

Abstract
We validate a practical methodology for the rapid profiling of small molecule inhibitors of protein–protein interactions. We find that a well known BH3 family inhibitor can potently inhibit the p53/hDM2 interaction.
Protein–protein interactions are involved in almost all biological processes.1 As a result, reagents capable of disrupting these interactions or stabilizing these interactions are highly sought after both as probes for dissecting biology and for therapeutic leads. A variety of elegant methods have been developed to disrupt protein–protein interactions (PPIs), including antibodies, peptides, and miniature proteins.2 Synthetic approaches have more recently found success, and include foldamers,3 terphenyl scaffolds,4 stabilized helices,5 small-molecule fragment engineering, in silico engineering, and compound library screening.6 Many of the small molecule approaches have honed in on helix–receptor PPIs and thus an important question arises: how specific are inhibitors for their intended targets? Answering a similar question in the field of protein kinase inhibition has resulted in a paradigm shift, where large scale profiling approaches have demonstrated unintended promiscuity or polypharmacology of known inhibitors.7 This promiscuity can be potentially beneficial by targeting several kinases of interest, or harmful. Herein we provide a potentially scalable methodology for the rapid and simple profiling for the helix/receptor class of PPIs and demonstrate that known small molecule inhibitors can display potent off-target effects.
Currently PPIs and their inhibitors are routinely interrogated by quantitative SPR or fluorescence based methods.8 These methods rely upon purified and often chemically modified components, which can be resource intensive and thus challenging for the routine profiling of larger panels of PPIs. We have recently described a split-protein methodology (also referred to as protein complementation) (Fig. 1a), which potentially provides an avenue for rapidly profiling PPIs and their inhibitors.9 This approach does not require cell culture, purified proteins, or chemical modification steps and perhaps mimics the complexity of a cellular environment to a first approximation. We explore the application of this methodology towards a set of relevant helix/receptor PPIs that have been targeted by small molecule inhibitors, leading to surprising results.
Fig. 1.

Specificity of helix–receptor interactions. (a) Schematic for the cell free interrogation of helix–receptor interactions resulting in interaction dependent luminescence. (b) (i) BIM (dark grey)/Bcl-xL (light grey) (ii) BIM (dark grey)/BFL (light grey), (iii) p53 (dark grey)/hDM2 (light grey), and (iv) Hif-1α (dark grey)/p300 (light grey). (c) Luminescence of co-translated helix–Nfluc and Cfluc–receptor interactions for all 18 pairs.
Helix–receptor interactions are generally characterized by the binding of an alpha-helical domain to a relatively hydrophobic groove of a larger protein domain (Fig. 1b). This small yet dense interface has been particularly amenable to the development of small-molecule inhibitors. Specifically, two similar yet unrelated groups of helix–receptor interactions were the first to yield to potent small molecule inhibitors; the interaction between hDM2 with the activation domain of p53 and interactions amongst pro-apoptotic BH3 only and anti-apoptotic members of the Bcl-2 family of proteins (Fig. 2).10 To interrogate this class of PPIs we chose the fragmented luciferase reporter over other split-proteins that also provide simple read-outs,11 as this reporter was found to be more sensitive than split-lactamase and split-GFP under cell free conditions.9,11 Appropriate fusions were created in which a series of helix/receptor pairs were appended to the N- and C-terminal fragments creating the fusions “helix-NFluc” and “CFluc-receptor” respectively (Table S1, ESI‡). Using this panel we first interrogated the interaction specificity of a set of 18 helix/receptor combinations (Fig. 1c). Importantly, we found the split-luciferase method recapitulated the affinity of BIM for the Bcl-2 family of receptors, whereas none of the four tested receptors bound p53 or Hif1-α. Similarly, neither hDM2 nor p300 were found to bind the BIM peptide, thus showing that the native helix/receptor pairs in our panel are orthogonal.
Fig. 2.
