

Abstract
Background
Alloimmune lung injury, characterized by perivascular lymphocytic inflammation, lymphocytic bronchiolitis (LB), and obliterative bronchiolitis (OB), causes substantial morbidity and mortality after lung transplantation and bone marrow transplantation (BMT), but little is known regarding its pathogenesis. We have developed and pursued the hypothesis that local activation of pulmonary innate immunity through toll-like receptor (TLR)-4 is critical to the development of posttransplant alloimmune lung injury.
Methods
We developed a fully major histocompatibility complex–mismatched murine BMT model without systemic graft-versus-host disease, and challenged mice with aerosolized lipopolysaccharide (LPS), a prototypic TLR4 agonist, to determine the effect upon pulmonary alloimmune lung injury.
Results
LPS-exposed allogeneic BMT recipient mice developed histological and biological features of LB and OB, which were not observed in non-LPS-exposed allogeneic controls or syngeneic LPS-exposed mice. LPS-induced lymphocytic lung inflammation was dependent upon intact TLR4 signaling in donor-derived hematopoietic cells but not recipient structural lung cells, demonstrating a distinct function for TLR4 on hematopoietic cells in mediating alloimmunity.
Conclusions
We demonstrate a critical role for localized, environmentally induced innate immune activation in promoting alloimmune lung injury. Local inhibition of TLR4 signaling in pulmonary resident hematopoietic cells represents a novel and potentially important therapeutic target to prevent posttransplant rejection.
Keywords: Bronchiolitis obliterans, Allograft rejection, Innate immunity, Toll-like receptor-4
The lung is vulnerable to alloimmune injury, both in the context of lung transplantation (host-versus-graft) and bone marrow transplantation (graft-versus-host) (1). Alloimmune lung injury has a spectrum of presentations: predominantly alveolar injury, such as idiopathic pneumonia syndrome (IPS) and diffuse alveolar hemorrhage; perivascular lymphocytic infiltrates and lymphocytic bronchiolitis (LB); or chronic alloimmune airway injury that appears histologically as obliterative bronchiolitis (OB) and clinically as progressive airflow obstruction called bronchiolitis obliterans syndrome (BOS). OB/BOS is the most common cause of morbidity and mortality after lung transplantation, and treatment options are very limited. More than 50% of lung transplant recipients develop BOS 5 years after transplantation (2). Similarly, up to 20% of bone marrow transplantation (BMT) patients develop BOS, although reports may underestimate the true disease prevalence due to less frequent pulmonary function testing after BMT (3).
Despite the high frequency of rejection after lung or bone marrow transplantation, little is known regarding the factors that contribute to the marked interindividual differences in the onset and progression of BOS. Systemic graft-versus-host disease and systemic innate immune activation are significant risk factors for some forms of alloimmune lung injury such as IPS (4). Unlike other solid organ transplants, however, the lung is also continuously exposed to environmental stimuli including microbial pathogen associated molecular patterns, which promote local activation of innate immunity. We have developed and pursued the hypothesis that local activation of pulmonary innate immunity promotes alloimmune lung injury, leading to acute rejection, LB and BOS. In support of this hypothesis, we have previously found that human lung transplant recipients with polymorphisms of toll-like receptor (TLR)-4 that blunt the response to lipopolysaccharide (LPS), a prototypic trigger of innate immunity, have fewer episodes of acute rejection and greater BOS-free survival (5, 6).
In the current analysis, we utilized a fully mismatched murine BMT model, titrating donor cells so as to avoid any systemic graft-versus-host disease (GVHD) at baseline, and then challenged these mice with aerosolize dLPS to test the hypothesis that local (but not systemic) innate immune activation through TLR4 is sufficient for the development of alloimmune lung injury. We demonstrate that repeated exposure to inhaled LPS in the presence of alloimmunity leads to perivascular and peribronchial accumulation of activated CD4+ T-lymphocytes in lung lavage fluid and tissue, even without systemic GVHD. The pattern of lung injury resembles histologically, biologically, and immunologically acute rejection, LB, and OB observed in human subjects. Furthermore, the absence of functional TLR4 in donor-derived hematopoietic cells abrogates development of pulmonary pathology. Collectively, these results suggest that local activation of pulmonary innate immunity though LPS and its receptor TLR4 is sufficient for the development of LB and OB after lung or bone marrow transplantation, even without systemic GVHD.
