

Abstract
Small-molecule probes have been playing prominent roles in furthering our understanding of the molecular underpinnings of the circadian clock. We previously discovered a carbazole derivative, KL001 (N-(3-(9H-carbazol-9-yl)-2-hydroxypropyl)-N-(furan-2-ylmethyl)methanesulfonamide), as a stabilizer of the clock protein cryptochrome (CRY). Herein we describe an extensive structure–activity relationship analysis of KL001 derivatives leading to the development of a highly active derivative: 2-(9H-carbazol-9-yl)-N-(2-chloro-6-cyanophenyl)acetamide (KL044). Subsequent 3D-QSAR analysis identified critical features of KL001 derivatives and provided a molecular-level understanding of their interaction with CRY. The electron-rich carbazole, amide/hydroxy linker, sulfonyl group, and electron-withdrawing nitrile moieties contribute to greater biological activity. The hydrogen bonding interactions with Ser394 and His357 as well as stronger CH–π interactions with Trp290 make KL044 a better binder than KL001. KL044 lengthened the circadian period, repressed Per2 activity, and stabilized CRY in reporter assays with roughly tenfold higher potency than KL001. Altogether, KL044 is a powerful chemical tool to control the function of the circadian clock through its action on CRY.
Keywords: 3D-QSAR, circadian clock, cryptochrome, protein degradation, small molecule
Introduction
In mammals, many physiological processes such as body temperature, hormone secretion, metabolism, and sleep/wake behavior are controlled by the circadian clock in a daily rhythmic manner. Cell-autonomous circadian rhythms are generated through clock genes, which are orchestrated by interconnected transcriptional and translational regulatory loops.1 The core clock loop is constituted by two transcriptional activators, CLOCK and BMAL1, and two repressors: Cryptochrome (CRY1 and CRY2) and Period (PER1 and PER2). The CLOCK–BMAL1 heterodimer activates the transcription of Cry and Per though binding to E-box enhancer elements. Translated PER and CRY proteins dimerize and translocate into the nucleus to inhibit CLOCK–BMAL1 function, resulting in rhythmic gene expression pattern. The loop regulates a large number of clock-controlled genes in circadian physiology. The stabilities of clock proteins are further controlled by posttranslational modification and ubiquitination.2 The phosphorylation of PER protein by casein kinase 1 (CK1δ/ε) promotes its degradation through the ubiquitin–proteasome pathway, dependent on F-box proteins β-TrCP1 and β-TrCP2.3 The stability of CRY protein is separately modulated by degradation through the ubiquitin–proteasome pathway. The SCFFBXL3 ubiquitin ligase complex plays an important role in CRY ubiquitination and degradation to regulate the negative feedback loop.4 Recent X-ray crystallographic analysis revealed details of CRY stability control at the atomic level. The F-box protein FBXL3 recognizes CRY2 by occupying the FAD binding pocket of CRY2 with its C-terminal tail and also covers the PER binding domain of CRY2.5
To develop novel small-molecule modifiers of the circadian clock, we previously screened a collection of 60 000 structurally diverse compounds assembled from an in-house compound library using a cell-based Bmal1-dLuc reporter assay. We identified carbazole derivatives KL001, KL002, and KL003 as period-lengthening compounds (Figure 1 A). Subsequent studies using pull-down and proteomic target identification revealed that KL001 interacts with the clock protein CRY. Interestingly, KL001 competes with FAD for CRY binding and stabilizes CRY by inhibiting FBXL3-dependent ubiquitination.6 The co-crystal structure of the KL001–CRY2 complex revealed that KL001 occupies the FAD binding site of CRY2 and interferes with the binding of FBXL3 C-terminal to CRY2.7 KL001 is the first compound to target CRY, and its further characterization is important to understand the regulation of CRY function. Herein we describe the structural requirements for KL001 activity and the molecular basis for a highly potent derivative that we discovered.
Figure 1.
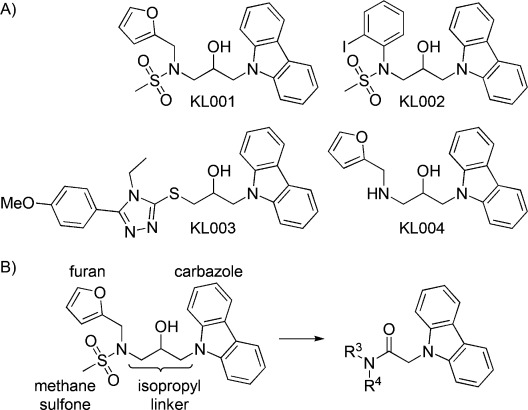
A) Structures of previously identified KL001 derivatives. B) Modification sites of KL001 for structure–activity relationship (SAR) studies.
Results and Discussion
KL001 consists of a cabazole structure linked to 2-hydroxypropyl and furan groups (Figure 1 B). Our preliminary SAR study identified a less active analogue, KL004, which lacks the terminal methanesulfonyl group (Figure 1 A).6 To systemically optimize each subsection of KL001, we designed and synthesized the KL001 derivatives considering four modification sites (Schemes 1 and 2). The period effect of these derivatives was analyzed using a cell-based Bmal1-dLuc circadian assay by testing 12 points of threefold dilution series to obtain EC50 values (Supporting Information Figure S1).
Scheme 1.
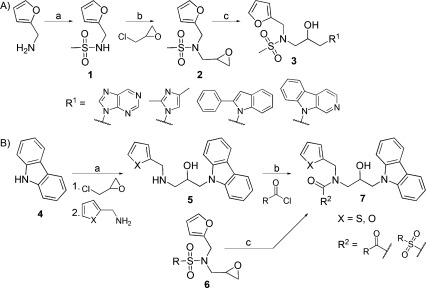
Modifications of KL001. A) Synthesis of modified carbazole. Reagents and conditions: a) MeSO2Cl, py, RT, 24 h, 85.3 %; b) NaH, DMF, 0–50 °C, ∼16 h, 52.1 %; c) NaH/DMF, 60 °C, 2 h. B) The synthesis of 3-acylation derivatives with acyl halide. Reagents and conditions: a) 1. NaH/DMF, 2. THF, RT, ∼16 h; b) DIEA, RT, ∼16 h; c) carbazole, NaH/DMF, 50 °C, 2 h.
Scheme 2.
