

Abstract
Information on the intestinal microbiota has increased exponentially this century because of technical advancements in genomics and metabolomics. Although information on the synthesis of bile acids by the liver and their transformation to secondary bile acids by the intestinal microbiota was the first example of the importance of the intestinal microbiota in biotransforming chemicals, this review will discuss numerous examples of the mechanisms by which the intestinal microbiota alters the pharmacology and toxicology of drugs and other chemicals. More specifically, the altered pharmacology and toxicology of salicylazosulfapridine, digoxin, l-dopa, acetaminophen, caffeic acid, phosphatidyl choline, carnitine, sorivudine, irinotecan, nonsteroidal anti-inflammatory drugs, heterocyclic amines, melamine, nitrazepam, and lovastatin will be reviewed. In addition, recent data that the intestinal microbiota alters drug metabolism of the host, especially Cyp3a, as well as the significance and potential mechanisms of this phenomenon are summarized. The review will conclude with an update of bile acid research, emphasizing the bile acid receptors (FXR and TGR5) that regulate not only bile acid synthesis and transport but also energy metabolism. Recent data indicate that by altering the intestinal microbiota, either by diet or drugs, one may be able to minimize the adverse effects of the Western diet by altering the composition of bile acids in the intestine that are agonists or antagonists of FXR and TGR5. Therefore, it may be possible to consider the intestinal microbiota as another drug target.
Introduction
This area of research probably began with studies on bile acids, when it was discovered that bile acids are excreted into bile, metabolized by the intestinal microbiota, and reabsorbed back into the blood to complete what is known as the enterohepatic cycle of bile acids. The intestinal microbiota consists mainly of anaerobic bacteria, which are difficult to culture. This area of research was limited by techniques to quantify numerous microbiota and chemicals rapidly. However, advancements in molecular biology and metabolomics this century resulted in a marked increase of research in this area. By examining the 16S rRNA of bacteria, scientists can determine the genus and species of bacteria. We now know that there are 500–1000 bacterial species in the intestinal microbiota. Over 90% of the intestinal microbiota is composed of the Bacteroides and Firmicutes families. Humans have 10-times more bacterial cells in their intestinal contents than all of the human cells in their bodies, but the bacterial cells are much smaller than human cells. It is said that we are born 100% human, but at death we are 90% bacterial. The amount of bacteria in the lumen of our intestine is two to three pounds and is about the same size as the brain. The bacterial content of our feces is approximately 60%. Most important for this area of research are the enzymes that these bacteria synthesize to biotransform chemicals. Although the main biotransformation pathway of drugs by humans involves oxidation and conjugation, the major pathways of metabolism by bacteria are reduction and deconjugation. The metabolites of drugs and other xenobiotics formed by the intestinal microbiota may be less or more toxic than those formed by humans. This review will describe a number of examples of how the intestinal microbiota, by metabolizing chemicals, can decrease or increase the health of the host.
Intestinal Microbiota Delivers Drug to the Large Intestine
Sulfonamides or sulfa drugs are antibacterial agents that were available before World War 2. Many derivatives of this class of antibiotics have been made, including salicylazosulfapyridine. This drug was synthetized at a time when it was thought that if a single drug combined the biologic activity of two drugs, it would produce the effects of both drugs. Thus, salicylazosulfapyridine was a sulfa drug, which has antibiotic activity, and a salicylate, which has anti-inflammatory activity. These two drug moieties were combined with an azo linkage (Fig. 1) but are therapeutically effective only after being cleaved.
Fig. 1.

Metabolism of salicylazosulfapyridine.
Salicylazosulfapyridine has been used to treat ulcerative colitis. When administered to rats or humans, the major excretory products were not the parent drug but rather the two component drugs and their metabolites. Others (Mueller and Miller, 1949; Fouts et al., 1957) had shown that the liver could catalyze the reductive cleavage of certain azo dyes and the sulfonamide prontosil, but the hepatic enzyme activity toward salicylazosulfapyridine was very low. Therefore, studies were done in germ-free rats to determine whether the drug was reduced by the intestinal bacteria. In germ-free animals the parent drug was excreted in the feces, and thus it was concluded that essentially all the reduction was performed by intestinal bacteria, rather than the liver (Peppercorn and Goldman, 1972b; Goldman et al., 1974). In addition, the oral antibiotic neomycin decreased the reductive cleavage of salicylazosulfapyridine in conventional rats. Because most of the bacteria are localized in the large intestine, the active drug to treat ulcerative colitis was produced precisely where it is needed. One might be able to take advantage of the intestinal microbiota-catalyzed metabolism of prodrugs to deliver to the colon drugs for preventing colon cancer. Sulfasalazine, another drug used in the treatment of ulcerative colitis, Crohn’s disease, and rheumatoid arthritis, is poorly absorbed by the gastrointestinal tract, because it is a substrate for the breast cancer resistant protein (ATP-binding cassette g2) transporter that effluxes chemicals from enterocytes back into the intestinal lumen (Zaher et al., 2006).
Intestinal Microbiota Decreases Drug Available for Absorption but Altered by Dietary Intervention
Cardiac glycosides were used as early as the time of the Egyptian and Roman empires and are still used today. Digoxin, which comes from the foxglove plant, is used for the treatment of atrial fibrillation and congestive heart failure. The three sugar molecules of digoxin are not necessary for its pharmacological actions, whereas the unsaturated lactone (a furanone) is about ten times more active than the saturated ring (butyrolactone or dihydrolactone) (Fig. 2).
Fig. 2.

Metabolism of digoxin.
In about 20% of people taking digoxin, a substantial amount is excreted with the lactone in the reduced form (dihydrolactone), and stool cultures from these people produce this same inactive metabolite from digoxin (Lindenbaum et al., 1981). Antibiotics, such as erythromycin and tetracycline, markedly decreased formation of the reduced-lactone metabolite and increased the serum concentration of the parent drug as much as twofold. Hundreds of human colonic isolates were examined, and it was concluded that Eubacterium lentum (now referred to as Eggerthella lenta) converted digoxin to the reduced metabolite (Saha et al., 1983). Surprisingly, individuals who did and did not produce the reduced-digoxin metabolite both had similar amounts of E. lenta in their intestinal microbiota, and thus the difference must not be due to the number of bacteria but the enzyme activity. When these bacteria were stimulated to grow with arginine, formation of the reduced-lactone of digoxin was diminished.
Thirty years later, using transcription profiling, comparative genomics, and culture- based assays, additional research was conducted to further understand the metabolism of digoxin by E. lenta. A cytochrome-encoding operon was identified in E. lenta, which is referred to as the “cardiac glycoside reductase” (cgr) operon. The transcription of the cgr operon is induced by digoxin, and its abundance predicts digoxin reduction by the human intestinal microbiota. Germ-free mice did not form dihydrodigoxin but when they were colonized with E. lenta, they formed the reduced lactone of digoxin. Feeding mice a high-protein diet decreased the formation of dihydrodigoxin and increased the serum concentrations of the active form of digoxin (Haiser et al., 2013).
Intestinal Microbiota Decreases Absorption of Drug by Metabolizing and Possibly Binding the Drug
Levodopa or l-dopa (l-3,4-dihydroxyphenylananine) is used for the treatment of Parkinson’s disease, which is a neurodegenerative disease characterized by four major clinical features: tremor, bradykinesia, rigidity, and disturbance of posture. Parkinson’s disease is the result of degeneration of the dopamine neurons in the basal ganglia. Therefore a major treatment of Parkinson’s disease is to increase dopamine levels in the basal ganglia. Because dopamine does not pass the blood-brain barrier, l-dopa is given and is decarboxylated by an amino acid decarboxylase in the brain to produce dopamine.
In the late 1960s, it was shown that the levels of m-hydroxyphenylacetic acid in patients being treated with l-dopa decreased markedly when they were given the antibiotic neomycin (Sandler et al., 1969) (Fig. 3). In fact, it was concluded the p-dehydroxylation of l-dopa to m-hydroxyphenylacetic acid was catalyzed entirely by intestinal microbiota. Similarly in rats, neomycin decreases the p-dehydroxylation of l-dopa to m-hydroxyphenylacetic acid (Bakke, 1971), and germ-free rats treated with l-dopa do not form m-hydroxyphenylacetic acid.
Fig. 3.

Metabolism of l-dopa.
Marshall and Warren received a Nobel Prize in 2005 for their discovery that inflammation in the stomach (gastritis) as well as ulceration of the stomach or duodenum (peptic-ulcer disease) is the result of infection of the stomach by the bacterium Helicobacter pylori. A number of clinicians have also noted an association of Parkinson’s disease and H. pylori infection. A clinical pharmacokinetic study was performed in Parkinson's disease patients and when the patients were pretreated with antibiotics to treat the H. pylori (amoxicillin, clarithromycin, and omeprazole), higher blood levels of l-dopa were observed than in patients not treated with the antibiotics (Pierantozzi et al., 2006). Subsequently, an in vitro study demonstrated that l-dopa can bind to H. pylori, and the authors suggested that this might be the reason for the lower absorption of l-dopa in Parkinson’s patients infected with H. pylori.
Intestinal Microbiota Increases Availability of Drug because Microbiota Produce a Chemical that Competes with Drug for Detoxification by the Host
Acetaminophen is one of the most used nonprescription drugs in the world. It is an analgesic and antipyretic, but with overdose produces liver injury and, if severe, may require liver transplantation. The importance of biotransformation of acetaminophen in producing hepatotoxicity has been known since the early 1970s (Mitchell et al., 1973; Klaassen ., 2013). Acetaminophen undergoes sulfation and glucuronidation, which are mediated by Phase II enzymes that conjugate the hydroxyl group of the drug with a sulfate or glucuronic acid, which detoxifies acetaminophen and enhances its excretion. These Phase II or conjugation reactions require not only an enzyme for the reaction but also the cosubstrate 3′-phosphoadenosine-5′-phosphosulfate for sulfation and uridine-diphospho-glucuronic acid for glucuronidation. Sulfation and glucuronidation of acetaminophen occurs in both the intestinal mucosa and liver, and both pathways are limited, apparently because of depletion of the cosubstrates (Hjelle and Klaassen, 1984; Hjelle et al., 1985; Goon and Klaassen, 1990; Boles and Klaassen, 2000). A small portion of acetaminophen (less than 10%) is activated by the cytochrome P-450 enzymes (CYP2E1, 1A2, and 3A4) to a reactive electrophile N-acetyl-p-benzoquinoneimine, which is detoxified by conjugation with glutathione. However, when glutathione levels are depleted, the reactive metabolites bind to macromolecules in hepatocytes, resulting in liver necrosis.
In an attempt to find biomarkers that might be used in personalized health care, a metabolomic analysis of chemicals in urine of people was quantified before and after a dose of acetaminophen. As anticipated, the major acetaminophen metabolites detected in urine were the sulfate and glucuronide conjugates; however, there were marked differences in the sulfate/glucuronide acetaminophen metabolite ratios among the various subjects. It was also noted that the people who excreted less acetaminophen-sulfate in urine had more cresol-sulfate in their urine before acetaminophen administration. Cresol-sulfate in urine originates from tyrosine, one of the amino acids in dietary proteins (Fig. 4). Tyrosine is metabolized by the intestinal microbiota to cresol (p-hydroxytoluene), which is then sulfated by the intestine and liver before excretion into urine. Therefore, it has been proposed that cresol and acetaminophen compete for sulfation by enterocytes, and thus the more cresol formed by the microbiota, the less acetaminophen is sulfated and detoxified (Clayton et al., 2009).
Fig. 4.