MATERIALS AND METHODS
Experimental Murine BMT
BALB/c, C3HeB/FeJ, or C3H/HeJ mice were used as BMT donors and C57BL/6 or TLR4−/− mice were used as recipients. Six- to 8-week-old male BALB/c (H2d), C3HeB/FeJ (H2k), C3H/HeJ(H2k), and C57BL/6 (H2b) mice were obtained from Jackson laboratories. Tlr4-deficient mice (TLR4−/−), backcrossed into C57BL/6 for >10 generations, are maintained in our laboratory. Recipient mice were lethally irradiated (10.5 Gy) and injected intravenously with 4×106 donor marrow cells and 106 donor splenocytes. Syngeneic control BMT was performed under identical conditions using C57BL/6 donors and recipients. Animals were housed in a pathogen-free facility and received acidified neomycin water. Experiments were approved by the Institutional Animal Care and Use Committees (IACUC) at Duke University and the National Institute of Environmental Health Sciences (NIEHS).
LPS Exposures
LPS exposure started 4 weeks after BMT. By the time of LPS challenge, mice had fully recovered from BMT, were gaining weight, and were free of any signs of pulmonary pathology or GVHD (Supplementary Fig. 1, available for viewing online only). Lyophilized LPS (Escherichia coli 0111:B4, Sigma, St. Louis, MO) was used. For LPS aerosol generation, we used a six-jet Atomizer (TSI Inc., Shoreview, MN) at 35 psi as previously described (7). LPS aerosol concentrations in these experiments were 6–8 μg/m3. Mice were exposed for 2.5 hr/day, for 7 or 14 days. Mice were sacrificed 3 days after last LPS exposure to avoid confounding effects of acute LPS exposure.
Whole Lung Lavage and Sera Cytokine Assays
Whole lung lavage was performed, and lavage cells were spun and stained as described previously (7). Serum was also obtained at the time of sacrifice. Supernatants were stored at −80°C until used. Levels of CCL5, tumor necrosis factor (TNF)-alpha, and interleukin (IL)-6 in serum and lavage fluid were quantitated using a Bio-Plex Mouse Panel (Bio-Rad, Hercules, CA).
Histology and Immunohistochemistry
Lungs were perfused with 0.9% saline; the right lung was removed, snap-frozen in liquid nitrogen, and stored at −80°C, while the left lung was instilled and fixed with 10% buffered formalin. Tissue was embedded in paraffin and 5-μm sections were stained as described previously (7). CD3 staining was performed using rabbit anti-CD3 antibody (Abcam, Cambridge, MA) and Vectastain ABC (Vector, Burlingame, CA). The pathological severity of lymphocytic lung inflammation was graded as described in the Supplemental Methods (available for viewing online only), similar to that previously described in human and murine transplant (8, 9). Samples were reviewed by two independent blinded investigators and scores were averaged for each sample. Interreviewer agreement was high (kappa=0.69).
Flow Cytometry
Undiluted lavage fluid or whole lung cell preparations were used for analysis. Where appropriate, whole lungs were homogenized and digested with collagenase A and DNAse I, filtered, red cell lysed, and washed prior to analysis. Cells were incubated and stained with fluorescein isothiocyanate–conjugated anti-mouse major histocompatibility complex class II, phycoerythrin-conjugated antimouse CD4, pacific blue conjugated antimouse CD8, antigen-presenting cell (APC)-conjugated antimouse CD3, and APC-Cy7 conjugated antimouse CD19 (all by BD Pharmingen, San Diego, CA). For foxp3 staining, cells were fixed, permeabilized, and stained with antimouse foxp3 antibody (clone PCH101, eBioscience, San Diego, CA). Cells were analyzed in a BD LSR II Flow Cytometer using BD FACSDiva software.
Mixed Lymphocyte Reaction
Responder lung lymphocytes from spleens or lungs were resuspended in Roswell Park Memorial Institute 1640 with 10% fetal calf serum plus 1% penicillin/streptomycin and 1% L-glutamine, added at 4×105 to mitomycin-treated allogeneic stimulator splenocytes (1×105) and cultured in a 96-well plate for 48 hr, then spiked with tritiated thymidine (50 μg/ml, Sigma, St. Louis, MO) and harvested (Tomtec Harvester 96, Hamden, CT) after 72 hr. Cell proliferation was measured in counts per minute (CPM) using a Wallac Microbeta Trilux 1450–022 (Perkin Elmer, Boston, MA).
