

Abstract
Propofol is a sedative and anesthetic agent that can both activate GABAA receptors and potentiate receptor activation elicited by submaximal concentrations of the transmitter. A recent modeling study of the β3 homomeric GABAA receptor postulated a high-affinity propofol binding site in a hydrophobic pocket in the middle of a triangular cleft lined by the M1 and M2 membrane-spanning domains of one subunit and the M2 domain of the neighboring subunit. The goal of the present study was to gain functional evidence for the involvement of this pocket in the actions of propofol. Human β3 and α1β3 receptors were expressed in Xenopus oocytes, and the effects of substitutions of selected residues were probed on channel activation by propofol and pentobarbital. The data demonstrate the vital role of the β3(Y143), β3(F221), β3(Q224), and β3(T266) residues in the actions of propofol but not pentobarbital in β3 receptors. The effects of β3(Y143W) and β3(Q224W) on activation by propofol are likely steric because propofol analogs with less bulky ortho substituents activated both wild-type and mutant receptors. The T266W mutation removed activation by propofol in β3 homomeric receptors; however, this mutation alone or in combination with a homologous mutation (I271W) in the α1 subunit had almost no effect on activation properties in α1β3 heteromeric receptors. We hypothesize that heteromeric α1β3 receptors can be activated by propofol interactions with β3–β3, α1–β3, and β3–α1 interfaces, but the exact locations of the binding site and/or nature of interactions vary in different classes of interfaces.
Introduction
The GABAA receptor (γ-aminobutyric acid type A receptor) is the principal target of propofol and several other sedative and anxiolytic agents (Rudolph et al., 1999; Low et al., 2000; Jurd et al., 2003; Ferguson et al., 2007). Propofol directly activates the GABAA receptor and can potentiate currents elicited by a low concentration of the transmitter GABA (Hales and Lambert, 1991; Sanna et al., 1995). Under physiologic conditions, this leads to hyperpolarization of cell or dampening of the effect of excitatory input.
Despite years of research, the location of the site(s) mediating the actions of propofol is unknown. A recent modeling study of the β3 homomeric receptor identified a hydrophobic cavity near the extracellular end of the membranous region, predicted to bind propofol with a submicromolar equilibrium dissociation constant (Miller and Aricescu, 2014; Franks, 2015). The cavity lies in the center of a column triangle formed by the M1 and M2 membrane-spanning domains of the subunit that contribute the “−” side of the interface (chain A in Fig. 1) and the M2 domain of the neighboring subunit that contributes the “+” side of the interface (chain B in Fig. 1). Molecular docking places the propofol molecule at the “−” side next to the β3(H267) residue (Franks, 2015). Incidentally, this residue is photolabeled by the photoreactive propofol analog, ortho-propofol diazirine (Yip et al., 2013). The key residue at the “+” side of the interface is T266, which points into the cleft toward the phenol ring of propofol. The cleft is capped from the extracellular side by the Y143 and F221 residues at the boundary between the extracellular and membrane-spanning domains (Franks, 2015).
Fig. 1.

Structures of the GABAA receptor and the putative propofol binding pocket. (A) A top (extracellular) view of the receptor. The five subunits surround a centrally located pore. By convention, the subunits have “+” and “−” sides that are ordered in the fashion shown in the figure. The putative propofol binding site is located at the interface between two neighboring subunits. In β3 homomeric receptors, there are five identical β–β interfaces. Heteromeric α1β3 receptors likely have a stoichiometry of two α and three β subunits, and assemble as β–β–α–β–α (top view, counterclockwise). These receptors contain two β–α interfaces, two α–β interfaces, and one β–β interface. (B) A side view of two subunits, viewed from the channel lumen in the direction of the arrow in (A). Chain A (yellow) supplies the “−” side of the interface; chain B (green) supplies the “+” side of the interface. In β3 receptors, both sides of the interface are β subunits. In α1β3 receptors, the yellow chain is a β subunit, and the green chain is a β subunit or an α subunit. (C) The intersubunit interface at a higher resolution showing the side chains of amino acid residues probed in this study.
Mutations to this region affect receptor properties. The β3(H267A) mutation shifts the GABA concentration-response curve of α1β3 receptors to higher agonist concentrations and reduces, albeit weakly, direct activation and potentiation of GABA-activated receptors by ortho-propofol diazirine (Yip et al., 2013). A tryptophan substitution at the β3(F221) site left shifts the GABA concentration-response curve, modestly reduces gating efficacy for ortho-propofol diazirine, and essentially eliminates potentiation by this propofol analog. Overall, we consider the existing functional data supportive of the photolabeling and modeling data, but inconclusive because of the relatively weak effect of the amino acid substitutions and absence of evidence that the effects are specific to the actions of propofol.
There are two caveats to prior functional data. The first concerns the heterogeneity of the intersubunit interfaces in the α1β3 receptor. The majority of studies now support the stoichiometry of two α subunits and three β subunits in αβ heteromeric receptors, assembled in the sequence of β–β–α–β–α (Tretter et al., 1997; Baumann et al., 2001; Horenstein et al., 2001). The α1β3 receptor thus has one β–β, two β–α, and two α–β interfaces. Mutations to the β3(H267) residue can be expected to modify and affect the β–β and α–β interfaces, but not the β–α interface, whose involvement in the actions of propofol has been suggested by previous photolabeling and functional studies (Krasowski et al., 2001a; Bali and Akabas, 2004; Jayakar et al., 2014). It is therefore plausible that the contribution from the unaltered β–α interfaces conceals the true effect exerted by the H267A mutation at the “−” side of the β subunit. The second caveat is that the experiments were conducted using the photolabeling reagent ortho-propofol diazirine. It remains unclear whether the mutations influenced activation by the parent compound, propofol.
Mutational studies in general can be ambiguous with regard to the underlying mechanism of effect. Changes in activation properties may result from the mutation interfering with the binding of ligand or signal transduction, with the distinction between the two not always being straightforward (Colquhoun, 1998). Photolabeling studies may present a more direct approach to identifying the regions involved; however, a mutational study can reveal the functional involvement of a site.
To gain further insight into the role of the cavity formed by the M1 and M2 membrane-spanning domains at the intersubunit interface, we tested the effects of mutations to selected residues in β3 homomeric receptors. The data show that tryptophan substitutions at β3(Y143), β3(F221), β3(Q224), and β3(T266) drastically reduce activation by propofol with relatively modest effects on activation by pentobarbital. The effects of mutations were dependent on the ortho substituents of the phenol backbone, implying an underlying steric nature. The β3(H267W) and the neighboring β3(L268W) mutations had minimal effect on receptor activation by propofol or pentobarbital. Comparison of the effects of β3(T266W) and the homologous mutation in the α1 subunit [α1(I271W)] suggests that propofol-receptor interactions at the α1–β3 and β3–α1 interfaces are not equivalent with regard to the structures involved.
Materials and Methods
The experiments were conducted on wild-type and mutant human β3 and α1β3 GABAA receptors, expressed in Xenopus oocytes. Harvesting of oocytes was conducted in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health. The protocol was approved by the Animal Studies Committee of the Washington University in St. Louis.
