

Abstract
Upon treatment with the pregnane X receptor (PXR) activator rifampicin (RIF), human hepatocellular carcinoma HepG2-derived ShP51 cells that stably express PXR showed epithelial-mesenchymal transition (EMT)–like morphological changes and migration. Our recent DNA microarrays have identified hepatocyte nuclear factor (HNF) 4α and insulin-like growth factor-binding protein (IGFBP) 1 mRNAs to be downregulated and upregulated, respectively, in RIF-treated ShP51 cells, and these regulations were confirmed by the subsequent real-time polymerase chain reaction and Western blot analyses. Using this cell system, we demonstrated here that the PXR-HNF4α-IGFBP1 pathway is an essential signal for PXR-induced morphological changes and migration. First, we characterized the molecular mechanism underlying the PXR-mediated repression of the HNF4α gene. Chromatin conformation capture and chromatin immunoprecipitation (ChIP) assays revealed that PXR activation by RIF disrupted enhancer-promoter communication and prompted deacetylation of histone H3 in the HNF4α P1 promoter. Cell-based reporter and ChIP assays showed that PXR targeted the distal enhancer of the HNF4α P1 promoter and stimulated dissociation of HNF3β from the distal enhancer. Subsequently, small interfering RNA knockdown of HNF4α connected PXR-mediated gene regulation with the PXR-induced cellular responses, showing that the knockdown resulted in the upregulation of IGFBP1 and EMT-like morphological changes without RIF treatment. Moreover, recombinant IGFBP1 augmented migration, whereas an anti-IGFBP1 antibody attenuated both PXR-induced morphological changes and migration in ShP51 cells. PXR indirectly activated the IGFBP1 gene by repressing the HNF4α gene, thus enabling upregulation of IGFBP1 to change the morphology of ShP51 cells and cause migration. These results provide new insights into PXR-mediated cellular responses toward xenobiotics including therapeutics.
Introduction
Pregnane X receptor (PXR, NR1I2), an orphan member of the nuclear steroid/thyroid receptor superfamily, is characteristically activated in response to numerous xenobiotics, including therapeutics (Kliewer et al., 1998). Upon activation, PXR regulates transcription of its target genes, playing roles in various liver functions from metabolism and excretion of therapeutics to energy metabolism (i.e., gluconeogenesis, lipogenesis, β-oxidation, and ketogenesis) (Kodama et al., 2004; Kodama et al., 2007; Nakamura et al., 2007). Through these regulations, PXR acts as a regulatory factor for various diseases, such as nonalcoholic steatohepatitis and fatty and cholestatic livers (Staudinger et al., 2001; Kakizaki et al., 2008; Konno et al., 2008; Wada et al., 2009; Zhou et al., 2009). In addition to these roles in metabolism, recent studies have indicated the regulation of cellular signals by PXR in various physiological and pathophysiological conditions. Treatment of pregnenolone 16α-carbonitrile, a potent rodent PXR activator, has been known to induce hepatocyte proliferation in rodents (Staudinger et al., 2001; Shizu et al., 2013). Drug-activated PXR has been shown to protect human and rat primary hepatocytes from drug-induced apoptosis (Zucchini et al., 2005). It has also been reported that PXR could alter both proliferative and apoptotic signal pathways in a cell type–specific manner in human colon cancer cell lines (Zhou et al., 2008; Ouyang et al., 2010). Meanwhile, studies using in vivo mouse models of inflammatory diseases and human samples have reported that PXR has anti-inflammatory activity wherein activated PXR interacts with nuclear factor κB–mediated inflammatory signals and suppresses expression of nuclear factor-κB target genes (Zhou et al., 2006; Shah et al., 2007b). Despite accumulating information, however, little is still known about PXR-mediated regulation of cellular signals.
Hepatocyte nuclear factor 4α (HNF4α, NR2A1), a member of the nuclear steroid/thyroid receptor superfamily, is one of the liver-enriched transcription factors (Sladek et al., 1990). HNF4α plays important roles in liver development and regulates various liver functions, cooperating with other hepatocyte nuclear factors such as HNF1 and HNF3 (Li et al., 2000; Hayhurst et al., 2001; Kyrmizi et al., 2006). Importantly, HNF4α plays a critical role in the development of liver cancer, such that the loss of HNF4α leads to increased cancer malignancy (Lazarevich and Alpern, 2008; Ning et al., 2010). Moreover, its cross-talk with PXR has been studied in the regulation of xenobiotic metabolism and energy metabolism in the liver (Tirona et al., 2003; Bhalla et al., 2004; Hwang-Verslues and Sladek, 2010). Whereas both HNF4α and PXR coordinately activate a number of genes in xenobiotic metabolism, recent findings have demonstrated that PXR could interfere with HNF4α-mediated expression of the key hepatic gluconeogenic genes (Bhalla et al., 2004; Kodama et al., 2007).
Our recent study has demonstrated that drug activation of PXR activates the immediate stress responsive growth arrest and DNA damage-inducible 45β (GADD45β) gene to elicit the p38 mitogen-activated protein kinase (MAPK) signaling pathways, resulting in epithelial-mesenchymal transition (EMT)–like morphological changes and migration in HepG2 cells that stably express human PXR (called ShP51 cells) (Kodama and Negishi, 2011). In the study, DNA microarray analyses were carried out to characterize gene expression in the cells during the PXR-induced cellular responses and identified GADD45β as one gene responsible for those cellular responses. There remains a possibility that PXR elicits cellular signals by activating additional unidentified genes that encode signaling molecules. Our DNA microarray analyses also identified HNF4α and insulin-like growth factor-binding protein (IGFBP) 1 as genes that are responsive to activation of PXR, with HNF4α being downregulated and IGFBP1 being upregulated.
Here, we characterized the PXR-HNF4α-IGFBP1 pathway as an additional cellular signal that facilitates morphological changes and causes migration of ShP51 cells after activation of PXR. First, we attempted to explore the molecular mechanism underlying the PXR-mediated repression of the HNF4α gene. Upon activation by a therapeutic rifampicin (RIF), PXR targeted the distal enhancer region and caused repressive changes in the chromatin structure of the HNF4α P1 promoter. After the elucidation of the molecular mechanism, we identified IGFBP1 to be another PXR-regulated signaling molecule that was upregulated as a consequence of the PXR-mediated downregulation of HNF4α and investigated the role of IGFBP1 in the PXR-induced EMT-like morphological changes and migration of ShP51 cells. Importantly, treatment with recombinant IGFBP1 augmented cell migration, whereas an anti-IGFBP1 antibody attenuated both induced EMT-like morphological changes and migration. As both IGFBP1 and GADD45β are known to regulate various cellular signals, PXR might enable cells to generate diverse cellular signals in response to xenobiotics, including therapeutics.
Materials and Methods
Rifampicin, SR12813 [[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]ethenylidene]bisphosphonic acid tetraethyl ester], phorbol 12-myristate 13-acetate (PMA), FLAG-M2 agarose beads, and anti–FLAG-M2 antibody were purchased from Sigma-Aldrich (St. Louis, MO); restriction endonucleases and DNA-modifying enzymes from New England Biolabs, Inc. (Ipswich, MA); mouse monoclonal antibodies to human PXR (H4417) and HNF4α (K9218 and H6939) from Perseus Proteomics Inc. (Tokyo, Japan); and mouse, goat, and rabbit normal IgGs and antibodies to HNF3β (M-20), HNF4α (H-171), retinoid X receptor α (C-20), IGFBP1 (H-5), IGFBP3 (C-19), and β-actin (C4) from Santa Cruz Biotechnology (Santa Cruz, CA); antibody to acetyl-histone H3 (K9/K14) from Merck Millipore (Billerica, MA); a recombinant IGFBP1 from R&D Systems (Minneapolis, MN); and ON-TARGETplus SMART pool HNF4α or ON-TARGETplus siCONTROL nontargeting pool from Thermo Fisher Scientific Inc. (Waltham, MA).