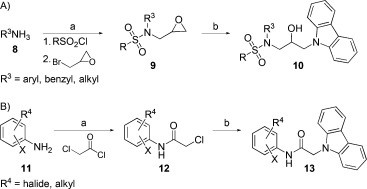
Modifications of KL001. A) Synthesis of furan-modified derivatives with aromatic and aliphatic amines. Reagents and conditions: a) 1. pyridine, RT, overnight, 2. NaH/DMF, 50 °C, overnight; b) carbazole, NaH/DMF, 60 °C, 2 h. B) Synthesis of acetamide linker derivatives. Reagents and conditions: a) DMF, RT, or 40 °C, or Et3N/CH2Cl2, RT, overnight; b) carbazole, NaH/DMF, 60 °C, overnight.
The synthetic routes followed for the preparation of carbazole derivatives 3, 7, 10, and 13 are depicted in Schemes 1 and 2. At first, the carbazole moiety was replaced with purine, imidazole, phenylindole, and pyridoindole. For the synthesis of intermediate 2, furfuryl amines were sulfonylated with methanesulfonyl chloride under basic conditions followed by alkylation with epichlorohydrin and sodium hydride in DMF (Scheme 1 A). The intermediate 2 was treated with carbazole substituents and sodium hydride to provide the derivatives 3. Replacement of the carbazole ring by purine (KL005), imidazole (KL006), and phenylindole (KL007) moieties led to the loss of effect of these compounds on the circadian period (Supporting Information Table S1). The pyridoindole derivatives KL008 and KL010, which are structurally similar to carbazole compounds KL001 and KL009, showed severely decreased period effect. Therefore, it is clear that the carbazole ring is essential for control of the circadian period.
We next modified the methanesulfonyl group by removing its sulfonyl group (compounds KL011 and KL012). Similar to KL004, the compounds showed significantly lowered period-lengthening activity (Table 1 and Supporting Information Table S1). The derivatives with an aliphatic and aromatic acyl group appeared to have decreased period effect (Tables 1 and S1). We therefore evaluated compounds with aliphatic and aromatic acyl groups at the nitrogen atom, or possessing other sulfonyl groups (Scheme 1 B). Intermediate 5 was obtained from carbazole using sodium hydride for deprotonation of a carbazole followed by reaction with furfuryl amine. The acylated derivatives KL013–KL019 were synthesized by acylation of intermediate 5 under basic conditions. KL013 and KL014, which were acylated with relatively short aliphatic and aromatic groups, showed tolerance to modification. However, derivatives with nicotinic groups (KL016 and KL017) showed a further decrease in activity. KL020 and KL021, whose sulfonyl moieties were modified with phenyl or cyclopropyl groups, showed reduced period effects.
Table 1.
Representative structures of KL001 derivatives and their period-lengthening effects.
| |||||
|---|---|---|---|---|---|
| ID | Structure | Period effect | ID | Structure | Period effect |
| log(EC50 [M]) | log(EC50 [M]) | ||||
| KL001 | 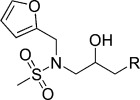 |
−6.16 | KL011 | 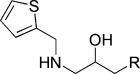 |
−4.82 |
| KL013 | 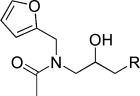 |
−5.57 | KL016 | 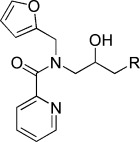 |
−4.73 |
| KL019 | 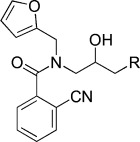 |
−5.61 | KL021 | 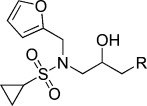 |
−5.49 |
| KL022 | 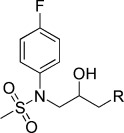 |
−5.70 | KL028 | 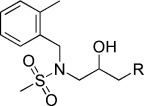 |
−4.94 |
| KL032 | 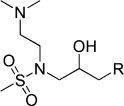 |
−4.25 | KL033 | 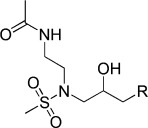 |
−4.23 |
| KL034 | 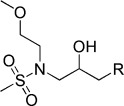 |
−6.01 | KL044 | 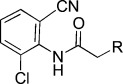 |
−7.32 |
| KL046 | 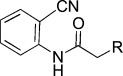 |
−6.46 | KL048 | 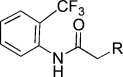 |
−6.12 |
We then replaced furan with phenyl, benzyl, or aliphatic moieties (Scheme 2 A). Intermediate 9 was obtained from reaction of aromatic or aliphatic amine with methanesulfonyl chloride followed by reaction with sodium hydride and epichlorohydrin. In the next step, intermediate 9 was linked with a carbazole moiety to generate derivatives KL022–KL035. The derivatives with benzyl and phenyl groups showed lower activity (Tables 1 and S1). Interestingly, KL034, which has a methoxyethyl group, exhibited potency similar to that of KL001. However, other derivatives with ethylaminoacetyl and dimethylaminoethyl groups (KL031–KL033) showed weaker activity. Therefore, a methoxyethyl group can be a replacement for furan. Further modification of the sulfonyl group did not improve potency (compounds KL036–KL038). Derivatives with phenyl and cyclopropylacyl or sulfonyl moieties (KL039–KL042) showed a decreased period effect.
To explore the role of the linker, we introduced an acetamide linker with a variety of anilines (KL043–KL064; Scheme 2 B). Intermediate 12 was obtained by reaction with various anilines and chloroacetyl chloride in DMF. Compound 13 was synthesized by nucleophilic reaction with chloroacetyl anilines 12 and carbazole using NaH/DMF. Derivatives with ortho-substituted phenyl groups (KL043–KL053) exhibited relatively high period effects, except KL045, with ortho- and para-substituted phenyl groups (Tables 1 and S1). We found that KL044 with an acetamide linker showed ∼10-fold higher potency than KL001. Interestingly, nitrile group substitution at the meta position of the phenyl group (KL055) results in complete loss in activity (Table S1). Therefore, ortho substitution at the phenyl group is important for the period-lengthening effect.
