

Abstract
Radiotherapy is a mainstay of cancer treatment, used in either a curative or palliative manner to treat approximately 50% of cancer patients. Normal tissue toxicity limits the doses used in standard radiation therapy protocols and impedes improvements in radiotherapy efficacy. Damage to surrounding normal tissues can produce reactions ranging from bothersome symptoms that negatively affect quality of life to severe life-threatening complications. Improved ways of predicting, prior to treatment, the risk for development of normal tissue toxicity may allow for more personalized treatment and reduce the incidence and severity of late effects. There is increasing recognition that the cause of normal tissue toxicity is multifactorial and includes genetic factors in addition to radiation dose and volume of exposure, underlying co-morbidities, age, concomitant chemotherapy or hormonal therapy and use of other medications. An understanding of the specific genetic risk factors for normal tissue response to radiation has the potential to enhance our ability to predict adverse outcomes at the treatment planning stage. Therefore, the field of radiogenomics has focused upon the identification of genetic variants associated with normal tissue toxicity resulting from radiotherapy. Innovative analytic methods are being applied to the discovery of risk variants and development of integrative predictive models that build on traditional normal tissue complication probability models by incorporating genetic information. Results from initial studies provide promising evidence that genetic-based risk models could play an important role in the implementation of precision medicine for radiation oncology through enhancing the ability to predict normal tissue reactions and thereby improve cancer treatment.
Introduction
Clinical Need for Radiogenomics Research
Approximately 50% of individuals diagnosed with cancer will receive radiation as part of their treatment, resulting in a large number of cancer survivors who are susceptible to development of treatment related toxicities1,2. A long-standing goal of research in radiation oncology has been to improve our ability to predict normal tissue toxicities to enable prevention of acute and late adverse effects without compromising treatment efficacy. In this context, it should be noted that despite the technological advances that have been made to conform the dose of radiation to the tumor, some amount of normal tissue is always irradiated during the course of radiotherapy and this exposure can lead to the development of adverse effects. For example, in a recent analysis of 20 publications that reported techniques and toxicity outcomes for 11,835 patients treated with radiotherapy for prostate cancer, it was estimated that either moderate or severe late gastrointestinal toxicity was observed in 15% and 2%, respectively, of these men3. In addition, either moderate or severe genitourinary complications developed in 17% and 3%, respectively, of these patients. Thus, overall about 25–30% and 4–5% of prostate cancer patients who received radiotherapy develop either moderate or severe, respectively, GI and GU complications. In addition, sexual functioning is affected for a substantial number of men who are treated with radiation for prostate cancer, and it has been estimated that approximately half of prostate cancer patients who receive radiotherapy develop erectile dysfunction4. Thus, the adverse effects resulting from radiotherapy have a large impact upon the quality of life for these patients, which is particularly important since the 10-year relative survival rate (which adjusts for the expected mortality from other causes of death) for all stages of prostate cancer combined approaches 100%5. Therefore, it is critical to consider the morbidity that may result from radiotherapy and steps that could be taken to prevent these complications, or exclude patients at the highest risk for their development.
Normal tissue complication probability (NTCP) models aim to estimate the risk of normal tissue toxicity based on dosimetric parameters6, but existing models are limited and could be improved, for example, by incorporation of patient-specific factors, such as age, gender, race, and genetics. The field of radiogenomics has emerged with the objective of identifying genetic variants, primarily single nucleotide polymorphisms (SNPs), associated with risk for the development of various normal tissue toxicities following treatment with standard radiotherapy protocols. In addition to understanding the biological underpinnings of radiosensitivity, one of the primary goals of radiogenomics is to develop an assay capable of predicting with a high level of sensitivity and specificity the likelihood that a particular cancer patient will develop complications from treatment with radiotherapy. The field envisions incorporating SNP information into predictive models along with dosimetric parameters, clinical risk factors, co-morbidities, and other patient-specific factors to significantly improve the predictive accuracy of such models.
The availability of a predictive assay may be of great help to medical providers, patients and their families if it allows an improved treatment decision to be reached for each individual. Information from such an assay, incorporated into a predictive model, could serve as a decision-making tool for patients by offering a genetically tailored approach for the prevention of radiation toxicity in the realm of precision medicine. In this paper, we provide both theoretical and empirical examples of predictive models. For patients predicted to be at high risk for the development of complications resulting from treatment with radiation, then alternative treatments with surgery and/or chemotherapy would be appropriate if they would provide similar efficacy. This decision making process would, of course, have to account for the risk of developing other adverse effects from these non-radiation treatment modalities. Alternatively, radiation dose parameters could be modified for the stratum of individuals at highest risk for toxicity. As increasingly targeted therapies emerge, along with associated increased cost, risk stratification based on genetic predisposition could help identify individuals who might be most appropriate candidates. It is also important to recognize that for those patients predicted to be at low risk for developing tissue injury from radiation, then possibly a more aggressive radiotherapy protocol using a higher dose could be considered, which may improve the chance for cure of their cancer. Such dose escalation would need to be carefully investigated through clinical trials to determine the safest possible maximum dose even for those at decreased risk of toxicity under current protocols.
Recent Progress in Radiogenomics Research
Researchers in the field of radiogenomics have made substantial progress in recent years towards the identification and validation in multiple cohorts of SNPs associated with the development of normal tissue toxicities resulting from radiotherapy. This is the first step towards achieving the goal of creating a predictive assay ready for implementation in the clinical setting. Much of the success of this field of research is due to the establishment of the Radiogenomics Consortium (RGC) in 2009 7,8, which is a National Cancer Institute/NIH-supported Cancer Epidemiology Consortium (http://epi.grants.cancer.gov/Consortia/single/rgc.html) consisting of 188 investigators at 110 institutions in 26 countries. The purpose of the RGC is to bring together collaborators to pool samples and data for increased statistical power of radiogenomic studies. Through the RGC, the number of radiogenomics cohorts has increased substantially, and it is now possible to perform large-scale studies that possess the statistical power needed to enable the discovery and validation of genetic markers that can be used to predict risk of adverse effects resulting from radiotherapy. The success of this effort is now being realized. Over the past 15 years, using approaches of candidate gene studies and, more recently, genome-wide association studies (GWAS), radiogenomics studies have so far identified seven SNPs that have been confirmed in replication studies as associated with one or more late effects of radiotherapy (Table 1): rs2868371 (esophagitis and pneumonitis following radiotherapy for lung cancer); rs1800469 (esophagitis following radiotherapy for lung cancer); rs1800629 (overall skin toxicity following radiotherapy for breast cancer); rs1139793 (fibrosis following radiotherapy for breast cancer); rs7120482 (rectal bleeding following radiotherapy for prostate cancer); rs264663 (overall toxicity following radiotherapy for prostate cancer); and rs1801516 (overall toxicity following radiotherapy for prostate or breast cancer) (9–16, reviewed in 17). There are many other candidates that have been reported and remain to be validated, and a recent GWAS provided evidence that many more common SNPs are associated with toxicity than have been so far discovered18. These SNPs can be identified definitively via larger, more statistically powerful studies that are currently underway.