Sulfation of acetaminophen and cresol.
Intestinal Microbiota Biotransform Common Food Constituents
Caffeic acid (3,4-dihydroxycinnamic acid) is a dietary constituent found in all plants, due to its key role in the biosynthesis of lignin, an integral part of plant cell walls. Caffeic acid is found in very high levels (140 mg/100 g) in black chokecherry, and high levels (20 mg/100 g) are present in herbs, especially thyme, sage, and spearmint, as well as in some spices such as Ceylon cinnamon.
Caffeic acid is biotransformed to more than 10 metabolites found in the urine of rats and humans, with dihydrocaffeic acid and m-hydroxyphenylproprionic acid being the primary metabolites formed, which can be further biotransformed by methylation and other reactions (Booth et al., 1957) (Fig. 5). By administration of neomycin to suppress the intestinal microbiota (Dayman and Jepson, 1969) or utilizing germ-free rats (Peppercorn and Goldman, 1972a), it was discovered that the reduction and dehydroxylation reactions were performed by the intestinal microbiota. More recently, the biotransformation of caffeic acid in chlorogenic acid, which is an ester of caffeic acid and quinic acid, is also largely dependent on the intestinal microbiota (Gonthier et al., 2003).
Fig. 5.

Metabolism of caffeic acid
Intestinal Microbiota Biotransform Tryptophan into Aryl-Hydrocarbon Receptor Agonists that Stimulate the Immune System to Maintain a Healthy Intestine
2,3,7,8-Tetrachlorodibenzo-p-dioxin is the most potent agonist of the aryl-hydrocarbon receptor (AhR) and an immunotoxicant (Holsapple et al., 1991; Kerkvliet, 2002). However, more recent data indicate that with less activation of the AhR, the receptor plays an important physiologic role in the immune system (Stockinger et al., 2011). The physiologic activator of the AhR in the gastrointestinal tract appears to be a metabolite of the amino acid tryptophan, as a diet lacking in tryptophan impairs intestinal immunity in mice and alters the microbial community (Hashimoto et al., 2012). By correlating changes in tryptophan catabolic profiles with microbiota metagenomic composition, it was concluded that indole-3-aldehyde might be the AhR ligand important for intestinal immunity (Zelante et al., 2013). When the AhR is activated in lymphoid cells, they release interleukin-22, which in turn induces the synthesis of lipocalin-2 and calprotectin by the host. These two proteins sequester essential metal ions (iron, zinc, and manganese) that reduces their availability to bacteria, which results in the inhibition and growth of opportunistic bacteria (Behnsen and Raffatellu, 2013).
Intestinal Microbiota Biotransforms Food Constituents into Chemicals that Promote Cardiovascular Disease
Cardiovascular disease is a leading cause of death and morbidity. In an attempt to determine pathways and/or a biomarker for cardiovascular disease, a metabolomics study of small molecules in the plasma was performed. Three chemicals in the blood, namely choline, trimethylamine N-oxide (TMAO), and betaine (N,N,N-trimethylgycine), all of which are metabolites of phosphatidylcholine, were shown to predict cardiovascular disease in a large clinical cohort (Wang et al., 2011). Phosphatidylcholine (also called lecithin) is a major component of biologic membranes, and foods rich in phosphatidylcholine include eggs, milk, liver, red meat, chicken, shellfish, fish, and soybeans. Choline has a number of functions in the body; it is a component of the neurotransmitter acetylcholine and betaine, a cofactor for one-carbon metabolism.
When mice were fed a diet supplemented with TMAO, an upregulation of multiple macrophage scavenger receptors linked to atherosclerosis was observed, as well as increased accumulation of lipids in macrophages, and atherosclerosis. However, in mice receiving antibiotics or in germ-free mice, the TMAO did not produce atherosclerotic lesions (Wang et al., 2011). Similarly in humans, plasma concentrations of TMAO were suppressed markedly after administration of antibiotics and then rebounded after withdrawal of the antibiotics (Tang et al., 2013). The anaerobic conversion of choline to trimethylamine (TMA) in sulfate-reducing bacteria is catalyzed by enzymes encoded by genes in the so-called choline utilization cluster (Fig. 6). One of the genes in this cluster encodes the glycyl radical enzyme choline TMA lyase, which converts choline to TMA by a glycyl radical reaction (Craciun and Balskus, 2012). TMA is a volatile compound with an ammonia-like odor. Although the intestinal microbiota are responsible for the formation of trimethylamine from choline, the flavin mono-oxygenases (FMOs) in the liver of the host are responsible for N-oxidation of TMA to TMA-N-oxide (TMAO). Some humans have a defective FMO and may develop “fish-odor syndrome.”
Fig. 6.

Metabolism of phosphatidylcholine.
Carnitine is abundant in red meats and is biologically important for the transport of long-chain fatty acids into mitochondria for their breakdown by β-oxidation in the generation of ATP. Carnitine has a quaternary nitrogen, similar to choline, and as described for choline, carnitine is catabolized by the intestinal microbiota to TMA, which is oxidized to TMAO by FMO in the liver of the host (Fig. 7).
Fig. 7.

Chemical structures of choline and carnitine.
Supplementation of the diets of mice with carnitine markedly enhanced the concentration of TMA and TMAO in the plasma as well as atherosclerosis but not in mice in which the intestinal microbiota were suppressed by antibiotics. Mice fed a diet enriched with carnitine, choline, or TMAO had reduced cholesterol transport in vivo. They also showed that humans convert carnitine to TMAO, and humans who eat carnivorous diets produce more TMAO than humans who consume vegetarian/vegan diets (Koeth et al., 2013).
Intestinal Microbiota Forms a Drug Metabolite that Inactivates a Host Enzyme that Biotransforms a Second Drug
In 1993, a drug-drug interaction resulted in the death of 18 Japanese cancer patients with herpes zoster, a viral disease, after treatment with a new antiviral drug Sorivudine, in addition to an anticancer prodrug of 5-fluorouracil (5-FU), namely tegafur or capecitabine, which are biotransformed to 5-FU. These deaths occurred within 40 days after Sorivudine was approved for clinical use.
5-FU and its prodrugs are known to have toxic effects to various tissues that have rapid cell proliferation, especially the intestinal mucosa and bone marrow. As a result, patients receiving high doses of 5-FU often experience diarrhea and decreases in white blood cells and platelets. The patients who died also experienced these signs and symptoms, suggesting that the toxicity was due to increased concentrations of 5-FU reaching the systemic circulation. 5-FU is detoxified by dihydropyrimidine dehydrogenase, the same enzyme that metabolizes the endogenous pyrimidines uracil and thymine.
Sorivudine and 5-FU were given to rats, and essentially identical drug-drug interactions were observed in rats as in humans, namely diarrhea, marked decrease in white blood cells and platelets, and death within 10 days (Okuda et al., 1997). However, Sorivudine did not inhibit in vitro either rat or human dihydropyrimidine dehydrogenase (Ogura et al., 1998). It had been demonstrated a decade earlier that bromovinyluracil was a potent inhibitor of dihydropyrimidine dehydrogenase and that it would increase the biologic half-life of 5-FU fivefold and increase the area under the curve by eightfold in rats (Desgranges et al., 1986). In patients, Sorivudine also decreased dihydropyrimidine dehydrogenase enzyme activity, and it took 4 weeks to return to normal (Yan et al., 1997).
There was minimal or no enzymatic activity in liver, kidney, or intestines of rats to convert Sorivudine to the bromovinyluracil metabolite, but the enzyme activity to produce the metabolite was high in the intestinal contents (Nakayama et al., 1997) (Fig. 8). When antibiotics were given to rats that also were receiving Sorivudine, very little bromovinyluracil was detected in the blood, in contrast to rats not receiving the antibiotics to decrease the intestinal microbiota. The metabolism of bromovinyluracil by dihydropyrimidine dehydrogenase (in the host) produces a reactive intermediate that irreversibly inhibits the enzyme, a process called suicide inhibition or mechanism-based enzyme inactivation (Nishiyama et al., 2000). When bromovinyluracil formed from Sorivudine by intestinal microbiota inactivates dihydropyrimidine dehydrogenase, 5-FU is not detoxified. Consequently, the metabolism of Sorivudine by intestinal microbiota played a key role in the deaths of patients prescribed both Sorivudine and 5-FU prodrugs.
Fig. 8.
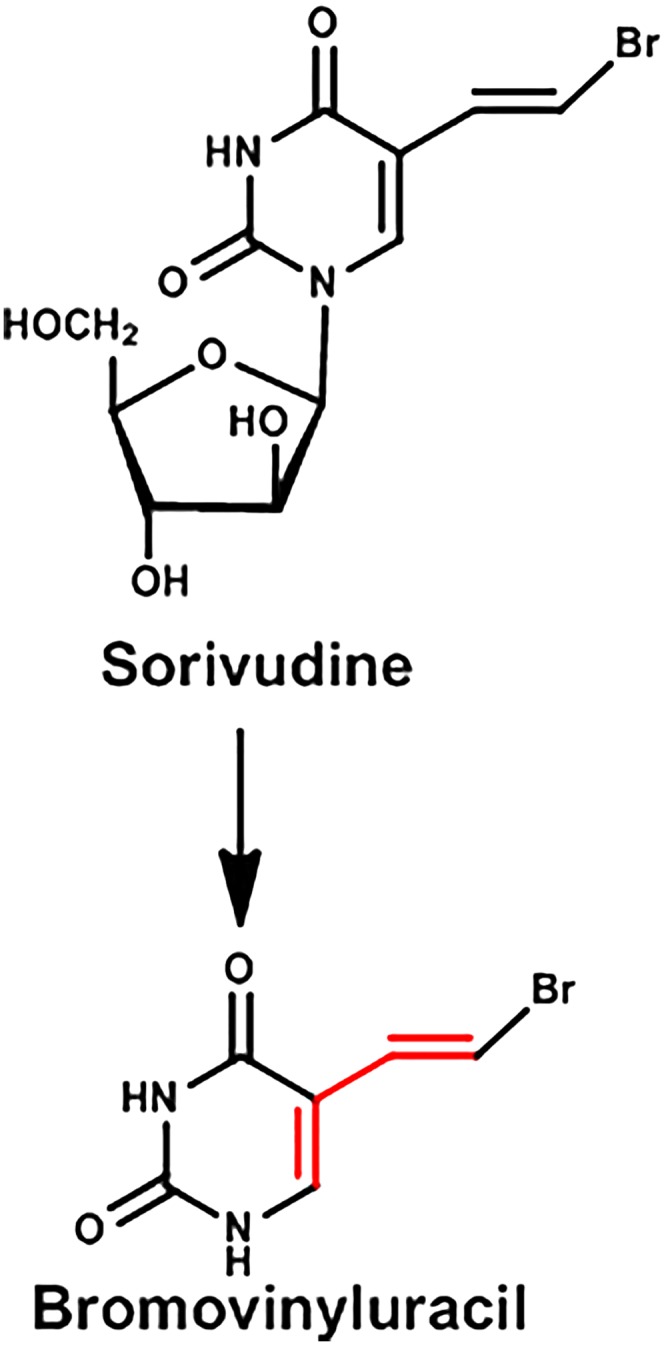
Metabolism of Sorivudine.
Intestinal Microbiota Responsible for Intestinal Toxicity of Cancer Chemotherapeutic Drug
Irinotecan is a common chemotherapeutic drug for colorectal cancer. The dose-limiting toxicities are neutropenia and late-onset diarrhea. The diarrhea is often sufficiently severe to require hospitalizations and dose reduction, which can lead to ineffective treatment. Irinotecan is biotransformed by carboxylesterase of the host to produce SN-38, the active form of the drug. SN-38 is detoxified in the host by glucuronidation (Fig. 9).
Fig. 9.

