

Abstract
Complete genome resequencing of populations holds great promise in deconstructing complex polygenic traits to elucidate molecular and developmental mechanisms of adaptation. Egg size is a classic adaptive trait in insects, birds, and other taxa, but its highly polygenic architecture has prevented high-resolution genetic analysis. We used replicated experimental evolution in Drosophila melanogaster and whole-genome sequencing to identify consistent signatures of polygenic egg-size adaptation. A generalized linear-mixed model revealed reproducible allele frequency differences between replicated experimental populations selected for large and small egg volumes at approximately 4,000 single nucleotide polymorphisms (SNPs). Several hundred distinct genomic regions contain clusters of these SNPs and have lower heterozygosity than the genomic background, consistent with selection acting on polymorphisms in these regions. These SNPs are also enriched among genes expressed in Drosophila ovaries and many of these genes have well-defined functions in Drosophila oogenesis. Additional genes regulating egg development, growth, and cell size show evidence of directional selection as genes regulating these biological processes are enriched for highly differentiated SNPs. Genetic crosses performed with a subset of candidate genes demonstrated that these genes influence egg size, at least in the large genetic background. These findings confirm the highly polygenic architecture of this adaptive trait, and suggest the involvement of many novel candidate genes in regulating egg size.
Keywords: experimental evolution, egg size, polygenic adaptation, pooled sequencing, complex traits, evolution, adaptation
Introduction
The advent of whole-genome sequencing and inexpensive genotyping has reinvigorated strategies for identifying genes undergoing adaptive change. One approach involves scanning along the genome to identify regions with patterns or signatures of polymorphism indicative of recent selection (Sabeti et al. 2006; Voight et al. 2006; Nielsen et al. 2007; Rubin et al. 2010). Genomewide scans of reduced heterozygosity, for example, have identified selective sweeps in many species including humans (Tishkoff et al. 2007; Pickrell et al. 2009), domesticated chickens (Rubin et al. 2010), Drosophila melanogaster (Karasov et al. 2010; Cassidy et al. 2013; Garud et al. 2013), and Arabidopsis (Hancock et al. 2011). In only a few cases, however, has it been possible to directly associate a phenotypic trait with molecular adaptations. Moreover, polygenic adaptations that draw on standing variation may not be detectable by scanning the genome to identify regions with low heterozygosity (Hermisson and Pennings 2005; Przeworski et al. 2005; Pritchard and Di Rienzo 2010; Pritchard et al. 2010; Hernandez et al. 2011; Burke 2012). Natural populations may also experience selective forces that are locale-specific, further obscuring the adaptive phenotype to which selection is responding. Thus, the inverse problem of relating polymorphism data to a population genetic model of adaptation does not easily lend itself to the identification of the actual trait(s) under selection.
Experimental evolution in laboratory populations followed by whole-genome sequencing, commonly called “Evolve and Resequence (E&R)” (Turner et al. 2011), is an attractive alternative for investigating the genetic basis of a selected trait (Kawecki et al. 2012). The key to this approach lies in the control an investigator has in specifying the phenotype under selection. The approach is seeing a resurgence in interest because it is now possible to affordably resequence samples from evolved and control populations. Because nearly every single nucleotide polymorphism (SNP) in the population is surveyed by whole-genome population sequencing, all targets of selection are potentially identifiable.
Evolve and Resequence has been applied in Drosophila with varying degrees of success to investigate the genetic basis of longevity and aging (Burke et al. 2010; Remolina et al. 2012), body size (Turner et al. 2011), hypoxia tolerance (Zhou et al. 2011), courtship song (Turner and Miller 2012), bristle development (Cassidy et al. 2013), adaptation to novel environments (Orozco-terWengel et al. 2012), temperature (Tobler et al. 2014), and diet (Reed et al. 2014). Drawbacks of this approach include the introduction of “false positives” caused by 1) linkage and hitchhiking, which can be exacerbated by linkage disequilibrium (LD) created by the investigator in the establishment of the artificial populations; 2) genetic drift; and 3) unintended natural selection in the laboratory populations.
Oogenesis is a complex developmental process involving a large number of genes and pathways in Drosophila (Klusza and Deng 2011). Egg size varies heritably within and between fly species (Azevedo et al. 1997; Lott et al. 2007; Markow et al. 2009) and has long been recognized as an adaptive trait. It is positively correlated with many aspects of offspring fitness—egg hatchability, embryonic viability, and embryonic development rate and is responsive to thermal selection in laboratory populations (Azevedo et al. 1997). Egg size exhibits geographic clines in many species (Azevedo et al. 1996), generally increasing with latitude (James et al. 1997). There can be little doubt, therefore, about egg size being an adaptive trait subject to selection within and between species.
Despite the obvious importance (and experimental tractability) of egg size in Drosophila, genetic mechanisms underlying variation in egg size remain poorly investigated (Azevedo et al. 1997), in part because of its polygenic nature. To the best of our knowledge only a single study, conducted at the dawn of Drosophila Mendelian genetics, has attempted to identify the genomic regions regulating egg size in flies (Warren 1924). This study revealed that egg size in D. melanogaster is governed by a large number of loci distributed throughout the genome. The resolution of the study, however, was limited to large chromosomal regions.
To identify genes and variants involved in regulating egg size, we resequenced thrice-replicated laboratory populations of D. melanogaster derived from wild-caught flies (in IL, USA) that were artificially selected for both large and small egg volume (Miles et al. 2011). The large- and small-selected lines diverged from the starting populations by an average of 1.5 and 1.2 σP, respectively; no detectable deviation from the starting population was observed in the control populations. Subsequent analysis showed the eggs to differ in the number of cells composing the blastoderm embryo, and a correlated effect on the spatial localization of pair-rule stripes along both anterior–posterior and dorsal–ventral axes (Miles et al. 2011).
The experimental protocol to evolve egg size employed a number of favorable attributes for follow-up resequencing—laboratory population established from a large fresh collection of a single natural population, a period of laboratory acclimation of the population for ten generations, artificial selection for both smaller and larger egg volume in triplicate populations, retention of an “ancestral” sample from the starting base population, artificial selection applied every other generation to enhance recombination, and triplicate control populations. Using whole-genome pooled sequencing on the divergently selected populations, we identified several thousand reproducibly diverged SNPs between the large and small egg-size treatments. We then applied additional filters to address linkage (and hitchhiking) of SNPs on selected haplotypes. Clusters of significantly diverged variants and mRNA expression data were used to identify potential targets of selection. Additional variants not organized into linked clusters also show evidence of both directional selection and hitchhiking effects. Taken together several hundred genes are conservatively identified as candidates affecting egg size. As expected, genetic crosses performed with flies carrying mutant candidate genes indicate that unlike monogenic traits, the effect size of each gene is likely to be modest in this complex trait.
Results
Selection for Egg Size and Resulting Phenotypes
The schematic representation of the selection regimen is shown in figure 1A. Nine replicate populations were established from a large starting base population generated by allowing the direct descendants of 120 wild-caught D. melanogaster isofemale lines to mate at random for ten generations. Three of these were used to select for large egg sizes (LEP1, LEP2, and LEP3), three for small egg sizes (SEP1, SEP2, and SEP3), and three were maintained by randomly selecting equal numbers of eggs (CP1, CP2, and CP3). Mean egg volume (±SEM) in LEP, CP (controls), and SEP were 12.1 ± 0.02, 10.0 ± 0.02, and 8.7 ± 0.02 (×10−3 mm3), respectively (Miles et al. 2011), indicating that egg size changed significantly in opposite directions in both LEP and SEP compared with the CP (fig. 1B). The differences in egg volumes in the evolved populations are comparable to those from 12 Drosophila species (Markow et al. 2009). For example, the LEP population produced a large egg that was second only to the large-egg producing D. sechellia and the egg sizes in SEP were comparable to those in D. simulans (supplementary fig. S1, Supplementary Material online). These species diverged from a common ancestor approximately 400,000 years ago (Cutter 2008). Isogenic lines derived from each of the divergent population cages retained their respective changes in egg sizes after 40 generations of inbreeding in the absence of further selection for egg size (supplementary fig. S1, Supplementary Material online). The artificial selection for egg size, therefore, appears to have targeted polymorphisms that are not deleterious under laboratory conditions (Warren 1924; Schwarzkopf et al. 1999).
Fig. 1.
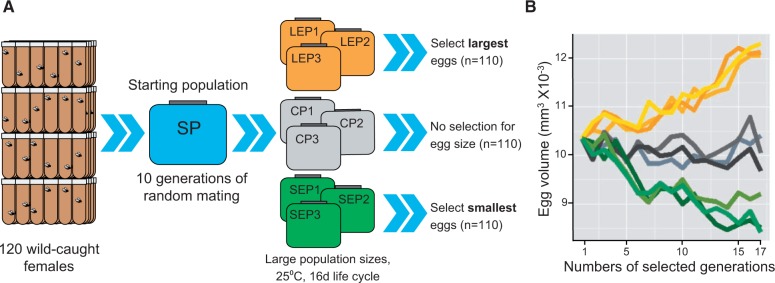
Experimental evolution in laboratory populations of Drosophila melanogaster. (A) Schematic representation of experimental evolution. (B) Changes in egg sizes due to artificial selection. On the x axis are number of selected generations. Mean egg volumes in each of the three large (yellow), small (green), and controls (gray) in each selected generations are shown. Compared with the controls, egg volumes increased and decreased significantly in the LEP and SEP, respectively, and no significant difference was observed in the CP.