Table 1.
Replicated SNPs associated with late radiotherapy toxicity.
| SNP | MAF | Location | Nearest Gene(s) | Cancer Site | Toxicity Endpoint | Study Type | Ref. |
|---|---|---|---|---|---|---|---|
| rs2868370 | 0.26 | promoter | HSPB1 | Lung Lung |
Esophagitis Pneumonitis |
Candidate Gene |
13 14 |
| rs1800469 | 0.31 | promoter | TGFB1 | Lung | Esophagitis | Candidate Gene | 11 |
| rs1800629 | 0.14 | promoter | TNF-α | Breast | Overall toxicity | Candidate Gene | 15 |
| rs1139793 | 0.28 | exonic (missense) | TXNRD2 | Breast | Fibrosis | Candidate Gene | 9 |
| rs7120482 | 0.30 | intergenic | MTNR1B, SLC36A4 | Prostate | Rectal bleeding | GWAS | 12 |
| rs264663 | 0.02 | intronic | TANC1 | Prostate | Overall toxicity | GWAS | 10 |
Here, we first describe some basic considerations for SNP discovery and model building. Next, the use of simulation data is explored to estimate the potential impact that SNPs could have on the ability to predict treatment toxicity. We then review various modeling efforts that are in progress to incorporate SNPs along with other risk factors. These methods draw on approaches developed for traditional NTCP modeling as well as ‘big data’ and machine learning approaches and highlight some of the exciting new directions in which the field is progressing.
Building validated models to predict the likelihood of post-therapy radiation toxicity using genomic, clinical and dosimetric variables
Pre-processing of SNP data is a critical first step to avoid introducing bias in SNP-toxicity association studies that are performed prior to predictive model development. Sources of variability and bias can arise due to different genotyping platforms across studies and ancestral differences of the populations studied. Neglecting the pre-processing step can increase the number of type-I or type-II errors. Several measures can be checked to test the quality of a SNP dataset: discordant sex information between genotype and phenotype, high rates of missing genotypes or elevated heterozygosity rates, duplicated samples or samples showing greater than expected relatedness, and discordant ancestry comparing self-report to SNP-based clustering19. The next step involves removing SNPs with high missing genotype rates, very low minor allele frequencies (MAF), significant deviation from Hardy-Weinberg equilibrium, and different missing genotype rates between cases and controls19. Genotype imputation, where missing genotypes are filled in based on haplotype structure in reference populations, such as those that are part of the 1000 Genomes project20, is now a standard, robust method that allows for harmonization of genetic datasets produced by different commercially available platforms.
It should be recognized at the initial model-building step that the interactions of radiation with tissue are biologically and temporally complex. Moreover, these interactions are affected by co-morbid conditions and adjuvant therapies. Robust multiparametric methods that incorporate genomic, clinical, and dosimetric parameters in a comprehensive model are more likely to succeed in identifying patients at high risk for radiation toxicity than consideration of single parameters alone. The development of such a model requires multivariable approaches to identify SNP-toxicity associations while additionally including clinical and dosimetric information. Andreassen et al. provided a recent review on the importance of accounting for clinical and dosimetric variables in genetic association studies21, and this is an important step if SNPs are to be combined in predictive models along with relevant clinical and dosimetric parameters to improve statistical power. The generalized linear regression model22 approach, which can be easily adapted to binary and categorical outcomes, is a powerful statistical method that can be used to introduce various clinical and dosimetric covariates along with genetic markers, while controlling for population stratification. Like any other area of research, combining radiogenomics data from different studies may introduce sources of variability and bias related to treatment types, dosimetric parameters, and reporting and grading toxicity. Meta-analysis across multiple GWAS datasets can address confounding by the so-called center-effect21, and identify SNPs that show consistent association with toxicity among differing treatment, clinical and patient-specific factors. Tests of heterogeneity can be helpful for deciding on methods for combining datasets and determining whether variability across datasets significantly impacts the SNP-toxicity association(s) of interest23,24.
As in any discipline, validation is a critical step in development of a predictive model for radiotherapy toxicity. Models developed and tested using a single dataset tend to be over-fit, and though they show good performance in the initial patient population from which they were derived, often they are not replicated when tested in an independent patient population. Standard approaches of internal and external validation can be used to combat this potential pitfall25. One internal validation approach is cross-validation, in which a sub-set of the data (the training set) is used in the model-building step and the remainder of the data (the test set) is used only for testing model performance. The advantage of this approach is the simplicity, but the disadvantage is that there is a loss in statistical power because the initial sample size is reduced when a portion of the data is removed for the test set. Leave-one-out cross-validation (LOOCV) is a special case of k-fold cross validation with k equal to the number of samples. A second validation approach uses bootstrapping analysis, sampling datapoints randomly with replacement. The advantage of this approach is that it can build relatively stable models for a small dataset, but the downside is that it is more computationally intensive than the simpler train-test cross validation approach when the size of dataset is large. A third approach is to use one or more completely independent external validation cohorts. External validation is the most rigorous approach to ensure that the model is generalizable outside of the initial study population, but the difficulty lies in having access to additional studies, particularly ones that were designed similarly to the first. Indeed this has been a challenge in radiogenomics, although successful validation studies have been accomplished in which SNPs associated with specific forms of toxicity have been identified, as outlined above. In addition, the Radiogenomics Consortium has worked towards bringing investigators together in the planning stages of studies so that, going forward, radiogenomics studies are conducted in a more uniform manner, allowing for easier external validation of predictive models.
