

Abstract
We previously reported that benzopyrimido-pyrrolo-oxazinedione BPO-27 [6-(5-bromofuran-2-yl)-7,9-dimethyl-8,10-dioxo-11-phenyl-7,8,9,10-tetrahydro-6H-benzo[b]pyrimido [4′,5′:3,4]pyrrolo [1,2-d][1,4]oxazine-2-carboxylic acid] inhibits the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel with low nanomolar potency and reduces cystogenesis in a model of polycystic kidney disease. We used computational chemistry and patch-clamp to show that enantiomerically pure (R)-BPO-27 inhibits CFTR by competition with ATP, whereas (S)-BPO-27 is inactive. Docking computations using a homology model of CFTR structure suggested that (R)-BPO-27 binds near the canonical ATP binding site, and these findings were supported by molecular dynamics simulations showing a lower binding energy for the (R) versus (S) stereoisomers. Three additional lower-potency BPO-27 analogs were modeled in a similar fashion, with the binding energies predicted in the correct order. Whole-cell patch-clamp studies showed linear CFTR currents with a voltage-independent (R)-BPO-27 block mechanism. Single-channel recordings in inside-out patches showed reduced CFTR channel open probability and increased channel closed time by (R)-BPO-27 without altered unitary channel conductance. At a concentration of (R)-BPO-27 that inhibited CFTR chloride current by ∼50%, the EC50 for ATP activation of CFTR increased from 0.27 to 1.77 mM but was not changed by CFTRinh-172 [4-[[4-oxo-2-thioxo-3-[3-trifluoromethyl)phenyl]-5-thiazolidinylidene]methyl]benzoic acid], a thiazolidinone CFTR inhibitor that acts at a site distinct from the ATP binding site. Our results suggest that (R)-BPO-27 inhibition of CFTR involves competition with ATP.
Introduction
Cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP-regulated epithelial cell chloride channel in which loss of function mutations causes the genetic disease cystic fibrosis. CFTR is an important target for drug discovery (Verkman and Galietta, 2009; Kim Chiaw et al., 2011). Correctors and potentiators of mutant CFTRs have been approved and are under development for treatment of cystic fibrosis (Davis et al., 2012; Amin and Ratjen, 2014). Inhibitors of CFTR have potential utility for therapy of enterotoxin-mediated secretory diarrheas such as cholera and for autosomal dominant polycystic kidney disease (ADPKD), as CFTR activation is required for intestinal fluid secretion in certain diarrheas (Thiagarajah et al., 2014) and for progressive fluid accumulation into renal cysts in ADPKD (Davidow et al., 1996; Li and Sheppard, 2009).
Our laboratory has identified several classes of small-molecule CFTR inhibitors by high-throughput screening, including thiazolidinones such as CFTRinh-172 [4-[[4-oxo-2-thioxo-3-[3-trifluoromethyl)phenyl]-5-thiazolidinylidene]methyl]benzoic acid] (Ma et al., 2002), glycine hydrazides such as GlyH-101 [N-(2-naphthalenyl)-((3,5-dibromo-2,4-dihydroxyphenyl)methylene)glycine hydrazide] (Muanprasat et al., 2004), and pyrimido-pyrrolo-quinoxalinediones such as PPQ-102 [6,7-dihydro-7,9-dimethyl-6-(5-methyl-2-furanyl)-11-phenylpyrimido[4′,5′:3,4]pyrrolo[1,2-a]quinoxaline-8,10(5H,9)-dione] (Tradtrantip et al., 2009) (Fig. 1). Proof-of-concept studies in tissue culture and rodent models have demonstrated the utility of several of these compounds, or analogs generated therefrom, in cholera toxin-induced intestinal fluid secretion (Thiagarajah et al., 2004; Sonawane et al., 2007) and renal cystic disease (Yang et al., 2008; Tradtrantip et al., 2009). CFTRinh-172 stabilizes the closed CFTR channel state (Taddei et al., 2004; Kopeikin et al., 2010) by binding at or near residue Arg-347 in the CFTR membrane-spanning domain (Caci et al., 2008), whereas GlyH-101 and its analogs directly occlude the CFTR pore near its extracellular entrance (Muanprasat et al., 2004; Sonawane et al., 2008). The CFTR inhibition mechanism of PPQ-102, and its more potent and water soluble analog, the benzopyrimido-pyrrolo-oxazinedione BPO-27 [6-(5-bromofuran-2-yl)-7,9-dimethyl-8,10-dioxo-11-phenyl-7,8,9,10-tetrahydro-6H-benzo[b]pyrimido [4′,5′:3,4]pyrrolo [1,2-d][1,4]oxazine-2-carboxylic acid] (Snyder et al., 2011), is not known.
Fig. 1.

Possible binding sites of (R)-BPO-27 on human CFTR suggested by docking computations. Chemical structure shown at the left. See text for further explanations.