Egg Size and Ovariole Numbers
Drosophila ovaries contain over a dozen ovarioles and each ovariole contains multiple developing oocytes (fig. 2A). Changes in egg size are thought to influence maternal fitness (Azevedo et al. 1997), which is correlated with ovariole number in adults (Boulétreau-Merle et al. 1982). Comparison of ovaries from adult females revealed that ovariole numbers differ significantly between isofemale lines derived from the selected populations for large and small eggs (P < 0.0001, analysis of variance [ANOVA]; fig. 2B). Furthermore, within each ovariole, egg chamber lengths differed significantly between a large and a small lines as early as stage 4 of oogenesis (P < 0.05, ANOVA; fig. 2C). Consistent with these observations, we also observed strikingly higher egg volumes in all of the LEP-derived lines relative to each of the SEP-derived lines (P < 0.0001, ANOVA; fig. 2D). In other words, compared with the flies derived from small egg populations, flies derived from large egg population not only have fewer ovariole numbers, but they also have larger eggs. Thus, these results suggest that egg size is correlated with ovariole numbers, at least in these laboratory selected lines and could potentially affect female reproductive fitness.
Fig. 2.

Ovariole numbers and egg chamber lengths. (A) Graphical representation of the Drosophila melanogaster ovaries with ovarioles extending out with egg chambers at various developmental stages. (B) Mean ovariole numbers per female (±SEM) in ten isofemale lines derived from the large (yellow) and small (green) selected populations (n = 10 pairs per line) are shown. P value was obtained from a nested ANOVA performed to compare ovariole numbers between large and small populations by nesting lines within the treatments. Both lines and treatments were significant. (C) Difference in mean (±SEM) egg chamber lengths between a large and a small line at various stages of oogenesis. (D) Egg volumes of isofemale lines derived from the large (yellow) and small populations (green, n = 50 per line). The boxes indicate the first and third quartiles and the whiskers indicate the interquartile ranges (1.5* IQR).
Sequencing Statistics and Genetic Diversity
To identify genetic variants for egg size, we performed pooled whole-genome sequencing on the starting population and the nine terminal populations: Three large egg populations, three small egg populations, and three control populations, using an Illumina HiSeq2000. Each of the samples had greater than 51× median sequence coverage; approximately 80% of the genome in each of the populations was sequenced greater than 40× (except CP2, 56% genome over 40×). Median sequencing depth in the starting population was 220× and 98% of the genome in the starting population was sequenced over 100× (supplementary table S1, Supplementary Material online). A total of 2,103,160 biallelic variants in the five major chromosome arms passed a series of stringent filters (see Materials and Methods). The median minor allele frequency in the starting population was approximately 10%. In total, 86% of these variants were also identified in 162 inbred lines comprising the Drosophila melanogaster Genetic Reference Panel (DGRP) (Mackay et al. 2012), indicating that a large percentage of the polymorphisms in our Illinois collection are common geographically widespread variants. The bulk of the genetic diversity (66–78%) was present in all nine samples and only 0.41% of the variants (n = 8,696) not detected in the starting population were found in a single population sample. Only 0.27% (5,623) variants were not found in any of the nine experimental populations and most of these lost variants (>80%) were low-frequency alleles in the starting population with allele frequency less than 10%. After removing these 5,623 variants 2,097,537 variants remained for further analysis. The allele frequency spectrum of these variants in each population is shown in supplementary figure S2, Supplementary Material online.
Allele Frequency Divergence
The experimental design allowed us to identify SNP frequency changes in the artificially selected populations that are unlikely to be the result of either genetic drift or natural selection in the laboratory. In particular, whereas inconsistent frequency changes across replicates may be attributable to drift, reproducible allele frequency changes across replicate populations exposed to the same selective pressure are likely to be enriched for natural or artificial selection (Pritchard and Di Rienzo 2010; Pritchard et al. 2010). As natural selection is expected to act consistently within the replicated populations but divergently between the two selection treatments, we attempted to identify SNPs whose frequencies differ consistently across replicates between the divergently selected populations. We first calculated allele frequency differences between the starting population and each of the nine terminal populations, and focused our attention on SNPs whose frequencies changed in the same direction in all three replicates. We reasoned these SNPs are likely to be enriched for sites under both natural and artificial selection in the laboratory populations. Small but significantly greater frequency differences were observed in both the SEP and LEP compared with the CP (median allele frequency = 7.9%, 8.3%, and 7.7%, respectively, P < 2.2e-16 for CP vs. SEP and CP vs. LEP, Wilcoxon rank sum test). Over 150,000 variants showed large and reproducible allele frequency divergence in opposite directions across all three replicates between the divergently selected populations (median difference = 13.77%).
To identify variants that have consistently diverged in response to egg-size selection, we compared allele frequency differences between the three independent populations of LEP and SEP at the 2,097,537 variants using a generalized linear-mixed model (GLMM, see Materials and Methods). This approach quantifies reproducible differences in allele frequencies between the two treatments while taking into account differences in sequencing depth and deviations of allele frequencies in each replicate population. Variants with extreme P values, that is, ones exhibiting consistent differences in allele frequencies between the large and small treatments, are putative targets of egg-size selection. After adjusting for genomewide multiple testing using a stringent empirical false discovery rate, FDR < 5e−6, we identified 4,137 polymorphisms (0.20% of all the variants in the genome) as significantly diverged variants, hereafter referred to as SDV (fig. 3A and supplementary table S2, Supplementary Material online). Only approximately 4% (n = 169) of the SDV were low frequency variants in the starting population (allele frequency ≤ 5%) and 98% were shared with DGRP. We compared the starting allele frequency from the approximately 4,137 SDV with all other non-SDV in the genome and with 1,000 sets of randomly sampled 4,137 non-SDV. In both cases, we found that the starting allele frequencies of the SDV are qualitatively higher than that of the non-SDV (supplementary fig. S3, Supplementary Material online), indicating adaptation occurred from common standing variants.
Fig. 3.
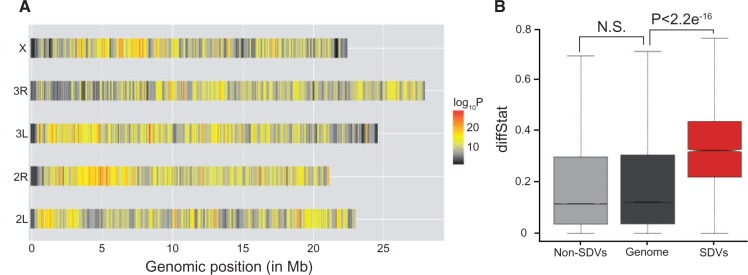
Distribution of SDV and diffStat statistic. (A) Distribution of significant variants in the five chromosome arms in the Drosophila melanogaster genome. (B) Comparison of the diffStat statistic between the significant variants (red), nonsignificant variants (light gray), and the genomewide background (dark gray).
We employed a conservative approach to further characterize each SDV by identifying the smallest allele frequency difference between all nine possible LEP–SEP comparisons, called “diffStat” (Turner et al. 2011). The median diffStat score for SDV, 32.22%, is 2.7 times higher than the genomewide median diffStat score (12.1% for SDV + non-SDV, P < 2.2e−16, Wilcoxon rank sum test; fig. 3B), demonstrating the consistency of the diverged variants between the two treatments. No significant increase in diffStat score relative to the genomewide background was observed at the non-SDV (P > 0.05). The higher diffStat scores in the SDV are robust even after taking starting allele frequencies and chromosomal locations into account (P < 0.0001, Permutation test; supplementary fig. S4A and B, Supplementary Material online).
False Positives, Genetic Hitchhiking, and Gene Expression
An unknown fraction of the SDV are expected to have diverged in frequencies not as a consequence of selection acting on these SNP but rather as a result of genetic hitchhiking with linked variants that are targeted by artificial selection (Nuzhdin and Turner 2013; Tobler et al. 2014). Although natural populations of D. melanogaster have relatively short spans of LD, generally measured in 10’s or 100’s of base pairs (Mackay et al. 2012), experimentally evolved populations are likely to have larger spans of LD that were created in establishing the founder laboratory population and expanded through the course of the artificial selection experiment. As individual haplotype information is not available from the pooled-sequencing data, we could not measure extent of LD in these populations. Instead, we attempted to identify genomic regions with clusters of SDV. To identify such genomic regions, we divided the genome into bins of 100-, 50-, and 10-kb sliding windows with 25-, 10-, and 2-kb step sizes, respectively, and performed a hypergeometric test comparing the proportion of SDV in each of the bins to the chromosomal background (see Materials and Methods). Applying a stringent genomewide FDR to minimize the false positives (FDR < 0.005), a total of 245, 342, and 415 windows were significant for 100-, 50-, and 10-kb windows respectively. There was, not surprisingly, a strong overlap of signals between the sliding windows of different sizes (supplementary fig. S5, Supplementary Material online). We chose to focus our attention on the 10-kb windows for further analysis because nearly all of the significant 100- and 50-kb windows also contain the significant 10-kb windows (fig. 4).
Fig. 4.