Metabolism of irinotecan.
In a study with rats, it was noted that the toxicity of irinotecan to various segments of the intestine was not correlated with intestinal tissue carboxylase activity, but rather with the β-glucuronidase activity of the contents in the lumen of the various segments. When antibiotics were given orally to rats to diminish their intestinal microbiota, the antibiotics completely inhibited the conversion of SN-38 glucuronide back to SN-38, and there was a marked amelioration of the diarrhea as well as reduced intestinal injury (Takasuna et al., 1996). In addition, irinotecan increases β-glucuronidase containing bacteria in the intestine of rats, further increasing the potential of conversion of SN-38 glucuronide back to the active and toxic metabolite SN-38 (Stringer et al., 2008). Inhibitors of bacterial β-glucuronidase also protected mice against the intestinal toxicity of irinotecan (Wallace et al., 2010). Thus the intestinal toxicity of irinotecan is due to the release of SN-38 in the colon after deconjugation of SN-38-glucurone by the intestinal microbiota. Thus either an inhibitor of intestinal β-glucuronidase or an antibiotic has the clinical potential to decrease irinotecan-induced intestinal toxicity.
Intestinal Microbiota Increases the Duration of Drug Action and Increases Drug Toxicity
There are many endogenous chemicals as well as drugs and other xenobiotics that are glucuronidated before being excreted into bile. A few examples include the sex hormones, thyroxine, morphine, cardiac glycosides, indomethacin, naphthol, propofol, and oxazepam. Once the drugs are glucuronidated by the intestine and/or the liver, they are often transported into the bile and excreted into the intestine. As noted for irinotecan, many glucuronidated chemicals can be de-conjugated by intestinal bacteria that express the β-glucuronidase gene. Thus when the drug glucuronide is hydrolyzed by intestinal microbiota, the parent drug can be reabsorbed into the systemic circulation and increase the duration of action of the drug.
It has been known for more than a half century that the enterohepatic circulation of drugs might be advantageous for the pharmacological effects of some drugs because it will prolong the action of the drug, but it is not desirable when a patient has developed toxicity from one of these drugs. For example, digitoxin has a very small margin of safety (a narrow therapeutic index), meaning that the difference between the blood concentrations of digitoxin that produces the desired effects on the heart and the blood concentration that produces toxicity is quite small. When the blood concentration of digitoxin reaches a toxic level, it would be advantageous to inhibit the enterohepatic circulation of digitoxin. Cholestyramine, an anion-binding resin prescribed for the treatment of hypercholesterolemia, will bind digitoxin and interfere with its enterohepatic circulation and decrease the toxic blood concentration to a safe-pharmacological concentration (Caldwell et al., 1971).
Intestinal Microbiota Responsible for Drug-Induced Intestinal Toxicity
Nonsteroidal anti-inflammatory drugs (NSAIDs) are used to treat inflammation, pain, and fever. Inflammation is a normal process in response to infections, as well as physical or chemical injury. The ability to mount an inflammatory response is essential to deal with pathogens and injury; however, the inflammatory response can be exaggerated and prolonged, apparently without benefit and with severe adverse consequences. The pharmacological actions of the NSAIDs are due to their ability to inhibit prostaglandin synthase enzymes, commonly known as the cyclooxygenases (COXs). Inhibition of COX-2 is considered responsible for producing the desired effects of NSAIDs, whereas inhibition of COX-1 is largely responsible for a major undesirable effect, gastrointestinal irritation and ulcers.
Many carboxylic acid-containing NSAIDs are conjugated with glucuronic acid in the liver before being transported into bile and excreted into the intestine (Fig. 10). In one study, mice were given the NSAID diclofenac at a dose that produces numerous large ulcers in the distal section of the small intestine. In a second group of mice, a bacteria-specific β-glucuronidase inhibitor was administered concurrently with the same dose of diclofenac, and this second group of mice had much less intestinal mucosal injury (LoGuidice et al., 2012). Similar studies have been done with two additional NSAIDs, indomethacin and ketoprofen, and again the intestinal lesions were reduced by an inhibitor of bacterial β-glucuronidase (Saitta et al., 2014). These data suggest the intestinal lesions produced by the NSAIDs are due to the β-glucuronidase in the intestinal microbiota that release the aglycone that is then free to enter the enterocytes to produce its toxic effects. Inhibition of β-glucuronidase of the intestinal microbiota prevents the formation of the aglycone and thus affords protection from NSAID-induced intestinal toxicity.
Fig. 10.

Metabolism of diclofenac.
Intestinal Microbiota Decreases and Increases the Mutagenicity of Food-Pyrolysis Products
Heterocyclic amines are formed during high-temperature cooking of meat and fish (food rich in protein, creatinine, and carbohydrates). Normally in such heating, desirable flavor compounds are formed; however, heterocyclic amines are also formed. Many heterocyclic amines are carcinogens.
There are more than 20 heterocyclic amines formed in high-temperature cooking of food. Noted is the structure of 2-amino-3-methylimidazo[4,5f]quinolone and 2-amino-1-methyl-6-phenylimidazo(4,5b)pyridine, more commonly known as IQ and PhIP (Fig. 11). Other heterocyclic amines in food are MeIQ, 2-amino-3,8-dimethyllimidazo(4,5)quinoxaline, 2-amino-6-methyldipyrido(1,2-a:3′,2′-d)imidazole, 2-amino-6-methyldipyrido(1,2-a:3′,2′-d)imidazole, 3-amino-1,4-dimethyl-pyrido(4,3-b)indole, and methyl-3-amino-5H-pyrido(4,3-b)indole. These chemicals are mutagenic in the Ames test and have been shown to be carcinogenic in rodent feeding studies. Although tumors are produced in many tissues, the most common is liver tumors. The carcinogenic properties of the heterocyclic amines are due to their metabolites, formed by N-hydroxylation by the cytochrome P450 enzymes, followed by O-sulfation and O-acetylation that results in reactive products that bind to DNA (Klaassen, 2013).
Fig. 11.

Chemical structures of PhIP and IQ.
An in vitro study showed that a number of these food-derived heterocyclic amines can bind to both Gram-positive and Gram-negative bacteria. Studies with Lactobacillus casei indicated that the binding was pH dependent (Morotomi and Mutai, 1986) and an additional study demonstrated that PhIP, a major mutagenic heterocyclic amine in the Western diet, bound better to lactic acid-producing strains than the other heterocyclic amines (Orrhage et al., 1994). A number of epidemiology and laboratory animal studies have shown that the consumption of cultured dairy products, such as yogurt, decreases the incidence of cancer. The lactic-acid producing bacteria in yogurt are usually Lactobacillus or Bifidobacterium. Therefore rats were fed a 5% diet of lyophilized cultures of Bifidobacterium, as well as 125 ppm IQ in the diet for 58 weeks. It was found that the Bifidobacterium in the diet inhibited IQ-induced colon and liver tumors in rats (Reddy and Rivenson, 1993). To determine whether the intestinal microbiota would also protect against heterocyclic amines, germ-free mice were inoculated with human fecal suspension. IQ was given to both germ-free mice and the humanized-microbiota mice, and binding of IQ in various tissues of the two groups of mice were quantified. Fewer DNA adducts of IQ were observed in the humanized-microbiota mice than in germ-free mice, indicating that both murine and human-microbiota are able to protect against IQ genotoxicity (Hirayama et al., 2000).
In contrast to the previous studies indicating that some bacteria can protect against food-derived heterocyclic amines, more recently, a study has been published indicating that other intestinal bacteria increase the genotoxicity of these chemicals. In this study, germ-free rats were inoculated with either an Escherichia coli strain that contains the uidA gene that codes for β-glucuronidase or with an identical strain that does not contain the uidA gene. The bacteria that expressed β-glucuronidase increased the genotoxicity of IQ in the intestine threefold over the rats that were inoculated with the bacteria that did not express β-glucuronidase (Humblot et al., 2007). Thus it has been proposed that glucuronidation of heterocyclic amines by the host decreases their mutagenicity, but the β-glucuronidase in the intestinal microbiota hydrolyze the glucuronide and release the toxic heterocyclic amine.
Intestinal Microbiota Metabolizes Substrate that Cocrystalizes with the Substrate and Precipitates in Kidney Tubules to Produce Toxicity
In 2007, both household pets and children were poisoned by melamine because it was added to cat and dog food as well as milk in China. The melamine was added to increase the apparent protein concentration of the food. The contaminated pet food was consumed by many cats and dogs in North America. The melamine in the pet food and milk resulted in acute kidney toxicity in cats, dogs, and humans. Pathologic evaluation of cats and dogs that had died indicated the presence of crystals in the kidney tubules. High-pressure liquid chromatography-mass spectrometry/mass spectrometry analysis identified melamine and cyanuric acid in the kidney, and microspectroscopy confirmed the presence of melamine-cyanuric acid cocrystals (Dobson et al., 2008) (Fig. 12). Thus the renal toxicity of two rather innocuous compounds resulted from their cocrystalizations, forming an insoluble precipitate in renal tubules leading to progressive tubular blockage and degeneration.
Fig. 12.

