P.)].
References
- Adler GK, Williams GH. (2007) Aldosterone: villain or protector? Hypertension 50:31–32. [DOI] [PubMed] [Google Scholar]
- Asterholm IW, Mundy DI, Weng J, Anderson RG, Scherer PE. (2012) Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin-1. Cell Metab 15:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batova S, DeWever J, Godfraind T, Balligand JL, Dessy C, Feron O. (2006) The calcium channel blocker amlodipine promotes the unclamping of eNOS from caveolin in endothelial cells. Cardiovasc Res 71:478–485. [DOI] [PubMed] [Google Scholar]
- Cain AE, Khalil RA. (2002) Pathophysiology of essential hypertension: role of the pump, the vessel, and the kidney. Semin Nephrol 22:3–16. [PubMed] [Google Scholar]
- Cohen AW, Razani B, Wang XB, Combs TP, Williams TM, Scherer PE, Lisanti MP. (2003) Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am J Physiol Cell Physiol 285:C222–C235. [DOI] [PubMed] [Google Scholar]
- Coutinho P, Vega C, Pojoga LH, Rivera A, Prado GN, Yao TM, Adler G, Torres-Grajales M, Maldonado ER, Ramos-Rivera A, et al. (2014) Aldosterone’s rapid, nongenomic effects are mediated by striatin: a modulator of aldosterone’s effect on estrogen action. Endocrinology 155:2233–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JI, Band M, Morris A, Struthers AD. (2004) Spironolactone impairs endothelial function and heart rate variability in patients with type 2 diabetes. Diabetologia 47:1687–1694. [DOI] [PubMed] [Google Scholar]
- Di Zhang A, Nguyen Dinh Cat A, Soukaseum C, Escoubet B, Cherfa A, Messaoudi S, Delcayre C, Samuel JL, Jaisser F. (2008) Cross-talk between mineralocorticoid and angiotensin II signaling for cardiac remodeling. Hypertension 52:1060–1067. [DOI] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, et al. (2001) Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293:2449–2452. [DOI] [PubMed] [Google Scholar]
- Drumm K, Kress TR, Gassner B, Krug AW, Gekle M. (2006) Aldosterone stimulates activity and surface expression of NHE3 in human primary proximal tubule epithelial cells (RPTEC). Cell Physiol Biochem 17:21–28. [DOI] [PubMed] [Google Scholar]
- Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. (1996) Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem 271:22810–22814. [DOI] [PubMed] [Google Scholar]
- Feron O, Saldana F, Michel JB, Michel T. (1998) The endothelial nitric-oxide synthase-caveolin regulatory cycle. J Biol Chem 273:3125–3128. [DOI] [PubMed] [Google Scholar]
- Ferrario CM, Strawn WB. (2006) Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol 98:121–128. [DOI] [PubMed] [Google Scholar]
- Fleming I, Busse R. (1999) NO: the primary EDRF. J Mol Cell Cardiol 31:5–14. [DOI] [PubMed] [Google Scholar]
- Funder JW. (2006) Mineralocorticoid receptors and cardiovascular damage: it’s not just aldosterone. Hypertension 47:634–635. [DOI] [PubMed] [Google Scholar]
- Funder JW. (2007) Why are mineralocorticoid receptors so nonselective? Curr Hypertens Rep 9:112–116. [DOI] [PubMed] [Google Scholar]
- Funder JW. (2009) Reconsidering the roles of the mineralocorticoid receptor. Hypertension 53:286–290. [DOI] [PubMed] [Google Scholar]
- Garg R, Rao AD, Baimas-George M, Hurwitz S, Foster C, Shah RV, Jerosch-Herold M, Kwong RY, Di Carli MF, Adler GK. (2015) Mineralocorticoid receptor blockade improves coronary microvascular function in individuals with type 2 diabetes. Diabetes 64:236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardina JB, Green GM, Rinewalt AN, Granger JP, Khalil RA. (2001) Role of endothelin B receptors in enhancing endothelium-dependent nitric oxide-mediated vascular relaxation during high salt diet. Hypertension 37:516–523. [DOI] [PubMed] [Google Scholar]
- Golestaneh N, Klein C, Valamanesh F, Suarez G, Agarwal MK, Mirshahi M. (2001) Mineralocorticoid receptor-mediated signaling regulates the ion gated sodium channel in vascular endothelial cells and requires an intact cytoskeleton. Biochem Biophys Res Commun 280:1300–1306. [DOI] [PubMed] [Google Scholar]
- Gros R, Ding Q, Liu B, Chorazyczewski J, Feldman RD. (2013) Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. Am J Physiol Cell Physiol 304:C532–C540. [DOI] [PubMed] [Google Scholar]
