

Abstract
Hundreds of copy number variants are complex and multi-allelic, in that they have many structural alleles and have rearranged multiple times in the ancestors who contributed chromosomes to current humans. Not only are the relationships of these multi-allelic CNVs (mCNVs) to phenotypes generally unknown, but many mCNVs have not yet been described at the basic levels—alleles, allele frequencies, structural features—that support genetic investigation. To date, most reported disease associations to these variants have been ascertained through candidate gene studies. However, only a few associations have reached the level of acceptance defined by durable replications in many cohorts. This likely stems from longstanding challenges in making precise molecular measurements of the alleles individuals have at these loci. However, approaches for mCNV analysis are improving quickly, and some of the unique characteristics of mCNVs may assist future association studies. Their various structural alleles are likely to have different magnitudes of effect, creating a natural allelic series of growing phenotypic impact and giving investigators a set of natural predictions and testable hypotheses about the extent to which each allele of an mCNV predisposes to a phenotype. Also, mCNVs’ low-to-modest correlation to individual single-nucleotide polymorphisms (SNPs) may make it easier to distinguish between mCNVs and nearby SNPs as the drivers of an association signal, and perhaps, make it possible to preliminarily screen candidate loci, or the entire genome, for the many mCNV–disease relationships that remain to be discovered.
Keywords: multi-allelic copy number variation, association, mCNV, CNV genotyping, optical mapping, ddPCR
Introduction
Human genomes have thousands of deletion and duplication polymorphisms larger than 1 kb. These so-called copy number variations (CNVs) cause many segments (collectively spanning as much as 0.78% of base pairs [1]) to differ in copy number between any two individuals’ genomes and can impact phenotypes by causing gene dosage and structure to vary among individuals. Rare and de novo CNVs have well-known roles in disease; many associate to disease phenotypes with strong odds ratios (2–30) [2–4], though typically with partial penetrance and variable expressivity. However, most of the CNV in any individual’s genome arises from a reservoir of polymorphisms that are common, ancient and stably inherited [5]. A majority of these inherited CNVs are simple, bi-allelic CNVs originating from a single ancestral deletion or duplication. Analyses suggest that the majority of these are benign, with a subset appearing to have modest effects on phenotypes, similar to the effects of other common variants [6].
An intriguing and understudied subset of common CNVs consists of loci that have many structural alleles and have rearranged multiple (perhaps many) times in human ancestors. A recent genome-wide survey based on whole genome sequencing (WGS) data from Phase 1 of the 1000 Genomes Project [7] found 1356 of these CNVs, out of a total of 8659 CNVs found in the genome [8]. These multi-allelic CNVs (mCNVs) vary widely in copy number, in patterns that imply the existence of three, four, five or more segregating alleles. Of the 1356 mCNVs found, 121 appeared to have four or more alleles, and 45 appeared to have five or more [8]. When mCNVs have been visualized by fiber fluorescence in situ hybridization (FISH), they have often been found to involve tandem or inverted duplications of a genomic segment [9–12]. Some of these duplications have been estimated (from sequencing data) to have up to 50 copies, though the great majority appear to be present in copy numbers of 0–12 [6, 8, 13]. Though mCNVs are a minority of all structural variants, they account for 88% of human variation in gene dosage [8]. Furthermore, mCNVs are disproportionately likely to encompass genes, and the great majority of gene-encompassing mCNVs affect the RNA expression levels of the genes they contain [8].
Whereas the analysis of simple forms of CNV is today mature—measurements using molecular analysis (for rare CNVs) or statistical imputation (for common CNVs) are now routine in genetic studies [5, 14–17]—complex and multi-allelic forms of CNV represent a frontier in genome analysis. Not only are the relationships of mCNVs to phenotypes generally unknown, but also most mCNVs still need to be described at the basic levels—alleles, allele frequencies, molecular features—that support genetic study. Fundamental challenges lie in ascertaining the structural forms of each locus, defining the alleles that are present and developing molecular and computational strategies to accurately analyze them with the scale and precision required to conclusively infer their relationships with phenotypes.
Candidate gene studies of mCNV associations
To date, most reported disease-to-mCNV associations have been ascertained through candidate gene studies. As a result, a handful of genes have received most of the research attention, likely because of their already-known or hypothesized roles in diseases of interest. These genes include FCGR3B (binds the Fc region of gamma immunoglobulins), CCL3L1 [ligand of the co-receptor for the human immunodeficiency virus (HIV)], beta-defensins (cluster of microbicidal and cytotoxic peptides), HBA1/2 (α-chain of hemoglobin) and C4 (part of the complement pathway) [18–31]. The cohort sizes in these studies have ranged from 50 to 2807, with a trend toward the initial studies having fewer samples and the attempted replication studies having more (Table 1).
Table 1.