Statistical Analyses
Data are expressed as means ± SEM. Individual comparisons between groups were performed using one-way analysis of variance and Bonferroni’s Multiple Comparison Test. Statistical calculations were performed using SAS (version 9, Cary, NC).
RESULTS
Aerosolized LPS Challenge Induces Lymphocytic Bronchiolitis, Epithelial Injury, and Obliterative Bronchiolar Lesions in Allogeneic But Not Syngeneic BMT Recipient Mice
Seven days of repeated LPS exposure after allogeneic BMT (either Balb/c or C3HeB/FeJ donors into C57/BL6 recipients) resulted in an intense and diffuse mononuclear infiltrate localized around the vessels and airways (Fig. 1A, 2A, 2B). Importantly, no alveolar inflammation was noted in these mice. Minimal lymphocytic inflammation was present in unexposed time-matched allogeneic BMT control mice (Fig. 1B) and LPS-exposed syngeneic mice (Fig. 1C), and no lymphocytic inflammation was present in the unexposed syngeneic mice (Fig. 1D).
FIGURE 1.

(A) Lymphocytic perivascular (arrowheads) and peribronchial (arrow) inflammation in LPS-exposed allogeneic BMT recipient mice (H&E, 40× magnification). (B) Minimal mononuclear inflammation in time-matched allogeneic unexposed mice (H&E, 100× magnification). (C) LPS-exposed syngeneic BMT recipient mice (H&E, 200× magnification). (D) No mononuclear inflammation in syngeneic control mice (H&E, 200×). (E) Significant lymphocytosis in the lung fluid of LPS-exposed allogeneic BMT mice compared to unexposed allogeneic controls, syngeneic LPS-exposed or syngeneic unexposed controls. (F) Quantification of perivascular and peribronchial inflammation using the pathology index.
FIGURE 2.

Immunohistochemistry demonstrates peribronchial CD3-positive cells (arrows) in LPS-exposed allogeneic BMT recipient mice (A, 40×, and B, 400× magnification). (C) Epithelial disruption and squamous metaplasia (arrows) are evident throughout a bronchus affected diffusely with lymphocytic bronchiolitis (400×). (D) Intraluminal lesion comprised of loose connective tissue and lymphocytes (arrow) partially obstructs the lumen of a bronchus with disrupted and metaplastic epithelium throughout (400×). (E) α-smooth muscle actin staining demonstrates hyperplasia of myofibroblasts (arrowhead, bottom panel) adjacent to squamous metaplastic epithelium (arrow, bottom panel). Normal epithelium and myofibroblasts in isogeneic mice (top panel). (F) Quantification of perivascular and peribronchial inflammation (*P<0.001 compared to all other groups, **P<0.01 compared to isogeneic mice).
Using a histopathological index (Supplementary Table 1, available for viewing online only) for the extent and severity of perivascular and peribronchial inflammation, we quantified pathological lung inflammation among LPS-exposed allogeneic, LPS-exposed syngeneic, allogeneic unexposed controls, and syngeneic unexposed controls 3 days after the last LPS exposure. LPS-exposed allogeneic BMT mice developed significantly greater total histopathological inflammation than all the other groups (Fig. 1F).
We then prolonged the duration of innate immune activation using 14 days of daily LPS challenge. We found that allogeneic BMT recipient mice developed severe epithelial squamous metaplasia and epithelial denudation in regions of intense lymphocytic bronchiolitis (Fig. 2C). Furthermore, proliferative intraluminal obstructive lesions were evident in approximately 10% of the airways of LPS-exposed allogeneic recipient mice, resulting in luminal obstruction (Fig. 2D). These lesions strongly resemble the pattern of epithelial injury and airway obliteration observed in chronic human transplant rejection. Immunohistochemical staining for α-smooth muscle actin showed hypertrophied subepithelial myofibroblasts which often could be found adjacent to metaplastic epithelium (Fig. 2E). Quantification of histological changes also showed significantly higher scores in LPS-exposed allogeneic recipient mice (Fig. 2F).
