

Summary
The inability to predict long noncoding RNAs from genomic sequence has impeded the use of comparative genomics for studying their biology. Here, we develop methods that use RNA-seq data to annotate the transcriptomes of 16 vertebrates and the echinoid sea urchin, uncovering thousands of previously unannotated genes, most of which produce long intervening noncoding RNAs (lincRNAs). Although in each species >70% of lincRNAs cannot be traced to homologs in species that diverged >50 million years ago, thousands of human lincRNAs have homologs with similar expression patterns in other species. These homologs share short, 5′-biased patches of sequence conservation nested in exonic architectures that have been extensively rewired, in part by transposable element exonization. Thus, over a thousand human lincRNAs are likely to have conserved functions in mammals, and hundreds beyond mammals, but those functions require only short patches of specific sequences and can tolerate major changes in gene architecture.
Graphical Abstract
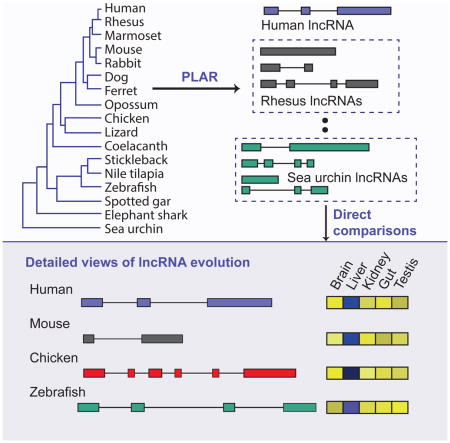
Introduction
Mammalian genomes are pervasively transcribed and encode thousands of long noncoding RNAs (lncRNAs) that are dispersed throughout the genome and typically expressed at low expression levels and in a tissue-specific manner (Clark et al., 2011). Long intervening noncoding RNAs (lincRNAs), lncRNAs that do not overlap protein-coding or small RNA genes, are of particular interest due to their relative ease to study and the poor understanding of their biology (Ulitsky and Bartel, 2013). The widespread dysregulation of lncRNA expression levels in human diseases (Wapinski and Chang, 2011; Du et al., 2013) and the many sequence variants associated with human traits and diseases that overlap loci of lncRNA transcription (Cabili et al., 2011) highlight the need to understand which lncRNAs are functional and how specific sequences contribute to these functions.
Comparative sequence analysis contributed greatly to our understanding of sequence-function relationships in classical noncoding RNAs (Woese et al., 1980; Michel and Westhof, 1990; Bartel, 2009). The study of lncRNA evolution may uncover important regions in lncRNAs and highlight the features that drive their functions. Shortly after the first widespread efforts for lncRNA identification, it became clear that lncRNAs generally are poorly conserved (Wang et al., 2004). Subsequent studies have refined the human and mouse lncRNA collections and used whole-genome alignments to show that lncRNA exon sequences evolve slower than intergenic sequences, and slightly slower than introns of protein-coding genes (Cabili et al., 2011). Nevertheless, lncRNA exon sequences evolve much faster than protein coding sequences or mRNA UTRs, suggesting that either many lncRNAs are not functional, or that their functions impose very subtle sequence constraints. We previously described lincRNAs expressed during zebrafish embryonic development (Ulitsky et al., 2011). Comparing the lincRNAs of zebrafish, human and mouse we found that only 29 lincRNAs were conserved between fish and mammals. Therefore, more intermediate evolutionary distances might be more fruitful for comparative genomic analysis.
In most vertebrates, direct lncRNA annotation has been challenging due to incomplete genome sequences, partial annotations of protein-coding genes and limitations of tools for reconstruction of full transcripts from short RNA-seq reads. Two recent studies looked at lncRNA conservation across mammals and across tetrapods (Necsulea et al., 2014; Washietl et al., 2014). These studies employed sequence conservation to predict genomic patches that may be part of a lncRNA and then used RNA-seq to seek support for their transcription. Such approach, however, introduces ascertainment bias into subsequent comparison of lncRNA loci. Other studies have directly compared lncRNAs within the liver and prefrontal cortex, respectively (Kutter et al., 2012; He et al., 2014), but focused only on closely related species.
To address these challenges we combined existing and newly developed tools for transcriptome assembly and annotation into a pipeline for lncRNA annotation from RNA-seq data (PLAR), applied it to >20 billion RNA-seq reads from 17 species and 3P-seq [poly(A)-position profiling by sequencing (Jan et al., 2011)] data from two species, and identified lincRNAs, antisense transcripts, and primary transcripts or hosts of small RNAs. This resource, along with a stringent methodology for identifying sequence-conserved and syntenic lncRNAs, allowed us to systematically explore features of lncRNAs that have been conserved during vertebrate evolution. We find that lncRNAs evolve rapidly, with >70% of lncRNAs having no sequence-similar orthologs in species separated by >50 million years of evolutionary divergence. Less than 100 lncRNAs can be traced to the last common ancestor of tetrapods and teleost fish, but several hundred were likely present in the common ancestor of birds, reptiles, and mammals. For the conserved lncRNAs, tissue specificity is conserved at levels comparable to that of mRNAs, suggesting control by conserved regulatory programs. In addition, we find that thousands of lncRNAs appear in conserved genomic positions without sequence conservation, including a group of lncRNAs that show sequence conservation only in mammals but have syntenic counterparts throughout vertebrates and another group that has conserved sequences throughout vertebrates and syntenic counterparts in sea urchin. The latter group contains candidates for the most conserved vertebrate lincRNAs identified to date. We also find that lncRNAs from distant species share short islands of sequence conservation, typically spanning only one or two exons and appearing closer to the 5′ end of the lncRNA. Furthermore, transposable elements have extensively rewired the architecture of conserved lincRNA loci, particularly in mammals. These findings support a model in which over a thousand lncRNAs have conserved functions in mammals, and hundreds beyond mammals, yet these functions require only short patches of specific sequences and can tolerate major changes in gene architecture.
Results
Pipeline for lncRNA annotation from RNA-seq data (PLAR)
To enable direct comparison of lncRNAs from different species, we first reconstructed lncRNA transcript models independently in each of 16 vertebrate species and the sea urchin. lncRNA identification is challenging due to a variety of factors, including limited ability to algorithmically reconstruct full-length transcripts from short-read RNA-seq data, incomplete genome sequence assemblies, and difficulties in distinguishing between coding and noncoding transcripts. The pipeline for lncRNA annotation from RNA-seq data (PLAR, Figure 1A, implementation available at http://webhome.weizmann.ac.il/home/igoru/PLAR/) presented here addressed these challenges by: (i) combining complementary datasets (RNA-seq and 3P-seq) in some of the species to tune thresholds and parameters and remove spuriously reconstructed models; (ii) combining multiple complementary filters for protein-coding potential to distinguish between coding and noncoding transcripts; (iii) combining the results of genome-assisted and de novo transcriptome assemblies to exclude artifacts due to gaps in genome sequences. Importantly, the application of PLAR to multiple species followed by inspection of loci harboring conserved orthologous lncRNA transcripts allowed us to leverage experience from one species to others and to tune both thresholds for calling lncRNAs and filters for exclusion of potential artifacts, which substantially improved overall catalog quality.