Chemical structures of melamine and cyanuric acid.
More recently, melamine was given to conventional rats and to rats that had their intestinal-microbiota suppressed by antibiotics. The renal toxicity of melamine was attenuated and melamine excretion was increased in the intestinal-microbiota-suppressed rats. It was also shown that normal rat feces can biotransform melamine to cyanuric acid. Klebsiella terrigena was shown to directly convert melamine to cyanuric acid, and rats inoculated with these bacteria showed exacerbated melamine-induced nephrotoxicity (Zheng et al., 2013). Therefore melamine-induced nephrotoxicity is dependent on the composition and metabolic activity of the intestinal microbiota.
Intestinal Microbiota Metabolizes Drug to a Teratogen
Nitrazepam is a benzodiazepine drug that is used for short-term relief of severe anxiety and insomnia. Nitrazepam is a teratogen when administered to pregnant rats, resulting in exencephaly, cleft palate, limb reductions, and other external and skeletal malformations. Incubation of nitrazepam with bacterial suspensions from rat cecal contents resulted in extensive reduction to 7-aminonitrazepam (Fig. 13). Antibiotic treatment of the pregnant rats before nitrazepam resulted in decreased production of the reduced compound from 30 to 2% of the dose. The incidence of nitrazepam-induced malformations was also markedly reduced by the antibiotic treatment. This suggests that the intestinal microflora reduces nitrazepam to 7-aminonitrazepam, which is responsible for the teratogenicity (Takeno et al., 1990; Takeno and Sakai, 1991).
Fig. 13.

Metabolism of nitrazepam.
Intestinal Microbiota Converts Prodrug to an Active Drug
Lovastatin is a hypolipedemic drug used to lower cholesterol. The mechanism of action of all statin drugs is to inhibit the enzyme 3-hydroxy-3-methyl-glutaryl-CoA reductase, which converts 3-hydroxy-3-methyl-glutaryl-CoA to mevalonate. Lovastatin is a prodrug, which needs to be converted from the gamma-lactone closed ring to the β-hydroxy acid open ring form to be active (Fig. 14). Incubation of lovastatin with bacteria from rat and human feces produced the active hydroxy-acid metabolite. When lovastatin was given to rats after their intestinal microbiota was markedly decreased by antibiotics, the hydroxy-acid metabolite was decreased about 50% in the plasma. These results indicate that antibiotics given to patients can result in drug-drug interactions by altering the intestinal microbiota (Yoo et al., 2014).
Fig. 14.

Metabolism of lovastatin.
Intestinal Microbiota Biotransforms Additional Drugs and Xenobiotics
Table 1 indicates a number of chemicals that are biotransformed by the intestinal microbiota, the type of chemical reaction, as well as the function of the drug or chemical. Many biotransformations by the intestinal microbiota do not have marked pharmacological or toxicological effects on the host. There are numerous reasons for this. A major reason is that most drugs are absorbed in the small intestine, but most of the microbiota is in the large intestine. Therefore, the microbiota is likely to have greater effects on poorly absorbed drugs and drugs that are designed to be delayed release. The effect is also dependent on the fraction of the drug that is biotransformed by the intestinal microbiota as well as the nature of the metabolite formed.
TABLE 1.
Chemicals biotransformed by intestinal microbiome
Other
| Bacterial Biotransformation | Chemicals | Chemical Applications |
|---|---|---|
| Azoreduction | Prontosil | Antibacterial |
| Neoprontosil | Antibacterial | |
| Sulfasalazine | Antibacterial | |
| Balsalazide | Anti-inflammatory | |
| Olsalazine | Anti-inflammatory | |
| Reduction | Nitrazepam | Antianxiety |
| Clonazepam | Antianxiety | |
| Misonidazole | Radiation sensitizer | |
| Omeprazole | Antiheartburn and antiulcer | |
| Sulfinpyrazone | Uricosuric | |
| Sulindac | Anti-inflammatory | |
| Digoxin | Antifibrillation | |
| Zonisamide | Anticonvulsant | |
| Metronidazole | Antibacterial | |
| Hydrolysis | Lactulose | Anticonstipation |
| Sorivudine | Antiviral | |
| Succinate group removal | Succinylsulfathiazole | Antibacterial |
| Dehydroxylation | Levo-dopa | Anti-Parkinsons |
| Acetylation | 5-Aminosalicylic acid | Anti-inflammatory |
| Deacetylation | Phenacetin | Analgesic and antipyretic |
| N-oxide bond bond cleavage | Ranitidine | Antiheartburn and antiulcer |
| Nizatidine | Antiheartburn and antiulcer | |
| Proteolysis | Insulin | Decrease blood glucose |
| Calcitonin | Decrease blood calcium | |
| Denitration | Glyceryl trinitrate | Antiangena |
| Isosorbide dinitrate | Antiangena | |
| Amine formation and hydrolysis of amide bond | Chloramphenicol | Antibiotic |
| Deconjugation | Digitoxin | Antifibrillation |
| Indomethacin | Anti-inflammatory | |
| Estrogens | ||
| Bile acids | ||
| Thiazole ring opening | Levamisole | Anthelmic |
| Isoxazole scission | Risperidone | Antipsychotic |
| Deglycosylation | Quercetin-3-glucoside | Natural product |
| N-demethylation | Methamphetamine | CNS stimulant |
| Other chemical reactions | Azetirelin | Increases thyrotropin |
| Potassium oxonate | Part of antitumor drug | |
| Flucytosine | Antifungal | |
| Hesperidin | Natural product | |
| Daidzein | Natural product |
Based on Sousa et al., 2008
Intestinal Microbiota Affects Host Drug Metabolism
Very few studies have been published on the effect of intestinal microbiota on drug disposition by the host. However, it is well known that some Phase I enzymes (oxidation, reduction, and hydrolysis enzymes), Phase II enzymes (conjugation enzymes), and transporters (both uptake and efflux transporters) are inducible and inhibited by drugs and other chemicals. Therefore, chemicals normally produced by the intestinal flora might likewise induce or inhibit drug metabolizing enzymes and transporters (Klaassen, 2013).
Comparisons of the expression of the mRNAs of these drug-processing genes in conventional and germ-free mice have been reported (Bjorkholm et al., 2009; Toda et al., 2009; Selwyn et al., 2015). Quantification of the mRNA in conventional and germ-free mice indicates that 21 drug-processing genes were increased, and 34 were decreased by the germ-free condition. Probably the most important findings are that the level of mRNA encoding Cyp3a in the germ-free mice is much lower, and the level of mRNA encoding Cyp4a is much higher in livers of germ-free mice (Selwyn et al., 2015) (Fig. 15).
Fig. 15.

Messenger RNA of Cyp3a11 and Cyp4a14 in conventional and germ-free mice.
The reason why the decrease in Cyp3a is of such importance is because it metabolizes over 50% of drugs, and the quantity of mRNA decreased markedly (87%) in the germ-free mice. In addition to mRNA content, the levels of Cyp3a11 protein also decreased markedly (Selwyn et al., 2015). The mechanism for the marked reduction of Cyp3a11 is not known, but Cyp3a11 is a well-known target gene of the PXR transcription factor. Because there is less Cyp3a11 in germ-free mice, this could indicate that the intestinal microbiota normally produces a ligand for PXR, and thus in the germ-free mice there may be less activation of PXR. One possible PXR ligand in conventional mice that is not present in germ-free mice is lithocholic acid. Lithocholic acid is a secondary bile acid produced from chenodeoxycholic acid by the intestinal microbiota, and lithocholic acid has been shown to be a PXR activator (Staudinger et al., 2001). Another possibility is an intestinal-microbiota metabolite of tryptophan, namely indole-3-propionic acid, which is formed by Clostridium sporgenes (Wikoff et al., 2009) (Fig. 16) and is a PXR ligand (Venkatesh et al., 2014). In humans there are marked interindividual differences in the expression of CYP3A4, and it is possible that these differences are due in part to variation in the intestinal microbiota in humans.
Fig. 16.