BPO-27 is of potential interest for development as a therapeutic for secretory diarrheas and ADPKD because of its druglike properties and low-nanomolar potency for inhibition of CFTR chloride conductance. We have purified the two BPO-27 enantiomers and determined their crystal structure and biologic properties (Snyder et al., 2013). (R)-BPO-27 was found to be the active enantiomer and showed good metabolic stability in hepatic microsomes and in mice. Here, we used computational chemistry and patch-clamp methods to investigate the mechanisms of CFTR inhibition by (R)-BPO-27. Computational docking suggested several possible (R)-BPO-27 binding sites in CFTR, with the best at the canonical ATP binding site at the interface of the nucleotide binding domains (NBD) 1 and NBD2. This prediction was investigated experimentally by competition studies in inside-out membrane patches and theoretically by molecular dynamics simulations to quantify binding energies of the R and S enantiomers of BPO-27. The computational model was also validated on a small set of lower-potency analogs.
Materials and Methods
Compounds.
Racemic BPO-27 was synthesized as described (Snyder et al., 2011). Enantiomerically pure (>99.5%) (R)-BPO-27 and (S)-BPO-27 were separated by chiral supercritical chromatography, as described (Snyder et al., 2013). CFTRinh-172 was synthesized as described (Ma et al., 2002).
Cell Culture.
Human embryonic kidney cell line HEK-293T cells were grown in Dulbecco’s modified Eagle’s medium-HG (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 50 units/ml penicillin and 50 μg/ml streptomycin. CHO-K1 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Plasmids encoding human wild-type CFTR (and green fluorescent protein in some studies) were transiently transfected into HEK-293T and CHO-K1 cells using Lipofectamine Plus Reagent (Invitrogen).
Whole-Cell Recordings.
Whole-cell recordings were done on CFTR-expressing CHO-K1 cells, as reported (Namkung et al., 2005). The bath solution contained (in millimolar): 140 N-methyl d-glucamine chloride, 1 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.4). The pipette solution contained (in millimolar): 140 N-methyl d-glucamine chloride, 5 EGTA, 1 MgCl2, 1 Tris-ATP, and 10 HEPES (pH 7.2). Pipettes were pulled from borosilicate glass and had resistances of 3–5 MΩ after fire polishing. Seal resistances were between 3 and 10 GΩ. After establishing the whole-cell configuration, BPO-27 was added for 5 minutes, and then CFTR was activated by the addition of forskolin (10 μM) in the continued presence of BPO-27. Whole-cell currents were elicited by applying hyperpolarizing and depolarizing voltage pulses from a holding potential of 0 mV to potentials between +80 and −80 mV in steps of 20 mV. Recordings were made at room temperature using an Axopatch-200B (Axon Instruments, Union City, CA). Currents were digitized with a Digidata 1440A converter (Axon Instruments) and filtered at 5 kHz.
Single-Channel Recordings.
Single-channel measurements were done in excised, inside-out patches in HEK-293T cells, as reported (Lee et al., 2007). Briefly, glass coverslips containing cells cotransfected with CFTR and green fluorescent protein were transferred to a chamber on the stage of an inverted microscope (IX71; Olympus, Tokyo). CFTR currents were measured using an Axon 200B amplifier at a holding potential of −60 mV. The pipette solution contained (in millimolar): 148 HCl, 148 NMDG, 10 HEPES, 1 MgCl2, and 5 EGTA (pH adjusted to 7.2 with NMDG). The bath solution contained (in millimolar): 146 HCl, 146 NMDG, 10 HEPES, 1 MgCl2, 1 CaCl2, and 10 glucose (pH adjusted to 7.4 with NMDG). The bath solution also contained MgATP (3 mM), and CFTR was activated by addition of the catalytic subunit of protein kinase A (10 U/m; Promega Corporation, Madison, WI). Electrode resistance was kept from 15 to 20 MΩ for single-channel recordings. Currents were filtered at 1 kHz and sampled at 500 Hz. The traces were further filtered with a 100-Hz cutoff frequency for data analysis and presentation. Macroscopic currents of inside-out patches were used to study the competition between ATP and BPO-27. All experiments were performed at room temperature (20–23°C). Command potential and data acquisition were controlled by pClamp 10.2 software (Axon Instruments).
Data Analysis.
Single-channel current amplitudes were measured either by manually placing a cursor at the open and closed channel levels or by constructing all-point histograms and fitting with Gaussian functions. Data analysis of data was performed with Clampfit 10.2 (Axon Instruments). In some inhibitor studies, currents were normalized to peak current in the absence of inhibitor or with 10 mM ATP (I/Imax). The results of multiple experiments are presented as the means ± S.E. Statistical analysis was performed with Student’s t test or by analysis of variance followed by Tukey’s multiple comparison test, as appropriate. P < 0.05 was considered statistically significant.
Docking Computations.