Figure 1. Reconstruction of lncRNA transcripts in 17 species.
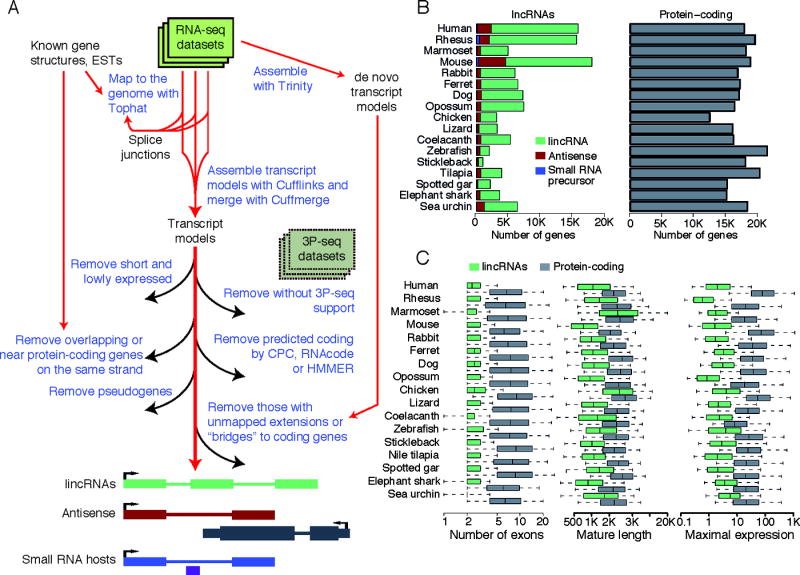
(A) PLAR pipeline. On the bottom, green, red and blue transcript models represent lincRNA, antisense RNA, and small RNA hosts, respectively. The gray and the purple models represent a coding gene and a small RNA, respectively. (B) Numbers of distinct lncRNA and protein-coding transcript models reconstructed in each species. (C) Features of lncRNA and protein-coding genes reconstructed in each species. Expression levels in each species are the maximum over all samples and computed in FPKM (fragments per kilobase per million of reads) units using CuffDiff (Trapnell et al., 2012). See also Tables S1,S2, and S3 and Figures S1 and S2.
In addition to lncRNA transcript models, PLAR provided improved models for annotated protein-coding genes and models for previously unannotated genes that have significant protein-coding potential. The lncRNA set in each species included: (a) antisense transcripts, defined as lncRNAs that overlap by at least one nucleotide a coding region on the other strand; (b) primary transcripts or hosts of short RNAs, defined as any lncRNA overlapping microRNA, snoRNAs, tRNA or other annotated small RNA (<200 nt) on the same strand; and (c) lincRNAs (defined as those lncRNAs that do not meet the other criteria). Most of analyses of this study focused on the third group, and thus when we use the term “lincRNAs” we refer to only this subset, as opposed to “lncRNAs,” which include the other two subsets. All lncRNAs, as well as the improved protein-coding models and the detailed implementation of PLAR, are provided to the community as resources for future studies.
PLAR identifies thousands of lncRNAs in each vertebrate species
We applied PLAR to RNA-seq data from 17 species, including the sea urchin and 16 vertebrates – three primates (human, rhesus macaque and marmoset), five non-primate mammals (mouse, rabbit, dog, ferret, and opossum), chicken, anole lizard, coelacanth, three teleost fish (zebrafish, stickleback, and nile tilapia), the non-teleost ray-finned fish spotted gar, and elephant shark (Table S1). In each species we used at least 250 million mapped paired-end RNA-seq reads from at least nine samples (mostly different adult tissues), totaling ~20 billion reads (Table S1). All libraries were of poly(A)-selected RNA, and most were strand-specific (all species except human and sea urchin). In chicken and zebrafish we also considered existing (Ulitsky et al., 2012) and newly collected 3P-seq data (Table S1), which mapped the 3′ termini of polyadenylated transcripts.
The first step of PLAR consists of assembly and initial annotation of the polyadenylated transcriptome in each species. This step produced 30,000–400,000 distinct transcript models per species that overlapped >80% of the protein-coding genes annotated in Ensembl in each species (Table S2). To focus on bona fide lncRNAs, after excluding transcripts overlapping coding genes, we retained only long and sufficiently expressed transcripts. For spliced transcripts, we required a length of >200 nt and an FPKM ≥0.1 in at least one sample, but for single-exon transcripts combined analysis of RNA-seq and 3P-seq data guided the use of more stringent cutoffs of >2 kb and FPKM >5, as only these more highly expressed and longer single-exon models were reasonably supported by 3P-seq data (Figure S1A). These filters retained 15,637–52,713 distinct transcripts as potential candidate lncRNAs in each species (Table S2). This set was further filtered using two or three different tools to identify protein-coding potential in each species. Transcripts ending close to annotated genes on the same strand were removed (as they were suspected to be fragments of 5′ or 3′ UTRs), and those overlapping predicted pseudogenes were also removed. Although some pseudogenes are transcribed and function as either lncRNAs or precursors for small RNAs (Khachane and Harrison, 2009; Rapicavoli et al., 2013; Watanabe et al., 2014), limitations in short-read assembly complicated determination of whether RNA-seq signals were coming from the pseudogene or its source gene, motivating our choice to exclude pseudogenes from consideration.
Another challenge was the variable completeness of genome sequence in different species, which varied from contig N50 of 9 Kb (coelacanth) to 33 Mb (human) (Table S2). The main concern with a fragmented genome is that a transcript model that appears to be a standalone lincRNA might be part of a longer protein-coding transcript that was fragmented due to gaps in genome assembly. To address this problem, in 13 of the species that had poorer assembly quality, we used Trinity (Grabherr et al., 2011) to reconstruct the transcriptome de novo. We then mapped the assembled transcripts to the reference genome and looked for assembled transcripts that overlapped a potential lncRNA and either had substantial additional unmapped sequence or also overlapped an annotated or reconstructed protein-coding gene (Figure S1B), and removed them from consideration. This procedure removed 320–3,003 lncRNA candidates (Table S2) that, as expected, were more likely than others to appear in proximity of genome assembly gaps (Figure S1C).
Conserved features of lncRNAs in vertebrate genomes
The application of these stringent filters retained 989–18,294 lncRNA genes per species (Figure 1B and Table S3), >70% of which were lincRNAs. We observed the general trend of a higher number of lincRNA loci in mammals, which also have the largest genomes of the species we studied. However, as in previous studies (Necsulea et al., 2014), directly comparing lincRNA numbers across species was difficult due to differences in genome sequence and RNA-seq data quality and quantity, as well as differences in the diversity of samples sequenced in different species (i.e., diverse embryonic samples were available only in mouse, ferret, chicken, zebrafish and sea urchin). Interestingly, while the genomes we studied differed by ~9-fold in their total size and in the number of lincRNAs, the genomic features of lincRNAs, including number of exons and mature sequence length were largely conserved throughout vertebrates (Figure 1C). lincRNAs were also consistently expressed at lower levels than mRNAs (Figure 1C), while always showing much higher tissue specificity (Figure S2A). Interestingly, lincRNA tissue specificity was comparable to that of mRNAs that had lincRNA-like expression levels, suggesting that similar mechanisms may drive tissue specificity of both lincRNAs and poorly expressed mRNAs (Figure S2A).