Vectors.
pCR3/hPXR, pCR3/FLAGhPXR, pcDNA3.1/hHNF3β, XREM-3A4-Luc, adeno-hPXR, and adeno-β-gal were described previously (Kodama et al., 2007, 2011; Kodama and Negishi, 2011). pCR3/hPXRΔAF2 and pCR3/FLAGhPXRΔAF2 were, respectively, generated from pCR3/hPXR and pCR3/FLAGhPXR by site-directed mutagenesis using a QuickChange mutagenesis kit (Agilent Technologies, Inc., Santa Clara, CA) and proper pairs of mutagenic oligonuleotides. Human PXRΔAF2 cDNA, digested from pCR3/hPXRΔAF2, was inserted into a pAdtrackCMV vector (American Type Culture Collection, Manassas, VA) to produce adeno-hPXRΔAF2. Human HNF4α P1 promoter containing the −7 kb/+67 bp region in a pGL3-basic vector (Promega, Madison, WI) was kindly provided by Dr. Iannis Talianidis (Biomedical Sciences Research Center Alexander Fleming, Greece), and we denoted it pGL3/7kb-hHNF4α-P1 in the present study. A series of mutants of the human HNF4α P1 promoter were generated by site-directed mutagenesis with the following mutagenic oligonuleotides: Δenhancer region, 5′-ACCGAGCTCTTACGCGGGTCTTAATCAGGCTAAGG-3′; HNF3 site, 5′-CCTTTATCTCTCTTTGGTAACGAGATCAATTTGCTCAGGACCCAGC-3′; DR1 site, 5′-GGGGGAACAAGCAGACTATGTCGACTTGAGCAAAGCCTCTTC-3′; C/EBP site, 5′-GGAGGCCAGCGGCCTGGATCCTAACCCTGGAGGCCTG-3′; HNF1 site, 5′-CGCAAACTCATGCCCAGTCTAGATTGGAAGGCAAAATCAACAGGC-3′.
Cell Culture, Drug Treatment, Transfection, and Infection.
Human hepatocellular carcinoma (HCC) HepG2 cells were maintained in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, penicillin (100 U/ml), and streptomycin (100 µg/ml) in an atmosphere of 5% CO2 at 37°C. ShP51, a stable cell line that expresses human PXR, was produced and established by transfection of HepG2 with pCR3/hPXR (Kodama and Negishi, 2011). Cells were seeded at a density of 3 × 105 cells per well of a six-well plate 48 hours before drug treatment. For real-time polymerase chain reaction (PCR) and Western blotting, respectively, total RNAs and whole-cell lysates were prepared from cells treated with 10 µM RIF in FBS-free MEM for a given time. For Luc reporter assays, cells were seeded at a density of 6 × 104 cells per well of a 24-well plate. After 24 hours, cells were transiently transfected with human HNF4α P1 promoter-firefly luciferase with or without a combination of expression plasmid as described in the figure legends, using FuGene6 (Roche, Indianapolis, IN). pRL-CMV for Renilla luciferase (Promega) was included in all transfection as a control. Luciferase reporter assays were performed as previously described (Kodama et al., 2004). For adenoviral infection, cells were seeded at a density of 3 × 105 cells per well of a six-well plate and cultured in MEM medium containing adeno-β-gal, adeno-hPXR, or adeno-hPXRΔAF2 at a multiplicity of infection of 10 for 30 hours. After being washed with FBS-free MEM, these cells were treated with 10 µM RIF in FBS-free MEM for a given time. For small interfering RNA (siRNA) knockdown, trypsinized cells (1.5 × 105 cells/well of a 12-well plate) were reverse-transfected with siRNA (50 pmol) in MEM medium, using Lipofectamine 2000 (Life Technologies, Carlsbad, CA). After 48 hours of incubation, total RNAs and whole-cell lysates were prepared for real-time PCR and Western blotting, respectively.
Western Blotting.
Cells were lysed and denatured in a fixed volume of NuPAGE LDS sample buffer (Life Technologies) and a fixed volume of the lysed cells was separated on a 10% SDS-PAGE gel and then transferred onto PVDF membrane. This membrane was blocked with 5% milk in tris-buffered saline with 0.05% (v/v) Tween 20 (TBS-T) for 1 hour at room temperature and then incubated with a given primary antibody in TBS-T containing 5% (w/v) bovine serum albumin for additional 16 hours at 4°C prior to incubation with secondary antibody in TBS-T with 5% milk for 1 hour at room temperature. Immunoreactive bands were visualized using ECL plus Western Blotting Detection Reagents (GE Healthcare, Piscataway, NJ).
Real-Time PCR.
Total RNAs were extracted using TRIZOL reagent (Life Technologies) to synthesize cDNA using a High Capacity cDNA Reverse Transcription Kit (Life Technologies). Real-time PCR was performed with an ABI Prism 7700 sequence detection system (Life Technologies). Assays-on-Demand probes (Life Technologies) were used for PCR with the TaqMAN PCR Master Mix (Life Technologies): Hs00430021_m1 for the human cytochrome P450 3A4 (CYP3A4) gene; Hs00230853_m1 for the human HNF4α gene; Hs00604431_m1 for the P1 promoter-driven human HNF4α gene; Hs01025522_m1 for the P2 promoter-driven human HNF4α gene. SYBR Green PCR Master Mix (Life Technologies) was also used for PCR using the following sets of primers: for the human IGFBP1 gene, 5′-GCCCTGCCGAATAGAACTCTAC-3′ and 5′-TCCATGGATGTCTCACACTGTCT-3′; for the human IGFBP3 gene, 5′-CGCCAGCTCCAGGAAATG-3′, and TGCCCTTTCTTGATGATGATTATC-3′; for the human heparin-binding epidermal growth factor–like growth factor (HB-EGF) gene, 5′-TCTGGACCTTTTGAGAGTCACTTTATC-3′ and 5′-CGTGCTCCTCCTTGTTTGGT-3′. The TaqMAN human β-actin control2 regents (Life Technologies) were used as the internal control.
Chromosome Conformation Capture Assays.
The chromosome confirmation capture (3C) assays were performed as previously described but with minor modifications (Hatzis et al., 2006; Saramaki et al., 2009; Kodama et al., 2011). Cells were seeded at a density of 1.2 × 106 cells on a 100-mm dish 48 hours before treatments. The cells were washed with FBS-free MEM, treated with RIF, dimethylsulfoxide (DMSO), or PMA for the indicated time in FBS-free MEM, and cross-linked by adding formaldehyde (final 2% (v/v)) for 10 minutes at room temperature. After being washed with ice-cold phosphate-buffered saline (PBS), the cells were incubated in the SDS lysis buffer for chromatin immunoprecipitation (ChIP) assays on ice for 10 minutes and then briefly sonicated on wet ice with intent to just disrupt the cellular membrane. After centrifugation, the supernatants were collected and diluted in restriction enzyme buffer 3 containing 1.8% (v/v) Triton X-100. After incubation at 37°C for 1 hour, aliquots containing approximate 1 × 106 nuclei were incubated with 500 U of BglII and 600 U of BclI at 37°C. Sixteen hours later, fresh enzymes were added to the reaction tubes and the digestion reaction was continued for another 4 hours. The reaction was terminated by adding SDS to a final concentration of 1.6% (v/v) and incubating for 20 minutes at 65°C. The digestion efficiency was confirmed by gel electrophoresis on a 2.0% agarose gel. An aliquot of digested chromatin was diluted with T4 DNA ligase buffer to 1% (v/v) final concentration Triton X-100 and 2.5 ng/μl of DNA and incubated at 37°C for 1 hour. The DNAs were ligated by using 1600 cohesive end units of T4 ligase for 4 hours at 16°C followed by 30 minutes at room temperature. Proteinase K, NaCl, and EDTA were added to the ligation mixture to final concentrations of 40 µg/ml, 0.2 M, and 1 mM, respectively. These mixtures were incubated at 65°C for 16 hours to reverse cross-linking and then treated with RNase A at 37°C for 30 minutes. The DNA was then purified by phenol extraction and ethanol precipitation. From the purified DNAs, ligated and control fragments were amplified by PCR using specific pairs of primers and resolved on a 2.0% (w/v) agarose gel. The PCR amplifications were quantified by densitometry, and the values were normalized by amplification of control products in each sample. The following primers were used for 3C assays: for the ligated fragment, 5′-CCAGCAGTTGTAATTAGCACC-3′ and 5′-TTAACTTCCAGGGTTGTCATG-3′ (Hatzis et al., 2006); for the control fragment, 5′-CGCTTCCCATCCCTGTTTGGA-3′ and 5′-CTCCAGGGTTATGCAAGAGGCC-3′.
ChIP Assays.