A molecular dynamics (MD)–refined homology model of CFTR in its inward and closed conformation (conformation 2) (Mornon et al., 2015), which includes membrane-spanning domains (MSD) 1 and MSD2, and NBD1, and NBD2 domains, was used as a starting point for docking computations. In this model of CFTR, the NBD1 and NBD2 domains are in a dimerized state conducive to small-molecule ligand binding. An unbiased search for putative BPO-27 binding sites on CFTR was done using the FRED-RECEPTOR program (Version 2.2.5; OpenEye Scientific Software, Santa Fe, NM; http://www.eyesopen.com). Structures for the (R)-BPO-27 and (S)-BPO-27 were drawn in ChemDraw (Cambridge Software, Cambridge, MA), converted to SMILES strings, transformed to three-dimensional conformations, and minimized using PIPELINE PILOT (Accelrys, San Diego, CA). The single conformations were passed through MOLCHARGE (Version 1.5.0; OpenEye Scientific Software) to apply MMFF charges and through OMEGA (Version 2.4.6; OpenEye Scientific Software) (Hawkins et al., 2010; Hawkins and Nicholls, 2012) to generate multiconformational libraries. The inhibitor conformational libraries were docked using FRED (Version 2.2.5; OpenEye Scientific Software) (McGann, 2012), free of pharmacophore constraint. FRED was configured to use consensus scoring, with seven scoring functions: ChemGauss3, ChemScore, OEChemScore, ScreenScore, ShapeGauss, PLP, and ZapBind. The lowest-energy consensus pose was compared with other low-energy poses to increase confidence on ligand orientation. The final protein-ligand complexes were visualized using PYMOL (Version 1.5.0.3; Schrödinger LLC, San Diego, CA).
Molecular Dynamics Simulations.
The MD simulations were performed using the AMBER14 and AMBER13TOOLS packages (Case et al., 2012) with relative binding energies estimated from computed trajectories using molecular mechanics Poisson-Boltzmann surface area (MM/PBSA) and molecular mechanics generalized Born surface area (MM/GBSA) algorithms (Srinivasan et al., 1998). Topology files for the protein-ligand complex at binding site C were constructed in TLEAP (Case et al., 2012) using the FF14SB and GAFF force fields. The docked ligand structures described herein were parameterized using ANTECHAMBER (Case et al., 2012). The complexes formed from the CFTR closed structure (Mornon et al., 2014), and the parameterized ligands were solvated in implicit TIP3 water 8 Å from the surface of the protein. Before minimization, the complexes were neutralized by the addition of 21 chloride anions.
The MD simulations were carried out on a server equipped with two NVIDIA GTX-690 graphical processor units (Santa Clara, Ca) using the particle mesh Ewald implementation of graphical processor units–accelerated MD with the particle mesh Ewald MD Compute Unified Device Architecture module of AMBER14 (Gotz et al., 2012). Before the production run, the complexes were subjected to a multistep preparation procedure: 1) minimization with restraint of the protein (1000 cycles); 2) unrestrained minimization (1000 cycles); 3) equilibration while warming from 50°K to 300 °K (30 picoseconds using a constant-volume periodic boundary condition); 4) equilibration with restraint of the protein at 310°K (20 picoseconds with a constant-pressure periodic boundary condition and using isotropic pressure scaling); and 5) unrestrained equilibration at 300°K (500 picoseconds). After minimization and equilibration the complexes were subjected to a production run at 300°K (50 nanoseconds). Preparation steps 1 and 2 comprised 500 steps of steepest descent, followed by 500 steps of conjugate-gradient descent. Steps 3–5 and the production runs used Langevin temperature regulation with the collision frequency of 2.0 picoseconds−1; bonds involving hydrogen were constrained by the SHAKE algorithm; and a 12-Å cutoff was used for nonbonded interactions calculated by the particle mesh Ewald method. The MD simulations were monitored by examination of the internal energy and root mean standard deviation (RMSD) of the resulting trajectories (Supplemental Figs. S3 and S4). Trajectories were either visualized using visual molecular dynamics (Humphrey et al., 1996) or converted to discrete time points with AMBPDB and visualized with PYMOL.
Binding Energy Computations.
After completion of the production runs, binding energies were estimated using the MMPBSA.py module of AMBER13TOOLS (Srinivasan et al., 1998). The simulations used the single trajectory method to reduce noise, and binding energies were calculated using the MM/PBSA and MM/GBSA algorithms (Gilson and Zhou, 2007). The GBSA simulation used generalized Born “method two” (igb = 5) as described by Onufriev et al. (2004) with 0.15 M salt concentration.
Results
Putative Binding Site for (R)-BPO-27 on CFTR Identified by Docking Computations.
Possible sites for binding of (R)-BPO-27 (Fig. 1, left) to CFTR were identified by docking computations using an MD-refined homology model of CFTR in its closed conformation (Mornon et al., 2014). Five potential binding sites were identified (sites A–E; Fig. 1). Binding sites A and B reside in the MSDs, whereas C–E reside in the NBDs. Binding site C was located at the canonical ATP binding site involved in channel gating, whereas binding site E was located at a nongating, degenerate ATP binding site.
To prioritize these putative binding sites, (R)-BPO-27 and (S)-BPO-27 were docked into binding sites A–E, with the lowest energy poses of (R)-BPO-27 shown in Fig. 1. The selective inhibition by (R)-BPO-27 versus (S)-BPO-27 was used to help establish the likely binding site, using results of multiple docking scoring functions (results in Supplemental Table 1). Based on these computations, we concluded that binding sites B, D, and E were unlikely, as they predicted similar or more favorable scoring of the inactive enantiomer, (S)-BPO-27. Binding site A showed a slight consensus of scoring functions favoring active (R)-BPO-27, whereas binding site C (the canonical ATP binding site) showed a perfect consensus of scoring functions. From these computations, we hypothesized that binding site C, the canonical ATP binding site involved for channel gating, is the most likely binding site for (R)-BPO-27, which was investigated experimentally below. We also examined an alternate homology model of CFTR in a closed (“inward”) conformation but with NBD1 and NBD2 nondimerized (Rahman et al., 2013). Docking (R) and (S)-BPO-27 was attempted at the degenerate and canonical ATP binding sites in this model, but neither compound successfully docked to those sites. Hence, this model was not further considered.