- Gros R, Ding Q, Sklar LA, Prossnitz EE, Arterburn JB, Chorazyczewski J, Feldman RD. (2011) GPR30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone. Hypertension 57:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Martinez-Vasquez D, Mendez GP, Toniolo MF, Yao TM, Oestreicher EM, Kikuchi T, Lapointe N, Pojoga L, Williams GH, et al. (2006) Mineralocorticoid receptor antagonist reduces renal injury in rodent models of types 1 and 2 diabetes mellitus. Endocrinology 147:5363–5373. [DOI] [PubMed] [Google Scholar]
- Guyton AC. (1991) Blood pressure control--special role of the kidneys and body fluids. Science 252:1813–1816. [DOI] [PubMed] [Google Scholar]
- Hall JE. (1986) Control of sodium excretion by angiotensin II: intrarenal mechanisms and blood pressure regulation. Am J Physiol 250:R960–R972. [DOI] [PubMed] [Google Scholar]
- Holtzman EJ, Braley LM, Williams GH, Hollenberg NK. (1988) Kinetics of sodium homeostasis in rats: rapid excretion and equilibration rates. Am J Physiol 254:R1001–R1006. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ. (2002) Nitric oxide as a unique signaling molecule in the vascular system: a historical overview. J Physiol Pharmacol 53:503–514. [PubMed] [Google Scholar]
- Jernigan NL, Drummond HA. (2006) Myogenic vasoconstriction in mouse renal interlobar arteries: role of endogenous beta and gammaENaC. Am J Physiol Renal Physiol 291:F1184–F1191. [DOI] [PubMed] [Google Scholar]
- Joffe HV, Adler GK. (2005) Effect of aldosterone and mineralocorticoid receptor blockade on vascular inflammation. Heart Fail Rev 10:31–37. [DOI] [PubMed] [Google Scholar]
- Khalil RA. (2006) Dietary salt and hypertension: new molecular targets add more spice. Am J Physiol Regul Integr Comp Physiol 290:R509–R513. [DOI] [PubMed] [Google Scholar]
- Khalil RA, van Breemen C. (1990) Intracellular free calcium concentration/force relationship in rabbit inferior vena cava activated by norepinephrine and high K+. Pflugers Arch 416:727–734. [DOI] [PubMed] [Google Scholar]
- Lee IH, Campbell CR, Cook DI, Dinudom A. (2008) Regulation of epithelial Na+ channels by aldosterone: role of Sgk1. Clin Exp Pharmacol Physiol 35:235–241. [DOI] [PubMed] [Google Scholar]
- Li L, Yang G, Ebara S, Satoh T, Nasu Y, Timme TL, Ren C, Wang J, Tahir SA, Thompson TC. (2001) Caveolin-1 mediates testosterone-stimulated survival/clonal growth and promotes metastatic activities in prostate cancer cells. Cancer Res 61:4386–4392. [PubMed] [Google Scholar]
- Li T, Sotgia F, Vuolo MA, Li M, Yang WC, Pestell RG, Sparano JA, Lisanti MP. (2006) Caveolin-1 mutations in human breast cancer: functional association with estrogen receptor alpha-positive status. Am J Pathol 168:1998–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez DV, Rocha R, Matsumura M, Oestreicher E, Ochoa-Maya M, Roubsanthisuk W, Williams GH, Adler GK. (2002) Cardiac damage prevention by eplerenone: comparison with low sodium diet or potassium loading. Hypertension 39:614–618. [PubMed] [Google Scholar]
- McEneaney V, Harvey BJ, Thomas W. (2008) Aldosterone regulates rapid trafficking of epithelial sodium channel subunits in renal cortical collecting duct cells via protein kinase D activation. Mol Endocrinol 22:881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. (2005) Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev 85:679–715. [DOI] [PubMed] [Google Scholar]
- Minshall RD, Sessa WC, Stan RV, Anderson RG, Malik AB. (2003) Caveolin regulation of endothelial function. Am J Physiol Lung Cell Mol Physiol 285:L1179–L1183. [DOI] [PubMed] [Google Scholar]
- Murad F. (2006) Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med 355:2003–2011. [DOI] [PubMed] [Google Scholar]
- Murphy JG, Khalil RA. (1999) Decreased [Ca(2+)](i) during inhibition of coronary smooth muscle contraction by 17beta-estradiol, progesterone, and testosterone. J Pharmacol Exp Ther 291:44–52. [PubMed] [Google Scholar]
- Oestreicher EM, Martinez-Vasquez D, Stone JR, Jonasson L, Roubsanthisuk W, Mukasa K, Adler GK. (2003) Aldosterone and not plasminogen activator inhibitor-1 is a critical mediator of early angiotensin II/NG-nitro-L-arginine methyl ester-induced myocardial injury. Circulation 108:2517–2523. [DOI] [PubMed] [Google Scholar]