On average, 9.6% (ranging from ~2% in sea urchin to 23% in chicken, Figure S2B) of lincRNAs in each species were divergently transcribed from a shared promoter with a protein-coding gene (<1Kb between transcription start sites). Similar fractions of conserved and non-conserved lincRNAs were divergent, which argued against the idea that a substantial fraction of conserved lincRNAs is conserved solely because they overlap promoter-proximal cis-regulatory elements. In mammalian genomes, where regions between protein-coding genes are typically large, on average ~40% of the lincRNAs shared such regions with at least one additional non-overlapping lincRNA gene. This fraction was <10% in the smaller zebrafish, coelacanth and stickleback genomes (Figure S2C).
2,869 clusters of orthologous lincRNAs from different species
To directly compare lncRNAs from different species and identify groups that likely share common ancestry, we used whole-genome alignments and BLASTN to construct a network of sequence similarities between lncRNAs. Sequence similarity was supported by synteny between at least one pair of species in 4,885 connected components in this network, and those were carried forward as groups of potentially orthologous lncRNAs. As the two closest species examined were human and rhesus, and any other two species were separated by >35 million years of parallel evolution, we focused the analysis on the 3,947 clusters that were not Catarrhini-specific, 2,869 of which were lincRNA clusters (Table S4). Each cluster contained lncRNAs from an average of 4.7 species. No significant sequence similarity was found between sea urchin and vertebrate lincRNAs.
Overall, most lincRNAs in each species were lineage-specific (Figure S2D). Consistent with our previous findings when studying zebrafish lincRNAs (Ulitsky et al., 2011), only 99 lincRNA genes, including 56 with annotated representatives in human (<3% of lincRNAs conserved between human and at least one non-primate mammal) could be traced to the last common ancestor of tetrapods and ray-finned fish, compared to >70% of protein-coding genes and >20% of small RNA primary transcripts (Figure 2A). Substantially more lincRNAs could be traced to more recent ancestors, with >280 lincRNAs shared between mammalian and non-mammalian amniotes and >200 additional lincRNAs conserved between marsupials and eutherian mammals. Interestingly, the number of lincRNAs shared between human and opossum (last common ancestor ~180 million years ago (Mikkelsen et al., 2007)) was much larger than that shared among any euteleosts (last common ancestor of the zebrafish, stickleback and tilapia lived ~110–160 million years ago (Wittbrodt et al., 2002)), suggesting that retention of lincRNAs that appeared 100–200 million years ago was more common in mammals than in some of the other lineages. Twenty four lincRNAs had orthologs in at least seven different species, allowing a detailed view into the evolution of lincRNA loci across >400 million years of evolution, which in many cases included multiple exon gain and loss events and dramatic changes in mature RNA size, as illustrated for the Cyrano (Ulitsky et al., 2011) lincRNA (Fig. 2B).
Figure 2. Conservation of lincRNAs in vertebrates.

(A) Phylogeny of the species studied with the numbers of lincRNAs that are estimated to have emerged at different times. The numbers shown next to each split are numbers of clusters with representatives in both lineages for the split and no representatives in more basally split groups. The bar plots present the fraction of all clusters with a representative from the human genome that are estimated to emerge before the adjacent split. (B) Evolution of the Cyrano lincRNA in vertebrates. Representative isoforms of the coding and lincRNA transcripts in each species are shown. Shaded boxes show magnification of splice junctions derived from transposable elements in dog, mouse and human. Cyrano is also annotated as OIP5-AS1 in human. (C) lincRNAs in the Sox21 locus in human and mouse. Representative isoforms are shown in each species. Sequence conservation computed by PhyloP (Pollard et al., 2010), EvoFold (Pedersen et al., 2006) predictions, CpG island annotations and whole genome alignments taken from the UCSC genome browser. (D) Numbers of human lincRNA genes that align to the indicated species are split based on the indicated categories. “Align to lincRNA” are lincRNAs that have sequences mapping to a lincRNA in the indicated species (and therefore are conserved lincRNAs by definition). “Conserved lincRNAs” have sequence-similar homologs in some other species, but the sequence they align to in the indicated genome does not overlap a lincRNA in that specific genome. “Align to coding” are lincRNAs that are not conserved and whose projection through the whole genome alignment overlapped with a protein-coding gene in the other species. “Align to transcribed” are those nonconserved lincRNAs that align to a transcribed region in our transcriptome reconstruction in the other species that was not classified as protein-coding or as lincRNA. “None” are those lincRNAs that have only sequences aligning to untranscribed portions of the corresponding genome. See also Table S4 and Figure S3.
Genomic sequence conservation often does not reflect conserved lincRNA production
Phylogenetic analysis of lncRNAs, such as computation of sequence conservation metrics and even identification of lncRNAs in different species (Necsulea et al., 2014), has relied on whole genome alignments that compare genomic sequences between species. The validity of such analyses depended on the assumption that corresponding sequences in other species are also part of lncRNA transcripts, or that if the sequence is transcribed in some species, all sequences homologous to it in other species are also transcribed (Necsulea et al., 2014). The lincRNAs in the 20 Kb region surrounding the Sox21 transcription factor (Figure 2C) illustrate the caveats in this approach. Three lincRNAs are currently annotated in this region in human (SOX21-AS1, linc-SOX21-B and linc-SOX21-C), the promoter of each overlapping a different CpG island. All three overlap DNA sequences alignable to other mammalian genomes. Most notable is linc-SOX21-B, which appears to be a highly conserved lincRNA as it overlaps a highly conserved element found in all vertebrates, and EvoFold (Pedersen et al., 2006) predicts on the basis of sequence alignments that linc-SOX21-B harbors several conserved secondary structures. However, we found no homologs of linc-SOX21-B or linc-SOX21-C in any of the other species, and did find homologs of SOX21-AS1 in four other amniotes. Thus, relying on genomic sequence conservation as a proxy for lincRNA conservation can lead to misleading results.
The number of human lincRNAs that had alignable sequences in other genomes was much larger than the number of conserved lincRNAs (i.e., those that aligned to a lincRNA transcript sequence identified in any other genome, Figure 2D). The majority of the other lincRNAs, which we refer to as “pseudo-conserved”, align to sequences that are not part of any transcript model, and therefore likely to have arisen de novo in their respective lineages. One potential cause for pseudo-conservation is overlap with tandem repeats. Due to the additive nature of the scoring schemes used in whole-genome alignments, tandem repetitions of slightly similar sequences can yield alignability scores that pass thresholds required for matching regions in a whole genome alignment. For example, the CDR1as transcript (Hansen et al., 2013; Memczak et al., 2013), which contains multiple sequence-similar repeats of the miR-7 binding site, appears in whole-genome alignment of the human genome as sporadically conserved throughout vertebrates (Figure S3A), but when the aligned sequences in other species are extracted from the whole-genome alignment they contain miR-7 sites and appear in syntenic positions only in mammals (data not shown). Another potential cause for pseudo-conservation is overlap with enhancer elements. For instance, the highly conserved element found in linc-SOX21-B overlaps a conserved brain and neural tube enhancer (VISTA (Visel et al., 2007) element hs488). In such cases, sequence conservation of lincRNA exons stems from the importance of the sequence as a DNA element rather than as part of the lincRNA.