Notable mCNV disease associations and their replication studies
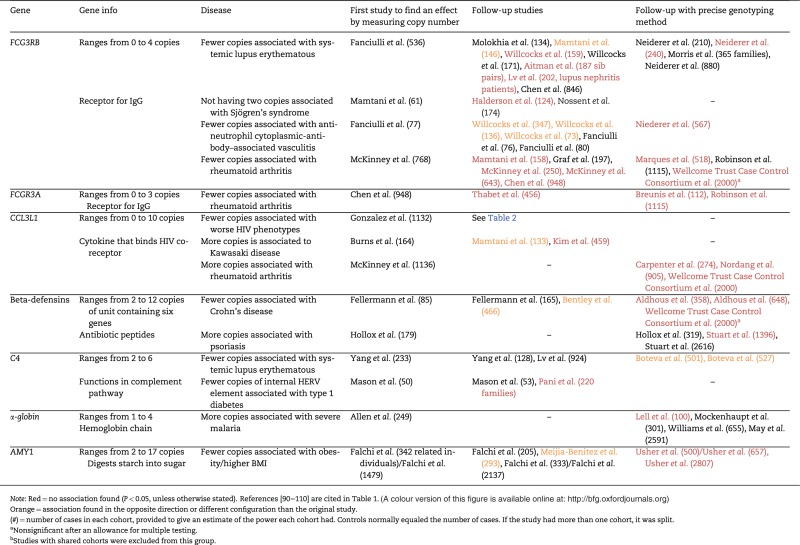 |
To date, the study of mCNV-to-disease associations has resembled the study of single-nucleotide polymorphism (SNP) associations in the pre-genome-wide association study (GWAS) era. Before about 2005, SNP studies focused on candidate genes and variants that were typed in small cohorts. Such studies had a sobering track record: thousands of associations were reported, yet only a handful replicated in other candidate-gene studies or in later well-powered genome-wide association studies [32–34]. In retrospect, science was not good at guessing which genes contribute to genetically complex phenotypes; it took unbiased genome-wide surveys to identify such genes. In the end, publication biases (particularly the increased likelihood of publication and visibility for positive results, relative to negative results), combined with modest statistical thresholds and large numbers of hypotheses being tested across the field, made it likely that many studies would find nominal levels of association—even in the absence of real underlying genetic relationships. These sobering lessons are worth considering when thinking about the trajectory of disease-mCNV analysis.
Like SNP candidate-gene studies a decade ago, only a handful mCNV-to-disease associations have reached the level of acceptance defined by replications in independent cohorts by independent groups of investigators [20, 23, 27, 28, 35]. A compelling example of the challenges of replicating can be found in the Wellcome Trust Case Control (WTCCC) study, which used an array-based CNV genotyping technology to perform a GWAS of thousands of CNVs in eight common diseases. Despite good copy number measurements (as discussed below) and a larger sample size than earlier studies (approximately 2000 cases), the WTCCC study did not replicate three previously published associations (FCGR3B on rheumatoid arthritis, CCL3L1 on rheumatoid arthritis and β-defensins on Crohn’s disease). Non-replication has also vexed other associations, another example being CCL3L1’s impact on HIV-related phenotypes [25].
Case study in replication: CCL3L1 and HIV
One of the most well-known mCNV associations was reported in 2005 in Science [25]. CCL3L1, a gene encoding a ligand to the co-receptor for the HIV virus, was found to range from 0 to 14 copies in diploid genomes. Having a below-average CCL3L1 copy number was found to associate with increased HIV susceptibility and faster progression from HIV+ status to acquired immune deficiency syndrome (AIDS) [25]. After publication, many follow-up studies sought to replicate and expand on the results (Table 2). New phenotypes were tested in the same cohorts and new cohorts were tested for the same associations, each having somewhat limited success, resulting in a complicated pattern of replication and non-replication that hinders final interpretation [11, 36–49]. Separate studies have attempted to track down the causes of the diverging results, concluding that certain analytical practices—such as genotyping cases and controls separately and rounding rough copy number measurements to the nearest integer—are likely to have generated false-positive associations [48–53]. Our own analysis suggests an additional pattern: studies finding positive associations have been published visibly and cited many times, while studies finding negative associations (when published at all) have been less visible. Debate about CCL3L1’s impact on HIV is likely to continue, and examples such as CCL3L1 highlight the need for experimental methods and designs that ensure durable association results.
Table 2.
Results of studies assessing whether CCL3L1 copy number affects HIV-related phenotypes
 |
Toward durable association results for mCNVs
Association analysis with precise molecular data
Negative replication studies reporting null results have often cited the imprecision or inaccuracy of the molecular methods used in the original positive-result study as a reason for non-replication [49, 54–56]. For mCNVs, molecular methods have often tended to yield a rough estimate (rather than a precise measurement) of a gene’s copy number, likely because counting copies is much more challenging than determining the presence or absence of an allele. The difference between a copy number call of 4 and 5 is only 20%, a difference that corresponds to a fraction of a polymerase chain reaction (PCR) cycle, making it difficult for real-time quantitative PCR (qPCR; the most frequently used method for analyzing mCNVs in studies to date) to detect these differences with the accuracy required for successful analysis [35, 50, 51, 54, 57, 58].
With the exception of unusual circumstances, such as somatic mosaicism, the number of copies of a genomic segment within an individual’s genome is always an integer. When measurements of copy number are sufficiently imprecise as to form a continuously varying distribution (i.e. a bell-shaped distribution, rather than a distribution with discrete peaks at integers), these measurements will usually hide technical confounds caused by experimental batch effects, DNA-isolation batch effects and other unknown factors [50, 51, 53, 54, 57] (Figure 1). The pitfalls of continuously varying measurements have long been recognized in SNP analysis [52], and poorly clustering SNP assays are systematically discarded during SNP QC. But the lack of better mCNV data has often meant that a similar level of fastidiousness would mean doing no mCNV study at all.
Figure 1.

