

Abstract
Computational chemistry has shown that backbone-alkylated imidazoles ought to be efficient ligands for transition metal catalysts with improved carbene-to-metal donation. In this work, such alkylated imidazoles were synthesized and complexed with silver(I) by means of an eight/nine-step synthetic pathway we devised to access a new class of biologically active silver complexes. The synthesis involves selective iodination of the imidazole backbone, followed by Sonogashira coupling to replace the backbone iodine. The installed alkyne moiety is then subjected to reductive hydrogenation with Pearlman’s catalyst. The imidazole N1 atom is arylated by the palladium-catalyzed Buchwald N-arylation method. The imidazole N3 position was then methylated with methyl iodine, whereupon the synthesis was terminated by complexation of the imidazolium salt with silver(I) oxide. The synthetic pathway provided an overall yield of ≈20 %. The resulting complexes were tested in vitro against HL60 and MOLM-13 leukemic cells, two human-derived cell lines that model acute myeloid leukemia. The most active compounds exhibiting low IC50 values of 14 and 27 μM, against HL60 and MOLM-13 cells, respectively. The imidazole side chain was found to be essential for high cytotoxicity, as the imidazole complex bearing a C7 side chain at the 4-position was four- to sixfold more potent than the corresponding imidazole elaborated with a methyl group.
Keywords: cytotoxicity, imidazoles, leukemia, metallodrugs, silver
Introduction
Numerous imidazoles are known for their potent biological activities,1 including analgesic,2 antibacterial,3 cytotoxic,4 and anticancer properties.5 The imidazole framework is also an integral part of alkaloids,6 and as a precursor for N-heterocyclic carbene (NHC) ligands7 in organometallic catalysis.8 Metallic silver has been known for centuries to possess bactericidal properties and has been used as a treatment for gonorrhea9 before the development of modern antibiotics. In recent years, silver has reemerged as a viable option in the treatment of infectious diseases,10 and silver-based products are currently used as topical antibacterial agents.11 One such example is silver sulfadiazine,12, 13 which has been associated with delayed wound-healing.14
Silver–NHC complexes15, 16 have been found to possess both antibiotic17 and anticancer properties18 and have an untapped potential as drug candidates. Furthermore, studies have shown that synergistic effects that involve both the silver and the NHC ligand play a profound role in the cytotoxicity of such complexes.19
Theoretical calculations performed at our laboratories revealed that substitution on the imidazole backbone is beneficial for ligand-to-metal donation.20 We envisioned that imidazoles elaborated with aliphatic groups on the backbone would have a less labile C–Ag bond and thus be able to afford slow release of silver, an effect that we believe could be beneficial for a potential metallodrug. Although a great number of imidazole-based silver complexes have been reported, only few bear backbone substitution such as bis-methylation,21a bis-chlorination,21b and alkenylation21c (Figure 1). However, the synthesis and biological activity of imidazoles elaborated with aliphatic alkyl chains on the backbone have not been previously reported. To date, such substitution patterns have been difficult to approach, due to lack of synthetic methodology. In fact, Ag–NHC complexes have been realized by elaboration of imidazoles with embedded functionality,21 or classical condensation reactions from linear precursors.22 A major drawback to these strategies is low synthetic flexibility and inferior structural diversity achieved in the target imidazoles.
Figure 1.
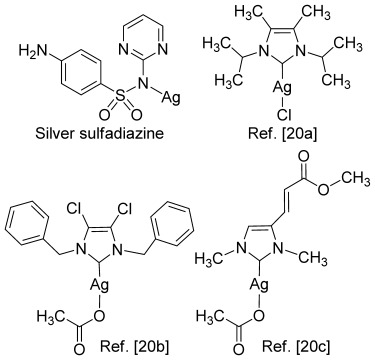
Previously disclosed biological active organosilver complexes.
To address these challenges, we have devised and developed new synthetic methods for the preparation of 4-alkylated imidazole–silver complexes. Along with this work, we have revealed new synthetic methods for imidazole functionalization that include selective halogenation,23 Suzuki cross-coupling,24 Stille coupling,25 and a method for Sonogashira coupling,26 the latter of which was used in the study described herein.
Results and Discussion
Chemistry
A retrosynthetic analysis to our target Ag–NHC is outlined in Scheme 1. We envisioned that the target (TM) could be produced by complexation of imidazolium salt A with a suitable silver salt. Two regioselective N-substitution reactions from 4-(5)-alkylated-1H-imidazole B might lead to the desired salt A. The backbone-substituted imidazole C could be produced from the iodoimidazole E and an appropriate alkyne D through a Sonogashira coupling reaction. The key intermediate E could be produced from commercially available imidazole F.
Scheme 1.
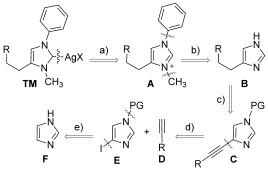
Retrosynthetic analysis leading to 4-substituted imidazolium silver complexes TM.