CFTR Cl− Current Measurements.
Figure 2A shows representative whole-cell patch-clamp recordings in CHO-K1 cells expressing human wild-type CFTR. The CFTR current was measured just after establishing the whole-cell configuration and then after activation by 10 µM forskolin. (R)-BPO-27 or (S)-BPO-27 was then added to bath solution for 5 minutes before measuring current again. Forskolin-induced CFTR Cl− current was inhibited by (R)-BPO-27 in a concentration-dependent manner at all voltages, whereas (S)-BPO-27 did not affect CFTR Cl− current. Current-voltage plots in Fig. 2B were linear without and with (R)-BPO-27, indicating a voltage-independent block mechanism. Figure 2C summarizes current densities measured at a holding potential of +80 mV. Near-complete inhibition was found for 1 µM (R)-BPO-27.
Fig. 2.
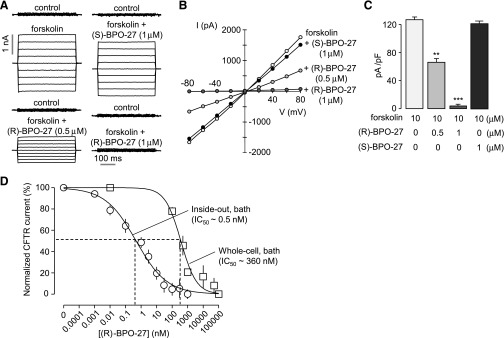
CFTR Cl− current measurements. (A) Whole-cell CFTR currents recorded in CHO-K1 cells at a holding potential of 0 mV and pulsing to voltages between ± 80 mV (in steps of 20 mV) in the absence and presence of (R)-BPO-27 (0.5 or 1 μM) or (S)-BPO-27 (1 µM). CFTR was stimulated by 10 μM forskolin. (B) Current/voltage (I/V) plot of mean currents at the middle of each voltage pulse. (C) Current density data measured at +80 mV (mean ± S.E., n = 5). (D) Whole-cell and macroscopic inside-out patch recordings were done in HEK-293T cells expressing wild-type CFTR. CFTR was stimulated by 10 μM forskolin in whole-cell recordings and by inclusion of 3 mM ATP and 10 U/ml protein kinase A catalytic subunit to the bath solution in inside-out patch recordings. Normalized CFTR current (mean ± S.E., n = 6–10) and IC50 values shown. **P < 0.01; ***P < 0.001.
Next, we measured the dose-response of (R)-BPO-27 for CFTR inhibition in HEK-293T cells. We compared the IC50 value of (R)-BPO-27 measured in whole-cell recordings with that in macroscopic inside-out patch recordings (Fig. 2D). In whole-cell recordings, the IC50 of (R)-BPO-27 was 0.36 μM, similar to that found in CHO-K1 cells. Remarkably, when (R)-BPO-27 was applied to the cytosolic side (bath) in inside-out patches, the CFTR current was inhibited by 50% at 0.53 nM (R)-BPO-27. This 700-fold difference in apparent IC50 indicates that (R)-BPO-27 acts from the cytoplasmic side and has low membrane permeability.
Single-Channel CFTR Recordings.
Single-channel patch-clamp experiments were done to further characterize the (R)-BPO-27 inhibitor mechanisms. After excision of inside-out membrane patches, the excised membrane patch was incubated for 5 minutes in a bath solution containing the protein kinase A catalytic subunit and ATP to activate CFTR, then (R)-BPO-27 or (S)-BPO-27 was added to the bath solution, and single-channel activity was recorded 5 minutes later. Figure 3A shows representative CFTR single-channel recordings obtained from a patch on HEK-293T cells treated with (R)-BPO-27 or (S)-BPO-27. Data from multiple experiments are summarized in Figs. 3, B–E. At a 5-nM concentration, (R)-BPO-27 reduced the channel open probability (NPo) from 0.29 ± 0.02 (n = 7) to 0.08 ± 0.01 (n = 6, P < 0.01), modestly reduced in mean channel open time, and strongly increased mean channel closed time; however, the same concentration of (S)-BPO-27 did not affect any of these parameters (n = 8). The CFTR unitary conductance was not affected by (R)-BPO-27 or (S)-BPO-27.
Fig. 3.

Single-channel recordings of CFTR in HEK-293T cells expressing human wild-type CFTR. (A) Representative recordings from inside-out patch-clamp experiments showing CFTR channel activity in the absence and presence of (R)-BPO-27 or (S)-BPO-27. The bath solution contained 3 mM ATP and 10 U/ml protein kinase A catalytic subunit to activate CFTR. The membrane potential was held at −60 mV. (B–E) Summary of BPO-27 effects on CFTR NPo (number of channels times open probability), mean open-time, mean closed-time, and single-channel conductance (mean ± S.E.). **P < 0.01.
(R)-BPO-27 Competes with ATP for CFTR Activation.