Estimating the contribution of SNPs to improve performance of NTCP models using simulation data
While SNP discovery is underway in radiogenomics, simulation experiments can provide a way to estimate the potential benefit that SNPs could have on predicting normal tissue toxicity in the clinic. In a simulation experiment using assumptions relevant to a variety of complex diseases, Janssens et al. reported the discriminatory ability of various predictive models that include hypothetical numbers, frequencies, and effect sizes of risk SNPs26. The findings offer some guidance as to what can be expected from predictive models for normal tissue toxicity that incorporate radiosensitivity SNPs. The simulation experiments asked: given the SNPs known to be associated with the disease, how well can predictive models discriminate between those individuals who will develop the disease and those who will not? In these simulation experiments, discriminatory ability, which is the model’s ability to separate those with events from those without events, is measured by the area under the receiver-operating characteristic curve (AUC). Random assignment of risks yields an AUC of 0.5, whereas perfect separation of events from nonevents yields an AUC of 1. The results of these simulation experiments demonstrate that: 1) increasing numbers of SNPs included in the risk model improves discrimination accuracy (measured by increased AUC), 2) inclusion of SNPs with larger effect sizes or higher risk allele frequency improve accuracy of risk models, and 3) relatively high AUCs (> 0.75) can be achieved with ~100 risk SNPs that each have an effect size of 1.05–1.5 and are relatively common 26.
Our group used this same approach (the methods of which have been described in detail26,27) to perform a set of simulations tailored to the normal tissue outcomes of interest in radiation oncology. In these simulations, we assumed that various baseline predictive models already exist based on dose-volume parameters, and SNPs are added to these models. Three different AUCs for baseline models were selected: 0.70, 0.75 and 0.80, based on published NTCP models for a variety of tumor types and normal tissues that are based on dose and volume parameters28–30. We assumed two different incidence rates for late effects: 5% for less common late effects (for example, severe rectal bleeding following prostate radiotherapy), and 30% for more common late effects (for example, dysphagia following head and neck radiotherapy). Because we don’t yet know all of the SNPs associated with normal tissue toxicities, we made educated assumptions about the likely distribution of risk SNPs with respect to MAFs and odds ratios (OR). One distribution was selected based on the distributions that have been seen for genetically well-characterized phenotypes such as cancer, heart disease, and type 2 diabetes in which approximately 100 risk SNPs have been identified with each individual SNP conferring a small increase in risk31,32. In this ‘low penetrance’ distribution, the majority of SNPs have small effect sizes (ORs, between 1.05 and 1.4) and are fairly common (risk allele frequencies ≥15%), with very few SNPs that have larger effects (OR ≥ 2) and are less common (Figure 1A). These diseases are, of course, very different from normal tissue toxicities of importance in the practice of radiation oncology. An important advantage in studying the genetics of radiotherapy toxicity is that the outcomes of interest occur in response to a defined exposure, the dose of which is known. Perhaps the most relevant example that can be used to guide assumptions about radiosensitivity SNPs is that of pharmacogenomics. Pharmacogenomics parallels radiogenomics with respect to the fact that the outcome of interest occurs in response to a specific, well-measured exposure: a drug, in the case of pharmacogenomics, or radiation, in the case of radiogenomics. SNPs that have been identified via pharmacogenomic studies show larger effect sizes. For example, the HLA-A*3101 variant is associated with adverse skin reactions to carbamazepine treatment, a drug commonly prescribed for epilepsy, with odds ratio > 5 33,34. Similarly, SNPs within the IL28B gene are associated with response to pegylated interferon and ribavirin used in the treatment of hepatitis C infection with odds ratios ranging from 5.6 to 7.3 35–38. In our radiogenomics simulation, we considered a second, ‘moderate penetrance’ distribution of SNPs in which a greater proportion of risk SNPs has larger effect sizes (Figure 1B).
Figure 1.
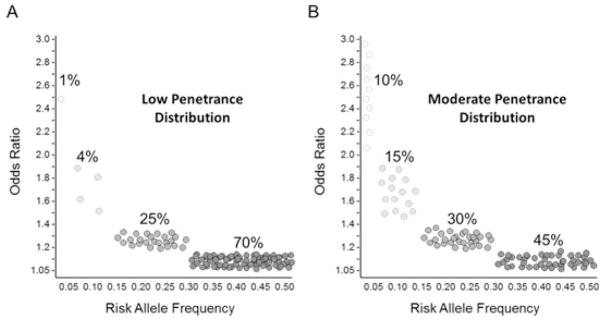
Hypothetical distributions of risk SNPs for radiotherapy late effects. A) low penetrance distribution, and B) moderate penetrance distribution. In both distributions, SNPs are selected in the given proportions from bins defined as follows: OR 1.05–1.2 and RAF 0.3–0.5, OR 1.2–1.4 and RAF 0.15–0.3, OR 1.4–2.0 and RAF 0.05–0.15, and OR 2.0–3.0 and RAF 0.01–0.05. OR, odds ratio; RAF, risk allele frequency.
Using the ‘low penetrance’ SNP distribution where the vast majority of radiosensitivity SNPs have small individual effect sizes, we found that relatively few SNPs, 118 for an effect with 30% incidence and 98 for an effect with 5% incidence, could achieve an AUC of 0.80, which is better than the performance of most of the existing NTCP models based on dosimetric parameters (Table 2A). Not surprisingly, however, a very large number of SNPs would be needed to achieve excellent discriminatory ability (AUC of 0.95) – 977 SNPs in the case of a common late effect and 557 SNPs in the case of a more rare late effect. When SNPs are added to an existing NTCP model (Table 2B) fewer SNPs are needed to achieve these AUCs. For example, just 78 SNPs would be necessary to improve the AUC of an existing NTCP model from 0.70 to 0.80 compared with 118 SNPs needed to achieve an AUC of 0.80 in a SNP-only model. Using the ‘moderate penetrance’ SNP distribution, even fewer SNPs are needed to achieve good discriminatory ability. For example, for an adverse effect with an incidence rate of 30%, 72 SNPs drawn from a moderate-penetrance distribution would be required to reach an AUC of 0.80, and for a toxicity with an incidence rate of 5%, just 53 SNPs would be needed (Table 3A). If SNPs from this distribution were added to an existing NTCP model with an AUC of 0.70, just 47 and 34 SNPs would be necessary to improve the model to an AUC of 0.80 for a common and rare late effect respectively (Table 3B).
Table 2.