To directly compare the effect on the activity of the complexes with increasing length of the side chains, we devised two different target molecules, NHC-1 (Scheme 2) and NHC-2 (Scheme 3), which differ by one feature, namely the substituent at the imidazole backbone position 4; NHC-1 contains a methyl group, and NHC-2 contains a heptyl group. The two NHC–silver complexes of this study can be prepared via two different pathways. Because 4-methylimidazole 1 is commercially available, a short pathway leading to NHC-1 was established, a synthesis that comprises a) N-arylation,27 b) N-methylation,28 and c) complexation with silver29 to obtain the NHC-1 silver complex. The overall sequence provides a yield of 30 %, which corresponds to a mean step yield of 67 %.
Scheme 2.
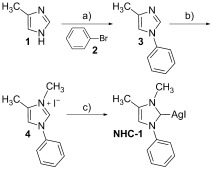
Synthesis of N-1-phenyl-N-3-methyl-4-methylimidazol-2-yliden silver iodide NHC-1. Reagents and conditions: a) PhBr (2), Pd2(dba)3, Me4tBuXPhos, K2HPO4, toluene, 120 °C, 5 h, 87 %; b) MeI, THF, reflux, 4 h, 62 %; c) Ag2O, CH2Cl2, RT, 3 h, 56 %.
Scheme 3.
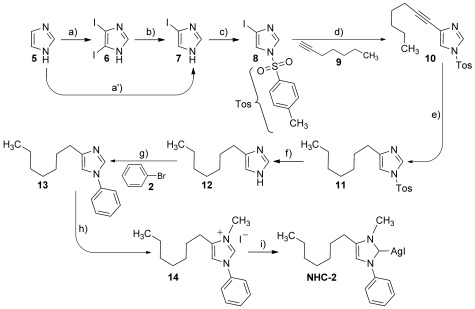
Synthesis of N-1-phenyl-N-3-methyl-4-heptylimidazol-2-yliden silver iodide NHC-2. Reagents and conditions: a′) DIH, H2SO4 (cat.), H2O, 0 °C, 81 %; a) I2, KI, NaOH, RT, 24 h, b) Pd(OAc)2 (0.15 %), XPhos, K2HPO4, MeOH, H2O, reflux, 90 min, (quant.); c) TosCl, NEt3, THF, RT, 24 h, 68 %; d) Pd(PPh3)4, CuI, NEt3, DMF, MW, 80 °C, 30 min, 79 %; e) H2 (1 atm), Pd(OH)2/C, MeOH, RT, 24 h, 92 %; f) HCl, MeOH, reflux, 2 h, 98 %; g) PhBr (2), Pd2(dba)3, Me4tBuXPhos, K2HPO4, toluene, 120 °C, 5 h, 78 %; h) MeI, THF, reflux, 4 h, 57 %; i) Ag2O, CH2Cl2, RT, 3 h, 90 %.
The devised synthesis leading to NHC-2 (Scheme 3) commenced with the preparation of N-toluenesulfonyl-4-iodoimidazole 4, which served as a key intermediate for the synthesis of backbone-alkylated imidazoles. The synthetic pathway 1→4 involves a di-iodination step (a) followed by selective de-iodination (b), or a selective mono-iodination step (a′). The 4-iodinated imidazole is then subjected to the introduction of an auxiliary group at the N-tosylation step (c).30 With the key substrate 4 in hand, we used our recently disclosed method for Sonogashira coupling26 to perform the desired C–C coupling reaction of step (d). Reduction of the alkyne bond of 10 was performed in excellent yield (92 %) by means of Pearlman’s catalyst (Pd(OH)2/C) in methanol under a hydrogen atmosphere. The following step, removal of the toluenesulfonyl auxiliary group, was performed by treatment with concentrated hydrochloric acid in methanol at reflux in excellent yield (98 %). The liberated imidazole derivative 12 was N-arylated [step (g)] by using bromobenzene in toluene with Pd2(dba)3 and Me4tBuXPhos as ligand27 in good yield (78 %). The last organic reaction step of the synthetic pathway involved conversion of the imidazole derivative into an imidazolium salt28 14 from the N-aryl-4-subsituted imidazole derivative 13 by reacting with methyl iodide at reflux in THF to obtain the N1-phenyl-N3-methyl-4-methylimidazolium iodine salt 14 (57 %). Finally, the target silver salts were prepared by using silver(I) oxide in dichloromethane.29 The nine-step synthesis providing NHC-2 afforded an overall yield of 19 %, which corresponds to a mean step yield of 81 %.