Conserved lincRNAs share short patches of sequence conservation
Conserved lincRNAs were on average longer, had more exons and were more highly and broadly expressed than both pseudo-conserved and lineage-specific lincRNAs (Figure S3B). Although these differences were each statistically significant, they were subtle, suggesting that presently it would be difficult to use them as indicators of functionality. The observation that conserved lincRNAs were generally more likely to be broadly expressed further argues that tissue-specificity by itself should not be considered a hallmark of lincRNA functionality.
Direct comparison of RNA from different species using BLASTN identified stretches of conserved sequence that we refer to as “conserved patches”, defined as regions within the human transcripts alignable with transcripts of the same type in other species. Conserved patches in lincRNAs are much shorter than those in mRNAs (Figure 3A–B), occupy a smaller fraction of the total transcript length (Figure S4A–B), and typically span just one or two exons (Figure 3C). Interestingly, conserved patches had a significant tendency to appear closer to the 5′ end of the lincRNA (P<10−15), with the distance from the middle of the conserved patch to the 3′ end being longer than its distance to the 5′ end by 42% on average. This 5′ bias resembled that observed in mRNAs, where the distance to the 3′ end was longer by 49%, consistent with the typically shorter lengths of 5′ UTRs compared to those of 3′ UTRs.
Figure 3. Conserved and paralogous patches in lincRNAs.
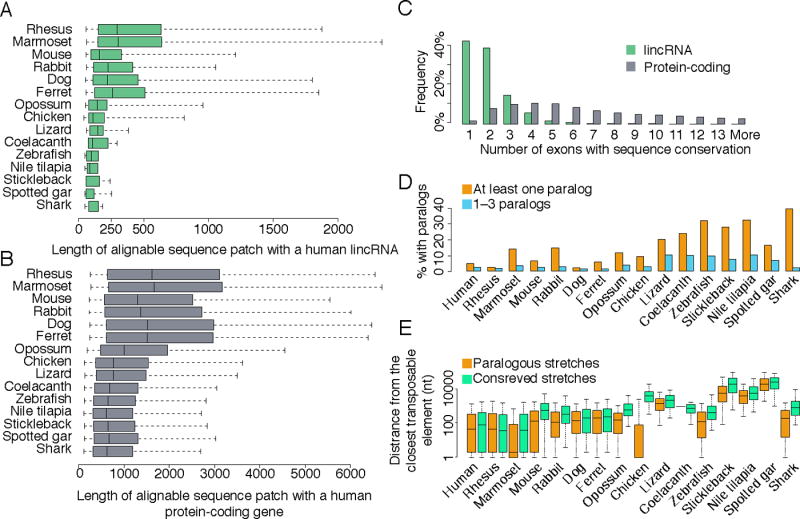
(A) Distributions of lengths of conserved patches, defined as the total length of the sequence alignable by BLASTN between a human lincRNA transcript and any lincRNA transcript in the indicated species. (B) Same as A, but for protein-coding gene reconstructions. (C) When considering patches of conservation of human lincRNAs with species except for rhesus, the distributions of the number of exons that overlap a conserved sequence patch. (D) Fraction of lincRNA genes that have a paralogous lincRNA (BLASTN E-value<10−5) within the same species. Fractions are shown either when including all paralogous pairs, or only considering lincRNAs that have less than four distinct paralogous lincRNAs. (E) Distributions of distances of paralogous and conserved sequence patches from the nearest annotated transposable element. See also Figure S4.
Short functional domains were previously reported in individual lincRNAs (Chureau et al., 2002; Pang et al., 2006; Ulitsky et al., 2011; Ilik et al., 2013; Quinn et al., 2014), and it was recently shown that as much as one tenth of a lincRNA sequence can be sufficient for recapitulating the function of the entire transcript (Quinn et al., 2014). Our findings generalized these cases and suggest that the overall locus architecture of lincRNAs could be quite flexible, much more so than that of protein-coding genes. Indeed, when we compared different genomic features of lincRNAs between human and other species, including number of exons, mature transcript length and genomic locus length, significant correlations were observed with Spearman’s r in the 0.1–0.3 range, but this range was much lower than the 0.6–0.8 range observed for analogous correlations in protein-coding genes (Figure S3C). We note that difficulties in precise reconstruction of the boundaries of the first and last exons might underlie some of the apparent divergence of mature lengths, as the “internal length” of lincRNAs and mRNAs, defined as the total length of non-terminal exons, was typically better conserved than the total mature length (Figure S3C). Still, when contrasted with the conservation of mRNA exon–intron structures, lincRNA loci clearly undergo more frequent rewiring of their architectures, rapidly losing and gaining exons, in part via adoption of new sequences from transposable elements (Figure 2B and see below).
We also used a similar BLASTN-based approach for comparing repeat-masked sequences of lincRNAs within each species, identifying “paralogous patches”, defined as regions alignable (after repeat masking) between the lincRNA transcript and other non-overlapping transcripts in the same species. Between 2% and 40% of lincRNAs in each species had such a paralogous patch (Figure 3D), with fractions generally higher in fish genomes. We suspect that, as previously suggested (Derrien et al., 2012), most of these patches correspond to presently unannotated fragments of transposable elements, as (i) paralogous patches rarely overlapped conserved patches (only 6.1% of conserved patches overlapped paralogous patches on average across species, Figure S4B), (ii) patches typically appeared in close proximity to annotated transposable elements (Figure 3E), and (iii) lincRNAs that had sequence similarity with other lincRNAs typically had sequence similarity with at least four other lincRNAs (Figure 3D), arguing against prevalence of specific duplications of functional RNAs.
Conservation of expression patterns of conserved lincRNAs
Some lincRNAs have tightly conserved spatial expression patterns (Chodroff et al., 2010), but recent reports disagree on the global extent of conserved lincRNA expression, which is estimated to be either as high as that of protein-coding genes (He et al., 2014; Washietl et al., 2014) or much lower (Necsulea et al., 2014). One difficulty in addressing this question is the sensitivity of expression-level estimates to the precision of isoform reconstruction, which, as already mentioned, is particularly inaccurate in first and last exons. Another difficulty is posed by cases in which the DNA sequence is conserved in distant species, but homologous lincRNAs are only found in proximal species. Consider gene X that has orthologs with virtually identical expression patterns in eutherian mammals, and one exon that is conserved on the DNA level in more distal vertebrates and those more distant pseudo-orthologs experience weak nonspecific transcriptional noise. When considering all species in which the DNA is conserved, the expression levels of X would be poorly conserved, but when considering only eutherian mammals they would be highly conserved. With these differences in mind, we directly compared expression levels and patterns between sequence-similar full-length reconstructions of lincRNAs expressed at FPKM>1, focusing on amniote species, for which sufficient numbers of lincRNAs were conserved. Within the same tissues from different species, the lincRNA expression levels correlated, with Spearman’s r ranging from 0.3 to 0.5, which were considerably lower than those of mRNAs, which ranged from 0.6 to 0.8 (Figure 4A). However, other analyses indicated less difference in expression conservation between lincRNAs and mRNAs. When we used cap analysis of gene expression (CAGE) data from the FANTOM5 project (Consortium et al., 2014) and compared human and mouse, conservation of absolute expression levels of lincRNAs and mRNAs were similar with Spearman’s r in the 0.5–0.6 range (Figure S5A). This apparent discrepancy between RNA-seq-based and CAGE-based estimates might be due to the relative robustness of CAGE-based estimates to accuracy of isoform reconstruction. Furthermore, when expression patterns of different tissues from four of the eutherian mammals (human, mouse, rabbit, and dog) were compared using hierarchical clustering of the Spearman’s correlations of RNA-seq profiles, the different tissues of each species clustered together when using either lincRNA or mRNA data, except for testis and brain, which formed clusters separate from the other tissues (Figure S5B). The comparable clustering again pointed to similarities between levels of expression conservation for lincRNAs and mRNAs. Lastly, when we normalized the expression values in each tissue to all the other tissues and then compared the resulting relative expression patterns between homologous lincRNAs, the distributions of the correlation coefficients of the lincRNAs where only slightly lower than those of mRNAs (Figure 4B). We conclude that the expression patterns of lincRNAs are almost as well conserved as those of mRNAs, suggesting that lincRNAs with conserved sequences have retained conserved regulatory programs and presumably conserved functions.