The ChIP assay was performed using a ChIP assay kit (Merck Millipore). Cells were seeded at a density of 2.5 × 106 cells on a 100-mm dish 48 hours before drug treatment. With the adenovirus infection, 24 hours after seeding, the cells were incubated for 30 hours with adenovirus at a multiplicity of infection of 10. These cells were then washed with FBS-free MEM and treated with RIF in FBS-free MEM for 6 hours, cross-linked by directly adding formaldehyde (final concentration, 1%) in medium, and incubated for 10 minutes at room temperature. After being washed with cold PBS, pellets of these cross-linked cells were sonicated to shear DNA in the SDS lysis buffer on wet ice. After being precleared by shaking with protein A or G, these lysates were incubated with 4 µg of antibodies or normal IgG at 4°C for 16 hours. The immunoprecipitated DNA was purified using a QIAquick PCR purification kit (Qiagen, Valencia, CA) according to the manufacture’s instruction. The purified DNA was used as a template for semiquantitative and real-time PCRs with specific pairs of primers. The amplicon was resolved on a 2.0% (w/v) agarose gel. The following primers were used for ChIP assays: for the HNF4α-enhancer, 5′-CGCTTCCCATCCCTGTTTGGA-3′ and 5′-CTCCAGGGTTATGCAAGAGGCC-3′; for the HNF4α-proximal, 5′-TGAGTCATGATGCCTGCCTTGTAC-3′ and 5′-CCTTCCTTTCAAACCGTCCTCTG-3′; for CYP3A4-XREM, 5′-ACTCATGTCCCAATTAAAGGTC-3′ and 5′-TGTTCTTGTCAGAAGTTCAGC-3′.
Immunoprecipitation Assays.
Trypsinized HepG2 cells (2.5 × 106) were reverse-transfected with expression plasmid as described in the figure legends, using FuGene6. After incubation for 30 hours, the cells were washed with FBS-free MEM and treated with 10 µM RIF in FBS-free MEM for 2 hours. Then, the cells were lysed in cold immunoprecipitation buffer (1% (v/v) Triton X-100, 150 mM NaCl, 10 mM Tris (pH 7.4), 1 mM EDTA, 1 mM EGTA (pH 8.0), 0.2 mM sodium ortho-vanadate, 0.2 mM PMSF, 0.5% NP-40, and 0.1% DMSO or 20 µM RIF) containing phosphatase inhibitor cocktail 1 and 2 (Sigma-Aldrich) for 20 minutes at 4°C. After centrifugation, the whole cell lysates were used for immunoprecipitation with FLAG-M2 agarose beads. After incubation for 2 hours at 4°C, the agarose beads were washed with immunoprecipitation buffer and subjected to Western blotting.
Cell Morphology and Migration.
Cells (48 hours after seeding; 1 × 105 cells/well in a 12-well plate) were incubated in FBS-free MEM with RIF for another 48 hours. These cells were fixed in PBS containing 5% (v/v) formaldehyde, followed by staining with a 0.1% (w/v) crystal violet solution. For siRNA knockdown, trypsinized cells were reverse-transfected with 40 μM of siRNA in MEM medium for 24 hours using Lipofectamine 2000 (Life Technologies). These cells were then maintained in FBS-free MEM for 48 hours before crystal violet staining. For antibody inhibition assays, control normal mouse IgG (1 µg/ml, Sigma-Aldrich), an anti-IGFBP1 (1 µg/ml), or an anti-IGFBP3 (1 µg/ml) antibody was added with RIF. Migration assays were performed using a 24-well transwell migration insert (Corning Incorporated, Corning, NY). For migration-stimulating and -inhibitory assays, a recombinant IGFBP1 (1 or 10 nM) and antibodies (normal mouse IgG, 1 µg/ml; anti-IGFBP1 antibody, 1 µg/ml; anti-IGFBP3 antibody, 1 µg/ml) were added to MEM in the lower chamber, respectively, in the presence of RIF or DMSO for 48 hours before crystal violet staining. PBS (1% (v/v)) was added in the corresponding control migrations.
Data Analysis.
All data are presented as mean ± S.D. Statistical analysis was performed by one-tailed Student’s t test or one-way analysis of variance followed by either Dunnett’s test or Tukey-Kramer’s test. A value of P < 0.05 was considered significant.
Results
Drug Activation of PXR Repressed the Expression of the HNF4α gene in HepG2 Cells.
Based on our previous DNA microarray analysis using the human HCC HepG2 cells and its clone that stably expresses human PXR, named ShP51, we had identified HNF4α as a gene that is downregulated by PXR under treatment with PXR activators (Kodama and Negishi, 2011). In the present study, we investigated the molecular mechanism for downregulation of the HNF4α gene by PXR. First, to confirm the DNA microarray data, we performed a time course analysis of the expression of the HNF4α gene in both parental HepG2 and ShP51 cells under RIF treatment by real-time PCR. As shown in Fig. 1A, a significant decrease in HNF4α mRNA was first observed at 4 hours after RIF treatment only in ShP51 cells. The levels of HNF4α mRNA gradually decreased for the duration of RIF treatment up to 24 hours. On the other hand, a typical PXR target CYP3A4 mRNA continuously increased for the duration of RIF treatment. SR12813, another human PXR activator we tested, was confirmed to decrease the levels of HNF4α mRNA and increase those of CYP3A4 mRNA in a PXR-dependent manner (unpublished data). We also tested other independent clones that stably express human PXR and obtained results similar to those obtained with ShP51 cells (unpublished data).
Fig. 1.

PXR downregulates transcription of the HNF4α gene in HepG2 cells. (A) Parental HepG2 and ShP51 cells were harvested at each time point after RIF treatment, and then total RNAs were prepared and subjected to real-time PCR. The levels of the total HNF4α and CYP3A4 mRNAs are expressed by taking their levels in the DMSO-treated parental HepG2 cells as one. Columns represent the mean ± S.D. from three independent experiments in triplicate. *P < 0.05 versus DMSO (Student’s t test); **P < 0.01 versus DMSO (Student’s t test). (B) Twenty-four hours after RIF treatment, total RNAs were prepared from cells and subjected to real-time PCR using specific probes for transcripts derived from the HNF4α P1 and P1 promoters. The levels of HNF4α transcripts are expressed by taking the HNF4α P1 promoter-derived transcripts in the DMSO-treated parental HepG2 cells as one. Columns represent the mean ± S.D. from three independent experiments in triplicate. *P < 0.05 versus DMSO (Student’s t test); **P < 0.01 versus DMSO (Student’s t test). (C) At time points of 12 and 24 hours after treatment with DMSO or RIF, whole-cell lysates were prepared and subjected to Western blotting using the following antibodies: total HNF4α, HNF4α-P1, HNF4α-P2, and actin. One representative of three independent experiments is shown. DM, DMSO.
The HNF4α gene is transcribed from either of two distinct promoters, P1 and P2 (Lazarevich and Alpern, 2008). The HNF4α P1 promoter regulates the expression of splicing variants 1−6 in the liver, kidney, and intestine/colon, and the HNF4α P2 promoter dictates that of splicing variants 7−9 in the intestine/colon, stomach, and pancreatic β cells and is also active in the fetal liver. The transcripts of the HNF4α gene derived from both promoters are reported to be expressed in several human HCC cells, including HepG2 cells. To assess which promoter is downregulated by PXR in HepG2 cells, we measured the levels of transcripts derived from both promoters by real-time PCR using probes specific to each promoter (Fig. 1B). In HepG2 cells, the HNF4α P1 promoter was predominantly active and the levels of HNF4α P2 promoter-derived transcripts were less than 10% of those of the HNF4α P1 promoter. In ShP51 cells, RIF treatment significantly decreased the levels of both promoter-derived transcripts (P1, about 80%; P2, about 55%) after 24 hours. These decreases were well correlated with the results of Western blotting using antibodies that specifically recognize the promoter-derived isoforms of HNF4α. In particular, the levels of the HNF4α P1–derived isoforms were dramatically decreased to the same extent as those of total HNF4α in ShP51 cells after treatment with RIF for 24 hours (Fig. 1C). We also found that ectopic PXR downregulated the transcription driven by the HNF4α P1 promoter in the human HCC cell line Huh7, which expresses human PXR adenovirally (unpublished data). Therefore, we next focused on the molecular mechanism of PXR-mediated downregulation of the HNF4α P1 promoter.
PXR Disrupts a Long-Range Interaction between the Distal Enhancer and Proximal Promoter Regions of the HNF4α P1 Promoter.
The HNF4α P1 promoter consists of the distal enhancer region extending around −6.5 kb from the transcription starting site and the proximal promoter region (Ladias et al., 1992; Parviz et al., 2003; Hatzis et al., 2006). A previous report demonstrated that the distal enhancer region stays close to the proximal promoter region by taking a looping structure to upregulate transcription from the HNF4α P1 promoter in HepG2 cells (Hatzis and Talianidis, 2001). To characterize the molecular mechanism by which PXR downregulates transcription from the HNF4α P1 promoter, we first performed 3C assays to determine whether PXR modulates the communication between the distal enhancer region and the proximal promoter region. After the previous report (Hatzis et al., 2006), we used the restriction enzymes BglII and BclI to digest the HNF4α P1 promoter. We amplified a 224-bp DNA fragment to assess intramolecular ligation of the BglII site located at −45 bp and the BclI site located at −7562 bp from the transcriptional starting site (Fig. 2A). RIF treatment significantly decreased PCR amplification by about 70% compared with DMSO treatment in ShP51 cells. On the other hand, no significant difference between the treatments was detected in parental HepG2 cells (Fig. 2B). The PMA-mediated MAPK activation has been reported to disrupt a loop between the distal enhancer region and the proximal promoter region to downregulate the HNF4α P1 promoter-derived HNF4α isoforms (Hatzis et al., 2006). As a quality control, cells were also treated with protein kinase C activator PMA, a potent inducer of the Ras-Raf-MEK-ERK signal, and subjected to 3C assays. As expected, PCR amplification was significantly decreased in both parental HepG2 and ShP51 cells after PMA treatment (Fig. 2B).