The mutations employed in the study were Y143W, F221W, Q224W, T263W, T266W, H267W, and L268W in the β3 subunit, and I271W in the α1 subunit. The β3(H267) residue was recently identified as a component of the propofol binding pocket using photolabeling with the propofol analog ortho-propofol diazirine (Yip et al., 2013). The β3(Y143), β3(F221), β3(Q224), β3(T263), β3(T266), β3(L268), and α1(I271) residues are located around the perimeter of the putative propofol binding pocket (Fig. 1). Although this putative binding pocket was identified (Franks, 2015) using a relatively low-resolution (3 Å) crystal structure (Miller and Aricescu, 2014), this was quite sufficient to highlight which residues might contribute to a possible propofol binding site.
All mutations were made using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). The β3 subunit contained the FLAG epitope in the amino terminus of the subunit (Ueno et al., 1996). The clones were fully sequenced before use. All mutated receptors, with the exception of β3(T263W) homomers, were functional.
The cDNAs, subcloned into the pcDNA3 vector, were linearized by digestion with XbaI (β3; NEB Laboratories, Ipswich, MA) or BglII (α1; Roche Diagnostics, Indianapolis, IN). The cRNAs were produced using mMessage mMachine (Ambion, Austin, TX). The oocytes were injected with a total of 3–18 ng cRNA in a final volume of 20–40 nl, and incubated in ND96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 2.5 mM Na pyruvate, 5 mM HEPES; pH 7.4) at 16°C. The ratio of cRNAs used for injection was 5:1 when α1 and β3 subunits were used, to reduce the fraction of β3 homomeric receptors. The oocytes were used within 1 to 3 days after injection.
Electrophysiologic experiments were conducted using the two-electrode voltage clamp technique. Voltage and current electrodes were patch-clamp electrodes that when filled with 3 M KCl had resistances of less than 1 MΩ. The oocytes were clamped at −60 mV. The chamber (RC-1Z; Warner Instruments, Hamden, CT) was perfused continuously at approximately 5 ml min−1. Bath solution (92.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, 10 mM HEPES; pH 7.4) was perfused between all test applications. Solutions were gravity-applied from 30-ml glass syringes with glass luer slips via Teflon tubing to reduce adsorption, and switched manually or by pClamp using a Warner Instruments VC-8T valve controller. A typical recording consisted of a 10-second baseline followed by a 10- to 60-second drug application and a bath application (up to 10 minutes) until full recovery. The duration of drug (propofol, pentobarbital, picrotoxin, or GABA) application was dependent on the nature of the drug and its concentration, and was aimed at reaching a saturated peak response without unnecessary further exposure to the drug, to facilitate washout and avoid accumulation of the drug in the cell. The current responses were amplified with an Axoclamp 900A amplifier (Molecular Devices, Sunnyvale, CA), digitized with a Digidata 1320 series digitizer (Molecular Devices) at a 100 Hz sampling rate, and stored using pClamp (Molecular Devices). The traces were subsequently analyzed with Clampfit (Molecular Devices) to determine the maximal amplitude of current response.
Concentration-response curves were fitted, individually for each cell, with the following equation:
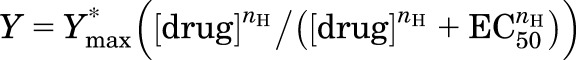 |
(1) |
where EC50 is the concentration of drug producing a half-maximal effect, nH describes the slope of the relationship, and Ymax is the high concentration asymptote. At high concentrations, both propofol (Adodra and Hales, 1995) and pentobarbital (Akaike et al., 1987) block current responses. This manifests as a suppressed initial peak response followed by a prominent tail or rebound response following termination of drug application. The rebound response reflects the transient repopulation of the conducting state(s) of the channel, which occurs during washout of the drug. We used the amplitude of the rebound response in curve fitting whenever it was greater than the initial response. Fitting was conducted using the NFIT software (Medical Branch, University of Texas at Galveston). Parameters of the fit are reported as mean ± S.E.M. Statistical analysis was conducted using two-sample t test (Excel; Microsoft, Redmond, WA).
The level of spontaneous activity was determined by comparing the effect of 10–100 µM picrotoxin to the response to saturating pentobarbital which, in all cases where β3 homomeric configuration was employed, produced a larger current response than saturating propofol. Spontaneous activity, expressed in units of estimated open probability (Poest), was calculated assuming that Po was 0 in the presence of saturating picrotoxin and 1 in the presence of saturating pentobarbital. This approach is similar to the one described previously for heteromeric GABAA receptors (Forman and Stewart, 2012; Eaton et al., 2014). We note, however, that this approach may result in overestimation of Po,spont, because the actual Po in the presence of saturating pentobarbital may be less than 1. In some cases [e.g., β3(F221W)], saturating pentobarbital was more effective at blocking spontaneous activity than 100 µM picrotoxin. For those receptors, Po,spontest was calculated assuming that the Po reached 0 during the initial blocking action of pentobarbital and 1 during the rebound response after termination of pentobarbital application.
Parameters for binding and gating in the presence of propofol or pentobarbital were derived from fitting the Poest from pooled data to the following equation (Chang and Weiss, 1999; Rusch et al., 2004):
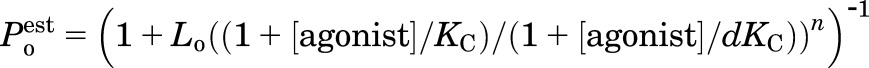 |
(2) |
where Lo is the ratio of the equilibrium occupancy of closed receptors to the equilibrium occupancy of open receptors in the absence of agonist, KC stands for the closed receptor equilibrium dissociation constant for a given agonist, d is a measure of efficacy expressed as the ratio of open receptor dissociation constant to closed receptor dissociation constant, and n is an integer, constrained to 2 to 5, corresponding to the number of agonist binding sites occupied to produce activation.
Fitting was conducted using NFIT. The fitting results we show here were obtained with n of 5 for propofol and 4 for pentobarbital. We note that changes in n had a relatively weak effect on the goodness of the fit. KC and d were free parameters. Lo was experimentally determined as (1 − Po,spontest)/Po,spontest, and equaled to 6.7, 13.3, 99, 0.7, 3.8, or 1.9 in β3 wild type, β3(H267W), β3(L268W), β3(F221W), β3(Y148W), and β3(T266W), respectively. The β3(Q224W) receptor showed no picrotoxin-sensitive spontaneous activity. To estimate the parameters for binding and gating in this mutant, we constrained Lo to arbitrarily chosen values of 100, 1000, 10,000, and 100,000 (corresponding to Po,spont of approximately 0.01, 0.001, 0.0001, and 0.00001, respectively). The best fit was obtained with Lo of 1000.
Potentiation of α1β3 receptors was estimated by examining the effect of a low concentration of propofol on currents elicited by a low concentration of GABA. The test GABA concentration was selected to produce a response of approximately 5% of the maximal response to GABA. Propofol was used at a concentration that elicited a response of less than 3% of the maximal response to GABA. The potentiating effect of propofol was calculated as I(GABA+propofol) / (IGABA + Ipropofol). Statistical analysis was conducted by comparing the potentiating effect to 1 (i.e., no effect), using a two-tailed paired t test (Excel). This test, equivalent to a one-sample t test with a hypothetical value of 1, is designed to determine whether the effect of propofol is statistically significant.