Figure 4. Expression patterns of conserved and lineage-specific lincRNAs.

(A) Correlation of absolute expression levels between human lincRNAs and mRNAs and their conserved homologs in indicated other species. (B) Distributions of correlations of relative expression levels, computed as Spearman’s correlations between expression patterns, between lincRNAs/mRNAs and their conserved homologs in the indicated species. (C) Fraction of all lincRNAs in the indicated species that are enriched in the indicated tissue. (D) Number of RNA-seq reads that mapped to a lincRNA out of all reads that could be mapped to any mRNA or lincRNA. “All tissues” is the median fraction across all tissues, and “Testis” is the fraction just in the testis samples. (E) Comparison of conservation levels of lincRNAs enriched in different tissues. The top part of the panel shows the fraction of the human lincRNAs enriched in the indicated tissue in human that are conserved in a non-mammalian species. The bottom part shows the absolute number of conserved lincRNAs enriched in each tissue, partitioned based on the conservation level of the lincRNA (the most distant species where homologs of the lincRNA can be found). “Ubiq.” are ubiquitously expressed genes. See also Figure S5.
Testis-bias of lincRNA expression in amniotes and hundreds of conserved testis-specific lincRNAs
A disproportionally large number of mammalian lincRNAs are specifically expressed in the testes (Cabili et al., 2011; Soumillon et al., 2013; Necsulea et al., 2014). Using RNA-seq data from testes in 11 species, we found that this disproportionate transcription occurs throughout amniotes, and to a lesser extent in other vertebrates, but not in the elephant shark or the sea urchin (Figure 4C). Furthermore, in most species and tissues, lincRNAs accounted for roughly 1% of the coding and noncoding polyadenylated RNA transcripts, but this fraction increased to ~4% in testes in amniotes (Figure 4D). Testes-specific transcripts have evolved rapidly, whereas brain-specific lincRNAs were better conserved than others (Figure 4E), consistently with previous reports (Soumillon et al., 2013; Necsulea et al., 2014). However, the numbers of testes-enriched lincRNAs are much higher than the numbers of lincRNAs enriched in other tissues, and thus in absolute numbers, there were more testes-enriched lincRNAs that were conserved to various depths than, for example, brain-enriched ones (Figure 4E), suggesting that multiple lincRNAs are likely to play functionally important, and still largely unknown roles in spermatogenesis.
Transposable elements globally rewire lincRNA transcriptomes
A large fraction of lincRNA exonic sequences in human and mouse is known to derive from transposable elements (Kelley and Rinn, 2012; Kapusta et al., 2013). The dramatic differences in transposable-element load across vertebrate genomes, ranging from at least 52% of the opossum genome to <2% of the tilapia and stickleback genomes, allowed us to evaluate the contribution of such elements to the evolution of lincRNA loci. As expected, protein-encoding sequences were highly depleted of repetitive elements, and depletion was slightly milder when all the mRNA sequence, including UTRs, was considered. Depletion was generally much weaker in lincRNA exons, and no depletion was observed in lincRNA or mRNA introns (Figure 5A). Although transcription start sites of conserved lincRNAs overlapped repetitive elements relatively rarely, those of lineage-specific lincRNAs often did overlap repetitive elements, particularly LTR elements (Figure 5B). A milder depletion of transposable elements was also evident in donor and acceptor splice sites of conserved lincRNAs, but no difference between conserved and nonconserved lincRNAs appeared in 3′ ends. Together with the general 5′ bias of the conserved patches, these results suggest that the position and sequence of the 3′ end of conserved lincRNAs are generally under less selection than those of either the promoter or of the processing signals, and are more amenable to rewiring. Evolution of the cancer-associated Pvt1 lincRNA illustrated 3′-end turnover (Figure 5C). The first exon and two of the seven or more internal exons of Pvt1 in mammals were conserved, but the predominant 3′ exon of Pvt1 mapped to different locations in primates, glires, dog and opossum, and in each of these species it derived from a different transposable element (Figure 5C). Our global analysis indicated that such trajectories — conservation of the promoter and short patches in the first few exons, alongside changes in the identity of the 3′ end of the lincRNA — were commonplace in lincRNAs during vertebrate evolution.
Figure 5. Transposable elements rewire lincRNA loci.

(A) Fraction of different genomic elements (bases, 5′ and 3′ ends of the transcript, and 5′ and 3′ splice sites (SSs)) overlapping a transposable element. (B) Same as (A), but showing only overlap with the transcription start sites and considering separately transposable elements of the indicated families. (C) Schematic representation of the Myc/Pvt1 locus in different vertebrates. Representative isoforms of Myc/Pvt1 are shown. Bars beneath exons represent their conservation and origin status. Shaded regions group together two groups of Pvt1 homologs that share alignable sequences, one in mammals and the other in fish. (D) Comparison of the fraction of lncRNA sequences in different subgroups of lincRNAs that overlap a transposable element. The number of conserved exons in a lincRNA gene is the maximum number of conserved exons across all its isoforms. See also Figure S6 and Table S5.
In human, we observed little difference in the fraction of lincRNAs that overlapped a transposable element when comparing conserved and primate- or human-specific lincRNAs, suggesting that even lincRNAs that rely on specific sequences for function can tolerate transposon insertions, as illustrated by the deeply conserved Cyrano lincRNA, which has two exons in most fish species, and four exons in most mammals, some of which were derived from transposable elements (Figure 2B). However, lincRNAs with two or more conserved exons were less likely to overlap repetitive elements than were either poorly conserved (human- or primate-specific) lincRNAs or those that have only one conserved exon (Figure 5D). This subset was presumably enriched for those lincRNAs that depend on multiple independent functional domains for function, which perhaps imparted stronger selection against drastic sequence changes.