Biology
The cytotoxic potential of metallodrugs may be influenced by their capacity to release metals from the associated auxiliary ligand. We hypothesized that the varied nature of the R group of the 4-substituted imidazoles may further impact the cytotoxic potential of the compounds. To compare the biological activity of the compounds, we incubated NHC-1 and NHC-2 with the human acute myeloid leukemia cell lines HL60 and MOLM-13. Following 24 h incubation, the WST1 viability assay was performed, revealing the estimated IC50 values of the compounds to vary as a function of both the side chain R group and the cell type used (Figure 2). The p53-null cell line, HL60, proved more sensitive to both compounds. NHC-1 showed an IC50 value of 78 μM in HL60 cells, compared with an IC50 value of 123 μM in MOLM-13 cells. A similar trend was observed for the more potent NHC-2 (HL60 IC50: 14 μM; MOLM-13 IC50: 27 μM). To confirm the compounds are truly cytotoxic and not only antiproliferative, we performed nuclear staining with Hoechst 33342 in both cell lines after 24 h incubation (NHC-1 at 100 μM, NHC-2 at 30 μM). Condensed and fragmented nuclei were observed in both cell lines and are characteristic of apoptosis (Figure 2).
Figure 2.
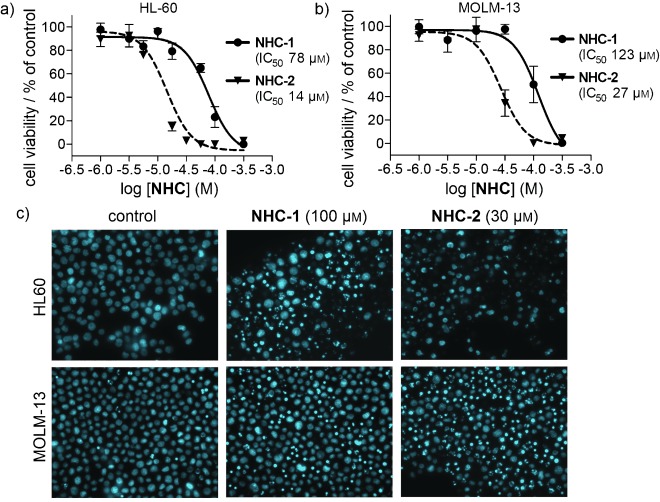
NHC-1 and NHC-2 are cytotoxic in leukemia cell lines. a) HL60 and b) MOLM-13 cells were treated with NHC-1 and NHC-2 for 24 h, and cell viability was determined by WST1 assay to generate dose–response curves. Experiments were performed in three independent replicates, and data are the mean±SD. c) HL60 and MOLM-13 cells were treated with NHC-1 (100 μM) and NHC-2 (30 μM) as indicated for 24 h, and nuclear morphology was determined by Hoechst 33342 staining to identify dead cells.
Using the WST1 assay to compare cell viability at 4, 12, and 24 h, we discovered the compounds (NHC-1 at 100 μM and NHC-2 at 20 μM) to induce rapid cell death. For both cell lines and complexes the majority of cell death was observed after 4 or 12 h (Figure 3). The rapidity of death was best exemplified in the HL60 cell line. Finally, we performed flow cytometry combined with Annexin V staining in HL60 cells treated with NHC-1, NHC-2, or the chemotherapeutic pyrimidine analogue cytarabine (arabinofuranosyl cytidine) for comparison. Cell viability was determined by forward and side scatter properties (Figure 4). Exposure of the lipid membrane component phosphatidylserine (PS) allows its staining with Annexin V; this is indicative of apoptosis. Furthermore, PS exposure aids to effectively eliminate large numbers of dying cells from the systemic circulation without releasing noxious intracellular material.31 Significant PS staining was observed exclusively in HL60 cells treated with NHC-1 and NHC-2, but was absent in cytarabine-treated cells. Our experiments confirm the cytotoxic potential of the silver–imidazole complexes and suggest that cell death in HL60 cells is mechanistically distinct from that which occurs in cells treated with the chemotherapeutic cytarabine.
Figure 3.
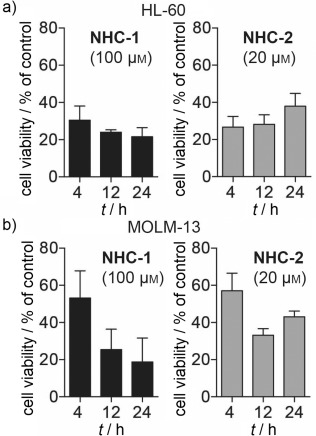
NHC-1 and NHC-2 induce rapid cell death in leukemia cell lines. a) HL60 cells (TP53-null, FLT3 wild-type) were treated with NHC-1 (100 μM) and NHC-2 (20 μM) for 4, 12, and 24 h, and viability was determined by WST1 cell viability assay. b) MOLM-13 cells (TP53 wild-type, FLT3-internal tandem mutation) were treated with NHC-1 (100 μM) and NHC-2 (20 μM) for 4, 12, and 24 h, and viability was determined by WST1 cell viability assay. Experiments were performed in three independent replicates, and data are the mean±SD.
Figure 4.