To obtain a molecular-level understanding of our SAR study, we further conducted a 3D-QSAR study which uses a ligand-field-based approach.8 The molecular fields include electrostatic, steric, hydrophobic, and van der Waals fields calculated by the interaction of the ligand with positive, negative, and neutral atoms as probes. These ligand fields can be used as molecular features, yielding a so-called “pharmacophore”, to identify structural regions in the ligands crucial for bioactivity. In our study, field points of KL001 and KL044 were used for generating a pharmacophore (Figure 2 A) in FieldTemplater available in the Forge software package,9 which was used to align a diverse range of KL001 derivatives. The data included a set of 60 compounds with their potency in terms of −log(EC50) in molar units (pEC50), ranging from 3.94 to 7.32. For 3D-QSAR studies, a random method was used for dividing the data set into two sets: 54 compounds in the training set, and six compounds in the test set (Table S1). Technical details of structure preparation, docking, pharmacophore template generation, and 3D-QSAR are given in Supporting Information Methods and Figure S2. Figure 2 B gives a representation of the electrostatic and steric coefficients of the 3D-QSAR model. The linear relationship between experimental and predicted activity of both the training and test sets (Figure 2 C) indicates the successful construction of a reliable model with high predictive power. We also performed leave-many-out cross-validation by leaving out 20 % of the molecules at each step for the same set of training and test set compounds as used for leave-one-out cross-validation. The values for leave-one-out (LOO) cross-validated correlation coefficient (q2), non-cross-validated correlation coefficient (r2), number of components (N), predicted non-cross-validated correlation coefficient (rpred2), and predicted RMSE were 0.639, 0.966, 6, 0.827, and 0.442, respectively. The respective analogous values for leave-many-out (LMO) parameters q2, r2, N, rpred2, RMSEpred were 0.606, 0.966, 6, 0.827, and 0.462. Hence, the stability of the 3D-QSAR model is robust, as indicated by the similar predictive values obtained by both LOO and LMO cross-validation methods. Based on the model, the higher activity corresponds to the following features: 1) negative electrostatics (cyan tetrahedrons) contributed by electron-rich carbazole, amide/hydroxy linker, and sulfonyl moieties; 2) positive electrostatics (red tetrahedrons) mediated by the electron-withdrawing nitrile and hydroxy groups; and 3) steric bulk associated with the furyl moiety (green tetrahedrons). The purple tetrahedrons show areas where it is best to avoid bulkier functional groups.
Figure 2.
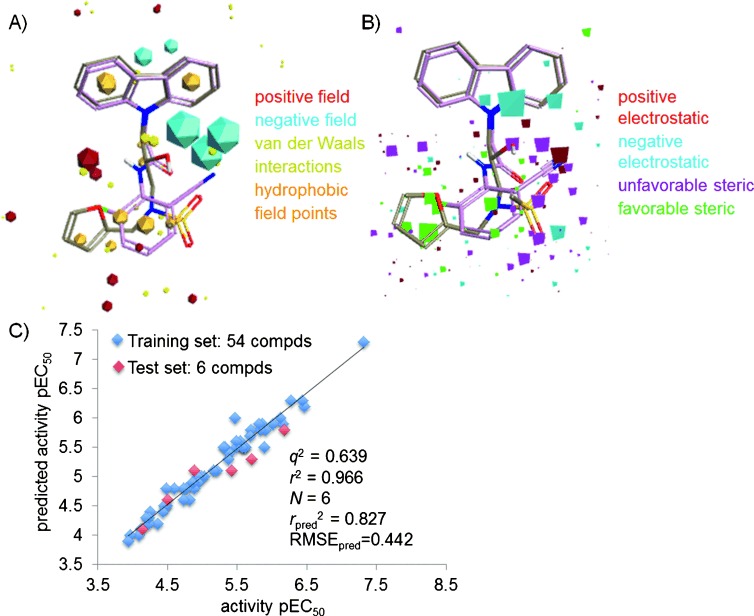
A) Model template generated by the conformational search of KL001 and KL044. B) 3D-QSAR model generated by using pEC50 values of 54 training compounds and six test set compounds. C) Plot of experimental and predicted pEC50 values.
For studying the complementarity of the binding pocket of the CRY protein, we further superimposed the selected field points with the higher coefficient values from the 3D-QSAR model and KL044 on the CRY protein structure (PDB ID: 4MLP7) as shown in Figure 3. The binding site analysis for CRY protein was carried out using Sitemap,10 and the superimposition was performed with Maestro.10 The surfaces were generated depicting the contour maps obtained by van der Waals repulsion, and hydrophilic and hydrophobic regions. The values for SiteScore, Dscore (druggability) and the ratio of hydrophobic and hydrophilic character were 1.109, 1.051, and 1.050, respectively, indicating that CRY protein has a druggable tight-binding pocket. The good ligand could be a better acceptor than a donor of hydrogen bonds, indicated by the ratio of donor verses acceptor as 0.692. In Figures 3 A and 3 A′, favorable steric field points (15–19) overlap with the empty pocket depicted by the green surface of the contour map drawn for the van der Waals repulsive term. In contrast, the unfavorable steric field points (9–14) are located at the edge of the contour map away from this empty pocket. In Figures 3 B and 3 B′, the amide moiety in KL044 overlaps with the hydrophilic map. The positive electrostatic field points (6–8) and a negative electrostatic field point (4) are within 3 Å of the ligand. In Figures 3 C and 3 C′, the hydrophobic surface map overlaps with the carbazole as well as the disubstituted benzene moiety of KL044. The negative electrostatic field points (1–3, 5) are within 3 Å of the ligand. This indicates that KL044 is a good fit in the pocket of the CRY binding site, and field points indicate the opportunities for obtaining additional interactions in the hydrophilic and hydrophobic pockets. Unfavorable steric interactions with residues Trp290, Trp397, Val390 and Phe294 should be avoided. Additionally, Figure S3 (Supporting Information) shows the superimposition with the CRY protein structure containing FBXL3 protein bound at the FAD binding site, (PDB ID: 4I6J5). The surface contour (Figure 3 A) extends in the space occupied by FBXL3 residues such as Trp428 and Thr427. Moreover, the position of FBXL3 P426 bound to CRY protein overlaps with a negative electrostatic field point number 5. Overall, we found that the 3D-QSAR results and the nature of the amino acid residues in the CRY ligand binding domain are very well correlated.
Figure 3.
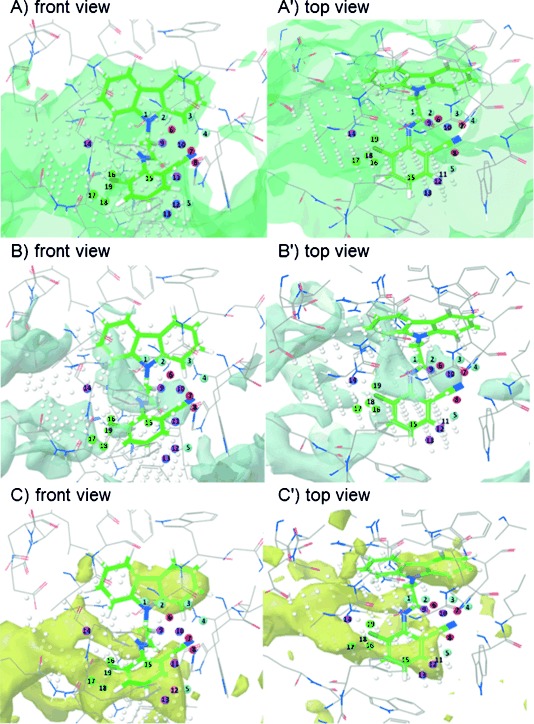
Overlap of QSAR field points on KL044 and surface maps generated for the binding pocket of CRY (PDB ID: 4MLP) by Sitemap depicted as: A) surface contour obtained by van der Waals repulsion, B) hydrophilic, and C) hydrophobic. Color code for field point coefficients represented as numbered spheres: negative electrostatic (field points 1–5, cyan); positive electrostatic (field points 6–8, red); unfavorable steric (field points 9–14, purple); favorable steric (field points 15–19, green).