Pentobarbital, propofol analogs, and inorganic salts used in buffers were bought from Sigma-Aldrich (St. Louis, MO). Propofol was from MP Biomedicals (Solon, OH). Stock solution of 5 mM pentobarbital was made in the bath solution. Stock solutions of 10 or 200 mM propofol or analogs were made in dimethylsulfoxide. Stock solutions were kept at room temperature and further diluted as needed on the day of the experiment.
Results
Activation of Wild-Type and Mutant Human β3 Homomeric Receptors by Propofol and Pentobarbital.
It was recently shown that ortho-propofol diazirine, a propofol analog photolabeling reagent, labels the β3(H267) residue in human β3 and α1β3 GABAA receptors expressed in Sf9 cells (Yip et al., 2013). The β3(H267) residue is located near the extracellular end (by convention, the 17ʹ residue) of the second membrane-spanning domain, pointing to the interior of the subunit where it flanks a hydrophobic cavity located between the TM2 and TM1 domains of two neighboring β subunits (Yip et al., 2013; Franks, 2015). Previous work has indicated a potential involvement of the 267 location in the actions of Zn2+ (Dunne et al., 2002; Trudell et al., 2008) and picrotoxinin (Carland et al., 2008). We mutated the β3(H267) residue to tryptophan. This substitution results in a more than 50% increase in bulkiness (a ratio of volume to length) of the side chain (Zimmerman et al., 1968).
Our data indicate that the H267W mutation does not affect propofol-activation of β3 receptors. The midpoint of the propofol concentration-response curve was at 9 ± 1 µM (mean ± S.E.M.; seven cells) in wild-type β3 receptors and at 10 ± 1 µM (eight cells) in β3(H267W). We also probed the effect of the mutation on activation by pentobarbital. In four cells expressing wild-type β3 receptors, the EC50 for pentobarbital was 59 ± 7 µM. In receptors containing the β3(H267W) mutation, the pentobarbital EC50 was 67 ± 20 µM (four cells). The difference was not statistically significant. To gain insight into the effect of β3(H267W) on gating efficacy, we compared responses to saturating (30–100 µM) propofol and saturating (300–1000 µM) pentobarbital.
In wild-type β3 receptors, propofol elicited a response that was 53 ± 3% of the response to pentobarbital (eight cells). In β3(H267W) receptors, the response to propofol was 27 ± 1% of the response to pentobarbital (five cells). The difference was statistically significant (P < 0.001; t test). We also examined the extent of spontaneous activity in wild-type β3 and β3(H267W). By comparing the effect of 10–100 µM picrotoxin on baseline current to the peak response to 300 µM pentobarbital, we calculated (see Materials and Methods for details and caveats) that Po,spontest is 0.13 ± 0.02 (eight cells) in wild type and 0.07 ± 0.02 in β3(H267W) (five cells; P > 0.05). The data are summarized in Fig. 2 and Table 1. Fitting eq. 2 to pooled Poest data yielded a KC of 8.8 µM and the parameter d (KO/KC) of 0.64 for wild-type β3 receptors and 7.1 µM and 0.67 for β3(H267W) (Fig. 3; Table 2). We infer based on these data that the histidine sidechain in position 267 does not contribute significantly to activation properties of β3 homomeric GABAA receptors.
Fig. 2.

Concentration-response properties of human β3 homomeric receptors. The figure shows propofol (A) and pentobarbital (B) concentration-response curves for wild-type and mutant receptors. The data points show normalized responses (mean ± S.E.M.) from at least four cells at each condition. Current responses were normalized to the response to the highest concentration of agonist used in the experiment. The curves show predictions of eq. 1 generated with the overall mean EC50 values from Table 1. In (A), the maximal fitted response and nH were 1.05 ± 0.03 and 1.6 ± 0.2 (wild type), 1.07 ± 0.02 and 1.7 ± 0.1 (H267W), 1.1 ± 0 and 1.6 ± 0.1 (L268W), and 0.99 ± 0.04 and 0.98 ± 0.16 (Y143W). Receptors containing the T266W, Q224W, or F221W mutations did not reliably produce responses in the presence of propofol. In (B), the maximal fitted response and nH were 1.03 ± 0.03 and 1.6 ± 0.04 (wild type), 1.14 ± 0.03 and 1.8 ± 0.3 (H267W), 1.0 ± 0.03 and 1.8 ± 0.2 (L268W), 1.05 ± 0.04 and 1.1 ± 0.04 (Y143W), 1.03 ± 0.03 and 0.9 ± 0.2 (T266W), and 0.98 ± 0.02 and 2.9 ± 0.04 (Q224W).
TABLE 1.
Propofol and pentobarbital concentration-response data for β3 homomeric receptors
The concentration-response data from each cell were fitted with eq. 1 (Materials and Methods). The table shows propofol and pentobarbital EC50 values (mean ± S.E.M.) from at least four cells under each condition. Propofol activation was minimal in receptors containing the Q224W, F221W, or T266W mutations; for these receptors the concentration-response relationship was not determined. Ipropofol/Ipentobarbital was calculated by measuring responses to saturating propofol and saturating pentobarbital in the same cell. Cells expressing β3(F221W) did not respond with inward current to application of propofol. Open probability of unliganded receptors (Po,spont) was calculated assuming that Po reached 0 in the presence of 10–100 µM picrotoxin and 1 in the presence of saturating pentobarbital. The β3(F221W) exhibited greater block during the initial application of 3 mM pentobarbital than in the presence of picrotoxin. Accordingly, we compared block by pentobarbital to the maximal rebound response to pentobarbital to calculate Po,spont in β3(F221W).
| Receptor | Propofol EC50 | Pentobarbital EC50 | Ipropofol/Ipentobarbital | Po,spont |
|---|---|---|---|---|
| µM | % | |||
| β3 wild type | 9 ± 1 | 59 ± 7 | 53 ± 3 | 0.13 ± 0.02 |
| β3(H267W) | 10 ± 1 | 67 ± 20 | 27 ± 1 | 0.07 ± 0.02 |
| β3(L268W) | 15 ± 2 | 272 ± 40 | 14 ± 1 | 0.01 ± 0.001 |
| β3(Q224W) | NA | 666 ± 21 | <3 | 0 |
| β3(F221W) | NA | 523 ± 45 | 0 | 0.62 ± 0.04 |
| β3(Y143W) | 22 ± 6 | 82 ± 24 | 13 ± 4 | 0.21 ± 0.02 |
| β3(T266W) | NA | 389 ± 81 | <4 | 0.34 ± 0.08 |
NA, not available.
Fig. 3.