Number of SNPs needed to achieve an AUC of 0.75–0.95 assuming a low-penetrance polygenic distribution. Numbers in each cell are the mean (range) of 100 iterations. A) Using a SNP-only model B) Adding SNPs to an existing NTCP model.
| A.
| |||||
|---|---|---|---|---|---|
| Incidence of late effects | AUC | ||||
| 0.75 | 0.80 | 0.85 | 0.90 | 0.95 | |
| 30% | 71 (60,80) | 118 (100,226) | 214 (196,228) | 397 (382,418) | 977 (928,1012) |
|
|
|||||
| 5% | 59 (52,64) | 98 (92,104) | 159 (150,168) | 269 (248,282) | 557 (520,580) |
| B.
| ||||||
|---|---|---|---|---|---|---|
| Incidence of late effects | NTCP model AUC | NTCP + SNP model AUC | ||||
| 0.75 | 0.80 | 0.85 | 0.90 | 0.95 | ||
| 30% | 0.70 | 28 (22,36) | 78 (72, 86) | 171 (152, 180) | 357 (325, 385) | 928 (905, 970) |
| 0.75 | 49 (40,62) | 145 (125, 164) | 335 (310, 348) | 868 (835, 900) | ||
| 0.80 | 75 (60, 90) | 272 (256, 296) | 821 (800, 855) | |||
|
| ||||||
| 5% | 0.70 | 24 (19, 28) | 61 (52, 72) | 118 (108, 126) | 233 (224, 250) | 529 (505–549) |
| 0.75 | 36 (29, 43) | 96 (82, 106) | 211 (200, 224) | 501 (475, 525) | ||
| 0.80 | 53 (45, 61) | 170 (165, 179) | 460 (438, 478) | |||
Table 3.
Number of SNPs needed to achieve the area under the receiver-operating characteristic curve (AUC) of 0.75–0.95 assuming a moderate-penetrance polygenic distribution. Each set of simulations was modeled for rarer forms of toxicity that occur at 5% incidence and more common types of radiation injury that occur at 30% incidence. Numbers in each cell are the mean (range) of 100 iterations. A) Using a SNP-only model B) Adding SNPs to an NTCP model that contains dosimetric risk variables.
| A.
| |||||
|---|---|---|---|---|---|
| Incidence of late effects | AUC | ||||
| 0.75 | 0.80 | 0.85 | 0.90 | 0.95 | |
| 30% | 41 (34,46) | 72 (67,77) | 124 (115,135) | 243 (240,252) | 566 (550,586) |
|
|
|||||
| 5% | 34 (26,36) | 53 (46,58) | 88 (78,96) | 152 (145,163) | 307 (284,326) |
| B.
| ||||||
|---|---|---|---|---|---|---|
| Incidence of late effects | NTCP model AUC | NTCP + SNP model AUC | ||||
| 0.75 | 0.80 | 0.85 | 0.90 | 0.95 | ||
| 30% | 0.70 | 17 (12, 20) | 47 (42, 51) | 100 (92, 112) | 207 (190, 230) | 539 (500, 570) |
| 0.75 | 30 (24, 36) | 82 (74, 90) | 192 (180, 212) | 522 (495, 548) | ||
| 0.80 | 50 (42, 56) | 155 (144, 172) | 492 (480, 510) | |||
|
| ||||||
| 5% | 0.70 | 13 (10,15) | 34 (30, 38) | 68 (64, 72) | 131 (120, 142) | 305 (285, 325) |
| 0.75 | 19 (14, 22) | 52 (46, 59) | 118 (108, 124) | 282 (260, 300) | ||
| 0.80 | 29 (24, 34) | 92 (83, 103) | 251 (232, 269) | |||
The results of this simulation exercise are encouraging because they suggest that a relatively small number of SNPs are required to achieve AUCs that are better than the AUCs of existing NTCP models, and they therefore set a benchmark in terms of the number of additional SNPs that need to be identified via future radiogenomics studies. As outlined earlier in this paper, substantial progress has already been made to identify and validate SNPs associated with various forms of toxicity and these simulations demonstrate that the number of additional SNPs needed to achieve reasonably good predictive models is within the range that we can expect to identify from large-scale, statistically powerful genetic association studies. By incorporating such SNPs into existing NTCP models, our ability to predict the likelihood that a given patient will develop complications from radiotherapy would improve substantially.
This data simulation study provides a theoretical picture of what we can expect to achieve in terms of predictive models in radiogenomics. It is important to note that the simulation approach used here was simplistic and thus conservative, in that it did not consider potential interaction between SNPs and other patient-specific, clinical, or dosimetric parameters, and so the improvement upon existing NTCP models could be greater than estimated. In the remainder of this review, we describe current efforts using patient data to build SNP-based predictive models of radiotherapy toxicity. These studies are yielding promising early results and demonstrate the variety of innovative modeling approaches that are being applied in radiogenomics.
Incorporation of SNPs as ‘dose modifying factors’ into NTCP models
A straightforward approach to using SNPs to predict normal tissue toxicity is to include them as dose modifying factors in NTCP models that have already been developed through years of research on the association between dosimetric parameters and toxicity. Studies have emerged in recent years suggesting that NTCP models can be improved by incorporating clinical risk factors39–42, so it seems logical that NTCP models could be improved further by incorporating SNP information as well. A recent study of radiation pneumonitis demonstrates empirically that SNPs can in fact be used to improve the performance of NTCP models for predicting toxicity43. In this study, five common SNPs within the TGFB1, VEGF, TNF, XRCC1 and APEX1 genes were incorporated as dose-modifying factors into the Lyman-Kutcher-Burman NTCP model, which was modified to account for duration of follow-up. The authors added various dose modifying factors to the model in a forward step-wise process and found that inclusion of these five SNPs significantly improved the model (based on results of a likelihood ratio test) compared with inclusion of mean lung dose alone. Though there was only a single dataset available for the modeling, a resampling procedure was used to reduce over-fitting.
The important need for refinement and validation of the model is acknowledged, but it serves as a preliminary assessment of the extent to which SNPs can improve prediction of toxicity using empirical data. Importantly, it hints at clinically actionable information that can be provided by SNP-based predictive models. For example, among patients with at least two risk alleles for these five SNPs, the incidence of pneumonitis is 10% at a mean lung dose of approximately 10 Gy, whereas a mean lung dose of approximately 25 Gy could be given to those with no risk alleles while maintaining the same 10% incidence of pneumonitis43. Though preliminary, these data support one of the goals described above, which is to enable dose escalation in the subset of the patient population that is at lower risk for toxicity. Further development of such a model, and evaluation in clinical trials of dose modification, could substantially improve the therapeutic index of radiotherapy in this setting.