Aerosolized LPS Challenge Promotes Lymphocytic Inflammation of Predominantly Activated (CD25+) T-Lymphocytes in the Lung of Allogeneic But Not Syngeneic BMT Recipient Mice
Total cell counts and cellular subsets were compared in the lung fluid between LPS-exposed and unexposed allogeneic and syngeneic BMT recipient mice. At the time of analysis (3 days after last LPS exposure), there were low neutrophil counts consistent with resolution of acute LPS-induced inflammation. No significant differences among the four groups in total neutrophil counts were detected (P=0.28, analysis of variance [ANOVA]).
In contrast, a significantly higher cell count was observed in allogeneic LPS-exposed mice compared to all other groups in the total number of lavage cells (P=0.0009, ANOVA), as well as in total lymphocyte counts (P=0.0004, ANOVA, Fig. 1E). Similar results were obtained when transplants were performed using BALB/c or C3HeB/FeJ donors into C57/BL6 recipients, demonstrating that this effect occurs across different allogeneic strain combinations. LPS exposure did not cause a statistically significant increase in lymphocyte counts in syngeneic mice (Fig. 1E). CD3 immunostaining of the peribronchial and perivascular mononuclear cells in LPS-exposed allogeneic BMT mice showed that they were predominantly T-cells (Fig. 2A and B). In flow cytometry, significant elevations in total numbers of CD3+ T-cells were present in whole lung tissue of LPS exposed allogeneic BMT mice compared to LPS-exposed syngeneic mice(566,600± 66,700 allogeneic vs. 325,300±75,800 syngeneic, P<0.05, Fig. 3A), with similar results in the bronchoalveolar lavage fluid.
FIGURE 3.
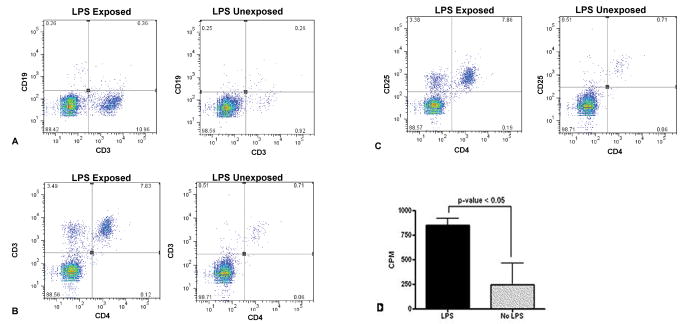
Recruitment of lymphocyte subpopulations into the BAL fluid of LPS exposed and unexposed BMT recipient mice. (A) Increased CD3+ T-cells are present in the lung fluid of LPS-exposed mice. (B) Increased CD4+ cells among the CD3+ population in LPS-exposed BMT mice. (C) Increased CD4+CD25+ T-cells among the CD4+ population in LPS-exposed BMT mice. (D) Increased proliferation of isolated lymphocytes from LPS-exposed lungs compared to non-LPS exposed lungs when stimulated with mitomycin-treated C57BL/6 splenocytes (P=0.03).
Furthermore, the T-cell population present in the lungs of the LPS-exposed allogeneic BMT recipient mice reflected predominately CD4+T-cells (282,700±37,100 in LPS-exposed allogeneic vs. 115,100±15,000 LPS-exposed syngeneic, P<0.01, Fig. 3B). Among these cells, there was high expression of the T-cell activation marker CD25 (Fig. 3C). There was no difference in the CD4+CD25+(high) populations among groups, indicating that regulatory T-cell numbers were not directly affected by inhaled LPS. To further corroborate this finding, we performed flow cytometry for foxp3. After LPS exposure, we found no difference in the percentage ratio of foxp3+ cells among all CD4+ cells, although the absolute numbers of foxp3+ cells increased in parallel with CD4. A small, nonsignificant elevation was observed in CD8+cells in LPS-exposed allogeneic recipient mice. No significant elevations were observed in B-cells in LPS-exposed as compared to unexposed mice. In aggregate, these results suggest that inhaled LPS exposure can lead to chemotaxis of activated CD4 lymphocytes into lungs, but only in the presence of allogeneic immunity.