It is important to understand how a field pattern of a particular molecule contributes to its predicted activity. Figure 4 A shows the medicinal chemistry evolution of KL044 based on the contribution of the field points to the predicted activity. When the isopropyl moiety is introduced to KL046, the activity is significantly decreased (KL058, ΔpEC50 2.2) due to unfavorable electrostatics (orange cubes) and steric clash (purple cubes). This emphasizes the importance of having a hydrogen bond donor such as the −NH moiety. By introducing Cl at the ortho position of the benzyl group, KL044 with favorable electrostatics and sterics can be obtained.
Figure 4.
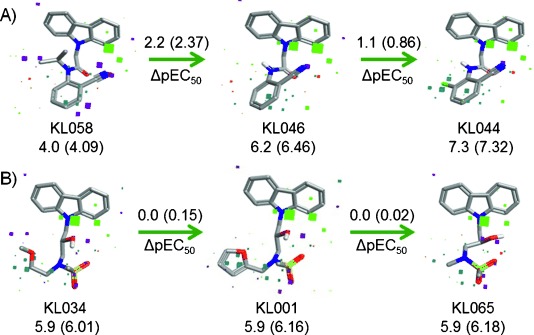
Evolution of medicinal chemistry based on the contribution of the field points to the predicted activity in 3D-QSAR model for A) KL044 and B) KL065. Experimental pEC50 values are shown in brackets. Color code for field point contributions: favorable electrostatic (green), unfavorable electrostatic (orange), favorable steric (cyan), unfavorable steric (purple).
For validating the QSAR model, we predicted the activities for five compounds, KL065–KL069 (Supporting Information, Methods). We synthesized these compounds and analyzed their activities. An excellent agreement between predicted and experimental bioactivity further supported the reliability of the model (Table S1). Figure 4 B shows a comparison of the predicted compound KL065 with KL001 and KL034. The van der Waals radii for chloro (1.8 Å) and methyl (2.0 Å) groups are similar, and these are well-known isosteres. The chloro moiety in KL044 and methyl group in KL065 overlap very well. Hence, we found that replacement of the furyl moiety (KL001) and methoxyethane group (KL034) with methyl retains the activity, as shown by the favorable electrostatic (cyan cubes) and decreased sterics (purple cubes). The log P values for KL001 and KL065 are 3.0 and 2.4, respectively, indicating that KL065 has higher solubility due to the less bulky substituent at the trigonal nitrogen atom. Figure S4 provides other examples for contribution of a field pattern on the predicted activity.
To further understand why KL044 provides the best activity among the tested compounds, differences in the electrostatic potential maps, which can be used to explain the differences in higher or lower activity of the compounds, were analyzed by using Activity Miner.11 The ligand binding site residues in CRY protein and electrostatic potential (ESP) map difference between KL044 and KL001 are shown in Figure 5. In KL044, the positive potential cloud (red) due to the −NH moiety, as well as the negative cloud (cyan) due to the disubstituted benzyl with the electron-withdrawing groups at the ortho position and −C=O correlated with increased activity over KL001. This highlights the importance of interactions such as the −NH and −C=O groups forming hydrogen bonds with Ser394 (3.06 Å) and His357 (2.35 Å), respectively. Moreover, the disubstituted benzyl group forms a CH–π interaction with Trp290 (3.71 Å). In KL001, the positive cloud due to trisubstituted nitrogen and the negative cloud due to the sulfonyl group result in decreased activity relative to KL044. The important interactions for KL001 are the hydrogen bond formed by the −OH group with Ser394 (2.75 Å) and CH–π interactions formed by the methanesulfonyl moiety with Trp290 (4.50 Å) and Trp397 (4.10 Å). Hence, these characteristics of the compound make KL044 a better fit than KL001 in the CRY binding pocket.
Figure 5.
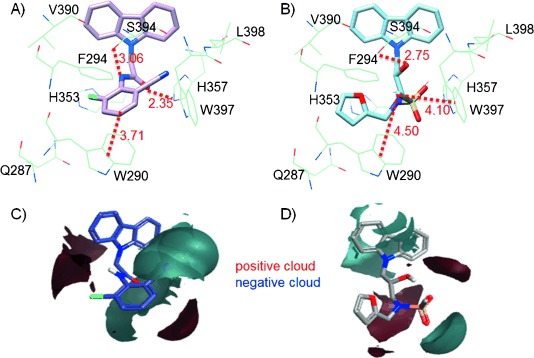
Ligand binding site residues in CRY protein interacting with A) KL044 and B) KL001 (distances given in Å). Electrostatic map difference between C) KL044 and D) KL001 based on field similarity.
In this experimental–theoretical study, we discovered a highly potent derivative KL044. We therefore characterized its biological activities by using Bmal1-dLuc and Per2-dLuc cell-based reporter assays. KL044 lengthened the circadian period and also suppressed Per2-dLuc activity much stronger than KL001 (Figures 6 A and 6 B), suggesting an activation of CRY, a repressor of the Per gene. Consistent with this observation, we revealed that KL044 potently stabilized the CRY1–LUC fusion protein in cell-based degradation assay without affecting LUC stability (Figure 6 C). The parallel relationship between KL044 and KL001 against CRY1 stability, Per2 repression, and period lengthening supports the mechanism of action of KL044 as a CRY stabilizer for circadian regulation.
Figure 6.