Interaction between ATP and the NBDs of CFTR is required for CFTR gating (Vergani et al., 2005). To analyze the effect of (R)-BPO-27 on ATP-mediated CFTR gating, macroscopic inside-out CFTR currents were measured at different ATP concentrations in the presence of 0.5 nM (R)-BPO-27 in the bath (Fig. 4), which inhibits the normalized CFTR current by ∼50% in the presence of 3 mM ATP (Fig. 2D). Inhibitor effects on ATP-mediated CFTR gating were analyzed by normalizing the CFTR currents measured at each ATP concentration to that of the maximum current at 10 mM ATP (Fig. 4A). Figure 4B shows normalized CFTR currents as a function ATP concentration in the presence of (R)-BPO-27 (0.5 nM), (S)-BPO-27 (0.5 nM), and CFTRinh-172 (0.5 μM); results are summarized in Fig. 4C. The EC50 of ATP for CFTR activation was significantly increased from 0.26 ± 0.05 mM to 1.77 ± 0.13 mM by 0.5 nM (R)-BPO-27 (P < 0.01), whereas the same concentration of (S)-BPO-27 did not affect the EC50 of ATP, indicating that (R)-BPO-27 specifically inhibits CFTR activation by ATP. As a control, similar experiments were performed with CFTRinh-172, a thiazolidinone CFTR inhibitor that acts at or near Arg-347, distant from ATP binding sites in the cytoplasmic nucleotide binding domains (Caci et al., 2008). As shown in Fig. 4, B and C, 0.5 μM CFTRinh-172, which inhibits CFTR current by ∼50% (Supplemental Fig. S1), did not significantly change the EC50 of ATP for CFTR activation.
Fig. 4.

CFTR inhibition by (R)-BPO-27 but not CFTRinh-172 alters the ATP concentration-dependence for CFTR activation. (A) Representative recordings from an inside-out patch-clamp experiment on HEK-293T cells expressing human wild-type CFTR showing normalized current at indicated ATP concentrations in the presence of 0.5 nM (R)-BPO-27 (mean ± S.E., n = 15). The bath contained 10 U/ml protein kinase A catalytic subunit to activate CFTR. The membrane potential was held at −60 mV. (B) ATP concentration-dependent CFTR activity (shown as normalized current) in the presence of 0.5 nM (R)-BPO-27, 0.5 nM (S)-BPO-27 or 0.5 μM CFTRinh-172 (mean ± S.E.). (C) EC50 values of ATP for CFTR activation. In some experiments, ATPγS (1 mM) was present with or without 0.5 nM (R)-BPO-27 (mean ± S.E.). *P < 0.05; **P < 0.01; n.s., not significant.
The effect of (R)-BPO-27 on ATP activation of CFTR was also studied in the presence of the nonhydrolyzable ATP analog adenosine 5′-[γ-thio]triphosphate (ATPγS). The addition of 1 mM ATPγS increased the EC50 of ATP activation of CFTR from 0.26 ± 0.05 mM to 1.12 ± 0.11 mM. Importantly, treatment with 0.5 nM (R)-BPO-27 further increased this value to 1.67 ± 0.14 mM (P < 0.05) (Fig. 4C), suggesting that (R)-BPO-27 competes with the nonhydrolyzable ATP analog at the CFTR ATP binding site.
Molecular Dynamics Simulations and Binding Energy Determination.
We conducted MD simulations for further validation of the docked conformations of (R)-BPO-27 and (S)-BPO-27 into binding site C of CFTR and to estimate binding energies. The topologies were prepared and solvated in water, omitting a simulated lipid bilayer due to the large distance (50–60 Å) of binding site C from the membrane. After equilibration, the complexes were subjected to 50-nanosecond MD production runs at 300°K. Notably, in the BPO-27–bound complexes, the extracellular α-helical domains that constitute the chloride channel remained in a closed (‘inward’) conformation.
Figure 5A shows a zoomed-in representation of (R)-BPO-27 bound to CFTR. After simulation, the ligand remained bound at the canonical ATP binding site, flanked by NBD1 residues I546, T547, L548, and S549, and NBD2 residues Y1219, T1220, T1246, G1247, S1248, K1250, and S1251. Notably, the (R)-BPO-27 carboxylate group was oriented within 2 Å of the T1246 and G1247 backbone NH atoms and S1248 side-chain hydroxyl group and likely to make favorable H-bond contacts.
Fig. 5.

Molecular dynamics computations of ligand binding to the canonical ATP binding site in human CFTR. Zoomed-in representation of (R)-BPO-27 (A) and (S)-BPO-27 (B) docked into CFTR and subject to a 50-nanosecond MD simulation. (C) For comparison, bound ATP shown based on the published homology model of CFTR (Mornon et al., 2015). Surrounding NBD1 (green) and NBD2 (orange) residues are indicated, and putative hydrogen-bond interactions (<3 Å) are shown by dashed yellow lines.