Metabolism of tryptophan.
The Cyp4 subfamily of Cyp enzymes is not as important for biotransforming drugs and other xenobiotics as are the Cyp1, 2, and 3 families. The Cyp4 enzymes are mainly involved in the metabolism of fatty acids and eicosanoids, and their mRNA and protein expression are regulated by the PPARα nuclear receptor. Because Cyp4a14 is regulated by PPARα, the increase in Cyp4a14 mRNA observed in germ-free mice might be a consequence of the absence of intestinal bacteria that catabolize chemicals that otherwise activate PPARα. It is known that PPARα is activated by lipids, as well as numerous drugs and other chemicals, such as the hypolipedemic fibrate drugs (clofibrate), NSAIDs, organic solvents (trichloroacetic acid), phthalate ester plasticizers, herbicides (2-4-dichlorophenoxyacetic acid), and perfluorodecanoic acid. At the present time it is not known what chemical, if any, is responsible for the activation of PPARα in germ-free mice, and the ultimate answer will require further studies to determine the mechanism for the increase in Cyp4a in germ-free mice.
Composition of the Intestinal Microbiota
The intestinal microbiota is thought to consist of 500 to 1000 species of bacteria. More than 90% of the species are Firmicutes and Bacteroidetes (Table 2). The various bacteria have numerous functions, but an essential function is energy harvest. Carbohydrates are important sources of energy for both host and bacteria. However, the host cannot degrade most complex carbohydrates and polysaccharides. These nondigestible carbohydrates are fermented in the colon by its microbiota to yield short-chain fatty acids, mainly acetate, propionate, and butyrate. Butyrate is an energy source for the colonic epithelium, whereas acetate and propionate are used by the peripheral tissues of the host. The types and amounts of short-chain fatty acids produced are determined by the amount of carbohydrate consumed and the composition of the gut microbiota. The short-chain fatty acids are an energy source for certain bacteria and the intestinal epithelial cells and are also important for intestinal morphology and function. The short-chain fatty acids produce their effects by binding to endogenous G protein receptors 41 and 43. A diet rich in fiber increases the proportion of Bacteroidaceae and Bifidobacteriaceae, whereas a low-fiber diet leads to microbiota that are dominated by Firmicutes, particularly those of the Erysipelotrichaceae family (Trompette et al., 2014). Thus there is a symbiotic relationship between the intestinal microbiota and the host. Furthermore the intestinal microbiota provides the host with vitamin B complex and vitamin K.
TABLE 2.
Common of the intestinal microbiota
| Intestinal Microbiota | Strains |
|---|---|
| Firmicutes (Gram positive) | Acetitomaculum spp. |
| Allisonella spp. | |
| Anaerofustis spp. | |
| Anaerotruncus spp. | |
| Anaerococcus sp. | |
| Bulleidia spp. | |
| Catenibacterium spp. | |
| Clostridium spp. | |
| Coprococcus spp. | |
| Dialister spp. | |
| Dorea spp. | |
| Entercoccus spp. | |
| Erysipelotricchus | |
| Eubacterium spp. | |
| Faecalibacterum spp. | |
| Gemella spp. | |
| Lactobacillus spp. | |
| Lachnospira spp. | |
| Peptococcus spp. | |
| Peptostreptococcus spp. | |
| Roseburia spp. | |
| Ruminococcus spp. | |
| Staphylococcus spp. | |
| Subdoligranulum spp. | |
| Actinobacteria (Gram positive) | Actinomyces spp. |
| Bifidobacterum spp. | |
| Collinsella spp. | |
| Corynebacterium spp. | |
| Bacteroidetes (Gram negative) | Alistipes spp. |
| Bacteroides spp. | |
| Porphyromonas gingivalis | |
| Prevotella spp. | |
| Rikenellaceae family | |
| Xylanibacter spp. | |
| Fusobacteria (Gram negative) | Fusobacterium spp |
| Proteobacteria (Gram negative) | Bilophilia wadsworthii |
| Desulfovibrio piger | |
| Enterobactericeae family | |
| Escherichia coli | |
| Haemophilus parainfluenziae | |
| Veillonellae (Gram negative) | Veillonella spp. |
| Verrucomicrobia | Akkermansia spp. |
| Archeae | Euryarchaeota spp. |
| Methanobrevibacter spp. | |
| Methanosphaera spp. |
Intestinal Microbiota and Obesity
In an attempt to determine whether the intestinal microbiota could play a role in the obesity epidemic, a high-fat, high-sugar (Western) diet was provided to germ-free and conventional mice. Surprisingly, the germ-free mice were protected from the obesity, insulin resistance, and hypercholesterolemia of the Western diet (Backhed et al., 2007; Rabot et al., 2010). The results of these landmark studies stimulated further research on the interactions of diet, intestinal microbiota, and obesity.
The intestinal microbiota of lean and obese mice littermates, as well as from lean and obese humans, differ in the proportion of Bacteroidetes and Firmicutes, with the obese mice and humans having an increase in Firmicutes (Turnbaugh et al., 2008, 2009). A similar change in the intestinal microbiota of mice was observed when mice were fed a high-fat and high-sugar (Western) diet in comparison with mice fed a low-fat and high-polysaccharide diet, with the Western diet increasing the Firmicutes and decreasing the Bacteroides, with a marked increase in Mollicutes, such as Eubacterium dolichum (Turnbaugh et al., 2008). When "obese microbiota" was transferred from obese mice to germ-free mice, the mice gained more weight than the mice that obtained the "lean microbiota" (Turnbaugh et al., 2006).
In a recent study, the microbiota from lean and obese twins were given to germ-free mice. The mice given the "obese microbiota" resulted in larger body weight and adiposity, increased metabolism of branched-chain amino acids, decreased fermentation of short-chain fatty acids, and changes in bile acid species with an accompanying decrease in intestinal FXR signaling. Cohousing the lean and obese mice transformed the intestinal microbiota of obese mice toward an intestinal microbiota more similar to that of lean mice (Ridaura et al., 2013). Diet is the dominate factor influencing the composition of the intestinal microbiota (Carmody et al., 2015). Numerous studies have shown that switching mice from a control to a high-fat (Western) diet will result in an increased Firmicutes/Bacteroidetes ratio in their intestinal microbiota, and the mice will exhibit metabolic syndrome, such as obesity, insulin resistance, dyslipidemia, and nonalcoholic steatohepatitis. This change in the intestinal microbiota by changing the diet can occur within a single day (Turnbaugh et al., 2009). Recently it has been shown that there is even a circadian rhythm in the intestinal microbiota of mice (Zarrinpar et al., 2014).
Prebiotics and Probiotics Alter the Intestinal Microbiota
For various perceived health benefits and because there is a correlation between the types of bacteria in the intestine and signs of metabolic syndrome, attempts have been made to alter the human intestinal microbiota by the use of probiotics and prebiotics. Probiotics are live microorganisms that, when consumed confer a health benefit to the host. Lactobacillis and Bifidobacterium are the most common strains used as probiotics. In contrast, prebiotics are nondigestible food ingredients that selectively stimulate the growth of some beneficial bacteria. Inulin and trans-galacto-oligosaccharide stimulate the growth of Bifidobacterium, known as a bifidogenic effect (Tremaroli and Backhed, 2012). Studies performed in both mice and humans using live organisms (probiotics) or fermentable carbohydrates (prebiotics) have demonstrated positive effects in altering obesity, hyperglycemia, and hepatic steatosis (Druart et al., 2014). Additional research is needed to determine the “dose response” of prebiotics and probiotics required to elicit desirable effects, undesirable effects, and the mechanism of both the desirable and undesirable effects of prebiotics and probiotics.
Bile Acids and the Intestinal Microbiota
Bile acids are toxic to microbiota. Bile acids are also metabolized by intestinal microbiota, which has consequences for the bacteria (decreased toxicity) and the host (altered homeostasis). Therefore, bile acid synthesis, transport, and regulation will be reviewed with emphasis on the role of intestinal microbiota.
Bile acids are synthesized in the liver from cholesterol. As depicted in Fig. 17, there are two pathways in the synthesis of bile acids, the classic and alternative pathway. The rate-limiting step in the synthesis of bile acids is catalyzed by Cyp7a1. The bile acids synthesized in liver are referred to as primary bile acids. The two primary bile acids synthesized by humans are cholic acid and chenodeoxycholic acid. Mice also produce these two primary bile acids, but in addition also synthesize α- and β-muricholic acid. Before excretion into bile, they are conjugated with glycine or taurine in humans, and in mice, primarily taurine (Fig. 17).
Fig. 17.

Synthesis and metabolism of bile acids.
In the intestine, the microbiota biotransform the primary bile acids synthesized in the liver into secondary bile acids. The conjugated bile acids excreted into bile are partially deconjugated, partially 7-dehydroxylated, and partially reduced/epimerized by the intestinal microbiota, which markedly increases the diversity of bile acids. The bile acids return to the liver, where some of the secondary bile acids can be biotransformed back into primary bile acids, for example, by 7-hydroxylation of deoxycholic acid to cholic acid (in mice) and reconjugating the bile acids that were deconjugated by the intestinal microbiota (Fig. 18).
Fig. 18.


Chemical structures of bile acids.
Humans have about eight grams of bile acids in their bodies, mainly in the liver and intestinal tract. Only about five percent of the bile acids are excreted in the feces each day. If a person eats three meals a day, the bile acids are excreted into and reabsorbed from the intestine at least three times a day, which suggests that at least 98% of the bile acids undergo enterohepatic circulation. This efficient system requires bile acid transporters in both the liver and intestine (Fig. 19). The bile acids in the liver are transported into the bile canaliculi by the bile salt export pump Bsep; in the intestine they are transported into enterocytes by the apical sodium bile acid transporter (Asbt) and effluxed out of the enterocytes by the heterodimer organic solute transporter α and β (Ostα/Ostβ). The bile acids then are delivered to the liver by the portal vein, where the hepatocytes have two transporters for the uptake of bile acids, one for the unconjugated bile acids, the organic anion transporting polypeptide 1b2 (Oatp1b2), and another for conjugated bile acids, the sodium taurocholate cotransporting polypeptide (Ntcp) (Klaassen and Aleksunes, 2010; Csanaky et al., 2011) (Fig. 19).
Fig. 19.

Uptake and efflux transporters of bile acids.
The intestinal microbiota is responsible for the conversion of primary bile acids synthesized in the liver to secondary bile acids in the intestine. The removal of taurine and glycine from conjugated bile acids is performed by the enzyme bile salt hydrolase, which is widespread in the intestinal microbiota. Deconjugation provides carbon, nitrogen, and sulfur for some bacteria. Unlike glycine, taurine contains a sulfonic acid moiety that is further catabolized to carbon disulfide. The 3-, 7-, and 12-hydroxyl groups of bile acids synthesized by livers are all in the alpha configuration, which results in the hydroxyl groups on one side of the steroid molecule, making the bile acids excellent detergents to aid in the solubilization and absorption of lipids and lipid-soluble vitamins. However, the intestinal bacteria can oxidize and epimerize the α-OH bile acids to β-OH bile acids and vice versa. The reason why bacteria convert α- to β-OH bile acids is probably to decrease the toxicity of the bile acid to the bacteria. The epimerization requires the formation of a stable oxo-bile acid intermediate, and thus the formation of the β-OH bile acid requires the combined effort of two specific hydroxysteroid dehydrogenases. Many Clostridia and a few other bacteria are known to perform these reactions. Deoxycholic and lithocholic acid are the two predominant bile acids in human feces; thus 7-dehydroxylation is an extremely common bile acid biotransformation reaction. The 7α/β-dehydroxylation pathway requires multiple oxidative and reductive steps with a net two-electron reduction, and thus is an energy benefit for the bacteria; however, more toxic secondary bile acids are formed (Ridlon et al., 2006). Thus, as a result of the host and the intestinal microbiota, a large diversity of bile acids is produced.
Bile Acids Regulate the Intestinal Microbiota
Bile acids are also a regulator of the intestinal microbiota. It has been known since the 1940s that bile acids have antimicrobial properties (Stacey and Webb, 1947). Most studies on the antimicrobial properties of bile acids have been done in vitro. However, a recent study in which rats were fed a diet containing cholic acid resulted in a marked decrease in Bacteroidetes from 37% to almost none and an increase in Firmicutes from 54 to 95% of the intestinal microbiota. Within the Firmicutes phylum, an increase in Clostridium and Ruminococcus occurred, which are 7α-dehydroxylating bacteria, resulted in an increased rate of conversion of cholic acid to deoxycholic acid (Islam et al., 2011), which is a better antimicrobial agent than cholic acid (Begley et al., 2005). Thus bile acids convert the intestinal microbiota to a higher Firmicutes/Bacteroidetes ratio, similar to what is observed after a high-fat diet.
Bile Acids as Signaling Molecules
Until recently, it was thought that the function of bile acids was twofold. One is to enhance the absorption of lipids and lipid soluble vitamins from the gastrointestinal tract, and the other is their secretion across the canaliculi to generate bile flow. At the turn of this century, it was discovered that there is a bile acid receptor in the liver, named the farnesoid-X-receptor (FXR) (Goodwin et al., 2000). This receptor appeared to be very important for the regulation of bile acid synthesis in the liver (Fig. 20, I). It was later discovered that the intestine also expresses FXR, which regulates the secretion of fibroblast growth factor 15 (Fgf15). Fgf15 is delivered by the portal blood to the liver, where it also regulates bile acid synthesis (Fig. 20, II) (Inagaki et al., 2005; Kim et al., 2007; Kong et al., 2012). The FXR receptor not only effects bile acid synthesis but also carbohydrate metabolism (Cariou et al., 2005; Duran-Sandoval et al., 2005; Ma et al., 2006) and inflammation (Gadaleta et al., 2011).
Fig. 20.