Shared sequence motifs enriched in lincRNAs across species
We next tested whether specific short sequence motifs were enriched in lncRNA exons in each species, and potentially shared between species. To do so, we counted the number of occurrences of all possible 6mers in lncRNA exons in each species, compared them to those in randomly shuffled sequences preserving the same dinucleotide frequencies, and identified those that were significantly enriched (P< 0.05 after Bonferroni correction, Table S5). As expected, in 12 of the species, the most enriched motif was AAUAAA (enriched ~2-fold over background), which corresponded to the consensus poly(A) sequence and was expected to be found in most polyadenylated transcripts. Interestingly, despite the generally low sequence conservation in lincRNA genes, many additional significantly enriched motifs were shared among multiple species, with 124 non-redundant motifs enriched in at least 5 species, and 31 enriched in at least 12 of the species (Figure S6A). The most enriched motifs had significant preference to appear close to the 3′ or the 5′ of the lincRNA (Figure S6A). A significant portion of the 124 non-redundant k-mers enriched within lincRNA exons in at least 5 species corresponded to exonic splicing enhancers (ESEs, taken from (Goren et al., 2006), P= 9.8×10−4), and included the purine-rich motifs bound by such factors as SF2/ASF (Tacke and Manley, 1995; Fairbrother et al., 2002). Four other commonly enriched 6-mers were combinations of CUG and CAG, (Figure S6A) forming binding sites of the splicing factors CUG-BP and Muscleblind. These splicing-related motifs had a general tendency to appear closer to the 5′ end of the lincRNA (Figure S6A). Two recent studies found that splicing-related motifs are preferentially conserved in lincRNA exons (Schuler et al., 2014; Haerty and Ponting, 2015). Here, we extend these observations to show that such motifs are over-represented in both conserved and lineage-specific lincRNAs. Strikingly, the overall motif enrichment profiles of conserved and lineage-specific lincRNAs were highly correlated (R2 = 0.95, Figure S6B), suggesting common building blocks and sequence biases within the exons of lincRNA sequences, regardless of conservation status. Examining k-mers of other lengths and allowing for imperfect matches to the k-mers resulted in similar observations (data not shown).
Synteny conservation without sequence conservation across distant vertebrates
We previously noted that many lincRNAs appear in conserved positions in zebrafish and in human or mouse without detectable sequence conservation (Ulitsky et al., 2011). Some might correspond to orthologous lincRNAs that depend on very short (<20 nt) conserved sequences that are difficult to align or fail to reach thresholds of statistical significance, whereas others might correspond to cases where transcription through a specific locus is important, but the sequence of the RNA product is not (Ulitsky and Bartel, 2013). Our previous approach for detecting synteny relies only on similarity of a neighboring protein-coding gene and thus has a high false positive rate in large regions containing multiple lincRNAs and no protein-coding genes. Therefore, we developed a more stringent approach for synteny identification, which used pairwise genome alignments and specifically handled cases in which lincRNAs in two species have an orthologous flanking protein-coding gene, but are unlikely to be orthologous based on their positions relative to other conserved noncoding elements (Figure 6A). This approach identified > 750 lincRNAs confidently syntenic with human lincRNA in each species (Figure 6B). As expected, most of the lincRNAs syntenic with human lincRNAs in rhesus also had conserved sequence with the human lincRNAs, whereas the vast majority of the syntenic lincRNAs in non-mammalian vertebrates did not (Figure 6B). Particularly interesting were cases in which the sequence of the lincRNA was conserved across multiple mammals and had syntenic but not sequence-similar counterparts in more distant species. For example, 174 human lincRNAs had a sequence-similar homolog in chicken, lizard, or opossum, but only syntenic counterparts in zebrafish and stickleback (Table S4). These included homologs of Pvt1 (Tseng et al., 2014) found near the Myc protein-coding gene (Figure 5C). Three of the exons of human PVT1 were conserved in sequence in other mammals, but none were conserved in sequence beyond mammals. Nonetheless, syntenic lincRNAs were found downstream of Myc in all studied vertebrate species, including elephant shark. Moreover, the putative Pvt1 homologs in stickleback and tilapia had sequence conservation to each other but not to mammalian homologs. Such lincRNAs are excellent targets for future experimental interrogation seeking to expose the functional meaning of syntenic conservation without sequence conservation.
Figure 6. Hundreds of lincRNAs appear in syntenic positions without sequence conservation.

(A) A cartoon illustrating our approach for identifying stringently syntenic lincRNAs between human and other genomes. (B) Number of lincRNAs appearing at syntenic positions with (“Synteny+Sequence”) and without (“Synteny only”) sequence conservation. Control numbers were obtained by randomly placing the human lincRNAs in intergenic regions and repeating the analysis ten times, averaging the numbers of observed synteny relationships. All numbers were obtained using the stringent procedure described in Experimental procedures, except for sea urchin. (C) Schematic representation of the Foxa2/Linc00261 locus in different species. (D) Schematic representation of the Fancl/Bcl11a locus in different species with lincRNA gene models collapsed into a single meta-gene. Transcripts on the left-to-right strand are in red and those on the right-to-left strand are in blue. See also Table S6.
Potential orthologs of vertebrate lincRNAs in the sea urchin
In our direct comparisons of lincRNA sequences, we found no significant sequence homology between vertebrate lincRNA and those of sea urchin. This was not unexpected, as the sequence homology regions between mammalian and fish lincRNAs were already very short and borderline in their statistical significance, and further divergence was expected in sea urchin. However, we did identify syntenic sea urchin lincRNAs for >2,000 of human lincRNA genes, which was ~600 more than the number expected by chance (Figure 6B), suggesting the potential existence of conserved functional homologs of vertebrate lincRNAs in sea urchin. Particularly interesting were those lincRNAs with sequence conservation observed between mammals and distant vertebrates, suggesting functions of the mature RNA, which also had syntenic homologs in sea urchin. One such lincRNA was LINC00261, transcribed from a large intergenic desert downstream of the Foxa2 transcription factor gene and found in multiple species, a subset of which is shown in Figure 6C. Homologs of LINC00261 were expressed in endodermal tissues in all vertebrates, and in the gut in sea urchin, further supporting functional conservation. Sequence homology was found between mammals and fish in the first exon of LINC00261, whereas other exons typically did not align, and as observed for Pvt1 and other lincRNAs, the 3′ end of this lincRNA mapped to drastically different regions in different species.
Another syntenic locus was found in the gene desert between Fancl and Bcl11a, spanning ~2 Mb in human and ~800 Kb in zebrafish (Figure 6D) and containing multiple lincRNAs predominantly expressed in the brain and reproductive tissues across vertebrates (partially annotated as LINC01122 and LOC101927285 in human). The syntenic sea urchin lincRNA spanned ~180 Kb upstream of the sea urchin Fancl homolog, and was specifically expressed in the adult ovary. Overall, such syntenic lincRNAs in sea urchin were found for 18 of the human lincRNAs conserved in sequence beyond amniotes (Table S6), making them the best current candidates for the most deeply conserved human lincRNAs.