Imprecise copy numbers can hide artifacts. When experimental measurements of a gene's copy number in each genome are a rough estimate (A) rather than a more precise, multi-modally distributed measurement (B), confounding technical influences are challenging to recognize. In these simulated data, Groups 1 and 2 (e.g. cases and controls) appear in the first analysis to exhibit different distributions of copy numbers (P = 4.9 × 10−13); the second, more precise analysis shows that the apparent difference between the groups is entirely technical in nature. A confound causing a 10% shift in the copy numbers of the cases is detectable with the precise copy numbers, but may be mistaken for a real effect with the imprecise calls. Note that this confounding occurs even though the measurements by the two methods are broadly correlated with each other (r2 = 0.90). (A colour version of this figure is available online at: http://bfg.oxfordjournals.org)
More precise molecular methods are becoming available, though they have not yet been widely adopted. Notably, the paralog ratio test (PRT), which uses paralogous, copy-number-invariant sequences elsewhere in the genome as embedded controls to carefully calibrate copy number measurements [59], appears to produce highly accurate copy number measurements. PRT has been used effectively in mCNV association studies, often giving copy number measurements with sufficient resolution to detect and remove batch effects [11, 28, 50, 51, 54–56, 58, 60, 61]; in fact, PRT was used to produce one of the most well-supported mCNV-disease results to date, the association of psoriasis with β-defensin gene copy number [28, 60]. We are surprised that PRT has not been more widely adopted, though its application is limited to loci that have copy-number-invariant paralogous sequences elsewhere in the genome.
Another emerging technique, droplet digital PCR (ddPCR) [62, 63], also appears to offer a profound improvement over real-time PCR for mCNV analysis and may be applicable to more genomic loci than PRT. In ddPCR, a PCR reaction with primers and fluorescent probes for each sequence of interest (e.g. a CNV and a two-copy-control locus) is partitioned into thousands of nanoliter-sized droplets at a sufficient dilution that most droplets contain just 0 or 1 copy of the locus of interest. After thermocycling, the number of fluorescent droplets is counted, supporting a calculation of the copy number of the target sequence in the DNA sample. The technique has been used to measure the precise integer copy number of copy-number-variable segments within the 17q21.31 inversion region and several other loci [8, 63–66]. Measurements of mCNVs by ddPCR appear to be strongly supported by analysis of the same samples using WGS: measurements from the two techniques exhibit not just a rough correlation (a standard that does not report on artifactual influences), but more importantly, a precise agreement on the integer copy number present in each genome [8, 66].
As large disease studies based on WGS are just beginning, it is simultaneously becoming possible to accurately detect and measure mCNVs genome-wide using new sequencing analysis methods [8, 67]. Though read depth of coverage has long been known to correlate roughly with the copy number of genomic segments [13], recent analytical innovations allow precise calibration of this signal to meet the exacting standards of mCNV genotyping [8, 67]. These methods can be used to measure any particular locus relatively quickly (after the sequencing has been done) and allow for genotyping refinement, in that certain parameters of the analysis can be adjusted and optimized until the copy numbers cluster at integers [8, 67]; this is sometimes necessary because mCNVs can contain elements that frustrate both computational and PCR-based approaches, such as stretches of extensive homology and varying breakpoints [68]. WGS remains expensive though, so it may be some time before WGS studies reach the sample sizes necessary to discover genetic influences on highly polygenic diseases at the significance thresholds required, given genome-wide multiple hypothesis testing.
Understanding the structural alleles
mCNVs are often complex, involving combinations of duplications, deletions, insertions and inversions [5, 10, 13, 66, 68, 69]. For example, the 17q21.31 inversion region, at which genetic markers associate with female fertility [70], recombination rates [70–72] and neurological diseases [73, 74], has nine structural forms that affect five genes through various numbers of duplications, sequence changes and a megabase-scale inversion [66, 69]. The 17q21.31 locus is one of the only complex CNVs for which a long series of complex structural alleles has been inferred; however, initial investigations into other loci, such as the amylase locus, FCGR3B/3A, CCL3L1 and C4 [9–11, 26, 55, 75], suggest that such complexity might be widespread (Figure 2).
Figure 2.

Examples of the alleles of complex loci. Boettger et al. [66] identified the common structural haplotypes of the 17q21.31 region using sequence analysis and ddPCR; similar conclusions were reached independently by Steinberg et al. [69]. Usher et al. [76] assembled the haplotypes of the amylase locus using ddPCR, sequence analysis and optical mapping; similar conclusions were reached independently by Carpenter et al. [58]. Both Perry et al. [9] and Aklillu et al. [11] performed fiber FISH experiments on the CCL3L1 locus, inferring the haplotypes displayed. (A colour version of this figure is available online at: http://bfg.oxfordjournals.org)
Designing an assay to a single gene within an mCNV without knowing all of the mCNV’s structural forms is analogous to flying blind. Sequence variants that are present on the haplotypes that are not in the human reference sequence can cause inaccurate gene measurements if an assay is in, or crosses a breakpoint of, one such variant. In the case of the amylase and C4 loci, insertions and deletions within the resident genes, as well as the extensive homology of the resident genes, can interfere with the genotyping of a single gene target [10, 27]. In the same vein, at the CCL3L1 locus, a CCL3L pseudogene may interfere with obtaining accurate copy number measurements [51].
Therefore, a challenging yet important first step of any association study will ideally be to identify the actual structural forms of the mCNV of interest. Though this is a challenging problem, it can be assisted by investigations of bacterial artificial chromosomes and cosmids [69, 77, 78], haplotype assembly from sequencing data [66, 79], techniques such as fiber FISH [9], optical mapping [80] or a combination of these approaches. Regardless of the method, knowing the alleles—the fundamental units of most genetic analysis—will be an important basis for conclusions about association.