Figure 5B shows the computed binding mode for (S)-BPO-27, which is surrounded by S549 and L548 in NBD1, and I1226, Y1219, T1220, and K1174 in NBD2. Interestingly, the (S)-BPO-27 carboxylate appears positioned to form a favorable salt bridge with the K1174 side-chain ammonium group. Notably, bound (S)-BPO-27 was ∼30 Å away from K1174 before the MD computations, showing significant reorganization of the neighboring residues to accommodate (S)-BPO-27. Also, the backbone RMSD of the ATP binding site was compared for (R)-BPO-27 and (S)-BPO-27 (Supplemental Fig. S2). (S)-BPO-27 was subject to ∼0.7 Å RMSD more reorganization of the ATP binding site residues than the R isomer. Binding of (S)-BPO-27 may thus be disfavored by structural perturbation of additional neighboring residues in NBD1 and NBD2. For reference, Fig. 5C shows ATP at the canonical binding site. Of note, several of the residues that surround ATP also interacted with (R)-BPO-27, including I546, T547, L548, S549, Y1219, T1220, T1246, and S1248.
After the MD production run, binding energies (ΔGbinding) of (R)-BPO-27 and (S)-BPO-27 with CFTR were estimated using two independent algorithms, MM/GBSA and MM/PBSA (Gilson and Zhou, 2007). Supplemental Table S2 provides computed ΔGbinding for 20 different time points in the simulation after 10 nanoseconds, a point at which the system appears equilibrated. The significantly lower ΔGbinding for (R)-BPO-27 versus (S)-BPO-27, determined by both MM/GBSA and MM/PBSA methods, is consistent with the subnanomolar CFTR inhibition potency of the (R) compound and the inactivity of the (S) compound. We noted that the computed binding energies are useful for relative rather than absolute binding affinity, as supported by earlier work showing that calculated ΔGbinding does not directly calibrate to absolute ΔGbinding for a series of compounds, but it does provide informative relative trends (Rastelli et al., 2010).
A similar docking/MD/ΔGbinding determination sequence was done for three inhibitors (compounds 31, 36, and 40) with lower CFTR inhibition potency than BPO-27 (Supplemental Table S3). The data correctly predicted more negative calculated ΔGbinding of the more potent (R)-isomers, both by the MM/GBSA and MM/PBSA methods. In terms of computed stereoselectivity, it is useful to consider ΔΔGRvsS, which is the difference between computed ΔGbinding for the (R) and (S) isomers (Supplemental Table S3). The most potent inhibitor (BPO-27) has the greatest computed stereoselectivity for the (R) stereoisomer as determined by both computational methods. The medium-potency compounds (36 and 40) are predicted to also have selectivity favoring the (R) isomer, but to a lesser extent. The least potent inhibitor (31) did not show any preference for the (R) isomer.
Discussion
We investigated the inhibition mechanism of CFTR inhibitor BPO-27, which is the most potent CFTR inhibitor identified to date and a potential development candidate for therapy of bacterial secretory diarrheas and ADPKD. In the absence of information about the binding site of (R)-BPO-27, the application of a molecular probe receptor identification technique on a homology model of CFTR in its closed conformation identified five possible binding sites. Subsequent docking, with analysis of multiple independent scoring functions, suggested that (R)-BPO-27 binds to the canonical ATP binding site, which was supported by experimental data showing (R)-BPO-27 competition with ATP and by MD simulations.
The conclusion that (R)-BPO-27 binds at the canonical ATP binding site makes it mechanistically unique from the other high-affinity inhibitors GlyH-101 and CFTRinh-172. Electrophysiology, computational studies and chemical modifications suggest GlyH-101 binds near Phe-337 in the extracellular CFTR pore (near binding site A in Fig. 1) and blocks the pore directly (Muanprasat et al., 2004; Sonawane et al., 2008; Norimatsu et al., 2012). Electrophysiology and mutagenesis studies suggest that CFTRinh-172 stabilizes the CFTR closed channel (Taddei et al., 2004; Caci et al., 2008; Kopeikin et al., 2010) by binding at or near Arg-347 in the MSD at a location roughly equidistant to the extracellular and cytoplasmic membrane surfaces. The distinct CFTR inhibition mechanism of (R)-BPO-27, which involves interference with ATP-mediated gating in the NBDs, might produce synergistic inhibition when combined with a thiazolidinone or glycine hydrazide CFTR inhibitor.
Additional validation of the docked conformation of (R)-BPO-27 was performed with MD simulations, showing that the inhibitor remained bound to the canonical ATP binding site. Similar computations done for (S)-BPO-27 showed significant spatial reorganization, and MM/PBSA and MM/GBSA calculations showed remarkably lower energy for CFTR binding of (R)-BPO-27 than of the S isomer. These results support the conclusion that the chiral surface of (R)-BPO-27 achieves a complementary low-energy fit with the canonical ATP binding site in a manner that (S)-BPO-27 cannot. Additionally, three other less potent analogs of BPO-27 were modeled in an analogous fashion and shown to have less negative computed binding energies. Ultimately, high-resolution structure determination of CFTR: (R)-BPO-27 cocrystals is needed to confirm the predictions here. Site-directed mutagenesis was not informative (unpublished data), as we found that mutations in the vicinity of the ATP binding site interfered with CFTR gating.