Modeling results from studies on human β3 homomeric receptors. (A) A simplified allosteric activation model based on Monod et al. (1965), Colquhoun (1998), Chang and Weiss (1999), and Rusch et al. (2004). The model describes receptor activation with five sites whose affinity to agonist (A) in the closed state is described by KC and in the open state by KO. LO (ratio of C to O) describes opening of unliganded receptors. Opening of fully liganded receptors is described by LO multiplied by d (= KO/KC) to the fifth power. (B) The data points show averaged values for open probability (Poest) of wild-type and mutant β3 receptors activated by propofol. The Poest values were obtained by comparing baseline current level and responses to propofol to a current range spanning from Po of 0 (determined in the presence of picrotoxin or blocking concentrations of pentobarbital) to Po of 1 (maximal inward current during or after application of pentobarbital). The curves were generated by fitting eq. 2 (with n = 5 sites) to the data. Fitting parameters are given in Table 3. (C) The data points show averaged values for open probability (Poest) of wild-type and mutant β3 receptors activated by pentobarbital. The Poest values were obtained by comparing baseline current level and responses to pentobarbital to a current range spanning from Po of 0 to Po of 1. The curves were generated by fitting eq. 2 (with n = 4 sites) to the Po data. Fitting parameters are given in Table 3.
TABLE 2.
Fitted propofol and pentobarbital binding and gating parameters for allosteric activation model
Pooled Po data from at least four cells were fitted with eq. 2 (Materials and Methods). Lo was calculated as (1 − Po,spont)/Po,spont, equaling 6.7 (for wild type), 13.3 (H267W), 99 (L268W), 0.7 (F221W), 3.8 (Y143W), or 1.9 (T266W). For Q224W, which showed no spontaneous activity, we used an arbitrary Lo value of 1000. KC is the equilibrium dissociation constant for closed receptors. Parameter d equals the ratio of open receptor and closed receptor dissociation constants (KO/KC), and is a measure of gating efficacy (large values denote poor efficacy). The goodness of the fit showed minimal changes when the number of propofol or pentobarbital binding sites varied between three and five. The parameters in the table are from fits with five propofol and four pentobarbital binding sites.
| Receptor | KC, propofol | d, propofol | KC, pentobarbital | d, pentobarbital |
|---|---|---|---|---|
| µM | µM | |||
| β3 wild type | 8.8 | 0.64 | 1022 | 0.07 |
| β3(H267W) | 7.1 | 0.67 | 3482 | 0.02 |
| β3(L268W) | 6.5 | 0.55 | 359 | 0.16 |
| β3(Q224W) | NA | NA | 2401 | 0.05 |
| β3(F221W) | NA | NA | >40 mM | 0.06 |
| β3(Y143W) | 14.4 | 0.91 | 118 | 0.34 |
| β3(T266W) | NA | NA | 1503 | 0.34 |
NA, not available (responses were too small for analysis).
We conducted further mutational analysis of selected residues in the putative propofol binding pocket. The β3(L268W) mutation had a minor influence over activation properties. The propofol activation curve had an EC50 of 15 ± 2 µM (four cells). The concentration-response curve for activation by pentobarbital was right-shifted and had an EC50 of 272 ± 40 µM (six cells). The mean peak current in the presence of saturating (100 µM) propofol was 14 ± 1% (five cells) of the response to saturating (5 mM) pentobarbital. Application of up to 100 µM picrotoxin had minimal (<1% of the maximal pentobarbital response; five cells) effect on the holding current, indicating a low level of spontaneous activity. The fitted KC and d for propofol were essentially unchanged (6.5 µM and 0.55) compared with the wild type.
Franks (2015) proposed that the main chain carbonyl oxygen of residue β3(Q224) forms a hydrogen bond with the oxygen atom of propofol. Although we were unable to make modifications to the peptide bond, we reasoned that addition of bulk to the side chain at this position may interfere with optimal positioning of the propofol molecule in the cavity and affect propofol activation properties. Receptors containing the β3(Q224W) mutation were essentially not responsive to propofol. Application of 500 µM propofol (a concentration over 50-fold higher than EC50 in β3 wild type) elicited a response that was only 2.7 ± 0.2% (four cells) of the response to saturating (3 mM) pentobarbital. We also probed activation in the presence of 50 µM propofol. In six cells, the peak response was 3.0 ± 0.4% of the response to 3 mM pentobarbital. The currents elicited by propofol are small, and the response at 500 µM propofol may be influenced by block, but these data suggest that the β3(Q224W) mutation may suppress gating rather than binding of propofol. The mutation also affected the pentobarbital concentration-response relationship. The pentobarbital activation curve had a midpoint at 666 ± 21 µM (four cells). Lack of effect of picrotoxin (three cells) on the holding current and inability of blocking concentrations of pentobarbital to elicit apparent outward current indicates minimal spontaneous activity in β3(Q224W).
The putative propofol-binding cavity is capped at the extracellular side by β3(Y143) and β3(F221) (Fig. 1). We probed the functional effects of tryptophan substitutions at these locations. In oocytes expressing β3(F221W) homomeric receptors, exposure to 20 µM propofol was without effect (seven cells) whereas application of 500 µM propofol elicited an apparent outward current (three cells), indicative of block of spontaneous activity. Exposure to 10–100 µM picrotoxin also resulted in apparent outward current. Application of 3 mM pentobarbital resulted in an initial outward current that we interpreted as block of spontaneous activity, followed by an inward rebound current upon the removal of the drug, which extended beyond the baseline level (a total of 22 cells). Incidentally, pentobarbital at 3 mM was a more efficacious blocker than 100 µM picrotoxin or 500 µM propofol. These findings indicate that the β3(F221W) mutant produces a high level of spontaneous activity. Using the current level during the application of 3 mM pentobarbital for Poest of 0 and the rebound current level in the end of pentobarbital application for Poest of 1, we estimate that Po,spontest equals 0.62 ± 0.04 (22 cells). The concentration-response relationship for pentobarbital was right-shifted in the mutant and had an EC50 of 523 ± 45 µM (nine cells).
The propofol activation curve in β3(Y143W) had an EC50 of 22 ± 6 µM (five cells; P < 0.05 versus wild type). The concentration-response relationship for pentobarbital was not affected by the mutation, and had an EC50 of 82 ± 24 µM (five cells). We estimate that the Po,spontest for β3(Y143W) is 0.21 ± 0.02 (four cells), which is a moderate increase (P < 0.05) over Po,spontest in wild type. By comparing maximal currents in the presence of propofol and pentobarbital, we calculate that the maximal Poest in the presence of propofol is only 0.3 ± 0.05 (four cells). Fitting pooled data to eq. 2 yielded a KC of 14.4 µM and a d (KO/KC) of 0.91.
We also tested the effect of placing a tryptophan residue in place of β3(T266). This residue is pointed toward the putative propofol-binding cavity; however, it is supplied by the neighboring subunit that contributes the “+” side to the intersubunit interface (Fig. 1). The β3(T266W) receptor showed minimal activation by propofol. In the presence of 10 µM propofol, the peak response was 4 ± 1% (seven cells) of the response to 3 mM pentobarbital. Exposure to higher concentrations of propofol resulted in outward current, which we interpreted as block of spontaneous activity. The concentration-response relationship for pentobarbital was shifted to higher agonist concentrations (EC50 = 389 ± 81 µM; four cells). By comparing block by 100 µM picrotoxin to the rebound response upon termination of the application of saturating (3 mM) pentobarbital, we estimate that the open probability of spontaneous activity is 0.34 ± 0.08 (five cells) in β3(T266W).
Activation of Wild-Type and Mutant β3 Receptors by Propofol Analogs.