Though identifying the structural forms of a complex CNV is a challenging problem, the scientific yield will likely reward the effort. Wherever an mCNV influences a disease phenotype, its various structural alleles are likely to have different magnitudes of effect (such as varying odds ratios), creating a natural allelic series of growing phenotypic impact. This could in principle be utilized to help determine whether an mCNV or the sequence variants around it are the true drivers of an association signal—a scientific opportunity that is not possible with most SNPs and bi-allelic CNVs, which often have near perfect linkage disequilibrium (LD) with many other variants that hinders fine-mapping and the evaluation of causality.
In addition, such an allelic series could give investigators a set of natural predictions about the direction of effect and testable hypotheses, about the extent to which each allele of an mCNV predisposes to a phenotype. These natural allelic series would most likely be based on the number of copies of a particular gene. However, a simple relationship to gene copy number may not be the only effect at an mCNV locus. For example, reduced FCGR3B copy number is associated with systemic lupus erythematosus, an effect that appears to be caused by a fusion gene created by the deletion of FCGR3B on one allele [81]. In this case, a lack of an allelic series of growing phenotypic impact based on FCGR3B copy number, which ranges from about 0 to 5, could have assisted in pinpointing the functional variant [81].
Using information in SNPs and haplotypes
The SNPs near mCNVs may, at many loci, offer substantial information that is mostly unexploited [8]. SNP genotyping is a mature, reliable technology that has already been applied to millions of genomes [82, 83]. While an individual SNP cannot serve as a good proxy for a multi-allelic variant, it is nonetheless likely that the individual structural alleles of an mCNV arose on specific SNP haplotypes. Depending on the mutation rate of the mCNV, the frequency of recombination near the mCNV and the age and number of structural alleles at the locus, the structural alleles may continue to bear relationships to surrounding genetic markers [8, 76].
For some mCNVs, it may be possible to impute their alleles from flanking SNP haplotypes; in other words, using the genotypes of the surrounding SNPs, one may be able to estimate the copy number or structural allele present at the mCNV for a given individual [8, 66]. Unlike imputing SNPs, imputing mCNVs tends to be only partially (rather than perfectly) predictive, and its efficacy depends on the mCNV’s evolutionary history—the more alleles, the higher the copy number, and the wider the copy number range, the more limited imputation’s efficacy appears to be [8]. Importantly, a SNP or SNP haplotype’s ability to capture an mCNV should not be thought of as a binary ‘true’ or ‘false’, but as a continuum. With statistical power arising from both r2 and sample size, and with some cohorts having SNP data from as many as 300 000 individuals [83, 84], even a SNP with a low r2 might be used to evaluate the plausibility of an mCNV’s association with a disease.
Case study in next generation association techniques: AMY1 and obesity
Humans have three amylase genes (AMY2B, AMY2A and AMY1) responsible for digesting starch into sugar. Each amylase gene varies widely in copy number, with AMY1 varying from 2 to 17 copies [10, 58, 76, 85], AMY2A from 0 to 4 [58, 76] and AMY2B from 2 to 6 [58, 76]. Higher AMY1 copy number has been observed in three populations with starch-rich ancestral diets [10], and two recent studies from the same group reported that increased copy number of AMY1 decreases the risk of obesity [75, 86], though in different ways: in the initial study, the association involved a shifting of the entire copy-number distribution, but the result in the follow-up study arose entirely from a small subset of samples (all lean) with extremely high AMY1 copy number. Combined, these studies used almost 5000 samples, much larger than the average candidate-gene study; however, they used qPCR, a technique that has been shown, with PRT and fiber FISH, to give imprecise copy numbers at this locus [58].
Two follow-up studies applying analytical principles similar to those outlined in this review, concluded that the pattern of copy-number variation at the locus was different from that reported in the earlier work [58, 76]. Whole genome sequence analysis, ddPCR and PRT each revealed an intriguing distribution of AMY1 copy number in which odd copy numbers are four times more common than even numbers. This distribution had never been detected with qPCR [10, 75], but optical mapping [76] and fiber FISH [58] confirmed the haplotypes inferred from the new analysis. Moreover, analysis using the data from the improved methods showed that some SNPs do correlate (modestly) with the copy number of AMY1, and that if AMY1 copy number influences body mass index (BMI), these SNPs would have been 99.9% likely to associate with BMI in the GIANT consortium GWAS of >300 000 individuals [84], yet did not [76]. In addition, association analyses using the improved molecular methods in three new cohorts with 99% power to detect the reported effect found no association [76].
An exciting future for mCNVs: toward genome-wide studies of mCNVs in disease
As the study of SNPs did over the past 10 years, the study of mCNVs might soon be able to move toward an effective genome-wide model. Two large-scale, array-based studies have addressed the challenges of obtaining genome-wide association information on mCNVs. The WTCCC analyzed approximately 2000 cases for each of eight common diseases, and Zanda et al. analyzed approximately 4000 families with type 1 diabetes [6, 87]. Though the sample sizes were much larger than earlier mCNV studies, they were smaller than what has been required to find most genetic influences on complex, polygenic phenotypes, and the studies found no novel associations. However, both studies identified CNVs at several loci already implicated in GWAS, which serves as an effective positive control [32, 88, 89].
The WTCCC and Zanda studies provide useful knowledge about how analysis methods can find and cope with potential artifacts. CNVs with duplications that had dispersed onto the sex chromosomes caused false associations when the sex ratio was not matched between cases and controls [6]. In addition, whether the DNA was isolated from blood or cultured cells affected the CNV measurement, causing false associations, particularly at the immunoglobulin heavy chain and T-cell receptor loci [6]. Zanda et al. found an additional artifact: age at sampling. Loci that were affected by somatic rearrangements had time in older people to accumulate mutations, thus skewing the result if there are differences in age between cases and controls [87].