Novel machine learning approaches to building multi-SNP predictive models of radiosensitivity
Radiogenomics studies are beginning to explore novel approaches to modeling SNP-toxicity association, highlighting the potential for use of a large number of SNPs to predict individual radiosensitivity. Preliminary studies that are outlined below, present proof-of-principle results to demonstrate the feasibility of these methods. These approaches aim to overcome the multiple-testing challenge of GWAS, which often results in failure of some potentially important SNPs to achieve genome-wide significance. Instead, they use machine learning–based methods to simultaneously investigate multi-SNP associations and predictions using many SNPs that do not individually reach statistical significance. In addition, these approaches allow for correlations or interactions among significant SNPs, something not often accounted for in single-SNP association tests due to statistical power limitations44,45. Several machine learning-based methods have been proposed to design predictive multi-SNP models for biological traits, and these can be applied to radiation response. The key point with these approaches is to recognize that only the final prediction has to be reliable, whereas inclusion of any particular SNP, among many, does not validate its causal importance. Thus, while the results of such models would not be ideal for guiding functional studies to discover new biology, they may have the potential to predict which patients are at high risk for developing toxicities based on a SNP profile.
Examples of penalized regression methods
Penalized regression methods are one approach to analyzing datasets with a very large number of potential predictors, as in genome-wide SNP studies, and these methods can be used in the development of predictive models in radiation oncology. Penalized regression methods eliminate as many terms as possible while still preserving predictive power, effectively performing feature selection and classifier construction (prediction) simultaneously. An L1-penalized support vector machine based on a sparse linear model is one type of penalized regression method, termed LASSO (for Least Absolute Shrinkage and Selection Operator). The Lasso method allows feature selection by constraining many features to have exactly zero coefficients and leaving only selective features that have strong collective impact on outcome. This type of approach has been used successfully to model other diseases and could be applied in radiogenomics studies. For example, this method was used to develop a predictive model for celiac disease using genome-wide SNP profiles, which led to a conclusion that several hundred SNPs are required to achieve an optimal predictive accuracy for this disease46. A novel method for ranking SNPs was proposed in which many bootstrap datasets were generated and GWAS p-values were calculated for all SNPs in each bootstrap dataset. The final SNP ranking was determined based on the median ranking of each SNP from a pool of p-values obtained from all bootstrap datasets47. This method increased the stability of SNP rankings across the cross-validation bootstrap datasets.
In another example of penalized regression, a two-step feature selection method was proposed to predict the risk of inflammatory bowel disease using GWAS data48. In the first step, single SNP association tests were performed. Irrelevant SNPs with a relatively liberal p-value were removed, which resulted in a computationally manageable group of SNPs. In a second step, penalized logistic regression was used with an absolute value (L1) penalty. This approach benefits from the ability of the L1-penalized model to choose SNPs with a similar effect.
L1 penalized regression to predict late rectal bleeding using genome-wide SNP data
L1 penalized regression was used in a recent radiogenomics study to design and develop a robust predictive multi-SNP model to predict late radiation-induced rectal bleeding49 among a set of 365 prostate cancer radiotherapy patients who were genotyped as part of a previously published GWAS12. Unlike standard modeling techniques, this machine learning method attempts to effectively determine a voting method, whereby a large number of SNPs ‘vote’ to decide if the patient is at risk or not. In this way, individual SNPs do not dominate the estimate, although some votes count more than others. The method does not aim to determine whether a given SNP is actually biologically causal; this type of modeling is focused on a different question, namely, maximizing the predictive accuracy of estimating overall sensitivity for a given patient, using as much relevant genomic information as possible. The challenge in this type of analysis is that it is attempting to find data features (SNPs) and a resulting model that predicts the outcome (toxicity), in a situation where the number of data features (SNPs) is vastly larger than the number of observed outcomes. This situation is common in the field of machine learning. Modeling is a balance between eliminating unimportant features, yet finding a way to integrate over the inherent uncertainty of the modeling process, in order to reduce bias in the final model.
A key challenge to this approach is to construct a model building process that does not over-fit to the dataset, but which also uses a majority of SNPs with small, actual, yet non-statistically significant correlations to the endpoint, in this case late rectal bleeding. This is precisely the challenge in many large scale predictive data mining/machine learning problems. For an unbiased assessment of the resulting model, one of the cross-validation approaches described above was used: the dataset was split into two groups: a training dataset (2/3 of samples) and a validation dataset (1/3 of samples). The modeling process can be briefly summarized as follows: (1) SNPs are ranked with respect to univariate correlation with the outcomes, (2) the general inter-patient genetic variation that relates to outcome is then modeled, through the PCA-logistic regression step, resulting in a further reduction of SNPs, (3) the LASSO method is then used to further filter SNPs by identifying SNPs that are important to estimating individual risk. By repeating the LASSO modeling with different random representations of the data the results become less sensitive to the noise in the data, and (4) SNPs are then ranked by frequency of appearance in the LASSO results. (5) Frequently-important SNPs are then used to build a predictive model using all the training data. Finally, (6) the resulting model is used to predict the risk in data that were not used in the model building process in any way (Figure 2).
Figure 2.
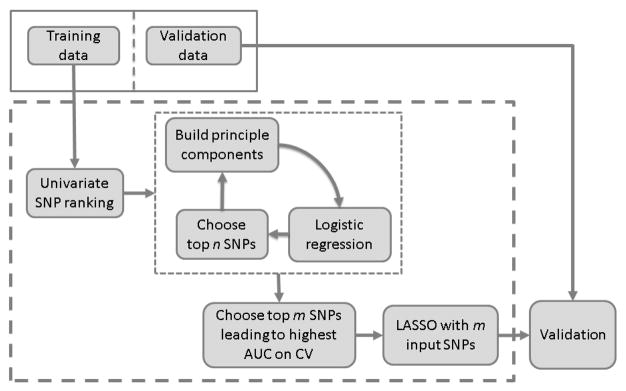
Diagram of the model building process. The dataset was split into a training dataset with 2/3 of samples and a validation dataset with 1/3 of samples. The validation dataset was used only for model validation. The LASSO model building process was performed 500 times using cross-validation datasets. A final LASSO model was built using the SNPs that are most frequently important for the cross-validation LASSO models.