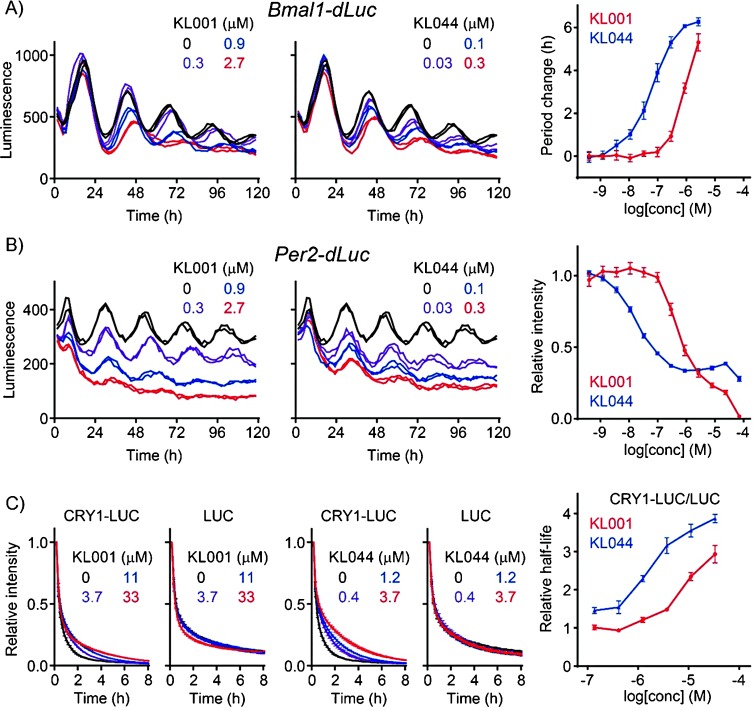
Effects of KL001 and KL044 in A) Bmal1-dLuc and B) Per2-dLuc cell-based circadian assays. C) Effects of KL001 and KL044 in CRY1-LUC and LUC degradation assays in the HEK293 stable cell line.
Conclusions
CRY is a key regulator not only for the circadian clock but also for the hepatic gluconeogenesis,12 suggesting a potential use of KL001 derivatives as circadian rhythm modulators as well as antidiabetics. KL044 provides a potent chemical probe for the regulation of CRY protein function. Our SAR and 3D-QSAR studies in combination with the published CRY–KL001 co-crystal structure aid in our understanding of the mechanism of CRY regulation in terms of molecule–protein interactions. Future diversification of the compounds by combining the pharmacophoric features of KL044 and KL001 will provide even better compounds for modulating circadian function and gluconeogenesis. Together with a recently published study in which period-shortening derivatives of KL001 through C−H activation were discovered,13 the development of KL001 analogues clearly demonstrates the power of chemical biology approaches in the research field of the circadian clock.
Experimental Section
Characterization of representative KL001 derivatives. 1H and 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer. Chemical shifts are reported relative to internal CDCl3 (CH4Si, δ=0.0), CD3OD (CH4Si, δ=0.0), and CD3CN (CH4Si, δ=0.0). All compounds were identified by LC–MS (Agilent Technology) using a C18 column (20×4.0 mm) with 20 min elution using a gradient solution of CH3CN/H2O (5:95 v/v containing 0.05 % trifluoroacetic acid (TFA)), with UV detector and an electrospray ionization source.
1-(9H-Carbazol-9-yl)-3-((thiophen-2-ylmethyl)amino)propan-2-ol (KL011). The reaction mixtures of 9-(oxiran-2-ylmethyl)-9H-carbazole (29.2 mg, 0.13 mmol) and thiophen-2-ylmethanamine (200 μL, 2.0 mmol) were heated at 55 °C. The crude material was purified by preparative HPLC (0.05 % TFA/H2O / 0.05 % TFA/CH3CN) to give 26 mg (81.3 %) of KL011; 1H NMR (CD3OD, 400 MHz): δ=8.16 (d, J=8 Hz, 2 H), 7.61 (d, J=8.5 Hz, 2 H), 7.54–7.51 (m, 3 H), 7.30 (t, J=8 Hz, 2 H), 7.17 (d, J=3 Hz, 1 H), 7.07 (dd, J=5, 3.5 Hz, 1 H), 4.54–4.46 (m, 5 H), 3.17 (dd, J=12.5, 10 Hz, 1 H), 3.10 ppm (dd, J=12.5, 2.5 Hz, 1 H); 13C NMR (CD3OD, 100 MHz): δ=141.0, 131.1, 128.6, 127.7, 127.6, 127.5, 126.0, 123.4, 120.2, 119.5, 109.1, 101.8, 66.1, 49.5, 48.0, 46.0 ppm; ESI-MS: 337.1 [M+H].
N-(3-(9H-Carbazol-9-yl)-2-hydroxypropyl)-N-(furan-2-ylmethyl)acetamide (KL013). A solution of 1-(9H-carbazol-9-yl)-3-((furan-2-ylmethyl)amino)propan-2-ol (50 mg, 0.16 mmol) in THF (2 mL) was treated with DIEA (25 μL, 0.16 mmol) followed by acetic anhydride (25 μL, 0.24 mmol). After stirring at room temperature overnight, the reaction mixture was extracted with EtOAc (50 mL) and H2O (20 mL). The organic layer was dried with MgSO4 and then evaporated under reduced pressure. The crude compound was purified by preparative HPLC (0.05 % TFA/H2O / 0.05 % TFA/CH3CN) to give 32 mg (56.6 %) of KL013; 1H NMR (CDCl3, 400 MHz): δ=8.10 (d, J=7.6 Hz, 2 H), 7.45 (t, J=8, 2 H), 7.39 (d, J=8, 2 H), 7.24 (t, J=7.6 Hz, 2 H), 7.07 (dd, J=2, 0.8 Hz, 1 H), 5.97 (dd, J=3.2, 1.6 Hz, 1 H), 5.55 (dd, J=3.2, 0.4 Hz, 1 H), 4.46 (br s, 1 H), 4.36–4.21 (m, 2 H), 4.17–4.10 (m, 3 H), 3.79 (dd, J=14.4, 8.8 Hz, 1 H), 3.17 (dd, J=14.4, 2 Hz, 1 H), 2.23 ppm (s, 3 H); 13C NMR (CDCl3, 100 MHz): δ=173.4, 148.8, 142.7, 140.6, 125.9, 123.0, 120.3, 119.2, 110.2, 109.0, 108.6, 71.0, 52.3, 47.4, 47.0, 21.7 ppm; ESI-MS: 363.1 [M+H], 385.1 [M+Na].