Directly measuring mCNVs need not be the only way to scan for phenotype associations genome-wide. With so much SNP information already available, it could be possible to build a genome-wide catalog of SNP-to-mCNV LD relationships and cross reference that with GWAS data. Querying this catalog for mCNV-associated SNPs with a nominal association to a phenotype could serve as preliminary genome-wide survey for mCNV associations. This would allow geneticists to make full use of already available SNP data sets while WGS data accumulates to an amount that enables systematic and well-powered analyses that reach a larger set of mCNVs.
We believe that a large number of mCNV–disease relationships remain to be discovered. Associations in complex, polygenic diseases tend to require very large cohorts (>10 000 samples) to discover novel relationships at genome-wide significance. Disease-mCNV studies on this scale have not been attempted yet. There is reason, though, to expect that such activity will be high-yield; with mCNVs accounting for 88% of human gene dosage variation and shaping RNA expression of the affected genes in almost all cases [8], it is reasonable to expect that there are many undiscovered influences still hiding in our genomes.
Key points
Multi-allelic CNVs (mCNVs) have the potential to affect phenotypes because of their large contribution to gene-dosage variation and their proclivity for recurrent mutation.
mCNVs are complex and have been challenging to measure and characterize experimentally.
The many structural forms of mCNVs and their complex relationships with single-nucleotide polymorphisms (SNPs) and SNP haplotypes can obscure their effects in genome-wide association studies.
Uncertain copy number measurements hide artifacts in association analyses and have likely contributed to false-positive mCNV association results.
New analytical methods, molecular and computational, are starting to enable precise measurements and an understanding of mCNVs that will facilitate more replicable associations and genome-wide scans for association.
Funding
This work was supported by a grant from the National Human Genome Research Institute (R01 HG006855).
Biographies
Christina Usher was a graduate student in Steven McCarroll’s lab, with her research focusing on identifying the structural haplotypes of the amylase locus and their relationship to SNPs and phenotypes. She is now a freelance science writer.
Steven McCarroll is an Associate Professor of Genetics at Harvard Medical School and is the Director of Genetics for the Broad Institute’s Stanley Center for Psychiatric Research. His research focuses on how genetic variation influences molecular-biological phenotypes in cells and clinical phenotypes in populations.
References
- 1.Conrad DF, Pinto D, Redon R, et al. Origins and functional impact of copy number variation in the human genome. Nature 2010;464:704–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med 2010;61:437–55. [DOI] [PubMed] [Google Scholar]
- 3.Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 2012;148:1223–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stankiewicz P, Lupski JR. The genomic basis of disease, mechanisms and assays for genomic disorders. Genome Dyn 2006;1:1–16. [DOI] [PubMed] [Google Scholar]
- 5.McCarroll SA, Kuruvilla FG, Korn JM, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet 2008;40:1166–74. [DOI] [PubMed] [Google Scholar]
- 6.Wellcome Trust Case Control Consortium, Craddock N, Hurles ME, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 2010;464:713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population-scale sequencing. Nature 2010;467:1061–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Handsaker RE, Van Doren V, Berman JR, et al. Large multiallelic copy number variations in humans. Nat Genet 2015;47:296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perry GH, Yang F, Marques-Bonet T, et al. Copy number variation and evolution in humans and chimpanzees. Genome Res 2008;18:1698–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perry GH, Dominy NJ, Claw KG, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet 2007;39:1256–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aklillu E, Odenthal-Hesse L, Bowdrey J, et al. CCL3L1 copy number, HIV load, and immune reconstitution in sub-Saharan Africans, BMC Infect Dis 2013;13:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollox EJ, Armour JA, Barber JC. Extensive normal copy number variation of a beta-defensin antimicrobial-gene cluster. Am J Hum Genet 2003;73:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sudmant PH, Kitzman JO, Antonacci F, et al. Diversity of human copy number variation and multicopy genes. Science 2010;330:641–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K, Li M, Hadley D, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 2007;17:1665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Handsaker RE, Korn JM, Nemesh J, et al. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet 2011;43:269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korn JM, Kuruvilla FG, McCarroll SA, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet 2008;40:1253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weischenfeldt J, Symmons O, Spitz F, et al. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet 2013;14:125–38. [DOI] [PubMed] [Google Scholar]
- 18.Pruitt KD, Brown GR, Hiatt SM, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 2014;42:D756–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen JY, Wang CM, Chang SW, et al. Association of FCGR3A and FCGR3B copy number variations with systemic lupus erythematosus and rheumatoid arthritis in taiwanese patients. Arthritis Rheumatol 2014;66:3113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fanciulli M, Norsworthy PJ, Petretto E, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet 2007;39:721–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mamtani M, Anaya JM, He W, et al. Association of copy number variation in the FCGR3B gene with risk of autoimmune diseases. Genes Immun 2010;11:155–60. [DOI] [PubMed] [Google Scholar]