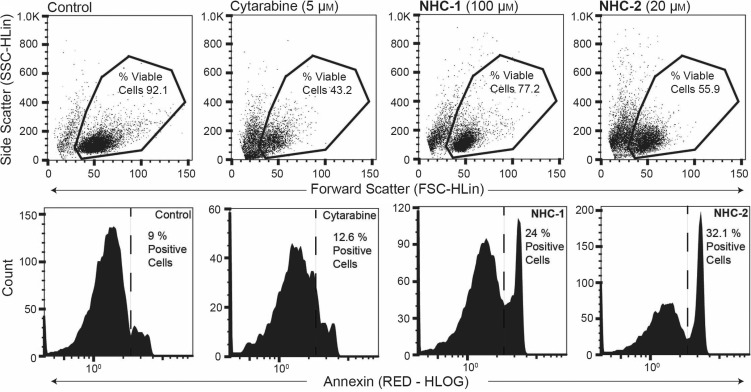
NHC-1 and NHC-2 induce expression of cell-surface phosphatidylserine (PS) in HL60 cells. Cells were treated with NHC-1 (100 μM), NHC-2 (20 μM), or cytarabine (5 μM) for 24 h. Cell viability was determined by comparing the forward scatter and side scatter properties with those of untreated control cells using flow cytometry, as illustrated by figures in the upper row. Gated events represent viable cells. PS expression was determined by Annexin V staining and analysis by flow cytometry as illustrated by histograms in the lower row. Events to the right of the dashed lines are considered positive for Annexin V as determined by comparison with unstained control cells. All analyzed cells are taken from events deemed viable.
Conclusions
A novel class of Ag–NHC complexes encompassing imidazoles furnished with alkyl side chains on the backbone 4-position have been realized by de novo synthesis using new methods developed in our research group. Two Ag–NHC complexes were prepared with different side chains: NHC-1 (methyl) and NHC-2 (heptyl). Both complexes were found to be potently cytotoxic against two human leukemia cell lines, HL60 and MOLM-13, in the micromolar range. IC50 values for NHC-2 were 14 and 27 μM, respectively, and are motivation for further development as an anticancer therapy. As the demand for novel alternative cancer therapeutics remains unmet, metallodrugs are an increasingly important compound class for investigation. The unique anti-leukemic properties of the Ag–NHC complexes described herein further underscore the value of exploring novel synthetic organometallic chemistry in drug development.
Experimental Section
Chemistry
GC analyses were performed on a capillary gas chromatograph equipped with a fused silica column (l: 25 m, i.d.: 0.20 mm, film thickness: 0.33 μm) at a helium pressure of 200 kPa, split less/split injector and flame ionization detector. DART-MS spectra were obtained using PEG as an internal standard under positive ionization mode with a ToF mass analyzer. 1H and 13C NMR spectra were recorded on instruments operating at 400 and 150 MHz, respectively. Chemical shifts were referenced to the deuterated solvent used in that experiment. All melting points are uncorrected. Synthesis of precursors 2, 3, 4, and 10 were reported previously. The microwave-assisted experiments were performed with a Biotage Initiator Sixty EXP Microwave System operating at 0–400 W at 2.45 GHz. The instrument operates in the temperature range 40–250 °C, a pressure interval of 0–20 bar (2 MPa, 290 psi) with reactor vial volumes of 0.2–20 mL. Multiple attempts to grow crystals of NHC-1 and NHC-2 suitable for X-ray analysis were performed in various solvent systems, but unfortunately only amorphous material was obtained in most cases.
N-Toluenesulfonyl-4-heptylimidazole 11. Imidazole (0.10 g) and Pd(OH)2/C (15 % w/w) were transferred to a round-bottom flask (50 mL) equipped with a magnetic stir bar. MeOH (25 mL) was added, and the flask was evacuated under reduced pressure and flushed with H2 from a balloon three times. The reaction mixture was stirred vigorously at room temperature for 24 h. The post-reaction mixture was filtered through a pad of Celite that was subsequently washed with multiple small portions of MeOH. The solvent was evaporated to give the product as a tan oil in 92 % yield (purity: ≥95 % as determined by GC) without need for further purification. 1H NMR (CDCl3): δ=7.91 (s, 1 H), 7.79 (d, 2 H, J=8.0 Hz), 7.34 (d, 2 H, J=7.0 Hz), 2.48 (t, 2 H, J=7.4 Hz), 1.57 (2 H, m), 1.23 (8 H, m), 0.86 ppm (t, 3 H, J=7.1 Hz); 13C NMR (CDCl3): δ=146.1, 136.3, 135.4, 130.5, 127.4, 112.9, 31.9, 29.3, 29.1, 28.7, 28.3, 22.7, 21.8, 14.2 ppm; HRMS (DART): m/z [M+H]+ calcd for C17H25N2O2S: 321.16367, found: 321.16367.