Fig. 2.

PXR disrupts a long-range interaction between the distal enhancer and proximal promoter regions of the HNF4α P1 promoter. (A) A schematic representation of the 3C assay for the P1 promoter of the HNF4α gene. Numbers indicate positions relative to the transcription starting site; arrows indicate the positions of the PCR primers. (B) Eight hours after treatment with DMSO or RIF, parental HepG2 and ShP51 cells were cross-linked by CH2O treatment, and then nuclei were prepared and subjected to 3C assays as described in Materials and Methods. From purified DNAs, formation of the 3C ligated fragment was detected by PCR amplification using TP1 and TP2 primers. A control fragment was also amplified using CP1 and CP2 primers to verify the quantity and quality of the DNA. The intensity of PCR amplification was quantified by densitometry, and the values were normalized by amplification of a control product in each sample and are expressed by taking the levels in the cells with DMSO treatment as one. Columns represent the mean ± S.D. from at least three independent experiments. *P < 0.05 (Dunnett’s test). DM, DMSO; nd, not detected. (C) Eight hours after treatment with DMSO or RIF, parental HepG2 and ShP51 cells were cross-linked by CH2O treatment and subjected to ChIP assays with normal IgG or anti–acetyl-histone H3 (K9/K14) antibody as described in Materials and Methods. The relative enrichment of the distal enhancer and proximal promoter regions of the HNF4α P1 promoter in the immunoprecipitated DNA fragments with anti–acetyl-histone H3 antibody was determined by real-time PCR. The XREM of the CYP3A4 promoter was also analyzed as a gene that PXR upregulates. Values are normalized by amplification of sample inputs and expressed by taking the values in the DMSO-treated parental HepG2 cells as one. Columns represent the mean ± S.D. from three independent experiments. *P < 0.05 (Tukey-Kramer’s test); **P < 0.01 (Tukey-Kramer’s test).
Subsequently, we performed ChIP assays to assess the levels of histone H3 acetylation in two regions of the HNF4α P1 promoter. In response to RIF treatment, acetylation at lysine 9 and lysine 14 was significantly reduced in both regions in ShP51 cells but not in parental HepG2 cells (Fig. 2C). Interestingly, a previous study reported that PMA treatment does not change the levels of acetylation of histones H3 and H4 in those two regions (Hatzis et al., 2006). On the other hand, the levels of histone H3 acetylation were significantly increased in the distal enhancer of the CYP3A4 promoter in RIF-treated ShP51 cells (Fig. 2C). Moreover, similar results were obtained from Huh7 cells that express PXR adenovirally (unpublished data). Based on these results, PXR appears to bring about changes in chromatin structure to downregulate the transcription from the HNF4α P1 promoter.
PXR Targets the Distal Enhancer Region to Repress Transcription from the HNF4α P1 Promoter.
Next, we performed cell-based Luc reporter assays using a 7 kb DNA fragment of the HNF4α P1 promoter (Hatzis and Talianidis, 2001) (Fig. 3A). RIF treatment repressed the activity of 7-kb HNF4α P1 promoter constructs by approximately 60% in ShP51 cells, but not in parental HepG2 cells. A deletion of the distal enhancer region (∆−7/−6.4 kb) decreased the activity of the 7-kb construct in both parental HepG2 and ShP51 cells and abrogated the response to RIF treatment in ShP51 cells. XREM-3A4-Luc reporter, a control for PXR activation, was strongly activated only in ShP51 cells in the presence of RIF. Similar results were obtained with Huh7 cells after cotransfecting the expression plasmid of PXR under the assay conditions (unpublished data). These results suggest that PXR targets the distal enhancer region to repress transcription from the HNF4α P1 promoter.
Fig. 3.
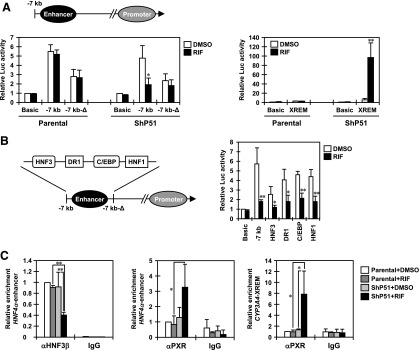
PXR targets HNF3β on the distal enhancer of the HNF4α P1 promoter. (A and B) Twenty-four hours after transfection, cells were treated with DMSO or RIF for another 24 hours in FBS-free MEM. pRL-TK was included in all transfections as a control. Relative Luc activity was calculated by taking the activity of the cells transfected with pGL3-Basic and treated with DMSO as one. Columns represent the mean ± S.D. from three independent experiments in triplicate. *P < 0.05 versus DMSO (Student’s t test); **P < 0.01 versus DMSO (Student’s t test). (A) Parental HepG2 and ShP51cells were transiently transfected with pGL3-basic, pGL3/7kb-hHNF4α-P1, pGL3/7kb-hHNF4α-P1-∆, or XREM-3A4-Luc reporter constructs. (B) ShP51cells were transiently transfected with pGL3-basic, pGL3/7kb-hHNF4α-P1, or the indicated mutant reporter constructs. (C) Parental HepG2 and ShP51 cells were treated with RIF or DMSO for 6 hours in FBS-free MEM and subjected to ChIP assays with normal IgG, an anti-HNF3β antibody, or an anti-PXR antibody using real-time PCR. Values are normalized by amplification of sample inputs and expressed by taking the values in the DMSO-treated parental HepG2 cells as one. Columns represent the mean ± S.D. from three independent experiments. *P < 0.05 (Tukey-Kramer’s test); **P < 0.01 (Tukey-Kramer’s test).
Previous studies have identified binding sites for HNF3β, HNF4α, C/EBPα, and HNF1α in the distal enhancer region of the HNF4α P1 promoter (Bailly et al., 2001; Hatzis and Talianidis, 2002; Hatzis et al., 2006).To identify the factor(s) responsible for the response to PXR activation, we constructed and used a series of 7-kb constructs harboring point mutations in each binding site for the transcription factors (Fig. 3B). Only a mutation introduced at the HNF3β-binding site strongly diminished the response to RIF treatment in ShP51 cells.
Given the results of Luc reporter assays, we performed ChIP assays to assess the occupation of the distal enhancer of the HNF4α P1 promoter by HNF3β (Fig. 3C). As reported previously, HNF3β occupancy was strongly detected in both parental HepG2 and ShP51 cells (Hatzis et al., 2006). RIF treatment significantly reduced the signal of HNF3β occupancy only in ShP51 cells (by about 60%), not in parental HepG2 cells. On the other hand, ChIP assays showed recruitment of PXR to the distal enhancer of the HNF4α P1 promoter (Fig. 3C), but not to the proximal promoter in RIF-treated ShP51 cells (unpublished data). As expected, RIF treatment significantly increased PXR occupancy in the CYP3A4 promoter. We obtained similar results from Huh7 cells that express PXR adenovirally, observing reduced HNF3β occupancy and recruitment of PXR in the distal enhancer (unpublished data). Further immunoprecipitation assays showed that PXR interacted with HNF3β in a ligand-dependent manner (Supplemental Fig. 1). Moreover, we confirmed that neither ectopic PXR nor RIF treatment had any effect on the levels of HNF3β protein for the duration of the assays (unpublished data). These results suggest that, upon activation by RIF, PXR targets the distal enhancer region of the HNF4α P1 promoter and stimulates dissociation of HNF3β from its binding site.
The Activation Function 2 Domain Determines the Repression Activity of PXR in the HNF4α P1 Promoter.
The activation function 2 (AF2) domain is essential for the ligand-dependent properties of nuclear receptors through interaction with coregulators. We constructed a PXR mutant in which helix 12 of the AF2 domain was deleted and used it for further analyses. First, we transduced HepG2 cells with adenoviruses expressing wild-type PXR and the AF2 mutant. Real-time PCR analyses showed that RIF treatment decreased the levels of HNF4α mRNA in HepG2 cells expressing wild-type PXR, but not the AF2 mutant (Fig. 4A). As expected, the wild-type PXR increased levels of CYP3A4 mRNA after RIF treatment and the AF2 mutant did not. Next, immunoprecipitation assays revealed functional defects caused by deletion of the AF2 domain (Supplemental Fig. 2). The AF2 mutant lost its ligand dependency for interaction with HNF3β, even though a weak interaction was maintained. We then conducted ChIP assays to further examine functional defects of the AF2 mutant in this repression. We found that the AF2 mutant was recruited to neither the distal enhancer of the HNF4α P1 promoter nor that of the CYP3A4 promoter under RIF treatment (Fig. 4B). These results indicate that the AF2 domain plays pivotal roles in this repression mechanism via interaction with HNF3β.