Schematic diagram of (I) FXR receptor in liver, (II) FXR receptor in intestine, (IIIA) TGR5 receptor in intestine, (IIIB) TGR5 receptor in gallbladder, and (IIIC) TGR5 receptor in fat and muscle.
The second major bile acid receptor is a G protein-coupled receptor that has been named TGR5 and is important in the regulation of energy metabolism (Watanabe et al., 2006). In contrast to FXR that is an intracellular receptor, TGR5 is a cell-surface bile acid receptor. As shown in Fig. 20, TGR5 has at least three important functions (Fig. 20, IIIA, IIIB, and IIIC). Figure 20, IIIA, illustrates that TGR5 is located in the intestine, and bile acids can promote glucagon-like peptide-1 (GLP-1) secretion upon activation by bile acids (Katsuma et al., 2005; Thomas et al., 2009). Patients treated with a bile acid-binding resin to reduce cholesterol levels also have improved glycemic control, and bile acid-binding resins also improve hyperglycemia in rodent models of obesity (Kobayashi et al., 2007; Chen et al., 2010), possibly because resins provide more bile acids to the lower intestine to activate TGR5. TGR5-overexpressing mice have enhanced GLP-1 secretion and insulin release in response to a glucose load, and TGR5-null mice have impaired glucose tolerance (Thomas et al., 2009). TGR5 activation by bile acids regulates energy expenditure and glucose homeostasis (Watanabe et al., 2006). Thus, through TGR5, bile acids regulate GLP-1 and subsequently regulate glucose homeostasis. A second function of TGR5, noted Fig. 20, IIIB, is its effects on the gall bladder and bile duct. In the gallbladder, TGR5 agonists increase cAMP levels and decrease intracellular chloride concentrations by increasing chloride secretion via the cAMP-regulated chloride channel, which is the cystic fibrosis transmembrane and conductance regulator (Keitel et al., 2009). TGR5-null mice have reduced gallbladder size. The TGR5 agonist INT-777 (a bile acid analog) increases gallbladder filling and gallbladder size in control mice but not in TGR5-null mice, illustrating that TGR5 activation promotes smooth muscle relaxation in gallbladder, leading to gallbladder filling. The third site where TGR5 has a major function is muscle and brown fat, as depicted in Fig. 20, IIIC. Bile acids bind to TGR5 expressed in brown adipose tissue and skeletal muscle and increase intracellular cAMP, which in turn increases type-2 iodothyronine-deiodinase activity. This enzyme is a thyroid hormone activating enzyme that converts thyroxine to tri-iodothyronine, which upregulates uncoupling proteins 1 and 3 and increases energy expenditure as heat (Watanabe et al., 2006). Treating diet-induced obese mice with the TGR5 agonist INT-777 leads to reduced body weight gain and increased energy expenditure in brown adipose tissue by upregulating the expression of mRNA for type-2 iodothyronine-deiodinase, uncoupling protein 1, and uncoupling protein 3 (Thomas et al., 2009). Therefore, activation of TGR5 influences energy expenditure in skeletal muscle and brown adipose tissue. Both bile acid receptors (FXR and TGR5) appear to be receptors that might be important in treating metabolic syndrome (Li and Chiang, 2014, 2015).
Bile Acids, Intestinal Microbiota, and Metabolic Syndrome
When mice are switched from a control to a high-fat diet, they gain considerable weight. However, when germ-free mice are placed on a high-fat diet, they do not exhibit such a marked increase in body weight (Backhed et al., 2007; Ding et al., 2010; Rabot et al., 2010). Germ-free mice also have higher bile acid concentrations in their plasma and liver than conventional mice. Mice fed antibiotics also have higher bile acid levels (Miyata et al., 2009; Zhang et al., 2014) as do Cyp8b1-null (12α-hydroxylase-null) mice. These three conditions have increased concentrations of tauro-β-muricholic acid (the taurine conjugate of β-muricholic acid), which acts as an antagonist of the FXR receptor, thus decreasing Fgf15, which decreases Cyp7a1, the enzyme that catalyzes the rate-limiting step of bile acid synthesis in the liver (Sayin et al., 2013; Hu et al., 2014). Thus some bile acids are FXR agonists and others are antagonists. It has been shown recently that Cyp8b1-null mice have improved glucose homeostasis due to elevated levels of GLP-1 (Kaur et al., 2015), which increases the biosynthesis and secretion of insulin by pancreatic beta cells, leading to improved glucose tolerance in Cyp8b1-null mice. This effect of GLP-1 is most likely due to the ability of various bile acids to activate the TGR-5 receptor. Unfortunately, at the present time there is little or no information on which bile acids are agonists and antagonists of either the FXR or TGR-5 receptor in vivo. However, both receptors appear to be drug targets that could improve human health.
Effect of a Chemical on Intestinal Microbiota, Bile Acids, and Metabolic Syndrome
Tempol, an antioxidant and radiation protector, prevents obesity in mice. To determine the mechanism of this phenomenon, a metabolomic analysis of chemicals in the intestine and feces of control and tempol-treated mice was investigated. A major difference between the two groups of mice was a marked increase in tauro-β-muricholic acid in the intestine of the tempol-treated mice. The tempol-treated mice have an altered intestinal microbiota with an increase in Bacteroidetes and decrease in Firmicutes. More specifically there was a marked decrease in Lactobacillis, which is a bile salt hydroxylase producing bacteria. Thus, in the mice treated with tempol, there is more tauro-β-muricholic acid, which is a FXR antagonist. Therefore less Fgf15, an inhibitor of bile acid synthesis, is secreted by the ileum to reach the liver, and thus Cyp7a1 is not inhibited and more bile acids are synthesized. Because FXR is not only involved in regulating bile acid synthesis, but also in glucose metabolism, these data suggest that FXR could be a target for antiobesity drugs (Li et al., 2013).
Effect of Environmental Chemicals on Intestinal Microbiota
The aryl hydrocarbon receptor (AhR) is a xenobiotic receptor that can be activated by numerous chemicals, such as the environmental pollutant 2,3,7,8-tetrachlorodibenzo-p-dioxin, polychlorinated biphenyls, and halogenated aromatic hydrocarbons. The toxicity of many of these chemicals appears to be due to binding and activating the AhR. As noted previously, tryptophan catabolites produced by the intestinal microbiota can also activate the AhR receptor. Recently, a persistent environmental contaminant, 2,3,7,8-tetrachlorodibenzofuran, an AhR agonist, was given to mice, and a marked increase in inflammation in the intestine and bile acids in the liver was detected. This was accompanied by an increase in Bacteroides and decrease in Firmicutes in the intestinal microbiota, more specifically an enrichment of Butyrivibrio and a depletion of Oscillobacter. This was accompanied by a decrease in FXR signaling. The authors concluded that the tetrachlorodibenzofuran altered the intestinal microbiota, which modulated the nuclear receptor signaling of FXR and subsequently altered metabolism of the host (Zhang et al., 2015).
In addition to organic chemicals, metals are also able to alter the intestinal microbiota. Germ-free and conventional mice were given cadmium or lead in their drinking water. Much more cadmium and lead were found in the tissues of the germ-free mice; thus the microbiota were protective for cadmium and lead (Breton et al., 2013). The mechanism for this protection is not known. Arsenic also perturbs the gut microbiota and its metabolomic profile. Mice were exposed to arsenic in the drinking water (10 ppm) for 4 weeks. The Bacteroidetes were not altered, but four Firmicutes families were decreased (Lu et al., 2014). A high-fat (Western) diet fed to mice will produce nonalcoholic hepatic steatosis, and the addition of arsenic to the water of mice fed a high-fat diet enhances the liver injury, which is characterized by enhanced inflammation (Tan et al., 2011). Feeding the prebiotic oligofructose protected mice against the enhanced liver damage caused by arsenic in the presence of a high-fat diet. These results indicate that prebiotics can protect against some toxicities (Massey et al., 2015).
Summary and Conclusion
In this review, we attempted to review the various effects of the intestinal microbiota on the biotransformation of drugs and other chemicals. As noted, the intestinal microbiota can deliver a drug to the large intestine, and it can also be responsible for the toxic effect of a chemical. Numerous examples have been given to demonstrate the mechanism by which the intestinal microbiota produces these toxic effects. However, as noted throughout the text, by understanding and altering the intestinal microbiota one can decrease the toxicity of a number of chemicals. Also by altering the intestinal microbiota one may be able to minimize the metabolic syndrome, which includes obesity, diabetes, atherosclerosis, fatty liver, and liver cancer. Thus one can consider the intestinal microbiota as another drug target.
Acknowledgments
The authors thank Andrew and Oliver Parkinson for proofreading this review and Dr. Feng Li for drawing the chemical structures.
Abbreviations
- AhR
aryl-hydrocarbon receptor
- cgr
cardiac glycoside reductase
- COX
cyclooxygenase
- Cyp
cytochrome P450
- Fgf
fibroblast growth factor
- FMO
flavin mono-oxygenases
- 5-FU
5-fluorouracil
- GLP-1
glucagon-like peptide-1
- H.
Helicobacter
- IQ
2-amino-3-methylimdazo[4,5f]quinolone
- mRNA
messenger-RNA
- NSAIDs
nonsteroidal anti-inflammatory drugs
- PhIP
2-amino-1-methyl-6-phenylimidazo(4,5b)pyridine
- PPARα
perixosomal proliferator activated receptor
- PXR
pregnane X receptor
- TGR5
transmembrane G protein-coupled receptor
- TMA
trimethylamine
- TMAO
trimethylamine N-oxide
- uidA
beta-d-glucuronidase
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Klaassen, Cui.
Footnotes
This work was supported by National Institutes of Health grants ES025708 and GM111381.
References
- Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. (2007) Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA 104:979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakke OM. (1971) Degradation of DOPA by intestinal microorganisms in vitro. Acta Pharmacol Toxicol (Copenh) 30:115–121. [DOI] [PubMed] [Google Scholar]