Increased Pulmonary But Not Systemic Proliferation of Lung Cells in Response to Recipient Antigens Occurs After Local LPS Challenge
Using a standard MLR assay, the proliferative response of cells isolated from the whole lung or spleen of LPS-exposed allogeneic BMT recipient mice were compared to unexposed allogeneic BMT recipient mice. Splenocytes from LPS-exposed vs. LPS-unexposed mice proliferated similarly in response to mitomycin-treated C57BL/6 stimulator cells (5546±634 vs. 6017±1557 counts per minute, P=0.68). However, there was a significant increase in the proliferative response of isolated lymphocytes from LPS-exposed lungs compared to the LPS-unexposed lungs (Fig. 3D). Therefore, although aerosolized LPS did not alter the degree of systemic lymphocyte proliferation in response to allogeneic recipient cells, there was a significant increase in the proliferation of lung lymphocytes in response to the allogeneic recipient cells.
LPS-Exposed Allogeneic BMT Recipient Mice Demonstrate Sustained Elevations of Lung CCL5 But Not TNF-α
RANTES and CCL5 have been repeatedly shown to play a significant role in the recruitment of lymphocytes in human and animal lung rejection (10–12). We therefore measured levels of (CCL5) in lavage fluid and serum of LPS-exposed mice. There was significant elevation of CCL5 in the lung lavage fluid after 14 days of LPS exposure in allogeneic BMT recipient mice as compared to controls (Fig. 4A). In contrast, among LPS-exposed vs. unexposed allogeneic BMT recipient mice after either 7 days or 14 days of LPS challenge (Fig. 4B). Concentrations of the proinflammatory cytokines TNF-alpha and IL-6 in lung lavage fluid and serum were very low, as expected 3 days after last LPS exposure, and not significantly different between the lavage fluid of either LPS exposed or LPS unexposed allogeneic BMT recipient mice (not shown). Collectively, these data demonstrate that aerosolized LPS exposure promotes adaptive immune activation through CCL5 locally in the absence of systemic GVHD.
FIGURE 4.
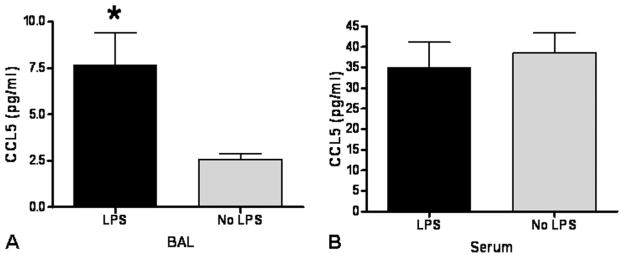
(A) Significant increase in CCL5 in the lung fluid of LPS-exposed allogeneic BMT recipient mice vs. time-matched allogeneic unexposed BMT mice after 2 weeks of LPS exposure (*P<0.05). (B) No difference in serum CCL5 levels in LPS-exposed vs. unexposed mice after 2 weeks of LPS exposure.
LPS Induced Lymphocytic Lung Inflammation Is Mediated Through TLR4 on Donor-Derived Hematopoietic Cells But Not Recipient Structural Lung Tissue
To determine if the effects of pulmonary innate immune activation were mediated through donor-derived hematopoietic cells (H+) versus recipient structural (S+) cells, a series of BMT experiments were performed to compare the effects of TLR4 deficiency in donor and/or recipient cells upon lymphocyte lung inflammation. C3H/HeJ (with a known TLR4 mutation that abrogates function) or C3HeB/ FeJ (with functional TLR4) were used as BMT donors and transplanted into C57BL/6 or TLR4−/− mice. C3H/HeJ mice are genetically closely related to and fully histocompatible with C3HeB/FeJ but lack LPS responsiveness. We therefore chose them as an appropriate allogeneic donor comparison to the LPS-responsive C3HeB/FeJ.
Deficiency of TLR4 in the donor marrow cells (H−/S+ or H−/S−) markedly reduced the degree of pulmonary inflammation (Fig. 5). Significant differences were observed among the groups with regards to the pathological index (P=0.003), the lymphocyte counts (P<0.0001), and production of CCL5 in the lung lavage fluid of mice (P<0.0001). In pairwise comparisons for each of these variables, the groups with intact donor leukocyte TLR4 (H+) were significantly elevated as compared to the groups with impaired donor leukocyte TLR4 signaling (H−). In contrast, deficiency of TLR4 in the recipient structural cells alone (H+/S−) did not diminish the inflammatory response compared to wild-type (H+/S+). Representative histology from each group is shown in Figure 6, demonstrating that donor-derived (H+) intact TLR4 signaling is necessary for the development of the intense and diffuse mononuclear peribronchial and perivascular inflammation.
FIGURE 5.