N-(3-(9H-Carbazol-9-yl)-2-hydroxypropyl)-N-(furan-2-ylmethyl)picolinamide (KL016). A solution of 1-(9H-carbazol-9-yl)-3-((furan-2-ylmethyl)amino)propan-2-ol (50 mg, 0.16 mmol) in DMF (4 mL) was treated with DIEA (35 μL, 0.22 mmol), HATU (76 mg, 0.2 mmol), and picolinic acid (25 mg, 0.2 mmol). After stirring at room temperature overnight, the reaction mixture was extracted with CH2Cl2 (50 mL) and H2O (30 mL). The organic layer was dried with Na2SO4 and evaporated under reduced pressure. The crude material was purified by column chromatography to give 41.5 mg (62.6 %) of KL016; 1H NMR (CDCl3, 400 MHz): δ=8.03 (d, J=7.6 Hz, 2 H), 7.65–7.62 (m, 2 H), 7.50–7.38 (m, 5 H), 7.24–7.19 (m, 3 H), 7.05–7.01 (m, 1 H), 6.22 (dd, J=3.2, 2.0 Hz, 1 H), 6.11 (d, J=2.8 Hz, 1 H), 5.13 (d, J=15.2 Hz, 1 H), 4.58–4.49 (m, 2 H), 4.38–4.27 (m, 2 H), 3.52 (dd, J=14.8, 12.0 Hz, 1 H), 3.19 ppm (dd, J=15.2, 3.2 Hz, 1 H); 13C NMR (CDCl3, 100 MHz): δ=167.8, 151.9, 149.9, 145.7, 142.4, 141.1, 138.0, 125.7, 125.5, 124.9, 122.9, 120.1, 119.1, 110.5, 109.5, 109.4, 68.0, 50.6, 47.5, 41.5 ppm; ESI-MS: 426.2 [M+H], 448.2 [M+Na].
N-(3-(9H-Carbazol-9-yl)-2-hydroxypropyl)-2-cyano-N-(furan-2-ylmethyl)benzamide (KL019). Same procedure as described for the synthesis of KL016. 1-(9H-Carbazol-9-yl)-3-((furan-2-ylmethyl)amino)propan-2-ol (50 mg, 0.16 mmol), DIEA (35 μL, 0.22 mmol), HATU (76 mg, 0.2 mmol), and 2-cyanobenzoic acid (29 mg, 0.2 mmol) were combined in DMF (4 mL) to give 38.1 mg (56.6 %) of KL019; 1H NMR (CDCl3, 400 MHz): δ=8.10 (d, J=8.0 Hz, 2 H), 7.65 (d, J=8.0 Hz, 2 H), 7.62 (d, J=8.0 Hz, 2 H), 7.47–7.37 (m, 4 H), 7.28–7.23 (m, 2 H), 7.13 (s, 1 H), 5.95 (s, 1 H), 5.46 (s, 1 H), 4.41–4.23 (m, 3 H), 4.17–4.08 (m, 2 H), 3.89 (dd, J=14.0, 8.4 Hz, 1 H), 3.36 ppm (d, J=14.4 Hz, 1 H); 13C NMR (CDCl3, 100 MHz): δ=171.9, 149.0, 147.7, 144.5, 143.1, 140.5, 139.5, 132.5, 128.0, 126.2, 126.0, 123.0, 120.4, 119.3, 114.0, 110.3, 109.7, 108.9, 70.5, 51.2, 47.7, 47.1 ppm; ESI-MS; 450.2 [M+H], 472.1 [M+Na].
General procedure for the synthesis of compound 10. We followed the previously described procedure for the synthesis of compound 9.6 A solution of carbazole (250 mg, 1.5 mmol) in DMF (5 mL) was treated with NaH (41 mg, 1.7 mmol, 1.2 equiv) and stirred for 30 min at room temperature, followed by compound 9 (242 mg, 0.85 mmol) in DMF (2 mL). The reaction mixture was stirred for 2 h at 60 °C. The reaction mixture was diluted with EtOAc, washed with 5 % HCl and brine. The crude material was purified by flash column chromatography to give the final products.
N-(3-(9H-Carbazol-9-yl)-2-hydroxypropyl)-N-(furan-2-ylmethyl)cyclopropanesulfonamide (KL021). N-(Furan-2-ylmethyl)-N-(oxiran-2-ylmethyl)cyclopropanesulfonamide (40 mg, 0.156 mmol), carbazole (52.1 mg, 0.321 mmol), NaH (8.2 mg, 0.34 mmol) were combined in DMF (5 mL) to give 41.5 mg (62.8 %) of KL021. 1H NMR (CDCl3, 400 MHz): δ=8.09 (d, J=7.6 Hz, 2 H), 7.46 (t, J=8 Hz, 2 H), 7.41 (d, J=7.6 Hz, 2 H), 7.25 (t, J=8 Hz, 2 H), 7.17 (dd, J=1.6, 0.8 Hz, 1 H), 6.13 (dd, J=3.2, 1.6 Hz, 1 H), 5.93 (d, J=3.2 Hz, 1 H), 4.40–4.31 (m, 4 H), 4.27–4.22 (m, 1 H), 3.47 (dd, J=14.8, 8 Hz, 1 H), 3.28 (dd, J=14.8, 3.2 Hz, 1 H), 2.28 (m, 1 H), 1.63–1.12 (m, 2 H), 0.93–0.89 ppm (m, 2 H); 13C NMR (CDCl3, 100 MHz): δ=149.2, 142.9, 140.6, 125.9, 123.1, 120.3, 119.3, 110.5, 110.1, 109.0, 69.2, 51.6, 46.8, 45.2, 29.0, 5.2, 5.0 ppm; ESI-MS; 425.1 [M+H], 447.1 [M+Na].
N-(3-(9H-Carbazol-9-yl)-2-hydroxypropyl)-N-(4-fluorophenyl)methanesulfonamide (KL022). N-(4-Fluorophenyl)-N-(oxiran-2-ylmethyl)methanesulfonamide (100 mg, 0.41 mmol), carbazole (102 mg, 0.6 mmol), and NaH (19.5 mg, 0.81 mmol) were combined in DMF (8 mL) to give 52.1 mg (44.6 %) of KL022. 1H NMR (CDCl3, 400 MHz): δ=8.07 (d, J=7.6 Hz, 2 H), 7.44 (t, J=8.4 Hz, 2 H), 7.37–7.31 (m, 4 H), 7.24 (t, J=8 Hz, 2 H), 7.07 (t, J=8 Hz, 2 H), 4.45–4.34 (m, 2 H), 4.32–4.25 (m, 1 H), 3.92 (dd, J=14.4, 7.6 Hz, 1 H), 3.76 (dd, J=14.4, 4.8 Hz, 1 H), 2.91 ppm (s, 3 H); 13C NMR (CDCl3, 100 MHz): δ=140.7, 130.4, 130.3, 126.0, 123.1, 120.4, 119.5, 116.9, 116.6, 108.8, 69.1, 55.2, 37.4 ppm; ESI-MS: 413.1 [M+H], 435.1 [M+Na].