Discussion
The importance of direct annotation and comparison of lncRNAs across species
Two recent studies examined lncRNA evolution by projecting sequences of human lncRNAs across whole genome alignments and testing whether the corresponding sequences are transcribed in the other species (Necsulea et al., 2014; Washietl et al., 2014). This approach has led to interesting insights but has several shortcomings. First, as we show here, most lncRNAs in each species are lineage-specific, and are thus missed by searching only for homologs of human lincRNAs. Second, and more important, is the phenomenon of pseudo-conservation. Among lncRNAs that do have sequence-similar regions in other genomes, many map to either untranscribed regions or regions for which transcription is supported with just a few RNA-seq reads and is thus much too rare for annotation as an lncRNA locus. For example, among the 171 lncRNAs recently reported to be conserved beyond mammals by Necsulea et al. (2014), only 59 overlapped our human lincRNA annotations (many others were projected to the human genome but lacked evidence for annotation as a lincRNA in human), and only 20 matched a lincRNA that we found conserved as a lincRNA in the other species. Further, the previous use of sequence similarity across species to identify lincRNAs creates an ascertainment bias that can artificially inflate measurements of lincRNA sequence conservation, which presumably led to unexpectedly high estimates of sequence conservation of lincRNAs conserved in tetrapods (Necsulea et al., 2014). These shortcomings are avoided when lincRNAs are independently reconstructed in each species and subsequently compared. As shown here, the latter approach also revealed conserved and paralogous patches and enabled the detailed study of how lincRNA architecture has evolved.
A resource for lincRNA sequence evolution
Our work generated an extensive set of full-length orthologous lincRNA sequences from diverse vertebrates, thereby providing an important resource for future studies. Existing methods for sequence comparison typically perform poorly in comparing lincRNA sequences, and we expect that our resource will contribute to the development of methods for identifying conserved sequence elements and RNA structures. Nonetheless, our collections of lncRNAs are by no means complete. The use of oligo-d(T)–based RNA purification for RNA-seq led us to focus on polyadenylated lncRNAs, which are typically more stable and abundant than non-polyadenylated transcripts but exclude some types of lncRNAs, such as enhancer RNAs (eRNAs) (Li et al., 2013). Gaps in genome assembly are one of the most prominent limiting factors for lncRNA identification, as they make it difficult to distinguish between fragmented protein-coding genes and bona fide lncRNAs. We expect that improvements in affordability and sequencing depth of long-read sequencing technologies will lead to better genome assemblies in non-model organisms and improve transcriptome assemblies that will enable more accurate isoform identification and quantification.
Differences in lincRNAs across vertebrates
Our decision to be as inclusive as possible in using RNA-seq data in transcript reconstruction resulted in differences in read depth between genomes, which together with differences in genome quality created difficulties for directly comparing the numbers of lincRNAs across species. Still, it is evident that a typical mammalian genome harbors more lincRNAs than do the much smaller teleost fish genomes or the intermediate-size chicken and lizard genomes. These differences are most likely explained by the differential prevalence of transposable elements that have heavily contributed to expansion of intergenic regions in mammals, parts of which are transcribed. Exonization of transposable elements has been studied extensively in mRNAs (Sela et al., 2010), but those events are quite strongly selected against, particularly in coding exons, and thus in absolute terms, transposons contributed to more exons in lincRNAs than in mRNAs. Specific transposon families, mostly LTR elements in mammals, also contributed promoters, thereby imparting expression to previously intergenic regions to help further expand the numbers of lincRNAs.
Evolutionary sources of lincRNAs and a model for lincRNA evolution
The observation that most lincRNAs in each of the studied vertebrates have emerged relatively recently implies a frequent birth of lincRNA genes. Most of the similarity between lincRNAs within a species can be attributed to transposable elements, and evolution of lincRNAs by whole-locus duplication appears to be rare. Instead, our results suggest that most of the new lincRNAs occur de novo from pre-existing intergenic regions that gained capacity to be transcribed into stable long RNAs, presumably by combining the ability to recruit transcription initiation, splicing and cleavage-polyadenylation complexes, all at favorable distances from each other. Each of these components relies on limited sequence information and either can arise by chance during neutral evolution or be adopted from transposable elements that bear sequences with such elements. Thus, the approximately thousand mammalian lincRNAs with detectable sequence conservation make up a small subset of the lincRNAs that emerged over the past 450 million years of vertebrate evolution. These conserved lincRNAs have presumably been co-opted for functional roles that are supported by short patches of conserved sequence and have tolerated substantial sequence rewiring by transposable elements after function acquisition. This model for lincRNA evolution and our catalogue of conserved lincRNAs will inform future experiments designed to verify and characterize the functional roles of these intriguing noncoding RNAs, their short patches of conserved sequence and the rapidly evolving linkers connecting these patches.
Experimental Procedures
Chick embryo transcriptome analysis using RNA-seq and 3P-seq
Chick embryos were staged as previously described (Hamburger and Hamilton, 1951) and total RNA was collected from whole embryos at Hamburger–Hamilton (HH) stages HH4, HH11, HH14/15, HH21/22, HH25/26, HH32, and HH36. All animal protocols were approved by the Harvard Medical Area Standing Committee on Animals. Embryonic tissue was disrupted manually using forceps, cells were lysed with guanidine isothiocyanate and β-mercaptoethanol, lysate was homogenized using a series of syringes decreasing in size, and total RNA was purified on silica-gel columns according to standard procedures (Qiagen RNeasy). RNA-seq libraries were prepared using the dUTP strand-specific library preparation protocol and sequenced on Illumina HiSeq with paired-end 100 nt or 80 nt reads. 3P-seq libraries were prepared as previously described (Nam et al., 2014). All sequencing data have been deposited in the SRA: SRP041863. Sources for RNA-seq and 3P-seq data from other species are described in the Supplemental Experimental Procedures.
PLAR – Pipeline for lncRNA annotation from RNA-seq data
PLAR is described in detail in the Supplemental Experimental Procedures. Briefly, RNA-seq reads were mapped to the genome in two rounds, followed by transcript assembly and merging of samples and expression-level quantification. Those transcripts with substantial expression levels and no overlap with protein-coding genes were carried forward and tested for their coding potential using three independent methods. Candidate lncRNAs with no detectable coding potential were further filtered against potential artifacts resulting from fragmented genomes or pseudogenic sequences. Full implementation of PLAR is available at http://webhome.weizmann.ac.il/home/igoru/PLAR/.
Clustering of gene expression patterns
To compare expression patterns of lincRNAs and protein-coding genes, hierarchical clustering of the Spearman’s correlations of expression levels was done using R. This analysis was restricted to four eutherian mammals (human, mouse, rabbit and dog). A correlation matrix was generated for lincRNAs or mRNAs that had shared clusters found in all of these species, and a heatmap was generated for this matrix based on row and column means.
Supplementary Material
HIGHLIGHTS.
Hundreds of lncRNAs have homologs with similar expression throughout amniotes.
Gene structure evolves rapidly, and conserved patches are short and have 5′ bias.
Transposable elements often contribute new sequence elements to conserved lncRNAs.
Syntenic counterparts of 100s of mammalian lncRNAs were found in fish and urchin.
Acknowledgments
We thank members of the Ulitsky lab and Alena Shkumatava for useful discussions and comments on the manuscript. I.U. is incumbent of the Robert Edward and Roselyn Rich Manson Career Development Chair and recipient of an Alon Fellowship. D.P.B is an Investigator of the Howard Hughes Medical Institute. This work was supported by grants to I.U. from the Israeli Science Foundation (1242/14 and 1984/14), the I-CORE Program of the Planning and Budgeting Committee and The Israel Science Foundation (grant no 1796/12) and Fritz-Thyssen Foundation and to D.P.B from the NIH (GM067031).