Fig. 4.
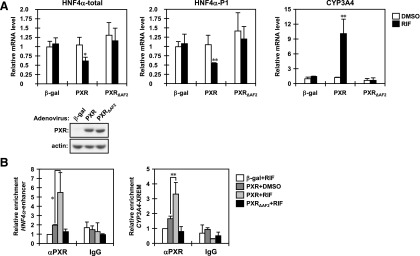
The AF2 domain determines the repression activity of PXR in the HNF4α P1 promoter. (A) HepG2 cells were infected with adeno-β-gal, adeno-hPXR, or adeno-hPXR∆AF2 for 30 hours and treated with DMSO or RIF for an additional 24 hours in FBS-free MEM. From those cells, total RNAs and whole-cell lysates were prepared and subjected to real-time PCR and Western blotting. The levels of total HNF4α, HNF4α P1 promoter-driven, and CYP3A4 mRNAs are expressed by taking the levels in the HepG2 cells infected with adeno-β-gal and treated with DMSO as one. Columns represent the mean ± S.D. from three independent experiments in triplicate. *P < 0.05 versus DMSO (Student’s t test); **P < 0.01 versus DMSO (Student’s t test). (B) After infection with the indicated adenovirus, HepG2 cells were treated with DMSO or RIF for 6 hours in FBS-free MEM and then subjected to ChIP assays with normal IgG or an anti-human PXR antibody as described in Materials and Methods. The relative enrichment of distal enhancer region of the HNF4a P1 promoter in the immunoprecipitated DNA fragments was determined by real-time PCR. The XREM of the CYP3A4 promoter was also analyzed as a gene that PXR upregulates. Values are normalized by amplification of sample inputs and expressed by taking the values in the HepG2 cells infected with adeno-β-gal and treated with DMSO as one. Columns represent the mean ± S.D. from three independent experiments. *P < 0.05 (Tukey-Kramer’s test); **P < 0.01 (Tukey-Kramer’s test).
PXR Downregulates HNF4α to Change Characteristics of HepG2 Cells.
ShP51 cells and HepG2 cells that express PXR adenovirally underwent EMT-like morphological changes and migrated when they were treated with RIF (Kodama and Negishi, 2011). Normally, ShP51 cells form an islet-like cell cluster and are indistinguishable from parental HepG2 cells. With RIF treatment, ShP51 cells became scattered and flattened (Fig. 5A). Our recent work demonstrated that PXR elicits the p38 MAPK signaling pathways by activating the GADD45β gene to lead HepG2 cells to change morphology and migrate (Kodama and Negishi, 2011). Given that HNF4α is a key regulator of hepatic gene expression and determines characteristics of hepatic cells (Parviz et al., 2003), we wondered about the consequence of loss of HNF4α on the morphological appearance of HepG2 cells. siRNA knockdown of HNF4α induced parental HepG2 cells to undergo morphological changes similar to those observed in ShP51 cells with RIF treatment (Fig. 5B). Likewise, the previous report showed that siRNA knockdown of HNF4α in HepG2 caused similar changes (Takagi et al., 2010). Taken together, these observations raise a possibility that, besides eliciting the p38 MAPK signaling pathways, PXR downregulates HNF4α to change the characteristics of HepG2 cells.
Fig. 5.

Loss of HNF4α induces morphological changes in HepG2 cells. (A) Parental HepG2 and ShP51 cells were treated with DMSO or RIF for 48 hours in FBS-free MEM and then stained with a crystal violet solution as described in Materials and Methods. Scale bar, 100 μm. One representative out of three independent experiments is shown. (B) Parental HepG2 cells were reverse-transfected with control or HNF4α siRNAs for 30 hours, treated with RIF or DMSO for another 48 hours in FBS-free MEM, and stained with a crystal violet solution. Scale bar, 100 μm. Whole cell extracts were also prepared and subjected to western blotting using anti-HNF4α or anti-actin antibodies. One representative out of three independent experiments is shown. cont, control.
PXR-Mediated Loss of HNF4α Is Responsible for IGFBP1 Induction.
Considering that HNF4α orchestrates the expression of a large number of genes to determine the characteristics of hepatic cells (Li et al., 2000; Hayhurst et al., 2001; Rhee et al., 2003; Kyrmizi et al., 2006; Lazarevich and Alpern, 2008), we hypothesized that PXR-mediated loss of HNF4α leads to broad changes in gene expression, so that PXR causes EMT-like morphological changes. Our DNA microarrays indicated that IGFBP1, IGFBP3, and HB-EGF mRNAs increase in RIF-treated ShP51 cells (Kodama and Negishi, 2011). Thus, real-time PCR was performed for confirmation. All three mRNAs were increased by RIF treatment in ShP51 cells but not in parental HepG2 cells, as observed with CYP3A4 mRNA (Fig. 6A). Several studies have demonstrated that these secreted proteins are either positively or negatively associated with cell migration in various cell lines (Jones et al., 1993; Madarame et al., 2003; Caceres et al., 2008; Chesik et al., 2010; Lin et al., 2011). Therefore, we used siRNA to examine whether the downregulation of HNF4α was the cause of their upregulation (Fig. 6A). Transfection of HNF4α siRNA decreased the levels of HNF4α mRNA to 50% of that of control siRNA in both parental HepG2 cells and ShP51 cells. Knockdown of HNF4α significantly increased mRNA levels of IGFBP1 and IGFBP3, but not HB-EGF, in both cell lines, and cotreatment with RIF further increased their mRNA levels only in ShP51 cells (Fig. 6A). As controls, we also confirmed that knockdown of HNF4α decreased mRNA levels of apolipoprotein C3 (APOC3) and sulfotranserase 1E1 (SULT1E1) in both cell lines and that RIF treatment further decreased their mRNA levels only in ShP51 cells (unpublished data). HNF4α has been reported to directly activate both the APOC3 and the SULT1E1 promoters (Ladias et al., 1992; Kodama et al., 2011).
Fig. 6.

PXR-mediated loss of HNF4α results in upregulation of IGFBP1. (A) Parental HepG2 and ShP51 cells were reverse-transfected with control or HNF4α siRNAs for 30 hours and were subsequently treated with DMSO or RIF for another 24 hours in FBS-free MEM. From those cells, total RNAs were prepared and subjected to real-time PCR using adequate PCR primers for each gene tested. The mRNA levels of the tested genes are expressed by taking the levels in the cells transfected with control siRNA and treated with DMSO as one. Columns represent the mean ± S.D. from three independent experiments in triplicate. **P < 0.01 (Tukey-Kramer’s test). (B and C) Forty-eight hours after treatment, whole-cell lysates were prepared and subjected to Western blotting using the following antibodies: IGFBP1, total HNF4α, and actin. (B) Parental HepG2 and ShP51 cells were treated with RIF or DMSO for 24 hours in FBS-free MEM. DM, DMSO. (C) ShP51 cells were reverse-transfected with control siRNA or HNF4α siRNA for 30 hours, and were subsequently treated with DMSO for another 24 hours in FBS-free MEM. One representative of three independent experiments is shown. cont, control.
Subsequently, we used Western blotting to determine the protein levels of IGFBP1 and HNF4α. IGFBP1 protein, which was expressed at low levels in ShP51 cells before RIF treatment, greatly increased after RIF treatment, whereas this protein was not detectable in HepG2 cells either before or after RIF treatment (Fig. 6B). Consistent with the increase in its mRNA, the IGFBP1 protein was significantly increased after knockdown of HNF4α in ShP51 cells without RIF treatment (Fig. 6C). On the other hand, the protein levels of IGFBP3 and HB-EGF appeared to be low and were not affected by RIF treatment and HNF4α knockdown (unpublished data). Therefore, IGFBP1 was selected for further investigation.
IGFBP1 Is Responsible for Cell Morphology and Migration.
As mentioned, ShP51 cells, but not parental HepG2 cells, underwent EMT-like morphological changes and migrated when they were treated with RIF. To examine whether IGFBP1 played any role in the RIF-induced morphological changes and migration, we carried out immune-neutralizing assays by cotreating RIF with normal IgG, an anti-IGFBP1 antibody, or an anti-IGFBP3 antibody. As expected, cotreatment with normal IgG caused the same morphological changes, as observed in our recent work (Kodama and Negishi, 2011). Treatment with an anti-IGFBP1 antibody, on the other hand, inhibited the morphological changes (Fig. 7A). No such inhibition was observed with an anti-IGFBP3 antibody.