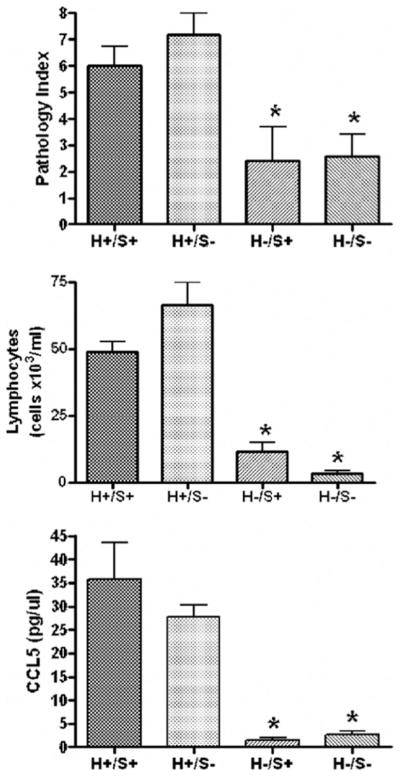
Development of BAL lymphocytosis, CCL5 production, and tissue mononuclear inflammation is dependent upon intact TLR4 signaling in donor-derived hematopoietic cells (H+) but not structural recipient tissues (S+).
FIGURE 6.

LPS-induced mononuclear peribronchial and perivascular inflammation is dependant upon intact hematopoietic (H+) donor-derived cells but not recipient structural (S+) cells. Extensive mononuclear inflammation (arrows) is present in mice with intact H+/S+(A, 40×) or H+/S−(C, 100×), but not in mice with deficiency of TLR4 in the donor cells, H−/S+ (B, 40×) or H+/S−(D, 200×). Hematoxylin and eosin staining.
DISCUSSION
In the present study, we demonstrate that local pulmonary activation of innate immunity by LPS, in the context of alloimmune sensitization, promotes the development of a robust alloimmune response that replicates biological, immunological, and pathological features of LB and OB in human transplantation. The response is characterized by accumulation of activated T-lymphocytes in the lung fluid and tissues, striking perivascular and peribronchiolar lymphocytic inflammation, airway epithelial injury, and heightened allospecific responsiveness to recipient antigens. Our results therefore provide considerable support for the hypothesis that local activation of pulmonary innate immunity can independently promote the development of alloimmune lung injury in lung transplantation and BMT.
Our BMT model system, which maintains environmental contact with fully vascularized lung tissue in the setting of circulating lung-reactive cells, provides new evidence for the importance of environmental exposures and resident pulmonary innate immunity in the development of alloimmune lung injury. Our approach contrasts with the commonly used murine models of orthotopic tracheal transplant, which lack vascularization, bronchi, and environmental contact (13, 14). In the context of our model, we were able to test the importance of environmental stimulation with lung cells and demonstrate that inhaled LPS exposure provokes local alloimmune responsiveness. The use of the BMT model potentially limits the applicability of our results for the field of lung transplantation, in that certain aspects of human lung transplant, such as ischemia-reperfusion injury or the presence of passenger leukocytes, are not present. However, we believe our results are more broadly relevant to pulmonary alloimmune injury given the underlying similarity of histopathology that develops in chronic lung injury after lung or bone marrow transplant. Furthermore, the lack of significant pathology in allogeneic unexposed mice or LPS-exposed syngeneic mice provide appropriate controls to ensure that our results are not an artifact of the process of bone marrow transplantation or irradiation.
Thus, our results suggest that alloimmune lung injury can develop as a form of environmentally mediated lung disease, a substantially different view than currently exists. Alloimmune lung injury after lung transplantation or BMT is generally thought to occur as a result of systemic sensitization of alloreactive T-cells to recipient antigens (15). Immunosuppression after lung transplant accordingly employs systemic T-cell based therapy (e.g., calcineurin inhibitors) for the prevention of rejection. Our findings, in contrast, demonstrate local but not systemic elevations of CCL5 and locally but not systemically heightened proliferation of lung immune cells in response to donor antigens. Collectively, these data imply that discrete foci of lung rejection can occur as a result of local pulmonary innate immune activation rather than primarily as a systemic global response to the allograft. This report may also help explain why noninfectious local lung injury, such as ischemia-reperfusion and gastroesophageal reflux, have been linked to rejection in some studies (16, 17). It has been recently reported that innate immune activation occurs in the lung during ischemia reperfusion injury (18). We also know that endogenous toll-receptor ligands such as hyaluronan are increased in lung transplant recipients (19, 20). We now show that such localized activation is sufficient for the generation of alloimmune lung injury. We have used LPS as a prototypic innate immune activator with a well-described chronic exposure model, but we hypothesize that other local innate immune activators may have similar effects in the context of alloimmunity. Consequently, strategies that inhibit local innate immune activation rather than systemic adaptive immune activation may prove most efficacious in preventing lung rejection.