Genetic hitchhiking in the genome. Overrepresentation of SDV in 10-kb sliding windows with 2-kb steps in the five major chromosome arms in the Drosophila melanogaster genome. Each dot represents a 10-kb window and the purple-dotted lines indicate the genomewide multiple testing threshold at FDR ≤ 0.005. Notice the x axis is in Mb scale; hence, multiple significant windows that are ≥50 kb apart may appear indistinguishable. Almost 50% of the significant windows are on chromosome 2 and 24% are on chromosome X.
We next investigated the genomewide heterozygosity in 10-kb sliding windows (with 2-kb step sizes) averaged across replicates of the three egg-size treatments (LEP, SEP, and CP) and in the starting population. Compared with the starting population, a modest reduction in genomewide heterozygosity was observed in all three egg-size treatments (median fold reduction relative to starting population = 13%), though this reduction was not significantly greater for the two selected treatments than for the control treatment. We then turned our attention to the 415 significant windows enriched for the divergent variants and found a more pronounced reduction in heterozygosity in them than in the genomewide background in the CP, and a stronger reduction in the LEP than SEP (mean fold reduction relative to starting population = 17.5%; P = 1.64e−06, one-sided Wilcoxon rank sum test). The heterozygosity in 415 significant windows in LEP, when contrasted with that in 10,000 sets of equal number of randomly sampled windows matched by chromosomal arm in CP, was significantly lower the vast majority of times (P = 6.6e−03, Permutation test; supplementary fig. S4C and D, Supplementary Material online). Some of the significant windows overlapped with one another and these comparisons do not take this dependence into account. We therefore compared the reduction in heterozygosity in significant windows with those in the exact same genomic regions in the CP and found stronger reduction in heterozygosity in the significant windows (P = 6.27e−08, one-sided Wilcoxon rank sum test). Comparison of all the SDV and only those in the 415 significant windows also revealed that the latter have lower starting ancestral allele frequencies in the starting population (P = 0.01, one-sided t-test). Low frequency variants are expected to have originated more recently than common variants on average, and as a consequence would be expected to be in stronger LD with other linked variants, including the SDV that are targeted by selection. Low frequency variants, therefore, may be more detectable as hitchhiking variants.
The 415 significant 10-kb windows could be further consolidated into a smaller number of distinct blocks. We first merged overlapping SDV within each block and then to be conservative, expanded these blocks to include other 10-kb blocks (or SDV) that were within 50 kb of one another. This procedure yielded 85 distinct genomic blocks (median size ∼18 kb) separated by a median distance of ∼480 kb. The relatively large distance between the 85 distinct SDV islands compared with the size of the islands raises the prospect that each is an independent response to selection for egg size. Individual blocks may contain more than one SDV targeted by selection in addition to SDV that hitchhiked due to LD with the targeted SDV (fig. 5A). The 85 differentiated blocks contain 206 annotated genes, each with at least one SDV. To test whether these genes are relevant to egg-size evolution, we quantified their expression in D. melanogaster ovaries using the RNA-seq data generated by the modENCODE consortium (Graveley et al. 2011) because egg size likely depends upon genes that are expressed in the ovaries (Azevedo et al. 1997). Compared with the genomic background, the differentiated blocks were enriched for genes expressed in the Drosophila ovaries (n = 174 with RPKM > 0, P = 0.011, hypergeometric test). As a control, we also tested whether the differentiated blocks are enriched for genes expressed in testis and found no enrichment (P = 0.91). To identify candidate genes influencing egg size, we identified genes within each differentiated block with higher expression in ovaries than the average of all adult tissues measured, or the gene with the highest expression in the ovaries (if none exceeded the average across tissues). This resulted in a total of 110 candidate genes in the differentiated blocks (fig. 5B and supplementary table S3, Supplementary Material online).
Fig. 5.

A representative differentiated block and expression of candidate genes in adult tissues. (A) A 64-kb hypergeometric region (orange) along with 50-kb flanking regions. Notice two distinct peaks separated by several kb regions in the shaded area (orange). As these two peaks are less than 50 kb apart, they are likely in long-range linkage and thus, are considered a single significantly diverged block. Purple line indicates genomewide significance threshold at FDR <0.005. Genes with at least one SDV are shown in black. Genes with at least one SDV and are expressed in the ovaries are shown in blue. (B) Gene expression of 110 candidate genes in adult tissues. Colors indicate relative expression in each tissue (RE) compared with the average of all the adult tissues. Rows are tissues and columns are genes.
Compared with all the genes in the data set, the 110 candidate genes are enriched for genes with known functions in Drosophila oogenesis (n = 12, P < 0.007, hypergeometric test). These 12 genes include: Cullin-4 (Cul-4), bazooka (baz), Cbl, closca (clos), female sterile (2) ltoPP43 (fs(2)ltoPP43), Mothers against dpp (Mad), midline (mid), Tao, cut (ct), division abnormally delayed (dally), eyes-absent (eya), and Leukocyte-antigen-related like (Lar). Although these genes have well-defined functions in Drosophila oogenesis, they have no previously described role in regulating egg size. As a control, we evaluated whether the candidate genes are enriched for genes with no obvious function in egg development—namely, genes associated with male gamete generation and male courtship behavior. No enrichment was observed (P > 0.05).
Sixty percent of SDV do not fall into the differentiated blocks; we refer to these as “orphan SDV.” With a conservative empirical FDR set at 5e−06, these orphan SDV are either enriched for additional targets of selection or differentiated by long-distance LD with targets of selection. The sample of flies used to find the starting population contained a reasonably large number of wild chromosomes (N ≥ 480) and almost all the SDV were common polymorphisms. The lack of LD between common variants in D. melanogaster may account for why SDV targeted by selection may be found as orphans.
Enrichment Analysis and Identification of Additional Candidate Genes
To elucidate the functional properties of genes for egg size targeted by selection, we investigated the enrichment of Gene Ontology (GO) terms for genes harboring SDV. We employed GOWINDA (Kofler and Schlotterer 2012), an unbiased analysis of gene set enrichment, to identify statistically significant GO terms (among 2,834 possible terms; supplementary table S4, Supplementary Material online). We carried out two analyses: 1) On the full set of approximately 4,100 SDV, and 2) on a selective set of 1,634 SDV, corresponding to the 40% most significant SDV in the GLMM analysis. The P-value threshold for the 40% selective cutoff was established by formulating an empirical null distribution of smallest P values for all annotated genes in the Drosophila genome (∼16,340) and determining the P-value threshold for the top 10% (see Materials and Methods for details).
Results of GOWINDA for the full set of SDV yielded no significant GO terms at FDR <5%, suggesting that a substantial fraction of SDV are variants that have diverged in frequencies as a result of genetic hitchhiking over short distances rather than as direct targets of selection. The more selective set of 1,634 SDV in contrast was enriched for genes in several GO categories associated with egg development (blue dots in fig. 6A), cell size regulation (orange dots in fig. 6A), and growth (red dots in fig. 6A, for complete GOWINDA analysis see supplementary table S4, Supplementary Material online). In addition to the 2,836 GO terms, we also separately tested whether all SDV or the selective set of SDV are enriched for maternally transcribed genes (276 genes compiled from Brody [2015]; supplementary table S5, Supplementary Material online) and found no enrichment (P < 0.05).
Fig. 6.

Enrichment of SDV for biologically relevant GO categories. (A) Selected set of 1,634 SDV is enriched for genes in several GO categories. On the x axis are number of genes with at least one SDV in each category and y axis indicates significance of enrichment after correcting for multiple testing. Dotted purple line indicates FDR <5%. Each dot is a GO category and size of the dots indicates the total number of genes in each GO category. Dots in gray: Broad GO categories containing several specialized GO terms; dots in blue, red, and orange: GO categories associated with reproduction (and egg development), growth, and cell size, respectively. (B) Overlap of genes harboring the enriched SDV in GO categories associated with cell size, egg development, and growth. Genes in differentiated blocks are largely distinct from those encompassed by the differentiated blocks. All of the genes in the differentiated blocks are expressed in the Drosophila ovaries (fig. 5B).
To further prioritize these genes, we focused on specialized GO terms (FDR < 5%) that are relevant for egg development. These include regulation of cell size, female gamete generation, oogenesis, ovarian follicle cell development, germ cell migration, cell growth, and growth because these biological processes are likely to influence egg size. Combining genes in these GO terms yielded 95 candidates that are largely distinct from candidate genes within the differentiated blocks (fig. 6B), indicating that SDV that do not fall in long-range linkage blocks can also be targets of egg-size selection. Combining all analyses, 193 genes can be considered candidates for influencing egg size (supplementary table S6, Supplementary Material online).
Genetic Crosses Reveal Complex Genetic Architecture of Egg Size
Many of the 193 candidate genes identified above (supplementary table S6, Supplementary Material online) have mutant alleles allowing genetic analysis of their effects on egg size. We chose to investigate three candidate genes, eya, procollagen lysyl hydroxylase (Plod), and longitudinals lacking (lola). All three regulate development in multiple tissues, including the egg chamber. eya is a regulator of follicle cell polarity in egg chamber formation during oogenesis (Bai and Montell 2002; Klusza and Deng 2011). eya[3]/eya[E(P)10] also has egg chambers which contain abnormal germline cells (Bai and Montell 2002). Procollagen lysyl hydroxylase (Plod) and longitudinals lacking (lola) are both highly expressed in the ovaries (Graveley et al. 2011). RNAi-mediated knockdown of Plod is reported to produce “round” eggs (Lerner et al. 2013). Germline clones of lola have numerous defects in egg chambers, including failure of nurse cells to transfer their cytoplasmic content to the egg and nurse cell nuclei failing to undergo programmed cell death (Bass et al. 2007).