The results of the first step, in which single-SNP chi-square tests were performed, show a large number of SNPs departing from the expected line on a Quantile-Quantile (Q-Q) plot for all SNPs (Figure 3), suggesting they are associated with radiotherapy-related rectal bleeding. A large fraction of these SNPs are incorporated into the subsequent model. Using the training dataset with the top n SNPs ranked by chi-square test, LASSO models were built 50 times with a 10-fold cross-validation approach, producing 500 models. SNPs were ranked based on the frequency in which they appeared in LASSO models. To build a final predictive model, an additional test was performed, splitting the training dataset into a sub-training dataset (2/3 of training samples) and a sub-testing dataset (1/3 of training samples). When predictive models were tested using an increasing number of SNPs in the ranked list, a model that obtained the best performance on the sub-testing dataset was used as a final model, and this final LASSO-based polygenic model was tested using the validation dataset. Models that used 500–700 SNPs had a similar performance on the validation data. The proposed method was iterated, changing the number of principal components used in the PCA step. When the first 2 components were used, a model with 484 SNPs reached the best performance using the sub-testing dataset. The final model with these SNPs obtained AUC = 0.63 on the validation dataset. This AUC was not improved further upon inclusion of additional principal components, implying that the first two components are enough to build a predictive model.
Figure 3.
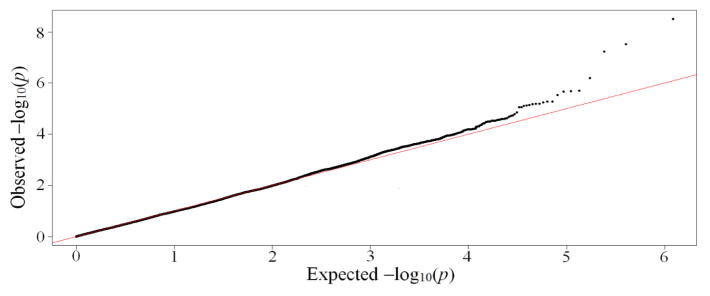
Quantile-Quantile (Q-Q) plot for all SNPs. The diagonal straight line indicates the distribution under the null hypothesis that there is no association between any of the SNPs and rectal bleeding. Strong deviation from the straight line indicates the significant association of SNPs with rectal bleeding.
A logistic regression model was applied to the predicted outputs obtained from the LASSO model with two principal components on the validation dataset with 484 SNPs that entered the LASSO model. Based on the newly generated predicted outputs from the logistic regression, the patients were binned into 6 groups, with 1 being the lowest toxicity group and 6 being the highest. A comparison of the predicted incidence of grade 2+ rectal bleeding and the actual incidence of grade 2+ rectal bleeding is shown in Figure 4. The ratio above each group represents the observed number of patients who experienced grade 2+ rectal bleeding and the total number of patients in the group.
Figure 4.

Comparison between the predicted incidence of grade 2+ rectal bleeding and the actual incidence of grade 2+ rectal bleeding. The predicted outcomes were produced after applying the logistic regression to outputs of the LASSO model using two principal components on the validation dataset with 484 SNPs that entered the LASSO. Based on the sorted predicted outcomes, the patients were binned into 6 groups, with 1 being the lowest toxicity group and 6 being the highest. The ratio above each group represents the observed number of patients who experienced grade 2+ rectal bleeding and the total number of patients in the group.
One of the major shortcomings of single-SNP models is that some potentially important SNPs may be excluded in the process of multiple-testing correction even though the predictive power could increase if such SNPs are included in a predictive model. Using an alternative approach based on machine learning, this limitation can be avoided, and accurate predictive models can be built for radiotherapy toxicity. The results of the approach are promising, and further modeling improvements are also possible through fine tuning using additional radiogenomics datasets that are now becoming available through the Radiogenomics Consortium7,8.
Combining Expectation-Maximization with LASSO to predict dysphagia using SNPs and clinical factors
In a related approach, an EMLasso model using stochastic expectation–maximization (EM) and Lasso 50 algorithms has been used in a radiogenomics study to identify patients at high risk for treatment-induced toxicity. The method used in this study aims at reducing the dimensionality of the high-dimensional data and then refitting by maximum likelihood (ML) estimation to produce accurate parameter estimates. Applying this approach to radiogenomics, De Ruyck et al. explored clinicopathological, dosimetric and genetic covariates for their ability to predict dysphagia following IMRT for head and neck cancer51. Using a dataset of 189 patients, the authors explored 41 potential predictive factors, including concurrent chemoradiotherapy, tumor site and stage, pre-treatment weight loss, smoking, dose–volume parameters for relevant anatomical structures, and 19 genetic polymorphisms selected on the basis of their location in genes involved in DNA damage repair. While this was not a high-dimensionality genome wide SNP study, the total number of potential predictors was high relative to the number of individuals in the study. They found that a model that included a SNP in XRCC1 improved the AUC. This approach was also used in a more recent study to develop multivariable predictive models of late genitourinary toxicity following high-dose intensity modulated radiotherapy for prostate cancer52. This study similarly showed that inclusion of SNPs improved predictive performance over inclusion of dosimetric parameters alone.
The results of these studies demonstrate that this modeling approach works well when applied to radiotherapy and genetics data, when there are often missing values in the predictors. Handling of missing values is a particular advantage of this approach over simply using LASSO53. Expansion to a larger, genome-wide SNP study could result in further improvement in model performance. Efforts are currently underway by several Radiogenomics Consortium members to build large prospective head and neck radiotherapy cohorts, which will serve as an invaluable resource for applying these types of modeling approaches for this disease site. Ultimately, this approach should be applicable to other disease sites.
Summary and Future Directions
In summary, the models presented in this paper represent the basis for the development of a clinically useful instrument to predict patient susceptibility for the development of adverse effects resulting from radiotherapy. The creation of a robust assay that is characterized by a high level of sensitivity and specificity to predict susceptibility for the development of adverse events arising from radiotherapy will serve as a powerful tool for cancer patients and their physicians to optimize treatment on an individual basis. This precision medicine approach has the potential to lead to reduced treatment-related toxicities and improved outcomes for individuals diagnosed with cancer.