CFTR is the only member of the C-subfamily of ATP-binding cassette proteins whose transmembrane domains form the pore of an ion channel (Sheppard and Welsh, 1999). It has been suggested that gating of the CFTR chloride channel involves a conformational change of the two cytoplasmic NBDs. Experimental evidence indicated that opening and closing of the gate in the CFTR transmembrane domains are coupled to the ATP binding-induced dimerization and the ATP hydrolysis-catalyzed partial separation of the two NBDs (Riordan, 2005; Guggino and Stanton, 2006; Mense et al., 2006). ATP thus increases CFTR open probability in a concentration-dependent manner by facilitating the conformation change cycles. Here, we found a 700-fold greater apparent potency of (R)-BPO-27 when applied to the cytosolic side than the extracellular side, supporting the results from modeling that (R)-BPO-27 acts from the intracellular side of CFTR. The ATP concentration-dependent activation of CFTR was shifted by (R)-BPO-27 but not by (S)-BPO-27 or CFTRinh-172. In addition, single-channel analyses revealed that (R)-BPO-27 reduced CFTR channel open probability (Po) and increased channel closed-time without affecting unitary channel conductance. Taken together, these results suggest that inhibition of ATP binding to the NBDs or the ATP-mediated conformational change of NBDs (which induces a delay in the closure to the open transition of the CFTR gate) is probably the major mechanism of CFTR Cl− channel inhibition by (R)-BPO-27.
In conclusion, (R)-BPO-27 appears to inhibit CFTR chloride conductance by binding in the region of the ATP canonical binding site in the CFTR cytoplasmic domain, thereby interfering with ATP gating. This putative site of action and the inhibition mechanism are distinct from those of thiazolidinone and glycine hydrazide CFTR inhibitors.
Supplementary Material
Acknowledgments
The authors thank OpenEye Scientific for its Academic Site License program.
Abbreviations
- ADPKD
autosomal dominant polycystic kidney disease
- BPO-27
6-(5-bromofuran-2-yl)-7,9-dimethyl-8,10-dioxo-11-phenyl-7,8,9,10-tetrahydro-6H-benzo[b]pyrimido [4′,5′:3,4]pyrrolo [1,2-d][1,4]oxazine-2-carboxylic acid
- CFTRinh-172
4-[[4-oxo-2-thioxo-3-[3-trifluoromethyl)phenyl]-5-thiazolidinylidene]methyl]benzoic acid
- GlyH-101
N-(2-naphthalenyl)-((3,5-dibromo-2,4-dihydroxyphenyl)methylene)glycine hydrazide
- MD
molecular dynamics
- MSD
membrane-spanning domain
- MM/PBSA
molecular mechanics Poisson-Boltzmann surface area
- MM/GBSA
molecular mechanics generalized Born surface area
- NBD
nucleotide-binding domain
- PPQ-102
6,7-dihydro-7,9-dimethyl-6-(5-methyl-2-furanyl)-11-phenylpyrimido[4′,5′:3,4]pyrrolo[1,2-a]quinoxaline-8,10(5H,9)-dione
- RMSD
root mean standard deviation
Authorship Contributions
Participated in research design: Kim, Anderson, Namkung, Verkman.
Conducted experiments: Kim, Anderson, Park.
Wrote or contributed to the writing of the manuscript: Kim, Anderson, Lee, Namkung, Verkman.
Footnotes
This work was supported by National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grants DK75302, DK72517, DK35124]; National Institutes of Health National Institute of Biomedical Imaging and Bioengineering [EB00415]; National Institutes of Health National Eye Institute [EY135740]; the Cystic Fibrosis Foundation [R613]; and the National Research Foundation, the Ministry of Science, ICT & Future Planning, Republic of Korea [2013R1A3A2042197].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Amin R, Ratjen F. (2014) Emerging drugs for cystic fibrosis. Expert Opin Emerg Drugs 19:143–155. [DOI] [PubMed] [Google Scholar]
- Caci E, Caputo A, Hinzpeter A, Arous N, Fanen P, Sonawane N, Verkman AS, Ravazzolo R, Zegarra-Moran O, Galietta LJ. (2008) Evidence for direct CFTR inhibition by CFTR(inh)-172 based on Arg347 mutagenesis. Biochem J 413:135–142. [DOI] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE. III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, et al. (2012) AMBER 12, University of California, San Francisco. [Google Scholar]
- Davidow CJ, Maser RL, Rome LA, Calvet JP, Grantham JJ. (1996) The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int 50:208–218. [DOI] [PubMed] [Google Scholar]
- Davis PB, Yasothan U, Kirkpatrick P. (2012) Ivacaftor. Nat Rev Drug Discov 11:349–350. [DOI] [PubMed] [Google Scholar]
- Gilson MK, Zhou HX. (2007) Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol Struct 36:21–42. [DOI] [PubMed] [Google Scholar]
- Götz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. (2012) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J Chem Theory Comput 8:1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guggino WB, Stanton BA. (2006) New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol 7:426–436. [DOI] [PubMed] [Google Scholar]
- Hawkins PC, Nicholls A. (2012) Conformer generation with OMEGA: learning from the data set and the analysis of failures. J Chem Inf Model 52:2919–2936. [DOI] [PubMed] [Google Scholar]
- Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. (2010) Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model 50:572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. (1996) VMD: visual molecular dynamics. J Mol Graphics 14:27–38. [DOI] [PubMed] [Google Scholar]
- Kim Chiaw P, Eckford PD, Bear CE. (2011) Insights into the mechanisms underlying CFTR channel activity, the molecular basis for cystic fibrosis and strategies for therapy. Essays Biochem 50:233–248. [DOI] [PubMed] [Google Scholar]