We carried out crosses with a strong (i.e., presumptive functional null) allele of each candidate gene. All three mutants are recessive, fully viable, and fertile as heterozygotes. Given the large numbers of genes involved in oogenesis, the contribution of a single gene is likely to be small. Nonetheless, a subtle effect in egg size can be expected in heterozygotes, if these genes contribute to egg-size evolution. To test this hypothesis, we crossed males bearing mutant alleles with females from three genetic backgrounds: The laboratory strain: w1118 (because isofemale lines from CP are not available), a small egg-size inbred line, and a large egg-size inbred line, each derived through repeated brother–sister mating of an SEP or LEP selected fly, respectively. The resulting F1 females carry one copy of the genome from their respective genetic backgrounds and the other from the mutant strain with either a balancer chromosome or a chromosome carrying the mutant allele. We then compared egg sizes from F1 flies carrying the mutant chromosome with those from sisters carrying the balancer chromosomes as well as the parental lines. These data provide a measure of control for the effect of genetic background.
Comparison of eggs from females with the balancers to those from parental lines showed that introduction of balancers increased egg length in w1118 and the small genetic background (P < 0.001, ANOVA, supplementary fig. S6, Supplementary Material online). This increase in egg length varied for the three different balancers, indicating that genetic variation in different balancers contributes differently to egg size, as expected for a polygenic trait. Interestingly, the effect of the balancer was far stronger than any effect from the mutant chromosome in these genetic backgrounds (supplementary fig. S6, Supplementary Material online), raising the possibility that genetic background alone might account for the effect in these genetic backgrounds. In the large genetic background, however, none of the balancers had any effect on egg length (gray bars in fig. 7), whereas chromosomes containing mutant Plod and lola alleles decreased egg length (P < 0.001, ANOVA, light yellow bars in fig. 7), consistent with the hypothesis that Plod and lola contribute to egg elongation in flies. In summary, the genetic crossing results demonstrate that the candidate genes have modest effects on egg size. Although their effects are indistinguishable from that of the genetic background in w1118 or small lines, they are appreciable in the large lines that we examined.
Fig. 7.
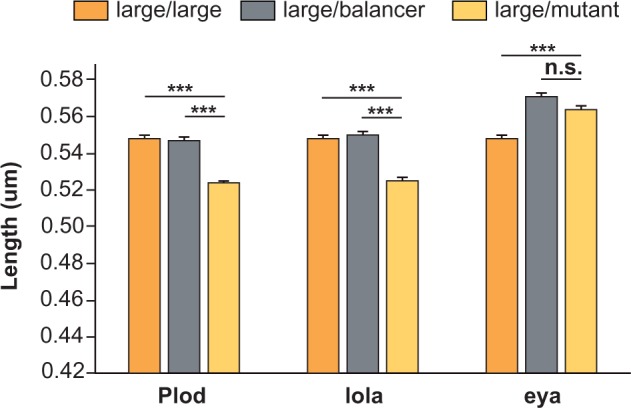
Genetic crosses show candidate genes affect egg size. Comparison of egg length from parental isogenic lines derived from large population cages (dark yellow) to those from F1 females carrying one copy of balancer (gray) or one copy of mutant chromosome of the indicated genes (light yellow). No detectable difference in egg length was observed between isogenic lines (dark yellow) and heterozygote females carrying balancer (gray). However, females carrying chromosomes with the mutant alleles of Plod and lola (light yellow) produced significantly shorter eggs. Y axis indicates mean length ± SEM. ***P < 0.0001.
Discussion
Insect ovaries are compound organs composed of multiple autonomously functioning subunits called ovarioles (Hodin 2009) and ovariole number has been the subject of extensive ecological and quantitative genetic studies. The number of ovarioles differs both within and between species (Büning 1998; Telonis-Scott et al. 2005; Markow and O’Grady 2007). Positive associations of ovariole numbers with female fecundity have been reported (Cohet and David 1978; Boulétreau-Merle et al. 1982), suggesting that ovariole number is related to reproductive success. Ovariole number varies along both latitudinal (Delpuech et al. 1995; Gibert et al. 2004; Schmidt, Matzkin, et al. 2005; Schmidt, Paaby, et al. 2005) and altitudinal clines (Klepsatel et al. 2014; but see Collinge et al. 2006), suggesting that it is an adaptive trait. Ovariole number increases with latitude (Delpuech et al. 1995; Gibert et al. 2004; Schmidt, Matzkin, et al. 2005; Schmidt, Paaby, et al. 2005), as does egg volume (Azevedo et al. 1996). Positive genetic correlations have also been reported between ovariole number and thorax length (Bergland et al. 2008). Thorax length, fecundity, and ovariole number are also positively correlated among various Dosophilid species (Kambysellis and Heed 1971; R’Kha et al. 1997). However, both Wayne et al. (1997) and Bergland et al. (2012) failed to detect a genetic correlation between ovariole number and various aspects of reproductive fitness. Clearly, the relationship among these life-history traits is a complicated one, also involving possible trade-offs with longevity (Wayne et al. 2001). Here we report a negative relationship between egg size and ovariole number in multiple isofemale lines derived from the populations that were directly, and rapidly, selected for egg volume (Miles et al. 2011). It may be that direct truncation selection on egg size over the short interval involved (18 months) simply did not allow enough time for other morphological traits to “catch up” with increasing egg size. The selected populations did not differ in body size at cessation of selection (Miles et al. 2011). Thus, body size itself may have presented a physical constraint resulting in reduced ovariole number in large lines. Lower ovariole numbers are correlated with larger egg size in some Drosophilds (and other flies), especially those that have become ecologically specialized (Kambysellis et al. 1995; R’Kha et al. 1997; Markow et al. 2009). We are continuing to examine sequence data in these inbred lines, as well as the process of ovary morphogenesis itself, and comparing these to cold-adapted laboratory evolving flies that came from the same base population.
Using experimental evolution followed by whole-genome sequencing, we investigated the genetic basis of egg-size evolution in a well-replicated experiment for both larger and smaller egg volumes. A notable feature of the experimental design was the establishment of a base population from a large and fresh collection of flies, which was then allowed to adapt to the laboratory environment as a large cage population for ten generations prior to establishment of nine treatment cages, three each for larger eggs, smaller eggs and controls. Importantly, a sample was also collected from the base population at the time the experimental cages were formed to compare with both the control and selected populations. The experimental populations were maintained at approximately 2,000 flies, although truncation selection reduced this number in alternating generations to 110 (selected from 500 measured eggs). Seventeen rounds of selection were imposed every other generation, allowing population size to rebound and recombination to further mix the selected genomes.
We gained considerable statistical power by comparing the two divergently selected treatments and found a large number of variants whose allele frequencies changed consistently across the thrice-replicated populations. Applying the stringent requirement that an SDV must consistently diverge from the starting population in all three replicates eliminated a number of targets of selection that diverged on average but almost certainly also reduced the number of false positive SDV. Comparison of LEP and SEP should also eliminate variants under background natural selection as all treatments were adapting in identical cage conditions. As predicted, SDV identified from the comparison of LEP and SEP treatments showed greater allele frequency divergence than controls, indicating egg-size-specific selection was stronger than background natural selection and affected several thousand variants. Within this embarrassment of riches, the more challenging problem is to estimate how many of these variants are targets of trait-specific selection and how many result from hitchhiking and/or long-range linkage in the laboratory populations.
Large numbers of significant variants due to pervasive hitchhiking spanning thousands of kilobases have been reported in experimental evolution studies (Zhou et al. 2011; Orozco-terWengel et al. 2012; Remolina et al. 2012; Turner and Miller 2012; Cassidy et al. 2013; Tobler et al. 2014). Our study is also consistent with hitchhiking effects acting over both short and long physical scales. Differentiated blocks contain clusters of several significant SNP (median = 7, min = 4, and max = 20) spanning approximately 18 kb on average, all of which are unlikely to be targets of selection for egg size. Hence, several variants included in each block must be hitchhiking with one or few variants that are targeted by selection. Similarly, on a larger physical scale, the complete set of approximately 4,100 SDV, which are separated by an average of approximately 28 kb, displayed a complete absence of GO enrichment. This lack of distinguishing characteristics may be evidence of the prevalence of genetic hitchhiking or long range associations acting over long physical distances (Nuzhdin and Turner 2013).
Nevertheless, hitchhiking or long-range association cannot be the sole reason for the large number of SDV in our study, as evidenced by enrichment of genes related to oogenesis found in the differentiated blocks. In fact, the majority of the SDV resides outside of the differentiated blocks and the selective set of approximately 1,600 most significant SDV (40% of all SDV) are enriched for GO terms that are functionally relevant for reproduction and egg development. Moreover, our genetic crosses demonstrate that chromosomes bearing mutant alleles of two candidate genes, Plod and lola, influence egg size, indicating that the candidate genes we have identified are functionally relevant for egg-size evolution. One may argue that other unknown variants on the chromosome carrying these mutant alleles, but not these mutant alleles themselves, may have contributed to the reduction in egg length. However, RNAi-mediated knockdown of Plod also results in reduced egg length in D. melanogaster (Lerner et al. 2013).