- Oliverio MI, Best CF, Smithies O, Coffman TM. (2000) Regulation of sodium balance and blood pressure by the AT(1A) receptor for angiotensin II. Hypertension 35:550–554. [DOI] [PubMed] [Google Scholar]
- Pojoga LH, Adamová Z, Kumar A, Stennett AK, Romero JR, Adler GK, Williams GH, Khalil RA. (2010a) Sensitivity of NOS-dependent vascular relaxation pathway to mineralocorticoid receptor blockade in caveolin-1-deficient mice. Am J Physiol Heart Circ Physiol 298:H1776–H1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pojoga LH, Romero JR, Yao TM, Loutraris P, Ricchiuti V, Coutinho P, Guo C, Lapointe N, Stone JR, Adler GK, et al. (2010b) Caveolin-1 ablation reduces the adverse cardiovascular effects of N-omega-nitro-L-arginine methyl ester and angiotensin II. Endocrinology 151:1236–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pojoga LH, Underwood PC, Goodarzi MO, Williams JS, Adler GK, Jeunemaitre X, Hopkins PN, Raby BA, Lasky-Su J, Sun B, et al. (2011) Variants of the caveolin-1 gene: a translational investigation linking insulin resistance and hypertension. J Clin Endocrinol Metab 96:E1288–E1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pojoga LH, Yao TM, Sinha S, Ross RL, Lin JC, Raffetto JD, Adler GK, Williams GH, Khalil RA. (2008) Effect of dietary sodium on vasoconstriction and eNOS-mediated vascular relaxation in caveolin-1-deficient mice. Am J Physiol Heart Circ Physiol 294:H1258–H1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnuchamy B, Khalil RA. (2009) Cellular mediators of renal vascular dysfunction in hypertension. Am J Physiol Regul Integr Comp Physiol 296:R1001–R1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, et al. (2001) Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276:38121–38138. [DOI] [PubMed] [Google Scholar]
- Rocha R, Stier CT, Jr, Kifor I, Ochoa-Maya MR, Rennke HG, Williams GH, Adler GK. (2000) Aldosterone: a mediator of myocardial necrosis and renal arteriopathy. Endocrinology 141:3871–3878. [DOI] [PubMed] [Google Scholar]
- Schiffrin EL. (2006) Effects of aldosterone on the vasculature. Hypertension 47:312–318. [DOI] [PubMed] [Google Scholar]
- Segal SS, Brett SE, Sessa WC. (1999) Codistribution of NOS and caveolin throughout peripheral vasculature and skeletal muscle of hamsters. Am J Physiol 277:H1167–H1177. [DOI] [PubMed] [Google Scholar]
- Seto SW, Krishna SM, Yu H, Liu D, Khosla S, Golledge J. (2013) Impaired acetylcholine-induced endothelium-dependent aortic relaxation by caveolin-1 in angiotensin II-infused apolipoprotein-E (ApoE-/-) knockout mice. PLoS One 8:e58481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L, Payne JA, Sedeek MH, Granger JP, Khalil RA. (2003) Endothelin-induced increases in Ca2+ entry mechanisms of vascular contraction are enhanced during high-salt diet. Hypertension 41:787–793. [DOI] [PubMed] [Google Scholar]
- Strawn WB. (2005) Eplerenone antagonizes atherosclerosis, but what is the agonist? Hypertension 46:1093–1094. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Schiffrin EL. (2000) Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev 52:639–672. [PubMed] [Google Scholar]
- Turchin A, Guo CZ, Adler GK, Ricchiuti V, Kohane IS, Williams GH. (2006) Effect of acute aldosterone administration on gene expression profile in the heart. Endocrinology 147:3183–3189. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Alexander RW. (2006) Caveolin-dependent angiotensin II type 1 receptor signaling in vascular smooth muscle. Hypertension 48:797–803. [DOI] [PubMed] [Google Scholar]
- Wunderlich C, Schober K, Lange SA, Drab M, Braun-Dullaeus RC, Kasper M, Schwencke C, Schmeisser A, Strasser RH. (2006) Disruption of caveolin-1 leads to enhanced nitrosative stress and severe systolic and diastolic heart failure. Biochem Biophys Res Commun 340:702–708. [DOI] [PubMed] [Google Scholar]
- Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV, Sessa WC. (2006) Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest 116:1284–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZH, Bubien JK. (2001) Nongenomic regulation of ENaC by aldosterone. Am J Physiol Cell Physiol 281:C1118–C1130. [DOI] [PubMed] [Google Scholar]