- Kopeikin Z, Sohma Y, Li M, Hwang TC. (2010) On the mechanism of CFTR inhibition by a thiazolidinone derivative. J Gen Physiol 136:659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Richter W, Namkung W, Kim KH, Kim E, Conti M, Lee MG. (2007) Dynamic regulation of cystic fibrosis transmembrane conductance regulator by competitive interactions of molecular adaptors. J Biol Chem 282:10414–10422. [DOI] [PubMed] [Google Scholar]
- Li H, Sheppard DN. (2009) Therapeutic potential of cystic fibrosis transmembrane conductance regulator (CFTR) inhibitors in polycystic kidney disease. BioDrugs 23:203–216. [DOI] [PubMed] [Google Scholar]
- Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. (2002) Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest 110:1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman KS, Cui G, Harvey SC, McCarty NA. (2013) Modeling the conformational changes underlying channel opening in CFTR. PLoS One 8:e74574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann M. (2012) FRED and HYBRID docking performance on standardized datasets. J Comput Aided Mol Des 26:897–906. [DOI] [PubMed] [Google Scholar]
- Mense M, Vergani P, White DM, Altberg G, Nairn AC, Gadsby DC. (2006) In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J 25:4728–4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mornon JP, Hoffmann B, Jonic S, Lehn P, Callebaut I. (2015) Full-open and closed CFTR channels, with lateral tunnels from the cytoplasm and an alternative position of the F508 region, as revealed by molecular dynamics. Cell Mol Life Sci 72:1377–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS. (2004) Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol 124:125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W, Kim KH, Lee MG. (2005) Base treatment corrects defects due to misfolding of mutant cystic fibrosis transmembrane conductance regulator. Gastroenterology 129:1979–1990. [DOI] [PubMed] [Google Scholar]
- Norimatsu Y, Ivetac A, Alexander C, O’Donnell N, Frye L, Sansom MS, Dawson DC. (2012) Locating a plausible binding site for an open-channel blocker, GlyH-101, in the pore of the cystic fibrosis transmembrane conductance regulator. Mol Pharmacol 82:1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onufriev A, Bashford D, Case DA. (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55:383–394. [DOI] [PubMed] [Google Scholar]
- Rastelli G, Del Rio A, Degliesposti G, Sgobba M. (2010) Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem 31:797–810. [DOI] [PubMed] [Google Scholar]
- Riordan JR. (2005) Assembly of functional CFTR chloride channels. Annu Rev Physiol 67:701–718. [DOI] [PubMed] [Google Scholar]
- Sheppard DN, Welsh MJ. (1999) Structure and function of the CFTR chloride channel. Physiol Rev 79(1, Suppl)S23–S45. [DOI] [PubMed] [Google Scholar]
- Snyder DS, Tradtrantip L, Battula S, Yao C, Phuan PW, Fettinger JC, Kurth MJ, Verkman AS. (2013) Absolute Configuration and Biological Properties of Enantiomers of Cftr Inhibitor Bpo-27. ACS Med Chem Lett 4:456–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder DS, Tradtrantip L, Yao C, Kurth MJ, Verkman AS. (2011) Potent, metabolically stable benzopyrimido-pyrrolo-oxazine-dione (BPO) CFTR inhibitors for polycystic kidney disease. J Med Chem 54:5468–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonawane ND, Zhao D, Zegarra-Moran O, Galietta LJ, Verkman AS. (2007) Lectin conjugates as potent, nonabsorbable CFTR inhibitors for reducing intestinal fluid secretion in cholera. Gastroenterology 132:1234–1244. [DOI] [PubMed] [Google Scholar]
- Sonawane ND, Zhao D, Zegarra-Moran O, Galietta LJ, Verkman AS. (2008) Nanomolar CFTR inhibition by pore-occluding divalent polyethylene glycol-malonic acid hydrazides. Chem Biol 15:718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan J, Miller J, Kollman PA, Case DA. (1998) Continuum solvent studies of the stability of RNA hairpin loops and helices. J Biomol Struct Dyn 16:671–682. [DOI] [PubMed] [Google Scholar]
- Taddei A, Folli C, Zegarra-Moran O, Fanen P, Verkman AS, Galietta LJ. (2004) Altered channel gating mechanism for CFTR inhibition by a high-affinity thiazolidinone blocker. FEBS Lett 558:52–56. [DOI] [PubMed] [Google Scholar]
- Thiagarajah JR, Broadbent T, Hsieh E, Verkman AS. (2004) Prevention of toxin-induced intestinal ion and fluid secretion by a small-molecule CFTR inhibitor. Gastroenterology 126:511–519. [DOI] [PubMed] [Google Scholar]
- Thiagarajah JR, Ko EA, Tradtrantip L, Donowitz M, Verkman AS. (2014) Discovery and development of antisecretory drugs for treating diarrheal diseases. Clin Gastroenterol Hepatol 12:204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradtrantip L, Sonawane ND, Namkung W, Verkman AS. (2009) Nanomolar potency pyrimido-pyrrolo-quinoxalinedione CFTR inhibitor reduces cyst size in a polycystic kidney disease model. J Med Chem 52:6447–6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Lockless SW, Nairn AC, Gadsby DC. (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433:876–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS, Galietta LJ. (2009) Chloride channels as drug targets. Nat Rev Drug Discov 8:153–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS. (2008) Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol 19:1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.