Furthermore, several genes in our list of candidates have been shown to be associated with ovariole number variation. The role of ecdysone signaling in establishment and maintenance of the stem cell niche in the Drosophila ovary has received considerable attention recently (Belles and Piulachs 2014). Two members of this pathway, ultraspiracle (usp) and Eip75B, are present in our candidate list. usp regulates the timing of ovarian morphogenesis and influences the number of functional niches (Hodin and Riddiford 1998; König et al. 2011). Overexpression of Eip75B, a nuclear hormone receptor and target of the ecdysone cascade, results in changes in timing of primordial germ cell differentiation into germline stem cells (Gancz et al. 2011). Another candidate gene Axin (Axn) is associated with ovariole numbers in D. melanogaster (Wayne and McIntyre 2002). Axn is a negative regulator of wg/Wnt signaling (Willert et al. 1999). Wingless is expressed in both terminal filament and cap cells and is known to regulate the maintenance of somatic stem cells in the ovary (Song and Xie 2003). Hedgehog, also a candidate gene in our populations, is required for proliferation and specification of ovarian somatic cells (Forbes et al. 1996), and controls interactions between the somatic and germline cells in the developing ovary (Besse et al. 2005). Similarly, another promising candidate in our list is Rassf, an antagonist of the Hippo pathway, which is involved in regulation of proliferation and cell death, thereby controlling cell number in many tissues (Polesello et al. 2006) and limiting organ size (Zhao et al. 2010). Hippo activity has been shown to influence the total number of terminal filament cells (Sarikaya et al. 2012) and regulate the growth of stem cell niche precursors in the developing ovary (Sarikaya and Extavour 2015). The presence among our candidates of key members of pathways known to influence ovariole number strengthens the idea that genes underlying the trait of egg size are also present. Unfortunately, many fewer studies attempting to identify the genes and pathways associated with variability in egg size have been completed. It is also noteworthy that many SDV are located in genes that have no annotated functions in flies (CG genes) or no known roles in egg development. For example, one of the differentiated blocks contains a micro-RNA, miR-280, which has no known functions in egg development, but has been implicated in the reduction of egg size in D. melanogaster (Nakahara et al. 2005). Future functional analysis of these candidate genes may reveal their effects on egg size.
Our results contrast with previous studies that have reported striking changes in allele frequencies between divergently selected populations coupled with marked reduction in heterozygosity after long-term experimental evolution in laboratory lines (>100 generations) (Burke et al. 2010; Turner et al. 2011; Zhou et al. 2011). Large reduction in heterozygosity was also observed in a short-term study (15 generations) when a few isogenized laboratory fly lines were used to find the initial base population (Cassidy et al. 2013). The lack of congruence between results from our study and those from previous studies may be attributed to three major differences in our experiment. First, artificial selection was applied for a relatively small number of generations. With a modest selection intensity (∼20% threshold), not enough time may have passed for beneficial alleles to approach fixation, thus limiting greater loss of heterozygosity. Second, selection was imposed in alternating generations allowing population sizes to rebound and recombination to reshuffle selected variants. Finally, our populations were derived from relatively large collection of wild-caught isofemales that were allowed to adapt to the lab prior to establishing the treatment populations. Unlike isogenic lines or strains that have been maintained in the laboratory for many generations, natural populations of flies have abundant genetic diversity and polymorphisms are likely to be distributed in multiple haplotypes by recombination. When variants such as these come under selection, appreciable allele frequency differences can occur without commensurate loss of genetic diversity at linked sites, as the theory of “soft sweeps” and “polygenic adaptation” shows (Hermisson and Pennings 2005; Burke 2012).
Such signatures in our selected populations—marked allele frequency difference accompanied by small reduction in heterozygosity—suggest that SDV are enriched for common polymorphisms. Most of the SDV in our evolved populations are also present in the DGRP lines, which provides additional evidence that adaptation likely occurred from common standing polymorphisms. However, it is noteworthy that the experimental design and method to identify SDV has greater power to detect common variants than rare variants for a number of reasons. First, even if they are beneficial, rare variants can be easily lost during laboratory selection due to genetic drift in the replicate cages. Second, artificial selection of a rare variant would only be effective in one phenotypic direction. Our study compares differentiated SNPs between large and small selected populations, and thus will be inherently biased toward identifying common polymorphisms. For these reasons, this study may not address the broader, historically important, question about the relative contributions of common versus rare mutations to complex fitness-related traits (Charlesworth 2015).
Interestingly even after including all the significant variants encompassed by the 193 candidate genes, a large proportion of the SDV remain unaccounted for. Some of these SDV might be false positives that have persisted for reasons other than hitchhiking but many are likely to be in genes that have yet uncharacterized functions in egg development that were subject to selection. However, the large number of significant variants may also be a result of compensatory adaptation in other developmental and physiological processes that must scale with changing egg size (Harshman and Hoffmann 2000). One of the genes with strong evidence of selection, for example, is Kruppel (Kr). Although Kr itself is not known to be associated with egg size, it plays a crucial role in embryonic patterning, which must scale with egg size (Lott et al. 2007, 2011; Miles et al. 2011). Furthermore, it is plausible that a proportion of the SDV could be under selection to ameliorate maladaptive correlated effects resulting from pleiotropy. Although it is difficult to pinpoint the numbers of variants that could be under selection in response to egg-size evolution, our results indicate that at least a few hundred variants in over 200 genes may have contributed to egg-size evolution. That a large number of variants are relevant to selection in our experimental populations is not unexpected: Such signatures of polygenic adaptation are well known in flies (Turner et al. 2011) as well as human populations (Turchin et al. 2012).
In addition to revealing candidate genes that affect egg size, our results also highlight the limitations of experimental evolution approaches that have emerged as a common theme in studies employing this technique. First, despite large population size (N = 2,000) truncation selection induced mild population reduction in alternating generations resulting in 110 flies (220 chromosomes). Although allowing population size to rebound allowed recombination to further mix the selected genomes, small population size exacerbates LD over short and long distances. To ameliorate the effects of hitchhiking, increasing population sizes and/or the number of replicate populations may aid future studies to identify the true targets of selection. Second, the complexity of experimental evolution studies allows sources of error to be introduced at several stages, all of which are difficult to take into account. For instance, we performed high depth sequencing and used read depth rather than allele frequencies in the GLMM to avoid read count induced variation in estimating allele frequencies. However, additional unaccounted sources of variability that persisted may have inflated the number of SDV. Third, whole-genome forward simulations, especially that account for all the events during an experimental evolution, are still a significant undertaking and theoretical and computational advancements in this area are highly desirable. Hence, our data may be useful in exploring designs to investigate genetic basis of complex traits using this powerful approach in future studies.
In summary, our results underscore the complexity of polygenic adaptation and molecular evolution of complex traits. First, our results demonstrate that fly populations have a large reservoir of common variation that can fuel their rapid response to a selective challenge. The signature of selection from these variants is different from that of new beneficial mutations—the reduction in heterozygosity may be relatively small in both scope and magnitude—and will not be detectable by methods identifying hard-selective sweeps. Methods to detect “soft” sweeps may be more relevant to detecting quantitative trait locus underlying polygenic traits such as egg size. Second, experimental evolution followed by pooled whole-genome sequencing can be an effective method for dissecting the genetic basis of complex traits such as egg-size evolution. With a well-structured experimental design, large numbers of variants responding to artificial selection can be identified with confidence, and integrating additional publicly available information can result in a refined list of candidate genes that can be tested for function. Our results indicate that a few hundred naturally existing common variants in at least 100 genes are likely to regulate egg-size differences in D. melanogaster.
Materials and Methods
Drosophila melanogaster Populations and Phenotype
The selection scheme and egg volume measurements have previously been described in detail (Miles et al. 2011). Briefly, a base population, created from a sample of 120 wild-caught nonvirgin female flies from IL, USA, was established. This base population was then maintained at large population size allowing random mating for ten generations to allow adaptation to laboratory conditions. This population served as the starting base population. Nine replicate populations were established by sampling individuals from the starting population. Three of these populations were used to select for large egg volumes and three were used to select for small egg volumes. The remaining three were maintained as CP in identical laboratory conditions but no artificial selection on egg volume was applied. In total, 19.1% truncation selection was applied in alternating generations by measuring 500 eggs and selecting 110 eggs to find the subsequent generation in each replicate population. We calculated egg volume using the following formula:
Selection was conducted for 16 generations. Each generation was 16 days and generations were nonoverlapping. Adult females from the 17th generation were used for whole-genome sequencing.
Ovariole Numbers, Egg Size, and Egg Chamber Length Measurements
After the 17th generation, selection was stopped and inbred fly lines were derived from the LEP and SEP population cages by isolating individual mothers and allowing brother–sister mating of offspring for 40 generations. Ten of the inbred lines, five for large and small eggs, were used to measure ovariole numbers. These flies were raised in low density cultures at 25 °C, over standard fly media. Nonvirgin females were kept over yeast for 3–4 days posteclosion before ovaries were dissected with forceps under a stereomicroscope. Ovaries (n = 10 females per line) were stained in a saturated solution of potassium dichromate for approximately 10 min and tungsten needles were used to tease apart the ovarioles. Ovariole number was manually counted using a stereomicroscope. Egg volumes were measured as described above using 50 eggs from each of the ten inbred lines (n = 500 eggs in total). ANOVA with lines nested within the treatments was used to assess difference between mean egg volumes between the LEP- and SEP-derived lines. To measure egg chamber size, ovarioles obtained from the dissected ovaries from one of the small and large lines (lines 2154 and 9314, respectively) were disrupted by pipetting through 10 µl tips. Ovaries were stained using Phalloidin (Phalloidin:TRITC, ECM Biociences, 1:200), which binds to actin, and DAPI (Invitrogen, 1:200) was used as the nuclear stain. Confocal microscopy (Olympus BX51) was used to image egg chambers in various stages of oogenesis and their dimensions were measured using ImageJ software.