Footnotes
Accession numbers
All sequencing data have been deposited in the SRA: SRP041863.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes & development. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodroff RA, Goodstadt L, Sirey TM, Oliver PL, Davies KE, Green ED, Molnar Z, Ponting CP. Long noncoding RNA genes: conservation of sequence and brain expression among diverse amniotes. Genome biology. 2010;11:R72. doi: 10.1186/gb-2010-11-7-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chureau C, Prissette M, Bourdet A, Barbe V, Cattolico L, Jones L, Eggen A, Avner P, Duret L. Comparative sequence analysis of the X-inactivation center region in mouse, human, and bovine. Genome research. 2002;12:894–908. doi: 10.1101/gr.152902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL, Ponting CP, Stadler PF, Morris KV, Morillon A, et al. The reality of pervasive transcription. PLoS biology. 2011;9:e1000625. doi: 10.1371/journal.pbio.1000625. discussion e1001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, F., the, R.P., Clst. Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Lassmann T, Itoh M, et al. A promoter-level mammalian expression atlas. Nature. 2014;507:462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome research. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Fei T, Verhaak RG, Su Z, Zhang Y, Brown M, Chen Y, Liu XS. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nature structural & molecular biology. 2013;20:908–913. doi: 10.1038/nsmb.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- Goren A, Ram O, Amit M, Keren H, Lev-Maor G, Vig I, Pupko T, Ast G. Comparative analysis identifies exonic splicing regulatory sequences--The complex definition of enhancers and silencers. Molecular cell. 2006;22:769–781. doi: 10.1016/j.molcel.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haerty W, Ponting CP. Unexpected selection to retain high GC content and splicing enhancers within exons of multiexonic lncRNA loci. RNA. 2015 doi: 10.1261/rna.047324.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. Journal of morphology. 1951;88:49–92. [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- He Z, Bammann H, Han D, Xie G, Khaitovich P. Conserved expression of lincRNA during human and macaque prefrontal cortex development and maturation. RNA. 2014;20:1103–1111. doi: 10.1261/rna.043075.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilik IA, Quinn JJ, Georgiev P, Tavares-Cadete F, Maticzka D, Toscano S, Wan Y, Spitale RC, Luscombe N, Backofen R, et al. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Molecular cell. 2013;51:156–173. doi: 10.1016/j.molcel.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan CH, Friedman RC, Ruby JG, Bartel DP. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature. 2011;469:97–101. doi: 10.1038/nature09616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, Yandell M, Feschotte C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS genetics. 2013;9:e1003470. doi: 10.1371/journal.pgen.1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Rinn JL. Transposable elements reveal a stem cell specific class of long noncoding RNAs. Genome biology. 2012;13:R107. doi: 10.1186/gb-2012-13-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachane AN, Harrison PM. Assessing the genomic evidence for conserved transcribed pseudogenes under selection. BMC genomics. 2009;10:435. doi: 10.1186/1471-2164-10-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutter C, Watt S, Stefflova K, Wilson MD, Goncalves A, Ponting CP, Odom DT, Marques AC. Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS genetics. 2012;8:e1002841. doi: 10.1371/journal.pgen.1002841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- Michel F, Westhof E. Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. Journal of molecular biology. 1990;216:585–610. doi: 10.1016/0022-2836(90)90386-Z. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Wakefield MJ, Aken B, Amemiya CT, Chang JL, Duke S, Garber M, Gentles AJ, Goodstadt L, Heger A, et al. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature. 2007;447:167–177. doi: 10.1038/nature05805. [DOI] [PubMed] [Google Scholar]
- Nam JW, Rissland OS, Koppstein D, Abreu-Goodger C, Jan CH, Agarwal V, Yildirim MA, Rodriguez A, Bartel DP. Global analyses of the effect of different cellular contexts on microRNA targeting. Molecular cell. 2014;53:1031–1043. doi: 10.1016/j.molcel.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grutzner F, Kaessmann H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014;505:635–640. doi: 10.1038/nature12943. [DOI] [PubMed] [Google Scholar]
- Pang KC, Frith MC, Mattick JS. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet. 2006;22:1–5. doi: 10.1016/j.tig.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS computational biology. 2006;2:e33. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome research. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JJ, Ilik IA, Qu K, Georgiev P, Chu C, Akhtar A, Chang HY. Revealing long noncoding RNA architecture and functions using domain-specific chromatin isolation by RNA purification. Nature biotechnology. 2014;32:933–940. doi: 10.1038/nbt.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge RM, Chang HY. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. eLife. 2013;2:e00762. doi: 10.7554/eLife.00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler A, Ghanbarian AT, Hurst LD. Purifying selection on splice-related motifs, not expression level nor RNA folding, explains nearly all constraint on human lincRNAs. Molecular biology and evolution. 2014;31:3164–3183. doi: 10.1093/molbev/msu249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela N, Kim E, Ast G. The role of transposable elements in the evolution of non-mammalian vertebrates and invertebrates. Genome biology. 2010;11:R59. doi: 10.1186/gb-2010-11-6-r59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthes P, Kokkinaki M, Nef S, Gnirke A, et al. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell reports. 2013;3:2179–2190. doi: 10.1016/j.celrep.2013.05.031. [DOI] [PubMed] [Google Scholar]
- Tacke R, Manley JL. The human splicing factors ASF/SF2 and SC35 possess distinct, functionally significant RNA binding specificities. The EMBO journal. 1995;14:3540–3551. doi: 10.1002/j.1460-2075.1995.tb07360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, Ronning P, Reuland B, Guenther K, Beadnell TC, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–86. doi: 10.1038/nature13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Bartel DP. lincRNAs: Genomics, Evolution, and Mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–1550. doi: 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Subtelny AO, Koppstein D, Bell GW, Sive H, Bartel DP. Extensive alternative polyadenylation during zebrafish development. Genome research. 2012;22:2054–2066. doi: 10.1101/gr.139733.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic acids research. 2007;35:D88–92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhang J, Zheng H, Li J, Liu D, Li H, Samudrala R, Yu J, Wong GK. Mouse transcriptome: neutral evolution of ‘non-coding’ complementary DNAs. Nature. 2004;431:1. p following 757; discussion following 757. [PubMed] [Google Scholar]
- Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Washietl S, Kellis M, Garber M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome research. 2014;24:616–628. doi: 10.1101/gr.165035.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Cheng EC, Zhong M, Lin H. Retrotransposons and pseudogenes regulate mRNAs and lncRNAs via the piRNA pathway in the germline. Genome research. 2014 doi: 10.1101/gr.180802.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittbrodt J, Shima A, Schartl M. Medaka--a model organism from the far East. Nature reviews. Genetics. 2002;3:53–64. doi: 10.1038/nrg704. [DOI] [PubMed] [Google Scholar]
- Woese CR, Magrum LJ, Gupta R, Siegel RB, Stahl DA, Kop J, Crawford N, Brosius J, Gutell R, Hogan JJ, et al. Secondary structure model for bacterial 16S ribosomal RNA: phylogenetic, enzymatic and chemical evidence. Nucleic acids research. 1980;8:2275–2293. doi: 10.1093/nar/8.10.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.