- Begley M, Gahan CG, Hill C. (2005) The interaction between bacteria and bile. FEMS Microbiol Rev 29:625–651. [DOI] [PubMed] [Google Scholar]
- Behnsen J, Raffatellu M. (2013) Keeping the peace: aryl hydrocarbon receptor signaling modulates the mucosal microbiota. Immunity 39:206–207. [DOI] [PubMed] [Google Scholar]
- Björkholm B, Bok CM, Lundin A, Rafter J, Hibberd ML, Pettersson S. (2009) Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS One 4:e6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles JW, Klaassen CD. (2000) Effects of molybdate and pentachlorophenol on the sulfation of acetaminophen. Toxicology 146:23–35. [DOI] [PubMed] [Google Scholar]
- Booth AN, Emerson OH, Jones FT, Deeds F. (1957) Urinary metabolites of caffeic and chlorogenic acids. J Biol Chem 229:51–59. [PubMed] [Google Scholar]
- Breton J, Daniel C, Dewulf J, Pothion S, Froux N, Sauty M, Thomas P, Pot B, Foligné B. (2013) Gut microbiota limits heavy metals burden caused by chronic oral exposure. Toxicol Lett 222:132–138. [DOI] [PubMed] [Google Scholar]
- Caldwell JH, Bush CA, Greenberger NJ. (1971) Interruption of the enterohepatic circulation of digitoxin by cholestyramine. II. Effect on metabolic disposition of tritium-labeled digitoxin and cardiac systolic intervals in man. J Clin Invest 50:2638–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk T, Grefhorst A, Bouchaert E, Fruchart JC, Gonzalez FJ, Kuipers F, Staels B. (2005) Transient impairment of the adaptive response to fasting in FXR-deficient mice. FEBS Lett 579:4076–4080. [DOI] [PubMed] [Google Scholar]
- Carmody RN, Gerber GK, Luevano JM, Jr, Gatti DM, Somes L, Svenson KL, Turnbaugh PJ. (2015) Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, McNulty J, Anderson D, Liu Y, Nystrom C, Bullard S, Collins J, Handlon AL, Klein R, Grimes A, et al. (2010) Cholestyramine reverses hyperglycemia and enhances glucose-stimulated glucagon-like peptide 1 release in Zucker diabetic fatty rats. J Pharmacol Exp Ther 334:164–170. [DOI] [PubMed] [Google Scholar]
- Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. (2009) Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci USA 106:14728–14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craciun S, Balskus EP. (2012) Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci USA 109:21307–21312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanaky IL, Lu H, Zhang Y, Ogura K, Choudhuri S, Klaassen CD. (2011) Organic anion-transporting polypeptide 1b2 (Oatp1b2) is important for the hepatic uptake of unconjugated bile acids: Studies in Oatp1b2-null mice. Hepatology 53:272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayman J, Jepson JB. (1969) The metabolism of caffeic acid in humans: the dehydroxylating action of intestinal bacteria. Biochem J 113:11P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgranges C, Razaka G, De Clercq E, Herdewijn P, Balzarini J, Drouillet F, Bricaud H. (1986) Effect of (E)-5-(2-bromovinyl)uracil on the catabolism and antitumor activity of 5-fluorouracil in rats and leukemic mice. Cancer Res 46:1094–1101. [PubMed] [Google Scholar]
- Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NM, Magness S, Jobin C, Lund PK. (2010) High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One 5:e12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson RL, Motlagh S, Quijano M, Cambron RT, Baker TR, Pullen AM, Regg BT, Bigalow-Kern AS, Vennard T, Fix A, et al. (2008) Identification and characterization of toxicity of contaminants in pet food leading to an outbreak of renal toxicity in cats and dogs. Toxicol Sci 106:251–262. [DOI] [PubMed] [Google Scholar]
- Druart C, Alligier M, Salazar N, Neyrinck AM, Delzenne NM. (2014) Modulation of the gut microbiota by nutrients with prebiotic and probiotic properties. Adv Nutr 5:624S–633S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran-Sandoval D, Cariou B, Percevault F, Hennuyer N, Grefhorst A, van Dijk TH, Gonzalez FJ, Fruchart JC, Kuipers F, Staels B. (2005) The farnesoid X receptor modulates hepatic carbohydrate metabolism during the fasting-refeeding transition. J Biol Chem 280:29971–29979. [DOI] [PubMed] [Google Scholar]
- Fouts JR, Kamm JJ, Brodie BB. (1957) Enzymatic reduction of prontosil and other azo dyes. J Pharmacol Exp Ther 120:291–300. [PubMed] [Google Scholar]
- Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen EC, Renooij W, Murzilli S, Klomp LW, Siersema PD, Schipper ME, Danese S, et al. (2011) Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 60:463–472. [DOI] [PubMed] [Google Scholar]
- Goldman P, Peppercorn MA, Goldin BR. (1974) Metabolism of drugs by microorganisms in the intestine. Am J Clin Nutr 27:1348–1355. [DOI] [PubMed] [Google Scholar]
- Gonthier MP, Verny MA, Besson C, Rémésy C, Scalbert A. (2003) Chlorogenic acid bioavailability largely depends on its metabolism by the gut microflora in rats. J Nutr 133:1853–1859. [DOI] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al. (2000) A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6:517–526. [DOI] [PubMed] [Google Scholar]
- Goon D, Klaassen CD. (1990) Dose-dependent intestinal glucuronidation and sulfation of acetaminophen in the rat in situ. J Pharmacol Exp Ther 252:201–207. [PubMed] [Google Scholar]
- Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. (2013) Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 341:295–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, Sigl V, Hanada T, Hanada R, Lipinski S, et al. (2012) ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 487:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama K, Baranczewski P, Akerlund JE, Midtvedt T, Möller L, Rafter J. (2000) Effects of human intestinal flora on mutagenicity of and DNA adduct formation from food and environmental mutagens. Carcinogenesis 21:2105–2111. [DOI] [PubMed] [Google Scholar]
- Hjelle JJ, Hazelton GA, Klaassen CD. (1985) Acetaminophen decreases adenosine 3′-phosphate 5′-phosphosulfate and uridine diphosphoglucuronic acid in rat liver. Drug Metab Dispos 13:35–41. [PubMed] [Google Scholar]
- Hjelle JJ, Klaassen CD. (1984) Glucuronidation and biliary excretion of acetaminophen in rats. J Pharmacol Exp Ther 228:407–413. [PubMed] [Google Scholar]
- Holsapple MP, Morris DL, Wood SC, Snyder NK. (1991) 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced changes in immunocompetence: possible mechanisms. Annu Rev Pharmacol Toxicol 31:73–100. [DOI] [PubMed] [Google Scholar]
- Hu X, Bonde Y, Eggertsen G, Rudling M. (2014) Muricholic bile acids are potent regulators of bile acid synthesis via a positive feedback mechanism. J Intern Med 275:27–38. [DOI] [PubMed] [Google Scholar]
- Humblot C, Murkovic M, Rigottier-Gois L, Bensaada M, Bouclet A, Andrieux C, Anba J, Rabot S. (2007) Beta-glucuronidase in human intestinal microbiota is necessary for the colonic genotoxicity of the food-borne carcinogen 2-amino-3-methylimidazo[4,5-f]quinoline in rats. Carcinogenesis 28:2419–2425. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, et al. (2005) Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2:217–225. [DOI] [PubMed] [Google Scholar]
- Islam KB, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. (2011) Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 141:1773–1781. [DOI] [PubMed] [Google Scholar]
- Katsuma S, Hirasawa A, Tsujimoto G. (2005) Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun 329:386–390. [DOI] [PubMed] [Google Scholar]
- Kaur A, Patankar JV, de Haan W, Ruddle P, Wijesekara N, Groen AK, Verchere CB, Singaraja RR, Hayden MR. (2015) Loss of Cyp8b1 improves glucose homeostasis by increasing GLP-1. Diabetes 64:1168–1179. [DOI] [PubMed] [Google Scholar]
- Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Häussinger D. (2009) The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology 50:861–870. [DOI] [PubMed] [Google Scholar]
- Kerkvliet NI. (2002) Recent advances in understanding the mechanisms of TCDD immunotoxicity. Int Immunopharmacol 2:277–291. [DOI] [PubMed] [Google Scholar]
- Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, Gonzalez FJ. (2007) Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res 48:2664–2672. [DOI] [PubMed] [Google Scholar]
- Klaassen CD, Aleksunes LM. (2010) Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol Rev 62:1–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen CD, Casarett LJ, Doull J. (2013) Casarett and Doull’s Toxicology: The Basic Science of Poisons, McGraw-Hill Education, New York. [Google Scholar]
- Kobayashi M, Ikegami H, Fujisawa T, Nojima K, Kawabata Y, Noso S, Babaya N, Itoi-Babaya M, Yamaji K, Hiromine Y, et al. (2007) Prevention and treatment of obesity, insulin resistance, and diabetes by bile acid-binding resin. Diabetes 56:239–247. [DOI] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. (2013) Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL. (2012) Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 56:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, Fabre KM, Mitchell JB, Patterson AD, Gonzalez FJ. (2013) Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun 4:2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chiang JY. (2014) Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev 66:948–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chiang JY. (2015) Bile acids as metabolic regulators. Curr Opin Gastroenterol 31:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbaum J, Rund DG, Butler VP, Jr, Tse-Eng D, Saha JR. (1981) Inactivation of digoxin by the gut flora: reversal by antibiotic therapy. N Engl J Med 305:789–794. [DOI] [PubMed] [Google Scholar]