DNA Extraction, Sequencing, and Alignment
Pooled DNA was extracted from 100 females per population using phenol–chloroform to create a total of ten pooled-DNA samples: 1 starting population, 3 LEP, 3 SEP, and 3 CP. Each pooled-DNA sample was fragmented to 500 bp using a Covaris sonicator. DNA fragments were size selected and Illumina libraries were generated from each of the ten populations at the High-throughput Genomic Analysis Core Facility of Institute of Genomic and Systems Biology at The University of Chicago. The 100-bp single and paired-end reads for each library were generated using an Illumina HiSeq2000. Data analysis was performed on the Bionimbus cloud of the Open Science Data Cloud (http://bionimbus.opensciencedatacloud.org, last accessed June 15, 2015). FastX toolkit was implemented to check the quality of each read. Reads were mapped to the D. melanogaster reference genome (dm3, BDGP Release 5) using BWA (Li and Durbin 2009). GATK (McKenna et al. 2010) was used for indel realignment and base quality recalibration. Alignments with mapping qualities less than 15 were discarded and Samtools –rmdup (Li et al. 2009) was used to remove potential polymerase chain reaction duplicates. Depth of coverage was calculated using BedTools (Quinlan and Hall 2010). A total of 1.34 billion reads passed QC and mapped to the Drosophila melanogaster reference genome. For each terminal population, total numbers of QC passed reads ranged between 78.2 and 155.2 million.
Genotyping and Identification of High-Confidence Variants
Bam files from all ten populations were merged and sequence reads with mapping quality less than 40 were discarded. The median genome coverage in this merged bam file (VarRef) increased to 903× allowing identification of variant positions with high confidence. Samtools –pileup (Li et al. 2009) was used to call SNP and small indels and additional information such as base quality, mapping quality, and SNP quality at each chromosomal position were extracted.
Over 40 M variant positions were initially identified as polymorphic in the VarRef, many of which appeared to be trialleleic positions reflecting potential sequencing errors. To determine the threshold for sequencing errors empirically, we considered only the triallelic positions. Assuming 90% of the third alleles are due to sequencing error, we set the sequencing error threshold at the 90th quantile in the read count distribution of the third allele position in the VarRef (read count = 13). Variant positions where the two nonreference allele counts were greater than 13 were considered true triallelic variants and removed from further analysis. Only those biallelic positions with nonreference allele count greater than 13 were further considered. Over 8.1 M biallelic positions passed these criteria. We further removed highly (>90th quantile biallelic sites, >995×) and lowly (<10× in each of the ten populations) covered biallelic positions and calculated 95% binomial confidence interval for the minor allele at each variant position in each population. Positions with lower bound 95%CI for the minor allele above zero in any one of the ten populations were considered high-confidence polymorphisms. A total of 2,149,584 high-confidence polymorphisms were identified of which 2,103,160 were in the five major chromosome arms and were used in subsequent analysis.
Annotation of Polymorphic Loci
Gene- and region-based annotation of high-confidence polymorphisms was performed using ANNOVAR (Wang et al. 2010). Polymorphisms in intronic, exonic, or untranslated regions were annotated to the single gene containing the variant. For intergenic variants, genes immediately upstream or downstream were considered.
Allele Frequency Divergence and diffStat
Allele frequency was estimated as a ratio of variant counts to coverage. To calculate allele frequency changes due to laboratory or egg-size selection, we first identified variants that have shifted in the same directions relative to the starting population in all three replicates in each treatment and then calculated the average allele frequency change relative to the starting population across each treatment, that is, (abs(average(p1-starting population, p2-starting population, p3-starting population)), where p = LEP, SEP, and CP). To calculate allele frequency difference between the LEP and SEP, we first identified variants that have shifted in the same direction relative to the starting population in all three replicate populations of the LEP and SEP. We then calculated difference in average allele frequencies between the two treatments.
To calculate diffStat statistic, we first calculated change in allele frequencies between all nine possible combinations of large and small treatments. We then identified variants that have changed in consistent directions in all nine comparisons. Then, we calculated the minimum absolute change in allele frequency difference between the two treatments as the diffStat score, that is, abs(min(LEP1-SEP1, LEP1-SEP2, LEP1-SEP3, LEP2-SEP1, LEP2-SEP2, LEP2-SEP3, LEP3-SEP1, LEP3-SEP2, LEP3-SEP3)).
Generalized Linear-Mixed Model
Although Fishers Exact Test has been used previously (Burke et al. 2010; Remolina et al. 2012) to quantify allele frequency changes between two populations, this method for quantifying selection from pooled-sequencing data has a high false positive rate (Turner et al. 2011). False positives could result because the variance in allele frequencies between populations is not accounted for and errors in allele frequency estimates due to varying sequencing depth in each sample are ignored.
In pooled sequencing, the basic statistics are the read counts at each variant position, treatment-groups, and the number of replicate populations within each treatment. As these types of data are not expected to be normally distributed, traditional normality based tests are also not useful but a GLMM is appropriate (Bolker et al. 2009). A GLMM can be used to identify highly correlated and systematic allele frequency changes between two treatments while simultaneously taking fluctuations in allele frequencies between replicate populations within each treatment into account. Systematic and reproducible changes in allele frequencies in multiple replicate populations can be attributable to selection, whereas random changes can be due to drift. As the input for the GLMM is read counts and not allele frequencies, it also takes into account differences in sequencing depth at each variant position in each replicate population. A P value for the fixed-effects can be calculated for each variant in the data set and the extreme P values represent highly correlated allele frequency differences between the two treatments.
Hence, we implemented a GLMM to quantify the allele frequency divergence due to selection between the large and small egg-size populations using the R package “lme4.” For each variant, read count (reference = 0, variant = 1) was the outcome variable. Read count at biallelic positions was assumed to be binomially distributed and treatment groups (large-eggs and small-eggs) were specified to have fixed effects whereas each replicate population was specified to have random effects. P values for each variant position were obtained using the default parameters. To assess genomewide multiple-testing threshold, FDR was calculated using q values using “pi0.method=smoother” (Storey and Tibshirani 2003). For a very stringent FDR of 0.0005% (FDR ≤ 5e−6), the P-value cutoff was 1.29e−8. Hence, any variant with P value below 1.29e−8 was considered statistically significant. These variants are among the most differentiated (extreme outliers) in the data set and are likely to be enriched for targets of selection.
Hypergeometric Test and Scans for Reduced Heterozygosity
To test whether the SDV are overrepresented in certain genomic windows, we performed a hypergeometric test for 100-, 50-, and 10-kb sliding windows with 25-, 10-, and 2-kb step size in each major chromosome arm as follows: The total number of variants (n) and SDV (s) in each window were assessed. Given the total numbers of variants (N) and total number of SDV (S) in each chromosome arm, we computed the probability (P) of observing s SDV when n variants are sampled. For multiple-testing adjustment, q values were estimated as above and FDR ≤ 0.005 was used as the genomewide multiple testing threshold.
Maximum-likelihood estimate of gene diversity (heterozygosity) in each replicate population was calculated using the following equation as described in Weir (1996):
Here is the observed allele frequency at each base and m is the nonoverlapping window-size where l polymorphic loci were identified. Genomewide pooled gene diversity (HP) was calculated for all ten populations in 100-, 50-, and 10-kb sliding windows with 25-, 10-, and 2-kb step sizes, respectively. For each window size, fold reduction in heterozygosity was calculated by averaging the HP scores across the three replicates in each treatment and dividing that by the HP in the starting population. The fold change in the terminal populations was also similar regardless of the window sizes.
Permutation Test for diffStat and Heterozygosity
To test whether the differentiated variants have higher diffStat scores compared with the nondifferentiated variants matched by chromosomal location, we compared the differentiated variants to 10,000 sets of equal numbers of randomly selected variants matched by the chromosome arm. We then asked whether it is possible to observe median diffStat scores as high as or higher than that observed with differentiated variants in the permuted set. We never observed higher median diffStat scores in the permuted sets (P = 0/10,000 < 0.0001). We also repeated the same procedure with variants matched by chromosome arm and ancestral allele frequency but did not observe higher median diffStat scores in the permuted sets (P = 0/10,000 < 0.0001).
We contrasted the heterozygosity in 415 significant windows in LEP with that in 10,000 sets of equal number of randomly sampled windows matched by chromosomal arm in CP. For each set, we asked whether as much as (15.29%) or stronger reduction in median heterozygosity relative to the starting population can be observed in the permuted set of windows and found that only 66 of 10,000 permuted sets showed ≥15.29% reduction in median heterozygosity (P = 66/10,000 = 6.6e−03).