N-(3-(9H-Carbazol-9-yl)-2-hydroxypropyl)-N-(2-methylbenzyl)methanesulfonamide (KL028). N-(2-Methylbenzyl)-N-(oxiran-2-ylmethyl)methanesulfonamide (120 mg, 0.53 mmol), carbazole (133.6 mg, 0.8 mmol), NaH (26.4 mg, 1.1 mmol) were combined in DMF (8 mL) to give 82 mg (36.4 %) of KL028. 1H NMR (CDCl3, 400 MHz): δ=8.07 (d, J=8.4 Hz, 2 H), 7.47 (td, J=8.4, 1.2 Hz, 2 H), 7.24–7.18 (m, 4 H), 7.06–6.99 (m, 2 H), 6.93 (d, J=7.2 Hz, 1 H), 6.87–6.83 (m, 1 H), 4.45 (d, J=13.6 Hz, 1 H), 4.19–4.04 (m, 3 H), 3.86 (m, 1 H), 3.45 (dd, J=15.2, 8.8 Hz, 1 H), 3.15 (dd, J=18, 3.2 Hz, 1 H), 2.94 (s, 3 H), 2.21 ppm (s, 3 H); 13C NMR (CDCl3, 100 MHz): δ=140.5, 137.2, 132.5, 130.9, 129.5, 128.4, 126.1, 125.8, 123.0, 120.3, 119.3, 108.8, 69.0, 52.1, 51.4, 46.9, 37.1, 19.0 ppm; ESI-MS: 423.1 [M+H], 445.1 [M+Na].
N-(3-(9H-Carbazol-9-yl)-2-hydroxypropyl)-N-(2-(dimethylamino)ethyl)methanesulfonamide (KL032). N-(2-(Dimethylamino)ethyl)-N-(oxiran-2-ylmethyl)methanesulfonamide (103 mg, 0.46 mmol), carbazole (153 mg, 0.92 mmol), and 60 % NaH (47 mg, 1.15 mmol) were combined in DMF (7 mL) to give 22 mg (12.3 %) of KL032. 1H NMR (CDCl3, 400 MHz): δ=8.05 (d, J=7.6 Hz, 2 H), 7.50 (d, J=8.0 Hz, 2 H), 7.45 (t, J=8.0 Hz, 2 H), 7.22 (t, J=8.0 Hz, 2 H), 4.45–4.34 (m, 3 H), 3.70–3.65 (m, 1 H), 3.53–3.46 (m, 1 H), 3.42–3.28 (m, 2 H), 3.24–3.11 (m, 2 H), 2.78 (s, 3 H), 2.74 ppm (m, 6 H); ESI-MS: 390.2 [M+H].
N-(2-(N-(3-(9H-Carbazol-9-yl)-2-hydroxypropyl)methylsulfonamido)ethyl)acetamide (KL033). N-(2-(N-(Oxiran-2-ylmethyl)methylsulfonamido)ethyl)acetamide (80 mg, 0.34 mmol), carbazole (114 mg, 0.68 mmol), and 60 % NaH (34 mg, 0.85 mmol) were combined in DMF (8 mL) to give 24 mg (17.5 %) of KL033. 1H NMR (CDCl3, 400 MHz): δ=8.06 (d, J=7.6 Hz, 2 H), 7.5–7.43 (m, 4 H), 7.28–7.20 (m, 2 H), 6.33 (s, 1 H), 4.48–4.38 (m, 1 H), 4.3 (t, J=6 Hz, 2 H), 3.45–3.35 (m, 2 H), 3.35–3.20 (m, 3 H), 3.18–3.10 (m, 1 H), 2.79 (s, 3 H), 1.87 ppm (s, 3 H); 13C NMR (CDCl3, 100 MHz): δ=171.3, 140.8, 126.0, 123.0, 120.3, 119.4, 109.1, 69.6, 54.3, 49.4, 47.3, 38.7, 37.1, 23.1 ppm; ESI-MS: 404.2 [M+H], 426.1 [M+Na].
N-(3-(9H-Carbazol-9-yl)-2-hydroxypropyl)-N-(2-methoxyethyl)methanesulfonamide (KL034). N-(2-Methoxyethyl)-N-(oxiran-2-ylmethyl)methanesulfonamide (100 mg, 0.48 mmol), carbazole (160 mg, 0.96 mmol), and NaH (43.2 mg, 1.08 mmol) were combined in DMF (10 mL) to give 82 mg (43.2 %) of KL034. 1H NMR (CDCl3, 400 MHz): δ=8.09 (d, J=7.6 Hz, 2 H), 7.49–7.46 (m, 4 H), 7.27–7.22 (m, 2 H), 4.43–4.32 (m, 3 H), 3.83 (d, J=3.6 Hz, 1 H), 3.5–3.47 (m, 2 H), 3.45–3.28 (m, 5 H), 3.23 (s, 3 H), 2.85 ppm (s, 3 H); 13C NMR (CDCl3, 100 MHz): δ=140.8, 125.9, 123.1, 120.3, 119.3, 109.1, 71.4, 69.8, 58.8, 53.9, 49.5, 37.7 ppm; ESI-MS: 377.1 [M+H], 399.1 [M+Na].
2-(9H-Carbazol-9-yl)-N-(2-chloro-6-cyanophenyl)acetamide (KL044). A solution of 2-amino-3-chlorobenzonitrile (50 mg, 0.33 mmol) in DMF (0.5 mL) was treated with chloroacetyl chloride (44.8 mg, 0.4 mmol, 1.2 equiv) and TEA (50.5 μL, 0.4 mmol, 1.2 equiv). The reaction mixture was stirred at 40 °C for 6 h. The reaction mixture was diluted with CH2Cl2 and washed with H2O. The organic layer was dried under Na2SO4, filtered, and concentrated under reduced pressure. The crude compound was purified by flash column chromatography (EtOAc/hexane 1:3) to give 2-chloro-N-(2-chloro-6-cyanophenyl)acetamide (54.0 mg, 71.8 %). A solution of carbazole (20 mg, 0.12 mmol) in DMF (1 mL) was treated with NaH (6 mg, 0.24 mmol, 2 equiv) and stirred at 0 °C for 30 min, followed by 2-chloro-N-(2-chloro-6-cyanophenyl)acetamide (27.4 mg, 0.12 mmol) in DMF (1 mL). The reaction mixture was stirred at 80 °C for 6 h. After reaction completion, the mixture was purified by preparative HPLC to give KL044 (22.1 mg, 51.4 %). 1H NMR (CD3CN, 400 MHz): δ=8.5 (br s, 1 H), 8.16 (d, J=7.6 Hz, 2 H), 7.74 (dd, J=8, 1.2 Hz, 1 H), 7.67 (dd, J=8, 1.6 Hz, 1 H), 7.59 (d, J=8 Hz, 2 H), 7.52 (t, J=8 Hz, 2 H), 7.41 (t, J=8 Hz, 1 H), 7.29 (t, J=7.6 Hz, 2 H), 5.25 ppm (s, 2 H); 13C NMR (CD3CN, 400 MHz): δ=168.3, 144.6, 137.2, 135.2, 133.6, 132.7, 129.8, 126.9, 121.0, 120.6, 116.4, 115.1, 110.0, 47.0 ppm; ESI-MS: 360.0 [M+H].