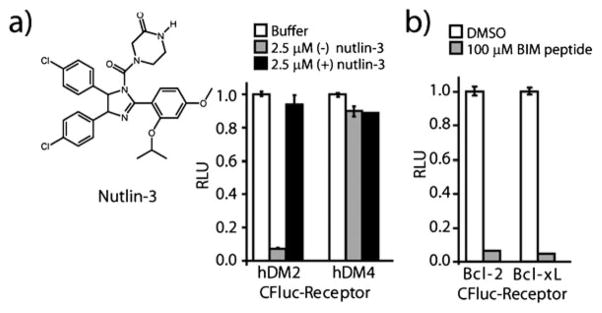
Interrogation of inhibitors of helix–receptor interactions. (a) Inhibition of the interaction of p53 with hDM2 and hDM4 by enantiomers of nutlin-3. (b) Inhibition of the interaction of Bcl-2 and Bcl-xL with the BIM BH3 domain upon addition of free BIM peptide.
Having a helix–receptor panel capable of reporting upon interaction specificity, we next sought to interrogate its suitability for PPI inhibitor profiling. This is particularly relevant as the constellation of residues implicated in binding their respective receptors are grossly similar for p53 (Phe19, Trp23, and Leu26) and BIM (Trp147, Ile155, Phe169), while those implicated for Hif1-α/p300 are primarily aliphatic (Leu795, Cys800, Leu818, and Leu822). To test whether differences in specificity could be evaluated for inhibitors of the p53/receptor interactions we first evaluated the ability of the (+) and (−) enantiomers of the p53/hDM2 specific inhibitor nutlin-3 to inhibit the interaction of p53 with hDM2 and hDM4 (Fig. 2a). Consistent with previous studies, addition of 2.5 μM (−) nutlin-3 resulted in the specific disruption of the reassembled p53/hDM2 complex while the same concentration of (+) nutlin-3 showed minimal inhibition for either the p53/hDM2 or p53/hDM4 interaction. Next, to test inhibitors of Bcl-2 family interactions, the inhibition of the interaction of BIM with Bcl-2 and Bcl-XL was evaluated following the addition of BIM BH3 peptide (residues 142–161) (Fig. 2b), demonstrating disruption of the interaction between BIM/bcl-2 and BIM/Bcl-XL interaction. The ability to competitively inhibit protein–protein interactions is an advantage for the split-luciferase based systems as split-GFP based systems result in an irreversible complex.
Thus with a viable method in hand to report upon the specificity of inhibitors of helix–receptor interactions we next investigated the specificity of nutlin as well as two well-studied inhibitors of interactions between the pro- and anti-apoptotic Bcl-2 family domains, specifically ABT-737 and its bioavailable analog ABT-263 (for synthesis details please see ESI‡). Each compound was tested for its ability to inhibit the panel of 6 helix–receptor interactions, along with the interactions between the coiled-coils Fos/Jun. In order to ensure that compounds do not inhibit luciferase activity, a tethered luciferase containing a covalent N- and C-terminal linkage designed to mimic post-reassembled split-luciferase was also included in the panel. The three compounds tested, significantly inhibited their known PPI targets. In addition to significant inhibition of p53/hDM2, (−)-nutlin-3 showed modest inhibition of the interaction between the BIM BH3 domain and Bcl-2. In the case of ABT-737 and its analogue ABT-263, both showed the most potency against interactions between BH3 with Bcl-2, Bcl-XL, and Bcl-w but not BFL6e,12,13 as previously observed. Interestingly, ABT-263 showed significant inhibition of the p300/Hif-1α interaction when compared to ABT-737, which we will interrogate further in future studies. Notably, modifications at positions remote fromthe pharmacophore for increasing bioavailability, may potentially lead to unanticipated changes in inhibition profiles when tested against larger PPI panels (Fig. 3).
Fig. 3.

Inhibition profile of (−) Nutlin, ABT-737, and ABT-263 against a panel of 6 helix/receptor interactions as well as the Fos/Jun leucine zipper and luciferase controls. All inhibition experiments were performed at 100 μM of the indicated compound.