4-(5)-Heptyl-1H-imidazole 12. Imidazole (2.94 mmol, 0.20 g) was dissolved in MeOH (20 mL) in a round-bottom flask (25 mL). HCl (concd, 1 mL) was added to this mixture in one portion. The reaction mixture was held at reflux for 2 h, whereupon the MeOH was evaporated and HCl (3 M, 10 mL) was added. The resulting mixture was extracted with Et2O (2×20 mL), and the organic phases discarded. The aqueous phase was made alkaline with NaOH (4 M) and again extracted with Et2O (3×20 mL). The organic extracts were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to provide the product at high purity. The product was isolated as a tan oil in 98 % yield. 1H NMR (CDCl3): δ=11.07 (1 H, m, br), 7.64 (1 H, s), 6.76 (1 H, s), 2.59 (t, 2 H), 1.61 (m, 2 H), 1.26 (m, 8 H), 0.85 ppm (t, 3 H); 13C NMR (CDCl3): δ=137.0, 134.2, 117.7, 31.9, 29.4, 29.4, 29.2, 26.6, 22.8, 14.2 ppm; HRMS (DART): m/z [M+H]+ calcd for C10H19N2: 167.15482, found: 167.15466.
General procedure for Pd-catalyzed N-arylation coupling reaction between 4-(5)-alkyl-1H-imidazoles and bromobenzene.27 To an oven-dried tube was transferred imidazole (1.2 mmol), K3PO4 (424 mg, 2.0 mmol), and bromobenzene (2.0 mmol). The vial was sealed with a septum and carefully flushed with argon through the septum. A second oven-dried tube was charged with Pd2(dba)3 (0.0125 mmol) and Me4tBuXPhos (0.025 mmol) before it was sealed with a septum and flushed with argon. The catalyst was dissolved in a mixture of anhydrous toluene (0.83 mL) and anhydrous 1,4-dioxane (0.17 mL), and the resulting dark-purple mixture was stirred at 120 °C for 3 min, at which point the color of the mixture turned to red–brown. The catalyst was then transferred to the first vial, and the reaction mixture was heated at 120 °C for 5 h. At the end of the reaction time, the mixture was cooled to room temperature, diluted with EtOAc (10 mL), washed with brine (2 mL), and dried over MgSO4. The drying agent was filtered off, and the organic solvent was removed under reduced pressure. The crude product was then purified by flash chromatography with the eluent systems specified below.
N-Phenyl-4-methylimidazole 3. Isolated as a yellow oil by silica gel column chromatography [EtOAc/hexanes (4:6)→(1:1)] in 87 % yield. 1H NMR (CDCl3): δ=7.74 (1 H, s), 7.44 (2 H, t, J=7.6 Hz), 7.33 (3 H, m), 6.99 (s, 1 H), 2.29 ppm (s, 3 H); 13C NMR (CDCl3): δ=139.6, 134.6, 134.7, 129.9, 127.1, 121.1, 114.7, 13.8 ppm.
N-Phenyl-4-heptylimidazole 13. The product was isolated by flash silica gel chromatography [EtOAc/hexanes (1:9)→(1:1)] as pale crystals in 78 % yield. Rf=0.58 [EtOAc/hexanes (1:1)]; mp: 53.9–55.0 °C; 1H NMR (CDCl3): δ=7.77 (s, 1 H), 7.45 (t, 2 H, J=7.5 Hz), 7.33 (m, 3 H, J=8.3 Hz), 7.00 (s, 1 H), 2.62 (t, 2 H, J=7.8 Hz), 1.69 (t, 2 H, J=7.4 Hz), 1.31 (m, 8 H), 0.87 ppm (t, 3 H, J=7.4 Hz); 13C NMR (CDCl3): δ=144.8, 137.7, 134.7, 129.9, 127.2, 121.3, 114.2, 320, 29.5, 29.5, 29.5, 29.3, 28.6, 22.8, 14.3 ppm; HRMS (DART): m/z [M+H]+ calcd for C16H23N2: 243.18558, found: 243.1860.
General procedure for N-3-methylation of N-1-phenyl-4-alkylimidazole.28 N-phenyl-4-alkylimidazole (2.0 mmol) was dissolved in THF (15 mL). Methyl iodide (4.00 mmol) was added in one portion, and the reaction mixture was held at reflux for 4 h. The post-reaction mixture was allowed to cool, resulting in crystallization of the product which was filtered, washed with small portions of hexane, and air-dried to furnish the product.
N-1-Phenyl-N-3-methyl-4-methylimidazolium iodide. Isolated as a white solid in 62 % yield. 1H NMR (CDCl3): δ=10.12 (s, 1 H), 7.65 (d, 2 H, J=7.8 Hz), 7.60 (s, 1 H), 7.42 (m, 3 H), 3.98 (s, 3 H), 2.37 ppm (s, 3 H); 13C NMR (CDCl3): δ=135.2, 134.5, 132.8, 130.7, 130.3, 122.0, 117.8, 34.9, 9.7 ppm; HRMS (ESI): m/z [M]+ calcd for C11H13N2: 173.10787, found: 173.10779; [M2I]+ calcd for C22H26N4I: 473.12021, found: 473.12594.