- 22.McKinney C, Fanciulli M, Merriman ME, et al. Association of variation in Fcgamma receptor 3B gene copy number with rheumatoid arthritis in Caucasian samples. Ann Rheum Dis 2010;69:1711–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allen SJ, O'Donnell A, Alexander ND, et al. alpha+-Thalassemia protects children against disease caused by other infections as well as malaria. Proc Natl Acad Sci USA 1997;94:14736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arason GJ, Kramer J, Blasko B, et al. Smoking and a complement gene polymorphism interact in promoting cardiovascular disease morbidity and mortality. Clin Exp Immunol 2007;149:132–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez E, Kulkarni H, Bolivar H, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 2005;307:1434–40. [DOI] [PubMed] [Google Scholar]
- 26.Mason MJ, Speake C, Gersuk VH, et al. Low HERV-K(C4) copy number is associated with type 1 diabetes. Diabetes 2014;63:1789–95. [DOI] [PubMed] [Google Scholar]
- 27.Yang Y, Chung EK, Wu YL, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet 2007;80:1037–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hollox EJ, Huffmeier U, Zeeuwen PL, et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet 2008;40:23–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burns JC, Shimizu C, Gonzalez E, et al. Genetic variations in the receptor-ligand pair CCR5 and CCL3L1 are important determinants of susceptibility to Kawasaki disease. J Infect Dis 2005;192:344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKinney C, Merriman ME, Chapman PT, et al. Evidence for an influence of chemokine ligand 3-like 1 (CCL3L1) gene copy number on susceptibility to rheumatoid arthritis. Ann Rheum Dis 2008;67:409–13. [DOI] [PubMed] [Google Scholar]
- 31.Fellermann K, Stange DE, Schaeffeler E, et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am J Hum Genet 2006;79:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007;447:661–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirschhorn JN, Lohmueller K, Byrne E, et al. A comprehensive review of genetic association studies. Genet Med 2002;4:45–61. [DOI] [PubMed] [Google Scholar]
- 34.Hirschhorn JN, Altshuler D. Once and again-issues surrounding replication in genetic association studies. J Clin Endocrinol Metab 2002;87:4438–41. [DOI] [PubMed] [Google Scholar]
- 35.Cantsilieris S, White SJ. Correlating multiallelic copy number polymorphisms with disease susceptibility. Hum Mutat 2013;34:1–13. [DOI] [PubMed] [Google Scholar]
- 36.Kulkarni H, Agan BK, Marconi VC, et al. CCL3L1-CCR5 genotype improves the assessment of AIDS Risk in HIV-1-infected individuals. PLoS One 2008;3:e3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahuja SK, Kulkarni H, Catano G, et al. CCL3L1-CCR5 genotype influences durability of immune recovery during antiretroviral therapy of HIV-1-infected individuals. Nat Med 2008;14:413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolan MJ, Kulkarni H, Camargo JF, et al. CCL3L1 and CCR5 influence cell-mediated immunity and affect HIV-AIDS pathogenesis via viral entry-independent mechanisms. Nat Immunol 2007;8:1324–36. [DOI] [PubMed] [Google Scholar]
- 39.Shostakovich-Koretskaya L, Catano G, Chykarenko ZA, et al. Combinatorial content of CCL3L and CCL4L gene copy numbers influence HIV-AIDS susceptibility in Ukrainian children. AIDS 2009;23:679–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meddows-Taylor S, Donninger SL, Paximadis M, et al. Reduced ability of newborns to produce CCL3 is associated with increased susceptibility to perinatal human immunodeficiency virus 1 transmission. J Gen Virol 2006;87:2055–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakajima T, Ohtani H, Naruse T, et al. Copy number variations of CCL3L1 and long-term prognosis of HIV-1 infection in asymptomatic HIV-infected Japanese with hemophilia. Immunogenetics 2007;59:793–8. [DOI] [PubMed] [Google Scholar]
- 42.Shao W, Tang J, Song W, et al. CCL3L1 and CCL4L1: variable gene copy number in adolescents with and without human immunodeficiency virus type 1 (HIV-1) infection. Genes Immun 2007;8:224–31. [DOI] [PubMed] [Google Scholar]
- 43.Kuhn L, Schramm DB, Donninger S, et al. African infants' CCL3 gene copies influence perinatal HIV transmission in the absence of maternal nevirapine. AIDS 2007;21:1753–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rathore A, Chatterjee A, Sivarama P, et al. Association of CCR5-59029 A/G and CCL3L1 copy number polymorphism with HIV type 1 transmission/progression among HIV type 1-seropositive and repeatedly sexually exposed HIV type 1-seronegative North Indians. AIDS Res Hum Retroviruses 2009;25:1149–56. [DOI] [PubMed] [Google Scholar]
- 45.Lee EY, Yue FY, Jones RB, et al. The impact of CCL3L1 copy number in an HIV-1-infected white population. AIDS 2010;24:1589–91. [DOI] [PubMed] [Google Scholar]
- 46.Huik K, Sadam M, Karki T, et al. CCL3L1 copy number is a strong genetic determinant of HIV seropositivity in Caucasian intravenous drug users. J Infect Dis 2010;201:730–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larsen MH, Thorner LW, Zinyama R, et al. CCL3L gene copy number and survival in an HIV-1 infected Zimbabwean population. Infect Genet Evol 2012;12:1087–93. [DOI] [PubMed] [Google Scholar]
- 48.Bhattacharya T, Stanton J, Kim EY, et al. CCL3L1 and HIV/AIDS susceptibility. Nat Med 2009;15:1112–15. [DOI] [PubMed] [Google Scholar]