Fig. 7.
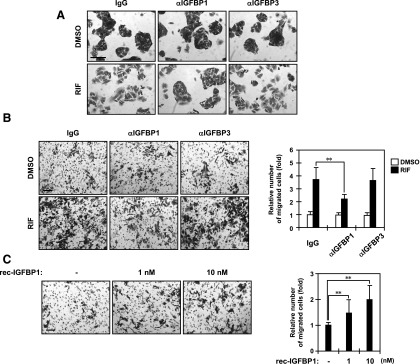
IGFBP1 is responsible for cell morphology and migration. (A) ShP51 cells were cotreated with normal IgG, an anti-IGFBP1 antibody, or an anti-IGFBP3 antibody and DMSO or RIF for 48 hours in FBS-free MEM and then stained with a crystal violet solution, as described in Materials and Methods. Scale bar, 100 μm. One representative out of three independent experiments is shown. (B and C) ShP51 cells were grown on the membrane of a transwell Boyden chamber and treated with the indicated stimuli for 48 hours in FBS-free MEM as described in Materials and Methods. Scale bar, 100 μm. The migrated cells were stained with a crystal violet solution and counted. Columns represent the mean ± S.D. from at least three independent experiments in triplicate. (B) Cells were cotreated with a normal IgG, an anti-IGFBP1 antibody, or an anti-IGFBP3 antibody and DMSO or RIF. **P < 0.01 (Dunnett’s test). (C) Cells were treated with PBS or a recombinant IGFBP1 protein at concentrations of 1 and 10 nM. **P < 0.01 (Dunnett’s test). rec, recombinant.
Using the Boyden chamber technique, we examined IGFBP1 for its effects on the RIF-induced migration of ShP51 cells. The number of staining cells that migrated to the bottom surface of the chambers decreased after cotreatment with an anti-IGFBP1 antibody compared with those that migrated after cotreatment with normal IgG or an anti-IGFBP3 antibody (Fig. 7B). RIF treatment increased migration approximately 4-fold in the presence of a normal IgG. Cotreatment with an anti-IGFBP1 antibody reduced this increase to approximately 2-fold, whereas an anti-IGFBP3 antibody did not reduce it. RIF, at concentrations greater than 100 µg/ml, has been reported to inhibit growth in various human cancer cells (Shichiri et al., 2009). No significant differences in cell growth were observed during treatments and cotreatments in our present experiments (unpublished data). Thus, an anti-IGFBP1 antibody inhibited the RIF-induced migration of ShP51 cells. Conversely, treatment of ShP51 cells with a recombinant IGFBP1 protein increased cell migration in a dose-dependent manner, with 50 and 100% increases at 1 and 10 nM IGFBP1, respectively (Fig. 7 C). No cell growth occurred after the IGFBP1 treatment (unpublished data). These results indicate that IGFBP1 is a PXR-induced factor responsible for cell morphological changes and migration in RIF-treated ShP51 cells, in which PXR represses the HNF4α gene, derepressing the IGFBP1 gene and leading the cells to induce IGFBP1.
Discussion
Xenobiotic exposure leads to diverse physiological consequences in the body through direct and indirect interactions with genes. PXR has been well characterized as a key mediator of xenobiotic action that interacts directly with a broad range of xenobiotics, including therapeutics, resulting in changes to gene regulation in the liver (Zhou et al., 2009). In particular, PXR is known to play the important roles in the various metabolic pathways in the liver. On the other hand, its roles in regulation of cellular signals are still far from being fully elucidated. Our recent study demonstrated that, upon activation by RIF, PXR activates the GADD45β gene to elicit the p38 MAPK-mediated cell migration signals in HCC HepG2 cells that stably express human PXR (called ShP51) (Kodama and Negishi, 2011). In the present study, IGFBP1 was demonstrated to be a PXR-regulated factor responsible for the RIF-induced EMT-like morphological changes and migration of ShP51 cells. PXR repressed the HNF4α gene by disrupting enhancer-promoter communication in the HNF4α P1 promoter and deacetylating histone H3. PXR-mediated downregulation of HNF4α resulted in upregulation of IGFBP1. Treatment with a recombinant IGFBP1 augmented migration of ShP51 cells, whereas an anti-IGFBP1 antibody attenuated the RIF-induced cell migration. The antibody treatment also inhibited the RIF-induced morphological changes. Thus, RIF activates the PXR-HNF4α-IGFBP1 pathway, signaling ShP51 cells to change morphology and migrate.
HNF4α, a member of the nuclear steroid/thyroid receptor superfamily, is an essential factor for the proper development and function of the liver (Sladek et al., 1990). Attenuation of HNF4α activity causes an EMT in human hepatocytes and promotes the progression of HCC (Lazarevich et al., 2004; Ning et al., 2010). Therefore, the repression of HNF4α by PXR may have serious implications in drug-induced liver injuries and tumor development and metastasis; for example, frequent intrahepatic recurrence and metastasis are considered critical reasons for poor prognosis of patients with liver cancer (Imamura et al., 2003; Llovet et al., 2003; Shah et al., 2007a). The present study has identified an upstream PXR response enhancer region within the HNF4α gene and has delineated the chromatin structure-based mechanism by which PXR represses transcription of the HNF4α gene. Not much is known about the direct HNF4α targets involved in EMT and/or migration at present; the Snail gene is the one that was characterized as the direct target of HNF4α to cause EMT (Cicchini et al., 2006). Therefore, IGFBP1 provides an alternative opportunity for further insight into the molecular mechanism of drug-induced EMT and cell migration via the nuclear xenobiotic receptor PXR.
Relatively limited information is available on the transcriptional regulation of the HNF4α gene compared with its physiological importance elucidated by in vivo studies using mouse models. Structurally, the HNF4α P1 promoter can be divided into two major components: the distal enhancer region and the proximal promoter region. Previous work has demonstrated that the distal enhancer region and proximal promoter region form a complex to trigger a critical nucleosome remodeling at the transcription start site to initiate activation of the HNF4α gene (Hatzis and Talianidis, 2002). In the present study, we found that, upon activation by RIF, PXR specifically targeted the distal enhancer to induce repressive changes in the chromatin structure of the HNF4α P1 promoter, such as disruption of enhancer-promoter communication and deacetylation of histone H3, so that PXR downregulated HNF4α in ShP51 cells. In this mechanism, HNF3β is considered a key molecule because a functional mutation at the HNF3-binding site in the distal enhancer significantly lowered the promoter activity and diminished the response to RIF treatment (Hatzis and Talianidis, 2002; Hatzis et al., 2006). PXR was also found to interact with HNF3β in a ligand-dependent manner, and deletion of the AF2 domain resulted in a weakened intensity of the interaction and loss of ligand-dependency. Our ChIP assays supported this hypothesis, showing that RIF treatment caused recruitment of PXR and dissociation of HNF3β at the distal enhancer region. Moreover, the AF2 mutant could no longer target the distal enhancer and repress the transcription of HNF4α gene under RIF treatment. However, the key questions remain unclear: what specifically targets PXR to the distal enhancer, in which no apparent PXR-binding sequence was found, and what enables PXR to dissociate HNF3β from the distal enhancer? Posttranslational modifications, including phosphorylation and acetylation, are known to regulate nuclear localization of HNF3β and its DNA binding activity (Wolfrum et al., 2003; van der Heide and Smidt, 2005; Howell and Stoffel, 2009; Kohler and Cirillo, 2010). The RIF-dependent PXR-HNF3β complex might be susceptible to these post-translational modifications and the resulting HNF3β dissociation might destabilize the active chromatin structure in the HNF4α P1 promoter. It will be of interest for us to define the molecular mechanisms that underline this PXR-mediated gene repression.