Enrichment Analysis
We used GOWINDA (Kofler and Schlotterer 2012) to perform GO enrichment analysis. GOWINDA calculates P values for enrichment of each GO term by simulating large sets of randomly sampled SNP and such permutation of the genome eliminates biases associated with gene length and reduces false positive GO terms (Kofler and Schlotterer 2012). All the GO terms in D. melanogaster were downloaded using FuncAssociate2.0 (Kofler and Schlotterer 2012). Gene sets with less than ten genes were excluded which resulted in 3,317 GO terms of which 2,836 terms had at least one gene with an SDV. We conducted 100,000 simulations for each of the 2,834 GO terms using gene annotations based on D. melanogaster reference genome version 3.0 and full coding region ±500-bp flanking regions were considered (-gene option in GOWINDA).
To generate a selected set of SDV, we first generated an empirical null distribution by identifying the variant with smallest P value from the GLMM for all genes in the entire data set (N ∼ 16,340). We then identified the variants in the top 10% tail of this distribution as the selected set of variants. As the 10% P-value threshold is at least an order of magnitude higher than the genomewide significance threshold (P = 1.29e−8 at FDR < 5e−6), the SDV in this analysis reflect the most divergent variants in the data set.
Drosophila Genetics
Three mutant lines, Plod (w; Plod [N26-5]FRT80/TM3 twi GFP), eya (eya [clift1]FRT40/CyO Act-GFP), and lola (cn[1] P{ry[+t7.2]=PZ}lola[00642]/CyO; ry[506], Bloomington Drosophila stock center number 10946) were gifts from Sally Horne-Badovinac. Crosses were raised at 25 °C. Five males from each mutant line were crossed with five females from each of the large, small, and w1118 lines. As the adult flies began eclosing, virgin F1 females carrying the mutant genes were identified and isolated in 6–8 h intervals. These females were backcrossed with males of their respective genotypic backgrounds (large, small or w1118) in three replicates each with five females and five males. In total, 20–25 eggs from each replicate for each mutant cross for each gene were measured as described above and previously (Miles et al. 2011). Eggs from mothers carrying a mutant copy of the gene were compared with sisters carrying balancer chromosomes as well as with parental lines in large, small, and w1118 backgrounds and statistical significance was measured using ANOVA. P values less than 0.05 were considered significant.
Supplementary Material
Supplementary figures S1–S6 and tables S1–S6 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Sally Horne-Badovinac for reagents and mutant fly lines and Michael Z. Ludwig, Matthew Stephens, Molly Przeworski, Dick Hudson, and John Reinitz for helpful suggestions. They are grateful to Jackie Gavin-Smith and Robert K. Arthur for constructive comments on the manuscript. This work was supported by the following grants from National Institute of Health (Training in Oxygen in Health and Disease Grant T32HL094282 to A.R.J. and P50 GM081892 to K.P.W.) and Institutional Development Award from the National Institute of General Medical Sciences of National Institute of Health (P20GM103443 to C.M.M.). The sequence files (.bam) for the ten populations sequenced in this study are available from SRA (BioProject PRJNA281953 and BioSamples SAMN03497949–SAMN03497958).
References
- Azevedo RB, French V, Partridge L. 1996. Thermal evolution of egg size in Drosophila melanogaster. Evolution 50:2338–2345. [DOI] [PubMed] [Google Scholar]
- Azevedo RB, French V, Partridge L. 1997. Life-history consequences of egg size in Drosophila melanogaster. Am Nat. 150:250–282. [DOI] [PubMed] [Google Scholar]
- Bai J, Montell D. 2002. Eyes absent, a key repressor of polar cell fate during Drosophila oogenesis. Development 129:5377–5388. [DOI] [PubMed] [Google Scholar]
- Bass BP, Cullen K, McCall K. 2007. The axon guidance gene lola is required for programmed cell death in the Drosophila ovary. Dev Biol. 304:771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belles X, Piulachs MD. 2014. Ecdysone signalling and ovarian development in insects: from stem cells to ovarian follicle formation. Biochim Biophys Acta 1849:181–186. [DOI] [PubMed] [Google Scholar]
- Bergland AO, Chae HS, Kim YJ, Tatar M. 2012. Fine-scale mapping of natural variation in fly fecundity identifies neuronal domain of expression and function of an aquaporin. PLoS Genet. 8:e1002631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergland AO, Genissel A, Nuzhdin SV, Tatar M. 2008. Quantitative trait loci affecting phenotypic plasticity and the allometric relationship of ovariole number and thorax length in Drosophila melanogaster. Genetics 180:567–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse F, Busson D, Pret A-M. 2005. Hedgehog signaling controls soma-germen interactions during Drosophila ovarian morphogenesis. Dev Dyn. 234:422–431. [DOI] [PubMed] [Google Scholar]
- Bolker BM, Brooks ME, Clark CJ, Geange SW, Poulsen JR, Stevens MH, White JS. 2009. Generalized linear mixed models: a practical guide for ecology and evolution. Trends Ecol Evol. 24:127–135. [DOI] [PubMed] [Google Scholar]
- Boulétreau-Merle J, Allemand R, Cohet Y, David JR. 1982. Reproductive strategy in Drosophila melanogaster: significance of a genetic divergence between temperate and tropical populations. Oecologia 53:323–329. [DOI] [PubMed] [Google Scholar]
- Brody TB. 2015. http://www.sdbonline.org/sites/fly/aimain/1aahome.htm The interactive fly (68th ed., 2013). Bethesda (MD): Society for Developmental Biology. Available from: [Google Scholar]
- Büning J. 1998. The ovariole: structure, type, and phylogeny. In: Harrison FW, editor. Microscopic anatomy of invertebrates. Wiley-Liss, Inc. p. 897–932. [Google Scholar]
- Burke MK. 2012. How does adaptation sweep through the genome? Insights from long-term selection experiments. Proc Biol Sci. 279:5029–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke MK, Dunham JP, Shahrestani P, Thornton KR, Rose MR, Long AD. 2010. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature 467:587–590. [DOI] [PubMed] [Google Scholar]
- Cassidy JJ, Jha AR, Posadas DM, Giri R, Venken KJ, Ji J, Jiang H, Bellen HJ, White KP, Carthew RW. 2013. miR-9a minimizes the phenotypic impact of genomic diversity by buffering a transcription factor. Cell 155:1556–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. 2015. Causes of natural variation in fitness: evidence from studies of Drosophila populations. Proc Natl Acad Sci U S A. 112:1662–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohet Y, David JR. 1978. Control of the adult reproductive potential by preimaginal thermal conditions. Oecologia 36:295–306. [DOI] [PubMed] [Google Scholar]
- Collinge JE, Hoffmann AA, McKechnie SW. 2006. Altitudinal patterns for latitudinally varying traits and polymorphic markers in Drosophila melanogaster from eastern Australia. J Evol Biol. 19:473–482. [DOI] [PubMed] [Google Scholar]
- Cutter AD. 2008. Divergence times in Caenorhabditis and Drosophila inferred from direct estimates of the neutral mutation rate. Mol Biol Evol. 25:778–786. [DOI] [PubMed] [Google Scholar]
- Delpuech J-M, Moreteau B, Chiche J, Pla E, Vouidibio J, David JR. 1995. Phenotypic plasticity and reaction norms in temperate and tropical populations of Drosophila melanogaster: ovarian size and developmental temperature. Evolution 49:670–675. [DOI] [PubMed] [Google Scholar]
- Forbes AJ, Lin H, Ingham PW, Spradling AC. 1996. Hedgehog is required for the proliferation and specification of ovarian somatic cells prior to egg chamber formation in Drosophila. Development 122:1125–1135. [DOI] [PubMed] [Google Scholar]
- Gancz D, Lengil T, Gilboa L. 2011. Coordinated regulation of niche and stem cell precursors by hormonal signaling. PLoS Biol. 9:e1001202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garud NR, Messer PW, Buzbas EO, Petrov DA. Recent selective sweeps in North American Drosophila melanogaster show signatures of soft sweeps. PLoS Genet. 11: e1005004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibert P, Capy P, Imasheva A, Moreteau B, Morin JP, Petavy G, David JR. 2004. Comparative analysis of morphological traits among Drosophila melangaster and D. simulans: genetic variability, clines, and phenotypic plasticity. Genetica 120:165–179. [DOI] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471:473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock AM, Brachi B, Faure N, Horton MW, Jarymowycz LB, Sperone FG, Toomajian C, Roux F, Bergelson J. 2011. Adaptation to climate across the Arabidopsis thaliana genome. Science 334:83–86. [DOI] [PubMed] [Google Scholar]
- Harshman LG, Hoffmann AA. 2000. Laboratory selection experiments using Drosophila: what do they really tell us? Trends Ecol Evol. 15:32–36. [DOI] [PubMed] [Google Scholar]
- Hermisson J, Pennings PS. 2005. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169:2335–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez RD, Kelley JL, Elyashiv E, Melton SC, Auton A, McVean G, Genomes P, Sella G, Przeworski M. 2011. Classic selective sweeps were rare in recent human evolution. Science 331:920–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodin J. 2009. She shapes events as they come: plasticity in insect reproduction. In: Whitman D, Ananthakrishnan TN, editors. Phenotypic plasticity of insects: mechanisms and consequences. Enfield (NJ): Science Publishers, Inc. p. 423–521. [Google Scholar]
- Hodin J, Riddiford LM. 1998. The ecdysone receptor and ultraspiracle regulate the timing and progression of ovarian morphogenesis during Drosophila metamorphosis. Dev Genes Evol. 208:304–317. [DOI] [PubMed] [Google Scholar]
- James AC, Azevedo RB, Partridge L. 1997. Genetic and environmental responses to temperature of Drosophila melanogaster from a latitudinal cline. Genetics 146:881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambysellis MP, Heed WB. 1971. Studies of oogenesis in natural populations of Drosophilidae. I. Relation of ovarian development and ecological habitats of the Hawaiian species. Am Nat. 105:31–49. [Google Scholar]