2-(9H-Carbazol-9-yl)-N-(2-cyanophenyl)acetamide (KL046). Same procedure as described for the synthesis of KL044. 2-Chloro-N-(2-cyanophenyl)acetamide (20 mg, 0.1 mmol), NaH (10 mg, 0.4 mmol), and carbazole (16.7 mg, 0.1 mmol) were combined in DMF (3 mL) to give 22.4 mg (68.9 %) of KL046. 1H NMR (CDCl3, 400 MHz): δ=8.21 (d, J=8.4 Hz, 1 H), 8.16 (d, J=7.6 Hz, 2 H), 7.73 (s, 1 H), 7.56–7.51 (m, 3 H), 7.46–7.41 (m, 3 H), 7.35 (t, J=8 Hz, 2 H), 7.14 (t, J=7.6 Hz, 1 H), 5.13 ppm (s, 2 H); 13C NMR (CDCl3, 100 MHz): δ=166.9, 140.0, 138.9, 133.9, 132.3, 126.7, 125.8, 123.9, 122.0, 121.0, 120.3, 119.4, 110.6, 108.4, 47.6 ppm; ESI-MS: 326.1 [M+H], 348.1 [M+Na].
2-(9H-Carbazol-9-yl)-N-(2-(trifluoromethyl)phenyl)acetamide (KL048). Same procedure as described for the synthesis of KL044. 2-Chloro-N-(2-(trifluoromethyl)phenyl)acetamide (40 mg, 0.17 mmol), 95 % NaH (10 mg, 0.4 mmol), and carbazole (28.4 mg, 0.17 mmol) were combined in DMF (4 mL) to give 26.8 mg (42.8 %) of KL048. 1H NMR (CDCl3, 400 MHz): δ=8.19 (d, J=8 Hz, 1 H), 8.15 (d, J=7.6, 2 H), 7.58 (s, 1 H), 7.52 (t, J=8, 3 H), 7.42 (d, J=8 Hz, 3 H), 7.34 (t, J=8 Hz, 2 H), 7.17 (m, 1 H), 5.11 ppm (s, 2 H); 13C NMR (CDCl3, 100 MHz): δ=167.0, 140.0, 132.8, 126.6, 125.0, 124.3, 123.7, 120.8, 120.7, 108.4, 47.6 ppm; ESI-MS: 369.1 [M+H], 391.1 [M+Na].
Acknowledgments
This research was supported by the U.S. National Institutes of Health (grant R01GM074868 to S.A.K.).
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Bass J, Takahashi JS. Science. 2010;330:1349–1354. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b.Asher G, Schibler U. Cell Metab. 2011;13:125–137. doi: 10.1016/j.cmet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Gallego M, Virshup DM. Nat. Rev. Mol. Cell Biol. 2007;8:139–148. doi: 10.1038/nrm2106. [DOI] [PubMed] [Google Scholar]
- 3a.Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, Vielhaber EL, Giovanni A, Virshup DM. Mol Cell Biol. 2005;25:2795–2807. doi: 10.1128/MCB.25.7.2795-2807.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b.Shirogane T, Jin J, Ang XL, Harper JW. J. Biol. Chem. 2005;280:26863–26872. doi: 10.1074/jbc.M502862200. [DOI] [PubMed] [Google Scholar]
- 3c.Vanselow K, Vanselow JT, Westermark PO, Reischl S, Maier B, Korte T, Herrmann A, Herzel H, Schlosser A, Kramer A. Genes Dev. 2006;20:2660–2672. doi: 10.1101/gad.397006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a.Busino L, Bassermann F, Maiolica A, Lee C, Nolan PM, Godinho SI, Draetta GF, Pagano M. Science. 2007;316:900–904. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- 4b.Godinho SI, Maywood ES, Shaw L, Tucci V, Barnard AR, Busino L, Pagano M, Kendall R, Quwailid MM, Romero MR, O′Neil J, Chesham JE, Brooker D, Lalanne Z, Hastings MH, Nolan PM. Science. 2007;316:897–900. doi: 10.1126/science.1141138. [DOI] [PubMed] [Google Scholar]
- 4c.Siepka SM, Yoo SH, Park J, Song W, Kumar V, Hu Y, Lee C, Takahashi JK. Cell. 2007;129:1011–1023. doi: 10.1016/j.cell.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xing W, Busino L, Hinds TR, Marionni SM, Saifee NH, Bush MF, Pagano M, Zheng N. Nature. 2013;496:64–68. doi: 10.1038/nature11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirota T, Lee JW, John PCSt, Sawa M, Iwaisko K, Noguchi T, Pongsawakul PY, Sonntag T, Welsh D, Brenner DA, Doyle FJ, III, Schultz PG, Kay SA. Science. 2012;337:1094–1097. doi: 10.1126/science.1223710. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nangle S, Xing W, Zheng N. Cell Res. 2013:1417–1419. doi: 10.1038/cr.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheeseright T, Mackey M, Rose S, Vinter A. J. Chem. Inf. Model. 2006;46:665–676. doi: 10.1021/ci050357s. [DOI] [PubMed] [Google Scholar]
- 9. Forge version 10.3.1, Cresset BioMolecular Discovery Ltd., 2014; www.cresset-group.com/products/forge/
- 10. SiteMap version 2.9, Schrödinger LLC, New York, NY (USA), 2013; www.schrodinger.com/SiteMap.
- 11. Activity Miner in Forge version 10.3.1, Cresset BioMolecular Discovery Ltd., 2014; www.cresset-group.com/products/activityminer/
- 12a.Zhang EE, Liu Y, Dentin R, Pongsawakul PY, Liu AC, Hirota T, Nusinow DA, Sun X, Landais S, Kodama Y, Brenner DA, Montminy M, Kay SA. Nat. Med. 2010;16:1152–1157. doi: 10.1038/nm.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b.Lamia KA, Papp SJ, Yu RT, Barish GD, Uhlenhaut NH, Jonker JW, Downes M, Evans RM. Nature. 2011;480:552–556. doi: 10.1038/nature10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oshima T, Yamanaka I, Kumar A, Yamaguchi J, Nishiwaki-Ohkawa T, Muto K, Kawamura R, Hirota T, Yagita K, Irle S, Kay SA, Yoshimura T, Itami K. Angew. Chem. Int. Ed. 2015;54:7193–7197. doi: 10.1002/anie.201502942. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2015;127 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information