IGFBP1 is one of six members within the IGFBP family that bind to insulin-like growth factor 1 and 2 (IGF1 and 2) and modulate various insulin-like growth factor (IGF) actions such as IGF-dependent cell growth (Shimasaki and Ling, 1991; Jones and Clemmons, 1995; Valentinis et al., 1995). In addition, IGFBPs can exhibit IGF-independent functions (Gleeson et al., 2001). IGFBP1 contains an integrin recognition motif (Arg-Gly-Asp) and binds to the α5β1 integrin receptor, stimulating cell migration in the absence of IGF activity (Jones et al., 1993; Chesik et al., 2010). During our cell morphology and migration assays, FBS was removed so that the conditions resembled the absence of IGF activity. Therefore, what we observed with IGFBP1 in RIF-treated ShP51 cells is consistent with the role of IGFBP1 in cell migration. In our recent work, we characterized a PXR-GADD45β-p38 MAPK pathway for the cellular signals that trigger morphological changes as well as migration of ShP51 cells (Kodama and Negishi, 2011). By identifying IGFBP1 as a PXR-regulated factor, we characterized the PXR-HNF4α-IGFBP1 pathway to be a second signal that regulates cell morphological changes and migration. The PXR-GADD45β-p38 MAPK pathway appears to act as an intracellular signal to stimulate cell migration, whereas the PXR-HNF4α-IGFBP1 pathway can be considered an autocrine/paracrine signal because IGFBP1 is a secreted protein (Jones et al., 1993; Chesik et al., 2010). The simultaneous activation of both signaling pathways may result in a maximal cellular response. However, whether and how these two signaling pathways are linked remain important questions for future investigation into understanding PXR-mediated morphological changes and migration of ShP51 cells.
In conclusion, PXR, which is expressed in various cancers—such as colon, ovary, breast, endometrial, and prostate cancers—has been suggested to be involved in tumor progression and drug resistance by inducing enzymes such as CYP3A4, thereby metabolizing therapeutics as well as steroid hormones such as estrogens (Masuyama et al., 2003; Chen et al., 2007; Gupta et al., 2008). PXR has been reported to specifically activate the fibroblast growth factor 19 signal only in colon tumor cells, but not in normal intestinal cells, thereby enhancing their neoplastic characteristics (Wang et al., 2011). Our recent study has demonstrated that PXR immediately elicits the p38 MAPK signaling pathways after ligand treatment to stimulate morphological changes and migration in well differentiated HepG2 cells. It is not understood, however, whether PXR activation initiates and/or promotes hepatocellular carcinoma development in either normal or injured livers. The PXR-HNF4α-IGFBP1 pathway can be activated by numerous xenobiotics, including therapeutics, providing an alternative mechanism by which PXR may become a risk factor for cancer development and treatment. Activation of this pathway may have diverse consequences in regulating cellular functions and fates, depending on the type and pathophysiological conditions of cells, such as drug toxicity and drug-drug interactions. Further work is needed to dissect the in vivo relevance of PXR activation in cancers, what the consequences of PXR activation are, and how PXR causes them.
Supplementary Material
Acknowledgments
The authors thank the microarray core and sequencing core at the National Institute of Environmental Health Sciences for excellent assistance in the microarray and sequencing analyses used in this study and Dr. Iannis Talianids for kindly providing human HNF4α promoter constructs.
Abbreviations
- 3C
chromosome conformation capture
- AF2
activation function 2
- ChIP
chromatin immunoprecipitation
- DMSO
dimethylsulfoxide
- EMT
epithelial-mesenchymal transition
- FBS
fetal bovine serum
- HCC
hepatocellular carcinoma
- HNF
hepatocyte nuclear factor
- IGF
insulin-like growth factor
- IGFBP
insulin-like growth factor-binding protein
- MAPK
mitogen-activated protein kinase
- MEM
minimum essential medium
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- PMA
phorbol 12-myristate 13-acetate
- PXR
pregnane X receptor
- RIF
rifampicin
- siRNA
small interfering RNA
- SR12813
[[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]ethenylidene]bis-phosphonic acid tetraethyl ester
Authorship Contributions
Participated in research design: Kodama, Yamazaki, Negishi.
Conducted experiments: Kodama, Yamazaki.
Performed data analysis: Kodama, Yamazaki, Negishi.
Wrote or contributed to the writing of the manuscript: Kodama, Yamazaki, Negishi.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health and National Institute of Environmental Health Sciences [Grant Z01-ES1005-01].
 This article has supplemental material available at mol.pharm.aspetjournals.org.
This article has supplemental material available at mol.pharm.aspetjournals.org.
References
- Bailly A, Torres-Padilla ME, Tinel AP, Weiss MC. (2001) An enhancer element 6 kb upstream of the mouse HNF4α1 promoter is activated by glucocorticoids and liver-enriched transcription factors. Nucleic Acids Res 29:3495–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla S, Ozalp C, Fang S, Xiang L, Kemper JK. (2004) Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1α: functional implications in hepatic cholesterol and glucose metabolism. J Biol Chem 279:45139–45147. [DOI] [PubMed] [Google Scholar]
- Cáceres M, Tobar N, Guerrero J, Smith PC, Martínez J. (2008) c-jun-NH2JNK mediates invasive potential and EGFR activation by regulating the expression of HB-EGF in a urokinase-stimulated pathway. J Cell Biochem 103:986–993. [DOI] [PubMed] [Google Scholar]
- Chen Y, Tang Y, Wang MT, Zeng S, Nie D. (2007) Human pregnane X receptor and resistance to chemotherapy in prostate cancer. Cancer Res 67:10361–10367. [DOI] [PubMed] [Google Scholar]
- Chesik D, De Keyser J, Bron R, Fuhler GM. (2010) Insulin-like growth factor binding protein-1 activates integrin-mediated intracellular signaling and migration in oligodendrocytes. J Neurochem 113:1319–1330. [DOI] [PubMed] [Google Scholar]
- Cicchini C, Filippini D, Coen S, Marchetti A, Cavallari C, Laudadio I, Spagnoli FM, Alonzi T, Tripodi M. (2006) Snail controls differentiation of hepatocytes by repressing HNF4α expression. J Cell Physiol 209:230–238. [DOI] [PubMed] [Google Scholar]
- Gleeson LM, Chakraborty C, McKinnon T, Lala PK. (2001) Insulin-like growth factor-binding protein 1 stimulates human trophoblast migration by signaling through α5β1 integrin via mitogen-activated protein Kinase pathway. J Clin Endocrinol Metab 86:2484–2493. [DOI] [PubMed] [Google Scholar]
- Gupta D, Venkatesh M, Wang H, Kim S, Sinz M, Goldberg GL, Whitney K, Longley C, Mani S. (2008) Expanding the roles for pregnane X receptor in cancer: proliferation and drug resistance in ovarian cancer. Clin Cancer Res 14:5332–5340. [DOI] [PubMed] [Google Scholar]
- Hatzis P, Kyrmizi I, Talianidis I. (2006) Mitogen-activated protein kinase-mediated disruption of enhancer-promoter communication inhibits hepatocyte nuclear factor 4α expression. Mol Cell Biol 26:7017–7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzis P, Talianidis I. (2001) Regulatory mechanisms controlling human hepatocyte nuclear factor 4α gene expression. Mol Cell Biol 21:7320–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzis P, Talianidis I. (2002) Dynamics of enhancer-promoter communication during differentiation-induced gene activation. Mol Cell 10:1467–1477. [DOI] [PubMed] [Google Scholar]
- Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. (2001) Hepatocyte nuclear factor 4α (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol 21:1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell JJ, Stoffel M. (2009) Nuclear export-independent inhibition of Foxa2 by insulin. J Biol Chem 284:24816–24824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang-Verslues WW, Sladek FM. (2010) HNF4α--role in drug metabolism and potential drug target? Curr Opin Pharmacol 10:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H, Matsuyama Y, Tanaka E, Ohkubo T, Hasegawa K, Miyagawa S, Sugawara Y, Minagawa M, Takayama T, Kawasaki S, et al. (2003) Risk factors contributing to early and late phase intrahepatic recurrence of hepatocellular carcinoma after hepatectomy. J Hepatol 38:200–207. [DOI] [PubMed] [Google Scholar]
- Jones JI, Clemmons DR. (1995) Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev 16:3–34. [DOI] [PubMed] [Google Scholar]
- Jones JI, Gockerman A, Busby WH, Jr, Wright G, Clemmons DR. (1993) Insulin-like growth factor binding protein 1 stimulates cell migration and binds to the alpha 5 beta 1 integrin by means of its Arg-Gly-Asp sequence. Proc Natl Acad Sci USA 90:10553–10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizaki S, Yamazaki Y, Takizawa D, Negishi M. (2008) New insights on the xenobiotic-sensing nuclear receptors in liver diseases--CAR and PXR--. Curr Drug Metab 9:614–621. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterström RH, et al. (1998) An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92:73–82. [DOI] [PubMed] [Google Scholar]