To gain insight into the mechanism by which tryptophan substitutions reduce activation by propofol, we examined the effects of mutations on activation by propofol analogs with different ortho substituents. The experiments were conducted by comparing responses to a saturating concentration of pentobarbital (300 µM in β3 wild type, 3 mM in mutants) and, typically, 500 µM propofol analog in the same cell. In some cases (e.g., 2-tert-butyl-6-methylphenol on F221W or T266W), only blockade of spontaneous activity was observed at 500 µM, so a lower concentration (10 µM) was employed to compare peak responses.
The data suggest that the deleterious effect of the β3(Y143W) mutation on propofol activation has a steric origin. Activation by compounds with compact ortho substituents (2,6-dimethylphenol) or a single ortho substituent (2-isopropylphenol) was not affected by the β3(Y143W) mutation. The ratio of responses to 2,6-dimethylphenol over pentobarbital was 0.74 ± 0.17 (four cells) in the mutant and 0.77 ± 0.07 (four cells) in the wild-type receptor. The relative response to 2-isopropylphenol was 1.44 ± 0.19 (four cells) in the mutant and 1.56 ± 0.25 (four cells) in the wild-type receptor. A compound with a relatively bulky ortho substituent (2,6-diethylphenol) was significantly worse at activating the mutant than wild-type receptors. The ratio of responses to 2,6-diethylphenol over pentobarbital was 0.14 ± 0.03 (four cells) and 0.78 ± 0.03 (four cells) in β3(Y143W) and β3 wild type, respectively. The β3(Y143W) mutation also reduced the relative current in the presence of 2-tert-butyl-6-methylphenol, which elicited a relative response of 0.53 ± 0.10 (five cells) in the wild-type and 0.25 ± 0.04 (four cells) in the mutant receptor.
The β3(Q224W) mutation diminished gating by 2,6-dimethylphenol (response ratio 0.29 ± 0.01; four cells) and 2,6-diethylphenol (0.14 ± 0.03; four cells) but not 2-isopropylphenol (1.37 ± 0.11; four cells) or 2-tert-butyl-6-methylphenol (0.66 ± 0.07; four cells). This indicates an asymmetric requirement for one bulky and one compact or missing ortho substituent.
Receptors containing the β3(F221W) or β3(T266W) mutation showed strongly diminished responses in the presence of all tested propofol analogs. In β3(F221W), the response ratios were 0.16 ± 0.03 (five cells), 0.03 ± 0.01 (five cells), 0.01 ± 0.01 (three cells), and 0.02 ± 0.02 (three cells) for 2-isopropylphenol, 2,6-dimethylphenol, 2,6-diethylphenol, and 2-tert-butyl-6-methylphenol, respectively. When the receptor contained the β3(T266W) mutation, the response ratios were 0.12 ± 0.03 (five cells), 0.13 ± 0.04 (six cells), 0.12 ± 0.02 (five cells), and 0.06 ± 0.02 (four cells) for 2-isopropylphenol, 2,6-dimethylphenol, 2,6-diethylphenol, and 2-tert-butyl-6-methylphenol, respectively. We propose that these residues are involved in signal transduction or located near an unaltered part of the propofol molecule (e.g., the hydroxyl group). The data are summarized in Fig. 4.
Fig. 4.

Effects of mutations on receptor activation by propofol and propofol analogs. The graph compares ratios (mean ± S.E.M.) of maximal responses to propofol or propofol analogs to responses to saturating pentobarbital in wild-type and mutant β3 receptors. Propofol was applied at 10–500 µM. Receptors containing the β3(F221W) mutation were not activated by propofol (#). Analogs were applied at 500 µM, except for 2-tert-butyl-6-methylphenol that was applied on β3(F221W) and β3(T266W) receptors at 10 µM. Pentobarbital was applied at 300 µM (wild type) or 3 mM (mutants). A value of 1 for the calculated parameter means that the compound/pentobarbital current ratio is the same in the mutant and wild type. The actual compound/pentobarbital current ratios for wild-type and mutant receptors are provided in the text. Statistical analysis (t test) was conducted by comparing the calculated parameter value to 1. The data show that the β3(Y143W) mutation does not affect the current ratio for 2-isopropylphenol or 2,6-dimethylphenol, and the β3(Q224W) mutation does not affect the current ratio for 2-isopropylphenol or 2-tert-butyl-6-methylphenol. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant.
Properties of α1β3 Receptors Containing the β3(T266W) or α1(I271W) Mutation.
In heteromeric α1β3 receptors, the residue homologous to β3(T266), supplied by the “+” side of the interface, is α1(I271). We examined the effect of the α1(I271W) mutation on the properties of α1β3. Our expectation was that this mutation imitates the effect of β3(T266W) in β3 homomeric receptors. However, α1(I271W)β3 receptors behaved, in most aspects, similar to wild-type α1β3 receptors. The mutation weakly, but statistically significantly (P < 0.05; t test), left-shifted the propofol concentration-response curve. The propofol EC50 was 13 ± 2 µM (six cells) in the wild type and 5 ± 1 µM (four cells) in the mutant. The concentration-response relationship for pentobarbital was not affected by α1(I271W). The EC50 was 87 ± 13 µM (five cells) and 68 ± 5 µM (four cells) in wild type and mutant, respectively. Unlike β3 homomeric receptors, the α1β3 receptors are activated by GABA. We found that the GABA concentration-response curve was modestly left-shifted in α1(I271W)β3. The midpoint of the curve was at 0.6 ± 0.1 µM (four cells) in the mutant and at 1.4 ± 0.1 µM (four cells) in the wild type (P < 0.01). The data are summarized in Fig. 5 and Table 3.
Fig. 5.

Concentration-response properties of human α1β3 receptors. The figure shows propofol (A), pentobarbital (B), and GABA (C) concentration–response curves for wild-type and mutant receptors. The data points show normalized responses (mean ± S.E.M.) from at least four cells at each condition. Current responses were normalized to the response to the highest concentration of agonist used in analysis. The curves show predictions of eq. 1 generated with the overall mean EC50 values from Table 3. In cases where no clear saturation was observed but higher drug concentrations produced block, Ymax was constrained to the value of the highest current response. In A, the maximal fitted response and nH were 1.07 ± 0.01 and 1.7 ± 0.3 (α1β3), 1.0 (constrained) and 4.3 ± 0.7 [α1β3(T266W)], 1 and 2.0 ± 0.1 [α1(I271W)β3], and 0.98 ± 0.02 and 2.9 ± 0.4 [α1(I271W)β3(T266W)]. In (B), the maximal fitted response and nH were 1 and 2.1 ± 0.3 (α1β3), 1.07 ± 0.03 and 0.9 ± 0.06 [α1β3(T266W)], 1.02 ± 0.01 and 2.4 ± 0.1 [α1(I271W)β3], and 1 and 2.9 ± 0.6 [α1(I271W)β3(T266W)]. In (C), the maximal fitted response and nH were 1.04 ± 0.01 and 1.5 ± 0.1 (α1β3), 1.07 ± 0.06 and 0.8 ± 0.1 [α1β3(T266W)], 1.0 and 2.0 ± 0.1 [α1(I271W)β3], and 1.04 ± 0.03 and 1.3 ± 0.2 [α1(I271W)β3(T266W)].