- 49.Urban TJ, Weintrob AC, Fellay J, et al. CCL3L1 and HIV/AIDS susceptibility. Nat Med 2009;15:1110–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carpenter D, Walker S, Prescott N, et al. Accuracy and differential bias in copy number measurement of CCL3L1 in association studies with three auto-immune disorders. BMC Genomics 2011;12:418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Field SF, Howson JM, Maier LM, et al. Experimental aspects of copy number variant assays at CCL3L1. Nat Med 2009;15:1115–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clayton DG, Walker NM, Smyth DJ, et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat Genet 2005;37:1243–6. [DOI] [PubMed] [Google Scholar]
- 53.Barnes C, Plagnol V, Fitzgerald T, et al. A robust statistical method for case-control association testing with copy number variation. Nat Genet 2008;40:1245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aldhous MC, Abu Bakar S, Prescott NJ, et al. Measurement methods and accuracy in copy number variation: failure to replicate associations of beta-defensin copy number with Crohn's disease. Hum Mol Genet 2010;19:4930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Niederer HA, Willcocks LC, Rayner TF, et al. Copy number, linkage disequilibrium and disease association in the FCGR locus. Hum Mol Genet 2010;19:3282–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nordang GB, Carpenter D, Viken MK, et al. Association analysis of the CCL3L1 copy number locus by paralogue ratio test in Norwegian rheumatoid arthritis patients and healthy controls. Genes Immun 2012;13:579–82. [DOI] [PubMed] [Google Scholar]
- 57.Fode P, Jespersgaard C, Hardwick RJ, et al. Determination of beta-defensin genomic copy number in different populations: a comparison of three methods. PLoS One 2011;6:e16768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carpenter D, Dhar S, Mitchell LM, et al. Obesity, starch digestion and amylase: association between copy number variants at human salivary (AMY1) and pancreatic (AMY2) amylase genes. Hum Mol Genet 2015;24:3472–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Armour JA, Palla R, Zeeuwen PL, et al. Accurate, high-throughput typing of copy number variation using paralogue ratios from dispersed repeats. Nucleic Acids Res 2007;35:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stuart PE, Huffmeier U, Nair RP, et al. Association of beta-defensin copy number and psoriasis in three cohorts of European origin. J Invest Dermatol 2012;132:2407–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson JI, Carr IM, Cooper DL, et al. Confirmation of association of FCGR3B but not FCGR3A copy number with susceptibility to autoantibody positive rheumatoid arthritis. Hum Mutat 2012;33:741–9. [DOI] [PubMed] [Google Scholar]
- 62.Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 2011;83:8604–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mazaika E, Homsy J. Digital Droplet PCR: CNV Analysis and Other Applications, Curr Protoc Hum Genet 2014;82:7.24.1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marques FZ, Prestes PR, Pinheiro LB, et al. Measurement of absolute copy number variation reveals association with essential hypertension. BMC Med Genomics 2014;7:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roberts CH, Jiang W, Jayaraman J, et al. Killer-cell Immunoglobulin-like Receptor gene linkage and copy number variation analysis by droplet digital PCR. Genome Med 2014;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boettger LM, Handsaker RE, Zody MC, et al. Structural haplotypes and recent evolution of the human 17q21.31 region. Nat Genet 2012;44:881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alkan C, Kidd JM, Marques-Bonet T, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet 2009;41:1061–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perry GH, Ben-Dor A, Tsalenko A, et al. The fine-scale and complex architecture of human copy-number variation. Am J Hum Genet 2008;82:685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Steinberg KM, Antonacci F, Sudmant PH, et al. Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat Genet 2012;44:872–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stefansson H, Helgason A, Thorleifsson G, et al. A common inversion under selection in Europeans. Nat Genet 2005;37:129–37. [DOI] [PubMed] [Google Scholar]
- 71.Chowdhury R, Bois PR, Feingold E, et al. Genetic analysis of variation in human meiotic recombination. PLoS Genet 2009;5:e1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fledel-Alon A, Leffler EM, Guan Y, et al. Variation in human recombination rates and its genetic determinants. PLoS One 2011;6:e20321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Skipper L, Wilkes K, Toft M, et al. Linkage disequilibrium and association of MAPT H1 in Parkinson disease. Am J Hum Genet 2004;75:669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 2009;41:1308–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Falchi M, El-Sayed Moustafa JS, Takousis P, et al. Low copy number of the salivary amylase gene predisposes to obesity. Nat Genet 2014;46:492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Usher CL, Handsaker RE, Esko T, et al. Structural forms of the human amylase locus and their relationships to SNPs, haplotypes, and obesity. Nat Genet 2015; 47:921–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Groot PC, Bleeker MJ, Pronk JC, et al. The human alpha-amylase multigene family consists of haplotypes with variable numbers of genes. Genomics 1989;5:29–42. [DOI] [PubMed] [Google Scholar]
- 78.Watson CT, Steinberg KM, Huddleston J, et al. Complete haplotype sequence of the human immunoglobulin heavy-chain variable, diversity, and joining genes and characterization of allelic and copy-number variation. Am J Hum Genet 2013;92:530–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huddleston J, Ranade S, Malig M, et al. Reconstructing complex regions of genomes using long-read sequencing technology. Genome Res 2014;24:688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Teague B, Waterman MS, Goldstein S, et al. High-resolution human genome structure by single-molecule analysis. Proc Natl Acad Sci USA 2010;107:10848–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mueller M, Barros P, Witherden AS, et al. Genomic pathology of SLE-associated copy-number variation at the FCGR2C/FCGR3B/FCGR2B locus. Am J Hum Genet 2013;92:28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ragoussis J. Genotyping technologies for genetic research. Annu Rev Genomics Hum Genet 2009;10:117–33. [DOI] [PubMed] [Google Scholar]