- LoGuidice A, Wallace BD, Bendel L, Redinbo MR, Boelsterli UA. (2012) Pharmacologic targeting of bacterial β-glucuronidase alleviates nonsteroidal anti-inflammatory drug-induced enteropathy in mice. J Pharmacol Exp Ther 341:447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Abo RP, Schlieper KA, Graffam ME, Levine S, Wishnok JS, Swenberg JA, Tannenbaum SR, Fox JG. (2014) Arsenic exposure perturbs the gut microbiome and its metabolic profile in mice: an integrated metagenomics and metabolomics analysis. Environ Health Perspect 122:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Saha PK, Chan L, Moore DD. (2006) Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest 116:1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey VL, Stocke KS, Schmidt RH, Tan M, Ajami N, Neal RE, Petrosino JF, Barve S, Arteel GE. (2015) Oligofructose protects against arsenic-induced liver injury in a model of environment/obesity interaction. Toxicol Appl Pharmacol 284:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Brodie BB. (1973) Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Ther 187:185–194. [PubMed] [Google Scholar]
- Miyata M, Takamatsu Y, Kuribayashi H, Yamazoe Y. (2009) Administration of ampicillin elevates hepatic primary bile acid synthesis through suppression of ileal fibroblast growth factor 15 expression. J Pharmacol Exp Ther 331:1079–1085. [DOI] [PubMed] [Google Scholar]
- Morotomi M, Mutai M. (1986) In vitro binding of potent mutagenic pyrolysates to intestinal bacteria. J Natl Cancer Inst 77:195–201. [PubMed] [Google Scholar]
- Mueller GC, Miller JA. (1949) The reductive cleavage of 4-dimethylaminoazobenzene by rat liver; the intracellular distribution of the enzyme system and its requirement for triphosphopyridine nucleotide. J Biol Chem 180:1125–1136. [PubMed] [Google Scholar]
- Nakayama H, Kinouchi T, Kataoka K, Akimoto S, Matsuda Y, Ohnishi Y. (1997) Intestinal anaerobic bacteria hydrolyse sorivudine, producing the high blood concentration of 5-(E)-(2-bromovinyl)uracil that increases the level and toxicity of 5-fluorouracil. Pharmacogenetics 7:35–43. [DOI] [PubMed] [Google Scholar]
- Nishiyama T, Ogura K, Okuda H, Suda K, Kato A, Watabe T. (2000) Mechanism-based inactivation of human dihydropyrimidine dehydrogenase by (E)-5-(2-bromovinyl)uracil in the presence of NADPH. Mol Pharmacol 57:899–905. [PubMed] [Google Scholar]
- Ogura K, Nishiyama T, Takubo H, Kato A, Okuda H, Arakawa K, Fukushima M, Nagayama S, Kawaguchi Y, Watabe T. (1998) Suicidal inactivation of human dihydropyrimidine dehydrogenase by (E)-5-(2-bromovinyl)uracil derived from the antiviral, sorivudine. Cancer Lett 122:107–113. [DOI] [PubMed] [Google Scholar]
- Okuda H, Nishiyama T, Ogura K, Nagayama S, Ikeda K, Yamaguchi S, Nakamura Y, Kawaguchi Y, Watabe T. (1997) Lethal drug interactions of sorivudine, a new antiviral drug, with oral 5-fluorouracil prodrugs. Drug Metab Dispos 25:270–273. [PubMed] [Google Scholar]
- Orrhage K, Sillerström E, Gustafsson JA, Nord CE, Rafter J. (1994) Binding of mutagenic heterocyclic amines by intestinal and lactic acid bacteria. Mutat Res 311:239–248. [DOI] [PubMed] [Google Scholar]
- Peppercorn MA, Goldman P. (1972a) Caffeic acid metabolism by gnotobiotic rats and their intestinal bacteria. Proc Natl Acad Sci USA 69:1413–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppercorn MA, Goldman P. (1972b) The role of intestinal bacteria in the metabolism of salicylazosulfapyridine. J Pharmacol Exp Ther 181:555–562. [PubMed] [Google Scholar]
- Pierantozzi M, Pietroiusti A, Brusa L, Galati S, Stefani A, Lunardi G, Fedele E, Sancesario G, Bernardi G, Bergamaschi A, et al. (2006) Helicobacter pylori eradication and l-dopa absorption in patients with PD and motor fluctuations. Neurology 66:1824–1829. [DOI] [PubMed] [Google Scholar]
- Rabot S, Membrez M, Bruneau A, Gérard P, Harach T, Moser M, Raymond F, Mansourian R, Chou CJ. (2010) Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J 24:4948–4959. [DOI] [PubMed] [Google Scholar]
- Reddy BS, Rivenson A. (1993) Inhibitory effect of Bifidobacterium longum on colon, mammary, and liver carcinogenesis induced by 2-amino-3-methylimidazo[4,5-f]quinoline, a food mutagen. Cancer Res 53:3914–3918. [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341:1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, Hylemon PB. (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47:241–259. [DOI] [PubMed] [Google Scholar]
- Saha JR, Butler VP, Jr, Neu HC, Lindenbaum J. (1983) Digoxin-inactivating bacteria: identification in human gut flora. Science 220:325–327. [DOI] [PubMed] [Google Scholar]
- Saitta KS, Zhang C, Lee KK, Fujimoto K, Redinbo MR, Boelsterli UA. (2014) Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica 44:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler M, Karoum F, Ruthven CR, Calne DB. (1969) m-Hydroxyphenylacetic acid formation from L-dopa in man: suppression by neomycin. Science 166:1417–1418. [DOI] [PubMed] [Google Scholar]
- Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. (2013) Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17:225–235. [DOI] [PubMed] [Google Scholar]
- Selwyn FP, Cui JY, Klaassen CD. (2015) RNA-Seq Quantification of Hepatic Drug Processing Genes in Germ-Free mice. Drug Metab Dispos, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa T, Paterson R, Moore V, Carlson A, Abrahamson B, Basta W. (2008) The gastrointestinal microbiota as a site for the biotransformation of drugs. Int J Pharm 363:1–25. [DOI] [PubMed] [Google Scholar]
- Stacey M, Webb M. (1947) Studies on the antibacterial properties of the bile acids and some compounds derived from cholanic acid. Proc R Soc Med 134:523–537. [DOI] [PubMed] [Google Scholar]
- Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, et al. (2001) The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA 98:3369–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger B, Hirota K, Duarte J, Veldhoen M. (2011) External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin Immunol 23:99–105. [DOI] [PubMed] [Google Scholar]
- Stringer AM, Gibson RJ, Logan RM, Bowen JM, Yeoh AS, Keefe DM. (2008) Faecal microflora and beta-glucuronidase expression are altered in an irinotecan-induced diarrhea model in rats. Cancer Biol Ther 7:1919–1925. [DOI] [PubMed] [Google Scholar]
- Takasuna K, Hagiwara T, Hirohashi M, Kato M, Nomura M, Nagai E, Yokoi T, Kamataki T. (1996) Involvement of beta-glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT-11) in rats. Cancer Res 56:3752–3757. [PubMed] [Google Scholar]
- Takeno S, Nakagawa M, Sakai T. (1990) Teratogenic effects of nitrazepam in rats. Res Commun Chem Pathol Pharmacol 69:59–70. [PubMed] [Google Scholar]
- Takeno S, Sakai T. (1991) Involvement of the intestinal microflora in nitrazepam-induced teratogenicity in rats and its relationship to nitroreduction. Teratology 44:209–214. [DOI] [PubMed] [Google Scholar]
- Tan M, Schmidt RH, Beier JI, Watson WH, Zhong H, States JC, Arteel GE. (2011) Chronic subhepatotoxic exposure to arsenic enhances hepatic injury caused by high fat diet in mice. Toxicol Appl Pharmacol 257:356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. (2013) Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 368:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, et al. (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 10:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T, Saito N, Ikarashi N, Ito K, Yamamoto M, Ishige A, Watanabe K, Sugiyama K. (2009) Intestinal flora induces the expression of Cyp3a in the mouse liver. Xenobiotica 39:323–334. [DOI] [PubMed] [Google Scholar]
- Tremaroli V, Bäckhed F. (2012) Functional interactions between the gut microbiota and host metabolism. Nature 489:242–249. [DOI] [PubMed] [Google Scholar]
- Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod LP, Harris NL, et al. (2014) Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 20:159–166. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. (2008) Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. (2009) A core gut microbiome in obese and lean twins. Nature 457:480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031. [DOI] [PubMed] [Google Scholar]
- Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, Qiu Z, Maher L, Redinbo MR, Phillips RS, et al. (2014) Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 41:296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, et al. (2010) Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330:831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. (2011) Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, et al. (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439:484–489. [DOI] [PubMed] [Google Scholar]
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. (2009) Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA 106:3698–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Tyring SK, McCrary MM, Lee PC, Haworth S, Raymond R, Olsen SJ, Diasio RB. (1997) The effect of sorivudine on dihydropyrimidine dehydrogenase activity in patients with acute herpes zoster. Clin Pharmacol Ther 61:563–573. [DOI] [PubMed] [Google Scholar]
- Yoo DH, Kim IS, Van Le TK, Jung IH, Yoo HH, Kim DH. (2014) Gut microbiota-mediated drug interactions between lovastatin and antibiotics. Drug Metab Dispos 42:1508–1513. [DOI] [PubMed] [Google Scholar]
- Zaher H, Khan AA, Palandra J, Brayman TG, Yu L, Ware JA. (2006) Breast cancer resistance protein (Bcrp/abcg2) is a major determinant of sulfasalazine absorption and elimination in the mouse. Mol Pharm 3:55–61. [DOI] [PubMed] [Google Scholar]
- Zarrinpar A, Chaix A, Yooseph S, Panda S. (2014) Diet and feeding pattern affect the diurnal dynamics of the gut microbiome. Cell Metab 20:1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’Angelo C, Massi-Benedetti C, Fallarino F, et al. (2013) Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39:372–385. [DOI] [PubMed] [Google Scholar]
- Zhang L, Nichols RG, Correll J, Murray IA, Tanaka N, Smith PB, Hubbard TD, Sebastian A, Albert I, Hatzakis E, et al. (2015) Persistent organic pollutants modify gut microbiota-host metabolic homeostasis in mice through aryl hydrocarbon receptor activation. Environ Health Perspect 123:679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Limaye PB, Renaud HJ, Klaassen CD. (2014) Effect of various antibiotics on modulation of intestinal microbiota and bile acid profile in mice. Toxicol Appl Pharmacol 277:138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Zhao A, Xie G, Chi Y, Zhao L, Li H, Wang C, Bao Y, Jia W, Luther M, et al. (2013) Melamine-induced renal toxicity is mediated by the gut microbiota. Sci Transl Med 5:172ra22. [DOI] [PubMed] [Google Scholar]