TABLE 3.
Propofol and pentobarbital concentration-response data for α1β3 receptors
The concentration-response data from each cell were fitted with eq. 1 (Materials and Methods). The table shows propofol, pentobarbital, and GABA EC50 values (mean ± S.E.M.) from at least four cells under each condition. Relative currents were estimated by comparing responses to saturating propofol, pentobarbital, and GABA in the same cell. Saturating GABA produces responses with the Po of near 1 because coapplication of 1 µM alphaxalone did not modify peak responses to saturating GABA (data not shown).
| Receptor | Propofol EC50 | Pentobarbital EC50 | GABA EC50 | Ipropofol/IGABA | Ipentobarbital/IGABA |
|---|---|---|---|---|---|
| µM | % | ||||
| α1β3 wild type | 13 ± 2 | 87 ± 13 | 1.4 ± 0.1 | 37 ± 5 | 21 ± 3 |
| α1(I271W)β3 | 5 ± 1 | 68 ± 5 | 0.6 ± 0.1 | 89 ± 3 | 91 ± 1 |
| α1(I271W)β3(T266W) | 9 ± 1 | 113 ± 30 | 3.3 ± 0.7 | 76 ± 3 | 87 ± 3 |
| α1β3(T266W) | 4 ± 1 | 166 ± 42 | 0.2 ± 0.04 | 74 ± 1 | 93 ± 4 |
We also conducted a test on receptor potentiation by propofol. For that, GABA at a concentration producing an EC5 response was applied in the absence and presence of a low concentration of propofol (see Materials and Methods for details). Proper comparison of potentiation across mutants may be complicated by differences in maximal open probability for GABA, so we used this experiment as a simple qualitative test of whether potentiation is present or absent in the particular receptor.
As expected, wild-type α1β3 receptors were potentiated by propofol. Coapplication of 1 µM propofol, which by itself elicited a response with a peak of <1% of the response to saturating GABA, with 0.3 µM GABA (EC5) resulted in 5- ± 1-fold potentiation (five cells). In α1(I271W)β3, coapplication of 0.5 µM propofol with 0.1 µM GABA led to 3.0- ± 0.1-fold (six cells) potentiation of the current response. Overall, the data demonstrate that the α1(I271W) mutation has minimal effect on activation and modulation of α1β3 receptors.
The α1(I271W) mutation is expected to influence propofol actions at the α(+)-β(−) interface. However, previous work has demonstrated that propofol (and other anesthetic drugs) may act via the β(+)-α(−) interface (Krasowski et al., 2001a; Bali and Akabas, 2004; Li et al., 2006; Jayakar et al., 2014), that remained unmodified in α1(I271W)β3 receptors. To additionally introduce the tryptophan substitution to the β–α interface, we combined α1(I271W) and β3(T266W) subunits. This double mutant receptor contains the tryptophan residue at homologous sites at “+” sides of α–β and β–α, and β–β interfaces, and can be considered a conceptual analog of the β3(T266W) homomeric receptor. To our surprise, the double mutation had minimal influence on channel properties. The propofol EC50 was not affected (9 ± 1 µM, eight cells versus 13 µM in wild type, P > 0.05). The concentration-response relationship for pentobarbital was unaffected (EC50 was 113 ± 30 µM; four cells). The activation curve for GABA was right-shifted by approximately 2-fold (midpoint at 3.3 ± 0.7 µM; four cells; P < 0.05). The α1(I271W)β3(T266W) receptors were potentiated by propofol. Currents elicited by 0.1 µM GABA (EC4) were enhanced 3.0- ± 0.1-fold (six cells) in the presence of 1 µM propofol.
As a negative control, we examined the properties of the α1β3(T266W) receptor. As expected, this mutation had a relatively small effect on activation and potentiation properties. The midpoint of the propofol concentration-response curve was left-shifted to 4.0 ± 0.5 µM (five cells; P < 0.01). In the presence of pentobarbital, there was a trend in EC50 to higher concentrations (EC50 = 166 ± 42 µM, five cells), but the effect did not reach statistical significance. The EC50 for GABA was left-shifted to 0.2 ± 0.04 µM (four cells). Receptors activated by low GABA (EC5) were potentiated by 3.5- ± 0.2-fold (five cells) in the presence of 1 µM propofol.
Discussion
A recent modeling study postulated a high-affinity propofol binding site at the intersubunit interface in β3 homomeric GABAA receptors (Franks, 2015). The putative binding site lies in the middle of a triangular structure formed by the M1 and M2 membrane-spanning domains of a subunit contributing the “−” side of the interface and the M2 domain of the neighboring subunit that contributes the “+” side of the interface (Fig. 1). The goal of the present study was to investigate the role of selected residues lining this cavity, using electrophysiology to gain functional evidence for its role in activation by propofol. We introduced tryptophan residues to Y143, F221, Q224, and H267 at the “−” side and T266 (in the β3 subunit) or I271 (α1 subunit) at the “+” side of the intersubunit interface. The major experimental finding is that mutations at some of these locations strongly reduce or eliminate activation by propofol with less significant effect on activation by pentobarbital.
We began this study with the null hypothesis that the cavity around the β3(H267) residue is not involved in activation of the β3 homomeric GABAA receptor by propofol. We reasoned that while a functional effect of a mutation is supportive of involvement but ultimately inconclusive, the lack of selective functional effect is an indication that a residue is not a critical component of the interaction site. We chose to make tryptophan substitutions. Tryptophan, due to its added bulkiness, is more likely to exclude the ligand, or, perhaps, mimic its presence in the binding pocket. Previous studies examining properties of putative binding sites for neurosteroids and the anesthetic etomidate have found that tryptophan substitution of key residues can mimic the effects of a bound modulator, such as increased spontaneous activity and left-shifted GABA concentration-response curves (Akk et al., 2008; Stewart et al., 2008).
Even though the β3(H267) residue is labeled by the propofol analog ortho-propofol diazirine (Yip et al., 2013) and is located adjacent to the modeled high-affinity propofol binding site (Franks, 2015), a tryptophan substitution at this position had minimal influence on activation properties. Specifically, there was no increase in spontaneous activity or shift in propofol EC50. Comparison of maximal currents in the presence of propofol and pentobarbital in β3(H267W) suggests a nearly 2-fold reduction in gating efficacy by propofol, but the caveat is that the difference may be caused by a change in Po for pentobarbital rather than propofol. In the case of heteromeric GABAA receptors, the estimate for maximal Po can be obtained by examining the ability of various potentiators to enhance the response to saturating agonist (e.g., Forman and Stewart, 2012; Eaton et al., 2014). We were, however, unable to observe potentiation of β3 homomeric receptors activated by pentobarbital in the presence of propofol or the steroid alphaxalone (data not shown). Our findings are in agreement with a previous work that found a 2-fold reduction in direct activation by the propofol analog ortho-propofol diazirine in α1β3 receptors containing the β3(H267A) mutation (Yip et al., 2013).