- Kodama S, Hosseinpour F, Goldstein JA, Negishi M. (2011) Liganded pregnane X receptor represses the human sulfotransferase SULT1E1 promoter through disrupting its chromatin structure. Nucleic Acids Res 39:8392–8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama S, Koike C, Negishi M, Yamamoto Y. (2004) Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol Cell Biol 24:7931–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama S, Moore R, Yamamoto Y, Negishi M. (2007) Human nuclear pregnane X receptor cross-talk with CREB to repress cAMP activation of the glucose-6-phosphatase gene. Biochem J 407:373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama S, Negishi M. (2011) Pregnane X receptor PXR activates the GADD45β gene, eliciting the p38 MAPK signal and cell migration. J Biol Chem 286:3570–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler S, Cirillo LA. (2010) Stable chromatin binding prevents FoxA acetylation, preserving FoxA chromatin remodeling. J Biol Chem 285:464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno Y, Negishi M, Kodama S. (2008) The roles of nuclear receptors CAR and PXR in hepatic energy metabolism. Drug Metab Pharmacokinet 23:8–13. [DOI] [PubMed] [Google Scholar]
- Kyrmizi I, Hatzis P, Katrakili N, Tronche F, Gonzalez FJ, Talianidis I. (2006) Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes Dev 20:2293–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladias JA, Hadzopoulou-Cladaras M, Kardassis D, Cardot P, Cheng J, Zannis V, Cladaras C. (1992) Transcriptional regulation of human apolipoprotein genes ApoB, ApoCIII, and ApoAII by members of the steroid hormone receptor superfamily HNF-4, ARP-1, EAR-2, and EAR-3. J Biol Chem 267:15849–15860. [PubMed] [Google Scholar]
- Lazarevich NL, Alpern DV. (2008). Hepatocyte nuclear factor 4 in epithelial development and carcinogenesis Mol Biol 42:699–709. [Google Scholar]
- Lazarevich NL, Cheremnova OA, Varga EV, Ovchinnikov DA, Kudrjavtseva EI, Morozova OV, Fleishman DI, Engelhardt NV, Duncan SA. (2004) Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology 39:1038–1047. [DOI] [PubMed] [Google Scholar]
- Li J, Ning G, Duncan SA. (2000) Mammalian hepatocyte differentiation requires the transcription factor HNF-4α. Genes Dev 14:464–474. [PMC free article] [PubMed] [Google Scholar]
- Lin WH, Martin JL, Marsh DJ, Jack MM, Baxter RC. (2011) Involvement of insulin-like growth factor-binding protein-3 in the effects of histone deacetylase inhibitor MS-275 in hepatoma cells. J Biol Chem 286:29540–29547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet JM, Burroughs A, Bruix J. (2003) Hepatocellular carcinoma. Lancet 362:1907–1917. [DOI] [PubMed] [Google Scholar]
- Madarame J, Higashiyama S, Kiyota H, Madachi A, Toki F, Shimomura T, Tani N, Oishi Y, Matsuura N. (2003) Transactivation of epidermal growth factor receptor after heparin-binding epidermal growth factor-like growth factor shedding in the migration of prostate cancer cells promoted by bombesin. Prostate 57:187–195. [DOI] [PubMed] [Google Scholar]
- Masuyama H, Hiramatsu Y, Kodama J, Kudo T. (2003) Expression and potential roles of pregnane X receptor in endometrial cancer. J Clin Endocrinol Metab 88:4446–4454. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Moore R, Negishi M, Sueyoshi T. (2007) Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J Biol Chem 282:9768–9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning BF, Ding J, Yin C, Zhong W, Wu K, Zeng X, Yang W, Chen YX, Zhang JP, Zhang X, et al. (2010) Hepatocyte nuclear factor 4 α suppresses the development of hepatocellular carcinoma. Cancer Res 70:7640–7651. [DOI] [PubMed] [Google Scholar]
- Ouyang N, Ke S, Eagleton N, Xie Y, Chen G, Laffins B, Yao H, Zhou B, Tian Y. (2010) Pregnane X receptor suppresses proliferation and tumourigenicity of colon cancer cells. Br J Cancer 102:1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, Kaestner KH, Rossi JM, Zaret KS, Duncan SA. (2003) Hepatocyte nuclear factor 4α controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet 34:292–296. [DOI] [PubMed] [Google Scholar]
- Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan M, Gonzalez FJ, Spiegelman BM. (2003) Regulation of hepatic fasting response by PPARgamma coactivator-1α (PGC-1): requirement for hepatocyte nuclear factor 4α in gluconeogenesis. Proc Natl Acad Sci USA 100:4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saramäki A, Diermeier S, Kellner R, Laitinen H, Vaïsänen S, Carlberg C. (2009) Cyclical chromatin looping and transcription factor association on the regulatory regions of the p21 (CDKN1A) gene in response to 1α,25-dihydroxyvitamin D3. J Biol Chem 284:8073–8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SA, Cleary SP, Wei AC, Yang I, Taylor BR, Hemming AW, Langer B, Grant DR, Greig PD, Gallinger S. (2007a) Recurrence after liver resection for hepatocellular carcinoma: risk factors, treatment, and outcomes. Surgery 141:330–339. [DOI] [PubMed] [Google Scholar]
- Shah YM, Ma X, Morimura K, Kim I, Gonzalez FJ. (2007b) Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am J Physiol Gastrointest Liver Physiol 292:G1114–G1122. [DOI] [PubMed] [Google Scholar]
- Shichiri M, Fukai N, Kono Y, Tanaka Y. (2009) Rifampicin as an oral angiogenesis inhibitor targeting hepatic cancers. Cancer Res 69:4760–4768. [DOI] [PubMed] [Google Scholar]
- Shimasaki S, Ling N. (1991) Identification and molecular characterization of insulin-like growth factor binding proteins (IGFBP-1, -2, -3, -4, -5 and -6). Prog Growth Factor Res 3:243–266. [DOI] [PubMed] [Google Scholar]
- Shizu R, Benoki S, Numakura Y, Kodama S, Miyata M, Yamazoe Y, Yoshinari K. (2013) Xenobiotic-induced hepatocyte proliferation associated with constitutive active/androstane receptor (CAR) or peroxisome proliferator-activated receptor α (PPARα) is enhanced by pregnane X receptor (PXR) activation in mice. PLoS One 8:e61802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek FM, Zhong WM, Lai E, Darnell JE., Jr (1990) Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev 4 (12B):2353–2365. [DOI] [PubMed] [Google Scholar]
- Staudinger J, Liu Y, Madan A, Habeebu S, Klaassen CD. (2001) Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab Dispos 29:1467–1472. [PubMed] [Google Scholar]
- Takagi S, Nakajima M, Kida K, Yamaura Y, Fukami T, Yokoi T. (2010) MicroRNAs regulate human hepatocyte nuclear factor 4α, modulating the expression of metabolic enzymes and cell cycle. J Biol Chem 285:4415–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirona RG, Lee W, Leake BF, Lan LB, Cline CB, Lamba V, Parviz F, Duncan SA, Inoue Y, Gonzalez FJ, et al. (2003) The orphan nuclear receptor HNF4α determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat Med 9:220–224. [DOI] [PubMed] [Google Scholar]
- Valentinis B, Bhala A, DeAngelis T, Baserga R, Cohen P. (1995) The human insulin-like growth factor (IGF) binding protein-3 inhibits the growth of fibroblasts with a targeted disruption of the IGF-I receptor gene. Mol Endocrinol 9:361–367. [DOI] [PubMed] [Google Scholar]
- van der Heide LP, Smidt MP. (2005) Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem Sci 30:81–86. [DOI] [PubMed] [Google Scholar]
- Wada T, Gao J, Xie W. (2009) PXR and CAR in energy metabolism. Trends Endocrinol Metab 20:273–279. [DOI] [PubMed] [Google Scholar]
- Wang H, Venkatesh M, Li H, Goetz R, Mukherjee S, Biswas A, Zhu L, Kaubisch A, Wang L, Pullman J, et al. (2011) Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice. J Clin Invest 121:3220–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrum C, Besser D, Luca E, Stoffel M. (2003) Insulin regulates the activity of forkhead transcription factor Hnf-3β/Foxa-2 by Akt-mediated phosphorylation and nuclear/cytosolic localization. Proc Natl Acad Sci USA 100:11624–11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Tabb MM, Nelson EL, Grün F, Verma S, Sadatrafiei A, Lin M, Mallick S, Forman BM, Thummel KE, et al. (2006) Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest 116:2280–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Verma S, Blumberg B. (2009) The steroid and xenobiotic receptor (SXR), beyond xenobiotic metabolism. Nucl Recept Signal 7:e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Liu M, Zhai Y, Xie W. (2008) The antiapoptotic role of pregnane X receptor in human colon cancer cells. Mol Endocrinol 22:868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchini N, de Sousa G, Bailly-Maitre B, Gugenheim J, Bars R, Lemaire G, Rahmani R. (2005) Regulation of Bcl-2 and Bcl-xL anti-apoptotic protein expression by nuclear receptor PXR in primary cultures of human and rat hepatocytes. Biochim Biophys Acta 1745:48–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.