Footnotes
This paper was developed under the auspices of the Radiogenomics Consortium
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Travis LB, Ng AK, Allan JM, et al. Second malignant neoplasms and cardiovascular disease following radiotherapy. J Natl Cancer Inst. 2012;104:357–70. doi: 10.1093/jnci/djr533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ringborg U, Bergqvist D, Brorsson B, et al. The Swedish Council on Technology Assessment in Health Care (SBU) systematic overview of radiotherapy for cancer including a prospective survey of radiotherapy practice in Sweden 2001--summary and conclusions. Acta Oncol. 2003;42:357–65. doi: 10.1080/02841860310010826. [DOI] [PubMed] [Google Scholar]
- 3.Ohri N, Dicker AP, Showalter TN. Late toxicity rates following definitive radiotherapy for prostate cancer. The Canadian journal of urology. 2012;19:6373–80. [PMC free article] [PubMed] [Google Scholar]
- 4.Zelefsky MJ, Chan H, Hunt M, Yamada Y, Shippy AM, Amols H. Long-term outcome of high dose intensity modulated radiation therapy for patients with clinically localized prostate cancer. J Urol. 2006;176:1415–9. doi: 10.1016/j.juro.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 5.American Cancer Society. Cancer Treatment and Survivorship Facts & Figures 2014–2015. Atlanta: American Cancer Society; 2014. [Google Scholar]
- 6.Bentzen SM, Constine LS, Deasy JO, et al. Quantitative Analyses of Normal Tissue Effects in the Clinic (QUANTEC): an introduction to the scientific issues. Int J Radiat Oncol Biol Phys. 2010;76:S3–9. doi: 10.1016/j.ijrobp.2009.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.West C, Rosenstein BS. Establishment of a radiogenomics consortium. Radiother Oncol. 2010;94:117–8. doi: 10.1016/j.radonc.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 8.West C, Rosenstein BS, Alsner J, et al. Establishment of a Radiogenomics Consortium. Int J Radiat Oncol Biol Phys. 2010;76:1295–6. doi: 10.1016/j.ijrobp.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 9.Edvardsen H, Landmark-Hoyvik H, Reinertsen KV, et al. SNP in TXNRD2 associated with radiation-induced fibrosis: a study of genetic variation in reactive oxygen species metabolism and signaling. Int J Radiat Oncol Biol Phys. 2013;86:791–9. doi: 10.1016/j.ijrobp.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 10.Fachal L, Gómez-Caamaño A, Barnett GC, Peleteiro P, Carballo A, Calvo-Crespo P, Kerns SL, Sánchez-García M, Lobato-Busto R, Dorling L, Elliott RM, Dearnaley D, Sydes MR, Hall E, Burnet NG, Carracedo A, Rosenstein BS, West CML, Dunning AM, Vega A. A three stage genome wide association study reveals susceptibility for late radiotherapy toxicity at the 2q24.1 (TANC1) locus. Nature Genetics. 2014 doi: 10.1038/ng.3020. In Press. [DOI] [PubMed] [Google Scholar]
- 11.Guerra JL, Gomez D, Wei Q, et al. Association between single nucleotide polymorphisms of the transforming growth factor beta1 gene and the risk of severe radiation esophagitis in patients with lung cancer. Radiother Oncol. 2012;105:299–304. doi: 10.1016/j.radonc.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 12.Kerns SL, Stock RG, Stone NN, et al. Genome-wide association study identifies a region on chromosome 11q14. 3 associated with late rectal bleeding following radiation therapy for prostate cancer. Radiother Oncol. 2013;107:372–6. doi: 10.1016/j.radonc.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez Guerra JL, Wei Q, Yuan X, et al. Functional promoter rs2868371 variant of HSPB1 associates with radiation-induced esophageal toxicity in patients with non-small-cell lung cancer treated with radio(chemo)therapy. Radiother Oncol. 2011;101:271–7. doi: 10.1016/j.radonc.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 14.Pang Q, Wei Q, Xu T, et al. Functional promoter variant rs2868371 of HSPB1 is associated with risk of radiation pneumonitis after chemoradiation for non-small cell lung cancer. Int J Radiat Oncol Biol Phys. 2013;85:1332–9. doi: 10.1016/j.ijrobp.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Talbot CJ, Tanteles GA, Barnett GC, et al. A replicated association between polymorphisms near TNFalpha and risk for adverse reactions to radiotherapy. Br J Cancer. 2012;107:748–53. doi: 10.1038/bjc.2012.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andreassen CN, Barnett GC, Kerns SL, et al. Analysis of 5434 patients shows a link between the ATM codon 1853 SNP and the risk of radiation-induced toxicity. European Society for Radiotherapy and Oncology (ESTRO); Vienna, Austria. 2014. [Google Scholar]
- 17.Kerns SL, CMLW, Andreassen CN, et al. Radiogenomics: the search for genetic predictors of radiotherapy response. Future oncology. 2014;10:2391–406. doi: 10.2217/fon.14.173. [DOI] [PubMed] [Google Scholar]
- 18.Barnett GC, Thompson D, Fachal L, et al. A genome wide association study (GWAS) providing evidence of an association between common genetic variants and late radiotherapy toxicity. Radiother Oncol. 2014 doi: 10.1016/j.radonc.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 19.Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP, Zondervan KT. Data quality control in genetic case-control association studies. Nature protocols. 2010;5:1564–73. doi: 10.1038/nprot.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genomes Project C. Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andreassen CN, Barnett GC, Langendijk JA, et al. Conducting radiogenomic research - Do not forget careful consideration of the clinical data. Radiother Oncol. 2012;105:337–40. doi: 10.1016/j.radonc.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Nelder J, Wedderburn R. Generalized linear models. Journal of the Royal Statistical Society. 1972;135:370–84. [Google Scholar]
- 23.Higgins J, Thompson S, Deeks J, Altman D. Statistical heterogeneity in systematic reviews of clinical trials: a critical appraisal of guidelines and practice. Journal of health services research & policy. 2002;7:51–61. doi: 10.1258/1355819021927674. [DOI] [PubMed] [Google Scholar]
- 24.Petitti DB. Approaches to heterogeneity in meta-analysis. Statistics in medicine. 2001;20:3625–33. doi: 10.1002/sim.1091. [DOI] [PubMed] [Google Scholar]
- 25.Good PI, Hardin JW. Common errors in statistics (and how to avoid them) 3. Hoboken, N.J: Wiley; 2009. [Google Scholar]
- 26.Janssens AC, Aulchenko YS, Elefante S, Borsboom GJ, Steyerberg EW, van Duijn CM. Predictive testing for complex diseases using multiple genes: fact or fiction? Genet Med. 2006;8:395–400. doi: 10.1097/01.gim.0000229689.18263.f4. [DOI] [PubMed] [Google Scholar]