Although photolabeling of H267 with ortho-propofol diazirine was the aegis for identifying the potential binding pocket at the top of β3-TM2, it appears not to be directly involved in propofol binding or effect. This may be due to its mechanism of photolabeling. After photolysis of ortho-propofol diazirine, an ortho-quinone methide would be predicted as a major photo product. Quinone methides have relatively long half-lives (Silva and Bozzelli, 2007) and, as strong electrophiles, will preferentially react with nearby nucleophilic amino acids (Modica et al., 2001). There are numerous water accessible nucleophilic amino acids in the β3 homomers (Miller and Aricescu, 2014). The fact that nucleophilic amino acids other than β3(H267) are not labeled suggests that ortho-propofol diazirine binding is concentrated near β3(H267) or may diffuse a short distance to photolabel it.
Mutations at the top of the putative binding cavity, near the boundary between the membrane-spanning and extracellular domains, had a major effect on activation by propofol. No propofol-elicited currents were observed in β3(F221W), and this mutation resulted in large (Po of 0.62) spontaneous currents. A tryptophan substitution at the nearby β3(Y143) residue had a modest effect on propofol affinity, but strongly reduced gating efficacy. At the β3(Q224) site, a tryptophan substitution essentially eliminated activation by propofol. The F221W, Y143W, and Q224W mutations had a relatively modest effect on activation of the β3 receptor by pentobarbital. We infer that the region between the membrane-spanning and extracellular domains occupied by the F221, Y143, and Q224 residues is a key determinant of activation by propofol.
Near the cytoplasmic end of the putative propofol binding site, we examined the effects of tryptophan substitutions at the β3(T263) and β3(T266) sites. The β3(T263W) receptor was not functional in the presence of propofol or pentobarbital at up to 1 mM (data not shown). Application of 100 µM picrotoxin had no effect on the holding current indicating the absence of spontaneous activity (data not shown). The β3(T266W) receptor showed increased spontaneous activity, a right-shifted pentobarbital concentration-response relationship, but minimal activity in the presence of propofol.
Fitting Poest data to eq. 2 revealed some unexpected information. The fitted gating efficacy parameter (d) indicated a surprisingly small difference in the affinity of closed and open β3 receptors to propofol. In wild-type β3, β3(H267W), and β3(L268W), propofol binds only ∼2 times (d−1) more tightly to open receptors than to closed receptors. This corresponds to a stabilization energy of 2 kcal/mol (assuming five binding sites). To put this in perspective, the estimate for d−1 for propofol in heteromeric α1β2γ2L receptors is 50, which corresponds to a stabilization energy of 6.9 kcal/mol in a receptor with three binding sites (Ruesch et al., 2012). In β3(Y143W), d−1 was 1.1 and the stabilization energy 0.3 kcal/mol. For pentobarbital, d−1 was 14, 50, and 3, corresponding to stabilization energies of 6.3, 9.2, and 2.5 kcal/mol in wild-type β3, β3(H267W), and β3(Y143W), respectively.
Equation 2 is derived from an allosteric activation model (Fig. 3) where the receptor can exist in multiple states with different affinities to ligand (Monod et al., 1965; Colquhoun, 1998). In this framework, β3(H267W) and β3(L268W), which show apparent decreased gating efficacy in the absence (Lo) and presence (dLo) of propofol but an unchanged KO/KC (d) for propofol, have a pure, albeit weak, gating effect. The β3(Y143W) with a d of 0.9 and a small effect on KC, predominantly affects the equilibrium dissociation constant of open receptors, suggesting that this residue may come in contact with the propofol molecule in the active state where the tryptophan side chain results in unfavorable interaction with the agonist. The deleterious effect of the mutation likely has a steric origin because propofol analogs 2,6-dimethylphenol and 2-isopropylphenol were efficacious activators of the mutant receptor.
Two other mutations (F221W and T266W) in β3 receptors exhibited large Po,spontest but no response to propofol. This can be accounted for by unchanged equilibrium dissociation constants in closed and open mutant receptors—that is, d near unity. We note that there was a wide range of effects on Po,spontest in mutant receptors, which perhaps is not surprising given the transmembrane location of most locations. We were, however, unable to find good correlation between changes in Po,spontest and effects on activation by propofol.
Overall, our mutational analysis strongly supports, although does not prove, the involvement of the cleft defined by the M2 and M1 membrane-spanning domains from neighboring subunits in the actions of propofol in β3 GABAA receptors. We have identified four residues (Y143, F221, Q224, and T266) where a tryptophan substitution strongly reduced activation or rendered the receptor inactive in the presence of propofol but not pentobarbital. Work with propofol analogs showing that the magnitude of effects of mutations is dependent on ortho substituents of propofol supports the notion that these residues are components of propofol binding site in the β3 receptor.
We employed β3 homomeric receptors to avoid complications arising from heterogeneity in subunit interfaces. The α1β3 receptor is expected to contain one β–β, two α–β, and two β–α interfaces. If we assume that mutations to the “−” side of β3 affect both β–β and α–β type interfaces, that still leaves two unmodified β–α interfaces. A recent study that employed the photoreactive propofol analog 2-isopropyl-5-[3-(trifluoromethyl)-3H-diazirin-3-yl]phenol demonstrated labeling at the β–α interface in human α1β3 receptors (Jayakar et al., 2014). Interestingly, the residues labeled [β3(M286), α1(M236), and α1(I239)] are located in a plane more cytoplasmic relative to the key residues in β3 homomers in the long axis of membrane-spanning domains. One possible conclusion is that propofol interaction sites at β–β and α–β versus β–α interfaces involve different structures. This is supported by our mutational data on the β3(T266) residue. The β3(T266W) mutation eliminates propofol activation in β3 homomeric receptors. Loss of propofol activation, however, is not reproduced in heteromeric α1β3 receptors containing homologous mutations at all interfaces [α1(I271W)β3(T266W)]. Complementary evidence comes from the finding that propofol activation is not eliminated in receptors containing mutations to the β2(M286) residue in α1β2γ2 receptors (Krasowski et al., 2001b). We hypothesize that propofol sites at the β–β and α–β interfaces involve the region between the β3(T266) and β3(F221) residues at the cytoplasmic and extracellular ends, respectively. The propofol site at the β–α interface may involve residues nearer to the cytoplasmic side of the membrane-spanning domains and include the β3(M286), α1(M236), and α1(I239) residues.
Acknowledgments
The authors thank Dr. Joe Henry Steinbach for insightful advice and valuable suggestions during the course of the project.
Authorship Contributions
Participated in research design: Eaton, Chen, Franks, Evers, Akk.
Conducted experiments: Eaton, Cao, Akk.
Performed data analysis: Eaton, Cao, Akk.
Wrote or contributed to the writing of the manuscript: Eaton, Franks, Evers, Akk.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grants R01-GM108580, R01-GM108799]; and the Taylor Family Institute for Innovative Psychiatric Research; and the Medical Research Council, UK [Grant G0901892].