- 83.Speliotes EK, Willer CJ, Berndt SI, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010;42:937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015;518:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Groot PC, Mager WH, Frants RR. Interpretation of polymorphic DNA patterns in the human alpha-amylase multigene family. Genomics 1991;10:779–85. [DOI] [PubMed] [Google Scholar]
- 86.Mejia-Benitez MA, Bonnefond A, Yengo L, et al. Beneficial effect of a high number of copies of salivary amylase AMY1 gene on obesity risk in Mexican children. Diabetologia 2015;58:290–4. [DOI] [PubMed] [Google Scholar]
- 87.Zanda M, Onengut-Gumuscu S, Walker N, et al. A genome-wide assessment of the role of untagged copy number variants in type 1 diabetes. PLoS Genet 2014;10:e1004367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zeggini E, Scott LJ, Saxena R, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 2008;40:638–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McCarroll SA, Huett A, Kuballa P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet 2008;40:1107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aitman TJ, Dong R, Vyse TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature 2006;439:851–5. [DOI] [PubMed] [Google Scholar]
- 91.Haldorsen K, Appel S, Le Hellard S, et al. No association of primary Sjogren's syndrome with Fcgamma receptor gene variants. Genes Immun 2013;14:234–7. [DOI] [PubMed] [Google Scholar]
- 92.Molokhia M, Fanciulli M, Petretto E, et al. FCGR3B copy number variation is associated with systemic lupus erythematosus risk in Afro-Caribbeans. Rheumatology (Oxford) 2011;50:1206–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morris DL, Roberts AL, Witherden AS, et al. Evidence for both copy number and allelic (NA1/NA2) risk at the FCGR3B locus in systemic lupus erythematosus. Eur J Hum Genet 2010;18:1027–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Willcocks LC, Lyons PA, Clatworthy MR, et al. Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J Exp Med 2008;205:1573–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Breunis WB, van Mirre E, Geissler J, et al. Copy number variation at the FCGR locus includes FCGR3A, FCGR2C and FCGR3B but not FCGR2A and FCGR2B. Hum Mutat 2009;30:E640–50. [DOI] [PubMed] [Google Scholar]
- 96.Marques RB, Thabet MM, White SJ, et al. Genetic variation of the Fc gamma receptor 3B gene and association with rheumatoid arthritis. PLoS One 2010;5:e13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thabet MM, Huizinga TW, Marques RB, et al. Contribution of Fcgamma receptor IIIA gene 158V/F polymorphism and copy number variation to the risk of ACPA-positive rheumatoid arthritis. Ann Rheum Dis 2009;68:1775–80. [DOI] [PubMed] [Google Scholar]
- 98.Bentley RW, Pearson J, Gearry RB, et al. Association of higher DEFB4 genomic copy number with Crohn's disease. Am J Gastroenterol 2010;105:354–9. [DOI] [PubMed] [Google Scholar]
- 99.Blasko B, Kolka R, Thorbjornsdottir P, et al. Low complement C4B gene copy number predicts short-term mortality after acute myocardial infarction. Int Immunol 2008;20:31–7. [DOI] [PubMed] [Google Scholar]
- 100.Boteva L, Morris DL, Cortes-Hernandez J, et al. Genetically determined partial complement C4 deficiency states are not independent risk factors for SLE in UK and Spanish populations. Am J Hum Genet 2012;90:445–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lv J, Yang Y, Zhou X, et al. FCGR3B copy number variation is not associated with lupus nephritis in a Chinese population. Lupus 2010;19:158–61. [DOI] [PubMed] [Google Scholar]
- 102.Lv Y, He S, Zhang Z, et al. Confirmation of C4 gene copy number variation and the association with systemic lupus erythematosus in Chinese Han population. Rheumatol Int 2012;32:3047–53. [DOI] [PubMed] [Google Scholar]
- 103.Pani MA, Wood JP, Bieda K, et al. The variable endogenous retroviral insertion in the human complement C4 gene: a transmission study in type I diabetes mellitus. Hum Immunol 2002;63:481–4. [DOI] [PubMed] [Google Scholar]
- 104.Mockenhaupt FP, Ehrhardt S, Gellert S, et al. Alpha(+)-thalassemia protects African children from severe malaria. Blood 2004;104:2003–6. [DOI] [PubMed] [Google Scholar]
- 105.Lell B, May J, Schmidt-Ott RJ, et al. The role of red blood cell polymorphisms in resistance and susceptibility to malaria. Clin Infect Dis 1999;28:794–9. [DOI] [PubMed] [Google Scholar]
- 106.May J, Evans JA, Timmann C, et al. Hemoglobin variants and disease manifestations in severe falciparum malaria. JAMA 2007;297:2220–6. [DOI] [PubMed] [Google Scholar]
- 107.Williams TN, Wambua S, Uyoga S, et al. Both heterozygous and homozygous alpha+ thalassemias protect against severe and fatal Plasmodium falciparum malaria on the coast of Kenya. Blood 2005;106:368–71. [DOI] [PubMed] [Google Scholar]
- 108.Kim HE, Kim JJ, Han MK, et al. Variations in the number of CCL3L1 gene copies and Kawasaki disease in Korean children. Pediatr Cardiol 2012;33:1259–63. [DOI] [PubMed] [Google Scholar]
- 109.Mamtani M, Matsubara T, Shimizu C, et al. Association of CCR2-CCR5 haplotypes and CCL3L1 copy number with Kawasaki Disease, coronary artery lesions, and IVIG responses in Japanese children. PLoS One 2010;5:e11458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nossent JC, Rischmueller M, Lester S. Low copy number of the Fc-gamma receptor 3B gene FCGR3B is a risk factor for primary Sjogren's syndrome. J Rheumatol 2012;39:2142–7. [DOI] [PubMed] [Google Scholar]