- 27.Kundu S, Aulchenko YS, van Duijn CM, Janssens AC. PredictABEL: an R package for the assessment of risk prediction models. Eur J Epidemiol. 2011;26:261–4. doi: 10.1007/s10654-011-9567-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong FM, Hayman JA, Griffith KA, et al. Final toxicity results of a radiation-dose escalation study in patients with non-small-cell lung cancer (NSCLC): predictors for radiation pneumonitis and fibrosis. Int J Radiat Oncol Biol Phys. 2006;65:1075–86. doi: 10.1016/j.ijrobp.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 29.Tucker SL, Dong L, Cheung R, et al. Comparison of rectal dose-wall histogram versus dose-volume histogram for modeling the incidence of late rectal bleeding after radiotherapy. Int J Radiat Oncol Biol Phys. 2004;60:1589–601. doi: 10.1016/j.ijrobp.2004.07.712. [DOI] [PubMed] [Google Scholar]
- 30.Marzi S, Iaccarino G, Pasciuti K, et al. Analysis of salivary flow and dose-volume modeling of complication incidence in patients with head-and-neck cancer receiving intensity-modulated radiotherapy. Int J Radiat Oncol Biol Phys. 2009;73:1252–9. doi: 10.1016/j.ijrobp.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 31.Eeles RA, Olama AA, Benlloch S, et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet. 2013;45:385–91. 91e1–2. doi: 10.1038/ng.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michailidou K, Hall P, Gonzalez-Neira A, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45:353–61. 61e1–2. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med. 2011;364:1134–43. doi: 10.1056/NEJMoa1013297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozeki T, Mushiroda T, Yowang A, et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet. 2011;20:1034–41. doi: 10.1093/hmg/ddq537. [DOI] [PubMed] [Google Scholar]
- 35.Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 36.Ochi H, Maekawa T, Abe H, et al. IL-28B predicts response to chronic hepatitis C therapy--fine-mapping and replication study in Asian populations. The Journal of general virology. 2011;92:1071–81. doi: 10.1099/vir.0.029124-0. [DOI] [PubMed] [Google Scholar]
- 37.Suppiah V, Gaudieri S, Armstrong NJ, et al. IL28B, HLA-C, and KIR variants additively predict response to therapy in chronic hepatitis C virus infection in a European Cohort: a cross-sectional study. PLoS Med. 2011;8:e1001092. doi: 10.1371/journal.pmed.1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanaka Y, Kurosaki M, Nishida N, et al. Genome-wide association study identified ITPA/DDRGK1 variants reflecting thrombocytopenia in pegylated interferon and ribavirin therapy for chronic hepatitis C. Hum Mol Genet. 2011;20:3507–16. doi: 10.1093/hmg/ddr249. [DOI] [PubMed] [Google Scholar]
- 39.Defraene G, Van den Bergh L, Al-Mamgani A, et al. The benefits of including clinical factors in rectal normal tissue complication probability modeling after radiotherapy for prostate cancer. Int J Radiat Oncol Biol Phys. 2012;82:1233–42. doi: 10.1016/j.ijrobp.2011.03.056. [DOI] [PubMed] [Google Scholar]
- 40.El Naqa I, Bradley J, Blanco AI, et al. Multivariable modeling of radiotherapy outcomes, including dose-volume and clinical factors. Int J Radiat Oncol Biol Phys. 2006;64:1275–86. doi: 10.1016/j.ijrobp.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 41.Appelt AL, Vogelius IR, Farr KP, Khalil AA, Bentzen SM. Towards individualized dose constraints: Adjusting the QUANTEC radiation pneumonitis model for clinical risk factors. Acta Oncol. 2013 doi: 10.3109/0284186X.2013.820341. [DOI] [PubMed] [Google Scholar]
- 42.Cella L, D’Avino V, Liuzzi R, et al. Multivariate normal tissue complication probability modeling of gastrointestinal toxicity after external beam radiotherapy for localized prostate cancer. Radiat Oncol. 2013;8:221. doi: 10.1186/1748-717X-8-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tucker SL, Li M, Xu T, et al. Incorporating single-nucleotide polymorphisms into the Lyman model to improve prediction of radiation pneumonitis. Int J Radiat Oncol Biol Phys. 2013;85:251–7. doi: 10.1016/j.ijrobp.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guy RT, Santago P, Langefeld CD. Bootstrap aggregating of alternating decision trees to detect sets of SNPs that associate with disease. Genet Epidemiol. 2012;36:99–106. doi: 10.1002/gepi.21608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y. A novel bayesian graphical model for genome-wide multi-SNP association mapping. Genet Epidemiol. 2012;36:36–47. doi: 10.1002/gepi.20661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abraham G, Tye-Din JA, Bhalala OG, Kowalczyk A, Zobel J, Inouye M. Accurate and robust genomic prediction of celiac disease using statistical learning. PLoS Genet. 2014;10:e1004137. doi: 10.1371/journal.pgen.1004137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manor O, Segal E. Predicting disease risk using bootstrap ranking and classification algorithms. PLoS computational biology. 2013;9:e1003200. doi: 10.1371/journal.pcbi.1003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei Z, Wang W, Bradfield J, et al. Large sample size, wide variant spectrum, and advanced machine-learning technique boost risk prediction for inflammatory bowel disease. Am J Hum Genet. 2013;92:1008–12. doi: 10.1016/j.ajhg.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oh JH, Kerns SL, Ostrer H, Rosenstein B, Deasy JO. A machine learning method demonstrates that a large number of SNPs contribute to clinical radiosensitivity. European Societry for Radiotherapy and Oncology (ESTRO); 2015; Barcelona, Spain. 2015. [Google Scholar]
- 50.Tibshirani R. Regression Shrinkage and Selection Via the Lasso. Journal of the Royal Statistical Society, Series B. 1996;58:267–88. [Google Scholar]
- 51.De Ruyck K, Duprez F, Werbrouck J, et al. A predictive model for dysphagia following IMRT for head and neck cancer: introduction of the EMLasso technique. Radiother Oncol. 2013;107:295–9. doi: 10.1016/j.radonc.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 52.De Langhe S, De Meerleer G, De Ruyck K, et al. Integrated models for the prediction of late genitourinary complaints after high-dose intensity modulated radiotherapy for prostate cancer: making informed decisions. Radiother Oncol. 2014;112:95–9. doi: 10.1016/j.radonc.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 53.Sabbe N, Thas O, Ottoy JP. EMLasso: logistic lasso with missing data. Statistics in medicine. 2013;32:3143–57. doi: 10.1002/sim.5760. [DOI] [PubMed] [Google Scholar]