References
- Adodra S, Hales TG. (1995) Potentiation, activation and blockade of GABAA receptors of clonal murine hypothalamic GT1-7 neurones by propofol. Br J Pharmacol 115:953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike N, Maruyama T, Tokutomi N. (1987) Kinetic properties of the pentobarbitone-gated chloride current in frog sensory neurones. J Physiol 394:85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Li P, Bracamontes J, Reichert DE, Covey DF, Steinbach JH. (2008) Mutations of the GABA-A receptor α1 subunit M1 domain reveal unexpected complexity for modulation by neuroactive steroids. Mol Pharmacol 74:614–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali M, Akabas MH. (2004) Defining the propofol binding site location on the GABAA receptor. Mol Pharmacol 65:68–76. [DOI] [PubMed] [Google Scholar]
- Baumann SW, Baur R, Sigel E. (2001) Subunit arrangement of γ-aminobutyric acid type A receptors. J Biol Chem 276:36275–36280. [DOI] [PubMed] [Google Scholar]
- Carland JE, Johnston GA, Chebib M. (2008) Relative impact of residues at the intracellular and extracellular ends of the human GABAC ρ1 receptor M2 domain on picrotoxinin activity. Eur J Pharmacol 580:27–35. [DOI] [PubMed] [Google Scholar]
- Chang Y, Weiss DS. (1999) Allosteric activation mechanism of the α 1 β 2 γ 2 γ-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys J 77:2542–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D. (1998) Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol 125:924–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne EL, Hosie AM, Wooltorton JR, Duguid IC, Harvey K, Moss SJ, Harvey RJ, Smart TG. (2002) An N-terminal histidine regulates Zn(2+) inhibition on the murine GABA(A) receptor β3 subunit. Br J Pharmacol 137:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton MM, Bracamontes J, Shu HJ, Li P, Mennerick S, Steinbach JH, Akk G. (2014) γ-aminobutyric acid type A α4, β2, and δ subunits assemble to produce more than one functionally distinct receptor type. Mol Pharmacol 86:647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson C, Hardy SL, Werner DF, Hileman SM, Delorey TM, Homanics GE. (2007) New insight into the role of the β3 subunit of the GABAA-R in development, behavior, body weight regulation, and anesthesia revealed by conditional gene knockout. BMC Neurosci 8:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman SA, Stewart D. (2012) Mutations in the GABAA receptor that mimic the allosteric ligand etomidate. Methods Mol Biol 796:317–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP. (2015) Structural comparisons of ligand-gated ion channels in open, closed, and desensitized states identify a novel propofol-binding site on mammalian γ-aminobutyric acid type A receptors. Anesthesiology 122:787–794. [DOI] [PubMed] [Google Scholar]
- Hales TG, Lambert JJ. (1991) The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. Br J Pharmacol 104:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horenstein J, Wagner DA, Czajkowski C, Akabas MH. (2001) Protein mobility and GABA-induced conformational changes in GABA(A) receptor pore-lining M2 segment. Nat Neurosci 4:477–485. [DOI] [PubMed] [Google Scholar]
- Jayakar SS, Zhou X, Chiara DC, Dostalova Z, Savechenkov PY, Bruzik KS, Dailey WP, Miller KW, Eckenhoff RG, Cohen JB. (2014) Multiple propofol-binding sites in a γ-aminobutyric acid type A receptor (GABAAR) identified using a photoreactive propofol analog. J Biol Chem 289:27456–27468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, et al. (2003) General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor β3 subunit. FASEB J 17:250–252. [DOI] [PubMed] [Google Scholar]
- Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL. (2001a) General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of γ-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility. J Pharmacol Exp Ther 297:338–351. [PubMed] [Google Scholar]
- Krasowski MD, Nishikawa K, Nikolaeva N, Lin A, Harrison NL. (2001b) Methionine 286 in transmembrane domain 3 of the GABAA receptor β subunit controls a binding cavity for propofol and other alkylphenol general anesthetics. Neuropharmacology 41:952–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. (2006) Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci 26:11599–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löw K, Crestani F, Keist R, Benke D, Brünig I, Benson JA, Fritschy JM, Rülicke T, Bluethmann H, Möhler H, et al. (2000) Molecular and neuronal substrate for the selective attenuation of anxiety. Science 290:131–134. [DOI] [PubMed] [Google Scholar]
- Miller PS, Aricescu AR. (2014) Crystal structure of a human GABAA receptor. Nature 512:270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modica E, Zanaletti R, Freccero M, Mella M. (2001) Alkylation of amino acids and glutathione in water by o-quinone methide. Reactivity and selectivity. J Org Chem 66:41–52. [DOI] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP. (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12:88–118. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Benke D, Brünig I, Benson JA, Fritschy JM, Martin JR, Bluethmann H, Möhler H. (1999) Benzodiazepine actions mediated by specific γ-aminobutyric acid(A) receptor subtypes. Nature 401:796–800. [DOI] [PubMed] [Google Scholar]
- Ruesch D, Neumann E, Wulf H, Forman SA. (2012) An allosteric coagonist model for propofol effects on α1β2γ2L γ-aminobutyric acid type A receptors. Anesthesiology 116:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüsch D, Zhong H, Forman SA. (2004) Gating allosterism at a single class of etomidate sites on α1β2γ2L GABAA receptors accounts for both direct activation and agonist modulation. J Biol Chem 279:20982–20992. [DOI] [PubMed] [Google Scholar]
- Sanna E, Mascia MP, Klein RL, Whiting PJ, Biggio G, Harris RA. (1995) Actions of the general anesthetic propofol on recombinant human GABAA receptors: influence of receptor subunits. J Pharmacol Exp Ther 274:353–360. [PubMed] [Google Scholar]
- Silva Gd, Bozzelli JW. (2007) Quantum chemical study of the thermal decomposition of o-quinone methide (6-methylene-2,4-cyclohexadien-1-one). J Phys Chem A 111:7987–7994. [DOI] [PubMed] [Google Scholar]
- Stewart D, Desai R, Cheng Q, Liu A, Forman SA. (2008) Tryptophan mutations at azi-etomidate photo-incorporation sites on α1 or β2 subunits enhance GABAA receptor gating and reduce etomidate modulation. Mol Pharmacol 74:1687–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V, Ehya N, Fuchs K, Sieghart W. (1997) Stoichiometry and assembly of a recombinant GABAA receptor subtype. J Neurosci 17:2728–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudell JR, Yue ME, Bertaccini EJ, Jenkins A, Harrison NL. (2008) Molecular modeling and mutagenesis reveals a tetradentate binding site for Zn2+ in GABA(A) αβ receptors and provides a structural basis for the modulating effect of the γ subunit. J Chem Inf Model 48:344–349. [DOI] [PubMed] [Google Scholar]
- Ueno S, Zorumski C, Bracamontes J, Steinbach JH. (1996) Endogenous subunits can cause ambiguities in the pharmacology of exogenous γ-aminobutyric acidA receptors expressed in human embryonic kidney 293 cells. Mol Pharmacol 50:931–938. [PubMed] [Google Scholar]
- Yip GM, Chen ZW, Edge CJ, Smith EH, Dickinson R, Hohenester E, Townsend RR, Fuchs K, Sieghart W, Evers AS, et al. (2013) A propofol binding site on mammalian GABAA receptors identified by photolabeling. Nat Chem Biol 9:715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman JM, Eliezer N, Simha R. (1968) The characterization of amino acid sequences in proteins by statistical methods. J Theor Biol 21:170–201. [DOI] [PubMed] [Google Scholar]