N-1-Phenyl-N-3-methyl-4-heptylimidazolium iodide. Isolated as a tan solid in 57 % yield. 1H NMR (CDCl3): δ=10.60 (s, 1 H), 7.74 (d, 2 H, J=7.9 Hz), 7.54 (m, 3 H), 4.12 (s, 3 H), 2.69 (t, 2 H, J=8.0 Hz), 1.71 (m, 3 H), 1.35 (m, 7 H), 0.87 ppm (t, 3 H, J=6.9 Hz); 13C NMR (CDCl3): δ=137.3, 135.9, 134.7, 130.8, 130.4, 122.1, 116.7, 34.9, 31.7, 29.3, 29.0, 27.3, 23.9, 22.8, 14.2 ppm; HRMS (DART): m/z [M+H−I]+ calcd for C17H25N2: 257.20177, found: 257.20179.
General procedure for the synthesis of imidazole-based silver complexes.29 Imidazolium salt (1.77 mmol) was dissolved in CH2Cl2 (15 mL), and silver(I) oxide (0.89 mmol) was added in one portion. The black reaction mixture was stirred for 2 h 25 min, at which point the mixture became pale brown. The solution was poured into a beaker containing hexanes (100 mL), resulting in precipitation of a crystalline solid. This was filtered and recrystallized from CH2Cl2 to furnish the silver complexes.
N-1-Phenyl-N-3-methyl-4-methylimidazol-2-yliden silver(I) iodide. Isolated as a white solid in 56 % yield after pooling three crops. 1H NMR (CDCl3): δ=7.52 (m, 2 H), 7.39 (m, 3 H), 6.98 (s, 1 H), 3.82 (s, 3 H), 2.29 ppm (s, 3 H); 13C NMR (CDCl3): δ=140.4, 131.3, 129.8, 1286, 124.1, 119.0, 36.9, 10.0 ppm; HRMS (DART): m/z [M+H−AgI]+ calcd for C11H13N2: 173.10732, found: 173.10215.
N-1-Phenyl-N-3-methyl-4-heptylimidazol-2-yliden silver(I) iodide. Isolated as a white solid in 90 % yield. 1H NMR (CDCl3): 7.54 (d, 2 H), 7.42 (m, 3 H), 6.95 (s, 1 H), 3.84 (s, 3 H), 2.58 (m, 2 H), 1.65 (m, 2 H), 1.30 (m, 8 H), 0.89 ppm (t, 3 H); 13C NMR (CDCl3): δ=182.6, 140.4, 136.0, 129.9, 128.7, 124.0, 118.1, 36.7, 31.8, 29.3, 29.1, 27.8, 24.5, 22.8, 14.2 ppm; HRMS (DART): m/z [M−AgI]+ calcd for C17H25N2: 257.20177, found: 257.20156.
Biology
The human cell lines MOLM-13 and HL60 were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in RPMI 1640 (Invitrogen), containing 10 % heat-inactivated fetal bovine serum (GE Healthcare, Life Sciences), 2 mm l-glutamine, and 50 U mL−1 penicillin/streptomycin (Sigma–Aldrich). Evaluation of viability/apoptosis was performed as described previously.32, 33 Cell analysis after drug treatment (2×105 cells per mL) was carried out by fixing cells in 8 % formaldehyde in PBS, DNA-specific staining with Hoechst 33342 (Invitrogen; 10 μg mL−1), followed by counting of normal and fragmented/condensed cell nuclei in an inverse fluorescence microscope (Zeiss Axio Vert.A1), or by flow cytometric analysis and Annexin staining. Annexin staining (Invitrogen) was performed in accordance with the manufacturer’s recommended procedure and run on the Guava easyCyte flow Cytometer (EMD Millipore). The WST1 assay cell proliferation reagent (Life Sciences) was used in accordance with the manufacturer’s procedure, followed by respective reading of luminescence and absorbance (Spectra Max Gemini EM, Molecular Devices). All cell viability assays were performed in flat-bottomed 96- or 24-well tissue culture test plates.