Finally, the well-studied Bcl-2 family inhibitor BH3I-16d,e was interrogated. BH3I-1, while inhibiting its reported target Bcl-2/Bim and Bcl-xL/Bim, showed significant inhibition of both the p53/hDM2 and p300/Hif-1α interactions (Fig. 4a). This surprising promiscuity, displayed by a well studied compound6d led us to further interrogate the p53/hDM2 interaction utilizing a standard fluorescence polarization (FP) assay with purified protein (Fig. 4b). The results from the FP assay validated the split-luciferase screen and demonstrated that BH3I-1 has a Kd = 5.3 μM against the p53/mDM2 pair, which is comparable to its low micromolar potency reported for the BH3 family of receptors.6e
Fig. 4.

(a) Inhibition profile of BH3I-1 (100 μM) against the PPI panel. (b) Fluorescence polarization experiment with fluorescein labeled p53-peptide and mDM2 with added BH3I-1 (400 μM to 391 nM) resulting in a Kd = 5.3 μM.
In conclusion, we have developed a methodology amenable for the rapid interrogation of the helix–receptor PPIs as an initial test for probing their specificity. Of particular note is the unanticipated inhibition of the p53/hDM2 interaction by BH3I-1 a well known inhibitor of the Bcl2 family further validated utilizing traditional fluorescence polarization experiments. These studies demonstrate that both beneficial and detrimental polypharmacology of existing compounds can be potentially uncovered when larger sets of helix–receptor pairs are interrogated. Future studies will aim to clarify the potential biological consequences of the observed polypharmacology as well as interrogate larger sets of PPI pairs against small molecule and peptide inhibitors. We anticipate that this simple approach for establishing selectivity profiles, whether for biological assays or for therapeutic leads, can help guide PPI inhibitor design.
Supplementary Material
Acknowledgments
This research was partially supported by NIH (GM077403) and NSF (CHE-0548264) to I. G and NIH (GM073943) to P. S. A. We thank Dr Neal Zondlo for the His-MDM2 construct.
Footnotes
This article is part of the ‘Enzymes and Proteins’ web-theme issue for ChemComm.
Electronic supplementary information (ESI) available: Details of experimental procedures, synthetic procedures, and FP assays. See DOI: 10.1039/c0cc02969f
Contributor Information
Paramjit S. Arora, Email: arora@nyu.edu.
Indraneel Ghosh, Email: ghosh@u.arizona.edu.
Notes and references
- 1.(a) Wells JA, McClendon CL. Nature. 2007;450:1001. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]; (b) Arkin MR, Whitty A. Curr Opin Chem Biol. 2009;13:284. doi: 10.1016/j.cbpa.2009.05.125. [DOI] [PubMed] [Google Scholar]
- 2.(a) Chin JW, Schepartz A. Angew Chem, Int Ed. 2001;40:3806–3809. [PubMed] [Google Scholar]; (b) Kritzer JA, Zutshi R, Cheah M, Ran FA, Webman R, Wongjirad TM, Schepartz A. ChemBioChem. 2006;7:29. doi: 10.1002/cbic.200500324. [DOI] [PubMed] [Google Scholar]; (c) Smith TJ, Stains CI, Meyer SC, Ghosh I. J Am Chem Soc. 2006;128:14456–14457. doi: 10.1021/ja065557e. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kritzer JA, Lear JD, Hodsdon ME, Schepartz A. J Am Chem Soc. 2004;126:9468. doi: 10.1021/ja031625a. [DOI] [PubMed] [Google Scholar]; (b) Sadowsky JD, Fairlie WD, Hadley EB, Lee HS, Umezawa N, Nikolovska-Coleska Z, Wang S, Huang DC, Tomita Y, Gellman SH. J Am Chem Soc. 2007;129:139. doi: 10.1021/ja0662523. [DOI] [PubMed] [Google Scholar]; (c) Lee EF, Sadowsky JD, Smith BJ, Czabotar PE, Peterson-Kaufman KJ, Kimberly J, Colman PM, Gellman SH, Fairlie DW. Angew Chem, Int Ed. 2009;48:4318. doi: 10.1002/anie.200805761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Orner BP, Ernst JT, Hamilton AD. J Am Chem Soc. 2001;123:5382. doi: 10.1021/ja0025548. [DOI] [PubMed] [Google Scholar]; (b) Yin H, Lee GI, Sedey KA, Kutzki O, Park HS, Orner BP, Ernst JT, Wang HG, Sebti SM, Hamilton AD. J Am Chem Soc. 2005;127:10191. doi: 10.1021/ja050122x. [DOI] [PubMed] [Google Scholar]; (c) Shaginian A, Whitby LR, Hong S, Hwang I, Farooqi B, Searcey M, Chen J, Vogt PK, Boger DL. J Am Chem Soc. 2009;131:5564. doi: 10.1021/ja810025g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Plante JP, Burnley T, Malkova B, Webb ME, Warriner SL, Edward TA, Wilson AJ. Chem Commun. 2009;5045 doi: 10.1039/b908207g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang D, Liao W, Arora PS. Angew Chem, Int Ed. 2005;44:6525–6529. doi: 10.1002/anie.200501603. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Henchey LK, Jochim AL, Arora PS. Curr Opin Chem Biol. 2008;12:692. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. Nature. 2005;435:677. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]; (b) Enyedy IJ, Ling Y, Nacro K, Tomita Y, Wu X, Cao Y, Guo R, Li B, Zhu X, Huang Y, Long YQ, Roller PP, Yang D, Wang S. J Med Chem. 2001;44:4313. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]; (c) Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]; (d) Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Nat Cell Biol. 2001;3:173. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]; (e) Zhai D, Jin C, Satterthwait AC, Reed JC. Cell Death Differ. 2006;13:1419. doi: 10.1038/sj.cdd.4401937. [DOI] [PubMed] [Google Scholar]
- 7.Knight ZA, Lin H, Shokat KM. Nat Rev Cancer. 2010;10:130. doi: 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Berg T. Angew Chem, Int Ed. 2003;42:2462. doi: 10.1002/anie.200200558. [DOI] [PubMed] [Google Scholar]; (b) Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, Boger DL, Vogt PK. Proc Natl Acad Sci U S A. 2002;99:3830. doi: 10.1073/pnas.062036999. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brooks PC, Silletti S, von Schalscha TL, Friedlander M, Cheresh DA. Cell (Cambridge, Mass) 1998;92:391. doi: 10.1016/s0092-8674(00)80931-9. [DOI] [PubMed] [Google Scholar]; (d) Owicki JC. J Biomol Screening. 2000;5:297. doi: 10.1177/108705710000500501. [DOI] [PubMed] [Google Scholar]
- 9.Porter JR, Stains CI, Jester BW, Ghosh I. J Am Chem Soc. 2008;130:6488. doi: 10.1021/ja7114579. [DOI] [PubMed] [Google Scholar]
- 10.(a) Chene P. Nat Rev Cancer. 2003;3:102. doi: 10.1038/nrc991. [DOI] [PubMed] [Google Scholar]; (b) Lessene G, Czabotar PE, Colman PM. Nat Rev Drug Discovery. 2008;7:989. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 11.(a) Johnsson N, Varshavsky A. Proc Natl Acad Sci U S A. 1994;91:10340. doi: 10.1073/pnas.91.22.10340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pelletier JN, Campbell-Valois FX, Michnick SW. Proc Natl Acad Sci U S A. 1998;95:12141. doi: 10.1073/pnas.95.21.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ghosh I, Hamilton AD, Regan L. J Am Chem Soc. 2000;122:5658. [Google Scholar]; (d) Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Proc Natl Acad Sci U S A. 2004;101:12288. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Cancer Cell. 2006;9:351. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 13.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW. Cancer Res. 2008;68:3421. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.