- Kambysellis MP, Ho K-F, Craddock EM, Piano F, Parisi M, Cohen J. 1995. Pattern of ecolgoical shifts in the diversification of Hawaiian Drosophila inferred from a molecular phylogeny. Curr Biol. 5:1129–1139. [DOI] [PubMed] [Google Scholar]
- Karasov T, Messer PW, Petrov DA. 2010. Evidence that adaptation in Drosophila is not limited by mutation at single sites. PLoS Genet. 6:e1000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawecki TJ, Lenski RE, Ebert D, Hollis B, Olivieri I, Whitlock MC. 2012. Experimental evolution. Trends Ecol Evol. 27:547–560. [DOI] [PubMed] [Google Scholar]
- Klepsatel P, Galikova M, Huber CD, Flatt T. 2014. Similarities and differences in altitudinal versus latitudinal variation for morphological traits in Drosophila melanogaster. Evolution 68:1385–1398. [DOI] [PubMed] [Google Scholar]
- Klusza S, Deng WM. 2011. At the crossroads of differentiation and proliferation: precise control of cell-cycle changes by multiple signaling pathways in Drosophila follicle cells. Bioessays 33:124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler R, Schlotterer C. 2012. Gowinda: unbiased analysis of gene set enrichment for genome-wide association studies. Bioinformatics 28:2084–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König A, Yatsenko AS, Weiss M, Shcherbata HR. 2011. Ecdysteroids affect Drosophila ovarian stem cell niche formation and early germline differentiation. EMBO J. 30:1549–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner DW, McCoy D, Isabella AJ, Mahowald AP, Gerlach GF, Chaudhry TA, Horne-Badovinac S. 2013. A Rab10-dependent mechanism for polarized basement membrane secretion during organ morphogenesis. Dev Cell. 24:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H Handsaker B Wysoker A Fennell T Ruan J Homer N Marth G Abecasis G Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott SE, Kreitman M, Palsson A, Alekseeva E, Ludwig MZ. 2007. Canalization of segmentation and its evolution in Drosophila. Proc Natl Acad Sci U S A. 104:10926–10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott SE, Ludwig MZ, Kreitman M. 2011. Evolution and inheritance of early embryonic patterning in Drosophila simulans and D. sechellia. Evolution 65:1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TF, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, et al. 2012. The Drosophila melanogaster Genetic Reference Panel. Nature 482:173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markow TA, Beall S, Matzkin LM. 2009. Egg size, embryonic development time and ovoviviparity in Drosophila species. J Evol Biol. 22:430–434. [DOI] [PubMed] [Google Scholar]
- Markow TA, O’Grady PM. 2007. Drosophila biology in the genomic age. Genetics 177:1269–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles CM, Lott SE, Hendriks CL, Ludwig MZ, Manu, Williams CL, Kreitman M. 2011. Artificial selection on egg size perturbs early pattern formation in Drosophila melanogaster. Evolution 65:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara K, Kim K, Sciulli C, Dowd SR, Minden JS, Carthew RW. 2005. Targets of microRNA regulation in the Drosophila oocyte proteome. Proc Natl Acad Sci U S A. 102:12023–12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Hellmann I, Hubisz M, Bustamante C, Clark AG. 2007. Recent and ongoing selection in the human genome. Nat Rev Genet. 8:857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzhdin SV, Turner TL. 2013. Promises and limitations of hitchhiking mapping. Curr Opin Genet Dev. 23:694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco-terWengel P, Kapun M, Nolte V, Kofler R, Flatt T, Schlotterer C. 2012. Adaptation of Drosophila to a novel laboratory environment reveals temporally heterogeneous trajectories of selected alleles. Mol Ecol. 21:4931–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, et al. 2009. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 19:826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesello C, Huelsmann S, Brown NH, Tapon N. 2006. The Drosophila RASSF homolog antagonizes the hippo pathway. Curr Biol. 16:2459–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Di Rienzo A. 2010. Adaptation—not by sweeps alone. Nat Rev Genet. 11:665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Pickrell JK, Coop G. 2010. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol. 20:R208–R215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M, Coop G, Wall JD. 2005. The signature of positive selection on standing genetic variation. Evolution 59:2312–2323. [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R’Kha S, Moreteau B, Coyne JA, David JR. 1997. Evolution of a lesser fitness trait: egg production in the specialist Drosophila sechellia. Genet Res. 69:17–23. [DOI] [PubMed] [Google Scholar]
- Reed LK, Lee K, Zhang Z, Rashid L, Poe A, Hsieh B, Deighton N, Glassbrook N, Bodmer R, Gibson G. 2014. Systems genomics of metabolic phenotypes in wild-type Drosophila melanogaster. Genetics 197:781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remolina SC, Chang PL, Leips J, Nuzhdin SV, Hughes KA. 2012. Genomic basis of aging and life-history evolution in Drosophila melanogaster. Evolution 66:3390–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, Jiang L, Ingman M, Sharpe T, Ka S, et al. 2010. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464:587–591. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen TS, Altshuler D, Lander ES. 2006. Positive natural selection in the human lineage. Science 312:1614–1620. [DOI] [PubMed] [Google Scholar]
- Sarikaya DP, Belay AA, Ahuja A, Dorta A, Green DA, 2nd, Extavour CG. 2012. The roles of cell size and cell number in determining ovariole number in Drosophila. Dev Biol. 363:279–289. [DOI] [PubMed] [Google Scholar]
- Sarikaya DP, Extavour CG. 2015. The hippo pathway regulates homeostatic growth of stem cell niche precursors in the Drosophila ovary. PLoS Genet. 11:e1004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt PS, Matzkin L, Ippolito M, Eanes WF. 2005. Geographic variation in diapause incidence, life-history traits, and climatic adaptation in Drosophila melanogaster. Evolution 59:1721–1732. [PubMed] [Google Scholar]
- Schmidt PS, Paaby AB, Heschel MS. 2005. Genetic variance for diapause expression and associated life histories in Drosophila melanogaster. Evolution 59:2616–2625. [PubMed] [Google Scholar]
- Schwarzkopf L, Blows MW, Caley MJ. 1999. Life-history consequences of divergent selection on egg size in Drosophila melanogaster. Am Nat. 154:333–340. [DOI] [PubMed] [Google Scholar]
- Song X, Xie T. 2003. wingless signaling regulates the maintenance of ovarian somatic stem cells in Drosophila. Development 130:3259–3268. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 100:9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis-Scott M, McIntyre LM, Wayne ML. 2005. Genetic architecture of two fitness-related traits in Drosophila melanogaster: ovariole number and thorax length. Genetica 125:211–222. [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, Silverman JS, Powell K, Mortensen HM, Hirbo JB, Osman M, et al. 2007. Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet. 39:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobler R, Franssen SU, Kofler R, Orozco-terWengl P, Nolte V, Hermisson J, Schlötterer C. 2014. Massive habitat-specific genomic response in D. melanogaster populations during experimental evolution in hot and cold environments. Mol Biol Evol. 31:364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchin MC, Chiang CW, Palmer CD, Sankararaman S, Reich D, Hirschhorn JN. 2012. Evidence of widespread selection on standing variation in Europe at height-associated SNPs. Nat Genet. 44:1015–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TL, Miller PM. 2012. Investigating natural variation in Drosophila courtship song by the evolve and resequence approach. Genetics 191:633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TL, Stewart AD, Fields AT, Rice WR, Tarone AM. 2011. Population-based resequencing of experimentally evolved populations reveals the genetic basis of body size variation in Drosophila melanogaster. PLoS Genet. 7:e1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. 2006. A map of recent positive selection in the human genome. PLoS Biol. 4:e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren DC. 1924. Inheritance of egg size in Drosophila melanogaster. Genetics 9:41–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne ML, Hackett JB, Dilda CL, Nuzhdin SV, Pasyukova EG, Mackay TF. 2001. Quantitative trait locus mapping of fitness-related traits in Drosophila melanogaster. Genet Res. 77:107–116. [DOI] [PubMed] [Google Scholar]
- Wayne ML, Hackett JB, Mackay TF. 1997. Quantitative genetics of ovariole number in Drosophila melanogaster. I. Segregating variation and fitness. Evolution 51:1156–1163. [DOI] [PubMed] [Google Scholar]
- Wayne ML, McIntyre LM. 2002. Combining mapping and arraying: an approach to candidate gene identification. Proc Natl Acad Sci U S A. 99:14903–14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS. 1996. Genetic Data Analysis II. Sunderland (MA): Sinauer Associates, Inc. [Google Scholar]
- Willert K, Shibamoto S, Nusse R. 1999. Wnt-induced dephosphorylation of Axin releases β-catenin from the Axin complex. Genes Dev. 13:1768–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Lei Q, Guan K-L. 2010. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 24:862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Udpa N, Gersten M, Visk DW, Bashir A, Xue J, Frazer KA, Posakony JW, Subramaniam S, Bafna V, et al. 2011. Experimental selection of hypoxia-tolerant Drosophila melanogaster. Proc Natl Acad Sci U S A. 108:2349–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.