Acknowledgments
A.H.S. is grateful to the University of Bergen Department of Chemistry for research fellowship funding. B.T.G. was supported by a grant from the Norwegian Cancer Society with Solveig and Ove Lund’s legacy. The students of the 2013 fall semester of our special topic course on organic synthesis and spectroscopy are acknowledged for reproducing steps of the devised synthetic plan.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Narasimhan B, Sharma D, Kumar P. Med. Chem. Res. 2011;20:1119. doi: 10.1016/j.ejmech.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Uçucu O, Karaburun NG, Işikdağ IS. Farmaco. 2001;56:285. doi: 10.1016/s0014-827x(01)01076-x. [DOI] [PubMed] [Google Scholar]
- 3.Antolini M, Bozzoli A, Ghiron C, Kennedy G, Rossi T, Ursini A. Bioorg. Med. Chem. Lett. 1999;9:1023. doi: 10.1016/s0960-894x(99)00112-2. [DOI] [PubMed] [Google Scholar]
- 4.Lindel T, Jensen PR, Fenical W, Long BH, Casazza AM, Carboni J, Fairchild CR. J. Am. Chem. Soc. 1997;119:8744. [Google Scholar]
- 5.Wanner MJ, Koom GJ. Chem. Soc. Perkin Trans. 1. 2002:1877. [Google Scholar]
- 6.Bjørsvik HR, Sandtorv AH. In: Marine Sponges. 2. Atta-ur-Rahman, editor. Vol. 42. Amsterdam: Elsevier; 2014. p. 33. [Google Scholar]
- 7a.Hu X, Tang Y, Gantzel P, Meyer K. Organometallics. 2003;22:612. [Google Scholar]
- 7b.Chen W, Wu B, Matsumoto K. J. Organomet. Chem. 2002;654:233. [Google Scholar]
- 7c.Nolan SP, editor. Synthesis. Weinheim: Wiley-VCH; 2006. [Google Scholar]
- 7d.Samantaray MK, Katiyar V, Pang K, Nanavati H, Ghosh P. J. Organomet. Chem. 2007;692:1672. [Google Scholar]
- 7e.Kuhn O. Functionalised N-Heterocyclic Carbene Complexes. Wiley; 2010. [Google Scholar]
- 7f.Kühl O. Chem. Soc. Rev. 2007;36:592. doi: 10.1039/b603765h. [DOI] [PubMed] [Google Scholar]
- 8.Wang F, Liu LJ, Wang W, Li S, Shi M. Coord. Chem. Rev. 2012;256:804. [Google Scholar]
- 9.Mirsattari SM, Hammond RR, Sharpe MD, Leung FY, Young GB. Neurology. 2004;62:1408. doi: 10.1212/01.wnl.0000120671.73335.ec. [DOI] [PubMed] [Google Scholar]
- 10.Barillo DJ, Marx DE. Burns. 2014;40:3–8. , S. [Google Scholar]
- 11.Atiyeh BS, Costagliola M, Hayek SN, Dibo SA. Burns. 2007;33:139. doi: 10.1016/j.burns.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 12.Fox CL. Modern Treatment. Harper & Row, New York: Hoeber Medical Division; 1967. [Google Scholar]
- 13.Miller AC, Rashid MR, Falzon L, Elamin EM, Zehtabchi S. J. Am. Acad. Dermatol. 2012;66:159. doi: 10.1016/j.jaad.2010.06.014. , e. [DOI] [PubMed] [Google Scholar]
- 14.Lee CAR, Leem H, Lee J, Park KC. Biomaterials. 2005;26:4670. doi: 10.1016/j.biomaterials.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 15.Liu A, Zhang X, Chen W, Qiu H. Inorg. Chem. Commun. 2008;11:1128. [Google Scholar]
- 16a.Garrison JC, Youngs WJ. Chem. Rev. 2005;105:3978. doi: 10.1021/cr050004s. [DOI] [PubMed] [Google Scholar]
- 16b.Hindi KM, Panzer MJ, Tessier CA, Cannon CL, Youngs WJ. Chem. Rev. 2009;109:3859. doi: 10.1021/cr800500u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16c.Patil S, Deally A, Gleeson B, Müller-Bunz H, Paradisi F, Tacke M. Appl. Organomet. Chem. 2010;24:781. [Google Scholar]
- 17a.Kascatan-Nebioglu A, Panzner MJ, Tessier CA, Cannon CL, Youngs WJ. Coord. Chem. Rev. 2007;251:884. [Google Scholar]
- 17b.Roland S, Jolivalt C, Cresteil T, Eloy L, Bouhours P, Hequet A, Mansuy V, Vanucci C, Paris JM. Chem. Eur. J. 2011;17:1442. doi: 10.1002/chem.201002812. [DOI] [PubMed] [Google Scholar]
- 17c.Streciwilk W, Cassidy J, Hackenberg F, Müller-Bunz H, Paradisi F, Tacke M. J. Organomet. Chem. 2014;749:88. [Google Scholar]
- 18. For selected examples of Ag(I)–imidazolium complexes with anticancer properties, see:
- 18a.Ray S, Mohan R, Singh JK, Samantaray MK, Shaikh MM, Panda D, Ghosh P. J. Am. Chem. Soc. 2007;129:15042. doi: 10.1021/ja075889z. [DOI] [PubMed] [Google Scholar]
- 18b.Medvetz DA, Hindi KM, Panzner MJ, Ditto AJ, Yun YH, Youngs WJ. Met.-Based Drugs. 2008:384010. doi: 10.1155/2008/384010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18c.Teyssot ML, Jarrousse AS, Manin M, Chevry A, Roche S, Norre F, Beudoin C, Morel L, Boyer D, Mahiou R, Gautier A. Dalton Trans. 2009:6894. doi: 10.1039/b906308k. [DOI] [PubMed] [Google Scholar]
- 18d.Wang CH, Shih WC, Chang HC, Kuo YY, Hung WC, Ong TG, Li WS. J. Med. Chem. 2011;54:5245. doi: 10.1021/jm101096x. [DOI] [PubMed] [Google Scholar]
- 18e.Eloy L, Jaarousse AS, Teyssot ML, Gautier A, Morel L, Jolivalt C, Cresteil T, Roland S. ChemMedChem. 2012;7:805. doi: 10.1002/cmdc.201200055. [DOI] [PubMed] [Google Scholar]
- 18f.Liu W, Gust R. Chem. Soc. Rev. 2013;42:755. doi: 10.1039/c2cs35314h. ; For a recent review see: [DOI] [PubMed] [Google Scholar]
- 18g.Budagumpi S, Haque RA, Endud S, Rehman GU, Salman AW. Eur. J. Inorg. Chem. 2013:4367. [Google Scholar]
- 18h.Banti CN, Hadjikakou SK. Metallomics. 2013;5:569. doi: 10.1039/c3mt00046j. [DOI] [PubMed] [Google Scholar]
- 19a.Monteiro DCF, Phillips RM, Crossley BD, Fielden J, Willans CE. Dalton Trans. 2012;41:3720. doi: 10.1039/c2dt12399a. [DOI] [PubMed] [Google Scholar]
- 19b.Padmaja P, Rao GK, Indrasena A, Reddy BVS, Patel N, Shaik AB, Reddy N, Dubey PK, Bhadra MP. Org. Biomol. Chem. 2015;13 doi: 10.1039/c4ob02015d. [DOI] [PubMed] [Google Scholar]
- 20.Occhipinti G, Bjørsvik HR, Jensen VR. J. Am. Chem. Soc. 2006;128:6952. doi: 10.1021/ja060832i. [DOI] [PubMed] [Google Scholar]
- 21a.de Frémont P, Scott NM, Stevens ED, Ramnial T, Lightbody OC, Macdonald CLB, Clyburne JAC, Aberneth CD, Nolan SP. Organometallics. 2005;24:6301. [Google Scholar]
- 21b.Patil S, Claffey J, Deally J, Hogan M, Gleeson B, Menéndez Méndez LM, Müller-Bunz H, Paradisi F, Tacke M. Eur. J. Inorg. Chem. 2010:1020. [Google Scholar]
- 21c.Hindi KM, Siciliano TJ, Durmus S, Panzner MJ, Medvetz DA, Reddy DV, Hogue LA, Hovis CE, Hilliard JK, Mallet RJ, Tessier CA, Cannon CL, Youngs WJ. J. Med. Chem. 2008;51:1577. doi: 10.1021/jm0708679. [DOI] [PubMed] [Google Scholar]
- 21d.Patil S, Dietrich K, Deally A, Gleeson B, Müller-Bunz H, Paradisi F, Tacke M. Helv. Chim. Acta. 2010;93:2347. [Google Scholar]
- 21e.Patil S, Deally A, Gleeson B, Hackenberg F, Müller-Bunz H, Paradisi F, Tacke M. Z. Anorg. Allg. Chem. 2011;637:386. [Google Scholar]
- 21f.Patil S, Dietrich K, Deally A, Hackenberg F, Kaps L, Müller-Bunz H, Schobert R, Tacke M. Helv. Chim. Acta. 2011;94:1551. [Google Scholar]
- 22.Liu W, Bensdorf K, Hagenbach A, Abram U, Niu B, Mariappan A, Gust R. Eur. J. Med. Chem. 2011;46:5927. doi: 10.1016/j.ejmech.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Sandtorv AH, Bjørsvik HR. Adv. Synth. Catal. 2013;355:499. [Google Scholar]
- 24.Sandtorv AH, Bjørsvik HR. Adv. Synth. Catal. 2013;355:3231. [Google Scholar]
- 25.Sandtorv AH, Törnroos KW, Bjørsvik HR. Eur. J. Org. Chem. 2015:3506. [Google Scholar]
- 26.Sandtorv AH, Bjørsvik HR. Eur. J. Org. Chem. 2015 doi: 10.1002/ejoc.201500520. [DOI] [Google Scholar]
- 27.Ueda S, Su M, Buchwald SL. J. Am. Chem. Soc. 2012;134:700. doi: 10.1021/ja2102373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.Berding J, van Paridon JA, van Rixel VHS, Bouwman E. Eur. J. Inorg. Chem. 2011:2450. [Google Scholar]
- 28b.Stringer BD, Quan LM, Barnard PJ, Wilson DJD, Hogan CF. Organometallics. 2014;33:4860. [Google Scholar]
- 29.Wang HMJ, Lin IJB. Organometallics. 1998;17:972. [Google Scholar]
- 30.Cliff MD, Pyne SG. Tetrahedron. 1996;52:13703. [Google Scholar]
- 31.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 32.McCormack E, Haaland I, Venås G, Forthun RB, Huseby S, Gausdal G, Knappskog S, Micklem DR, Lorens JB, Bruserud Ø, Gjertsen BT. Leukemia. 2012;26:910. doi: 10.1038/leu.2011.315. [DOI] [PubMed] [Google Scholar]
- 33.Gausdal G, Gjertsen BT, McCormack E, Van Damme P, Hovland R, Krakstad C, Bruserud Ø, Gevaert K, Vandekerckhove J, Døskeland SO. Blood. 2008;111:2866. doi: 10.1182/blood-2007-07-103242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information