Our model is similar in some aspects to a previously described model of idiopathic pneumonia syndrome (IPS) after BMT (21). Similar to these reports, we are utilizing a BMT model of allogeneic lung injury, although we are using a fully mismatched model in accordance to the usual situation in lung transplant recipients, as opposed to a minor mismatch used in the above studies. However, the two models have also several important differences. In our model, we titrated the donor splenocyte dose so as to avoid systemic GVHD, whereas in the IPS model, acute systemic GVHD is a hallmark of the presentation. Additionally, our model presents minimal alveolar histopathology, and at the time of sacrifice there is no elevation of TNF-α and IFN-γ in the alveolar lavage fluid, as is observed in the IPS murine model (22). We believe that this is due to the absence of systemic GVHD in our model. Cooke et al. have demonstrated that gut-origin LPS translocates into the circulation as a result of bowel GVHD, and results in systemic innate immune activation leading to alloimmune lung injury (23). Ultimately, we believe that similar mechanisms are responsible for the alloimmune lung injury in both models: innate immune activation in both instances leads to alloimmune injury. Our study, however, is novel in that we demonstrate that local activation of innate immunity in the lung through environmental exposure can lead to alloimmune airway injury in the absence of systemic immune activation or inflammation. The different histologic presentations in our model and the IPS model also support this concept. We observed significant bronchial pathology, mirroring the deposition kinetics of inhaled LPS. In contrast, the murine IPS model shows predominant alveolar pathology, in keeping with a systemic origin of alloimmune injury.
The present results also provide insight into the cells that regulate innate-adaptive cross-talk in the context of transplant alloimmunity. We demonstrate a critical role for TLR4 on hematopoietic cells distinct from TLR4 on structural lung tissue in mediating the development of alloimmune lung injury. These results are consistent with and validate our prior human genetic studies, in which we found that human lung transplant recipients with hyporesponsive TLR4 polymorphisms had diminished acute rejection and improved rejection-free survival (6). In these human lung transplant studies, impaired TLR4 signaling in recipient (i.e., hematopoietic derived) cells was protective, but impaired TLR4 signaling in the donor (i.e., structural lung tissue) was not protective against rejection, which is also consistent with the results of the current study. Our data also suggest functionally distinct roles for macrophage and epithelial TLR4 in regulating pulmonary inflammation. For example, while hyaluronic acid (HA) fragments interact with macrophages to promote inflammation, high molecular weight HA interacts with epithelial cell TLR4 to inhibit apoptosis and diminishes lung inflammation (24). The current report thus contributes to an emerging understanding for divergent functions of TLR4 on circulating immune cells and epithelial cells. The role of regulatory T-cells in our model is less clear. Published literature on the effect of TLR4 activation on regulatory T-cells (Treg) reports inhibition (25), activation (26), or no effect (27) on Treg function. We did not observe any relative changes in Treg populations after LPS exposure. However, we did find a net activation of T-lymphocytes in ex-vivo MLR after inhaled LPS exposure (Fig. 3D), so it is possible that in our model TLR4 activation affects Treg function, if not Treg recruitment into the lung. We are currently exploring this question in ongoing experiments.
Our data also confirm that CCL5 contributes to the development of lymphocytic lung inflammation in alloimmune lung injury. Previous reports using rat allograft models and studies in human transplant recipients have demonstrated elevation of CCL5 in the lung fluid during acute rejection (10, 11). CCL5 blockade was effective at reducing CD4+ T-cell recruitment in a rat heterotopic tracheal transplant model, and in reducing the severity of rejection in a rat allograft model (10, 12). One prior study identified alveolar macrophages as the primary source of CCL5 production in the development of acute rejection, as depletion of alveolar macrophages greatly reduced posttransplant CCL5 levels (11). We hypothesize that LPS promotes production of CCL5 in our model through donor alveolar macrophages and dendritic cells (e.g., pulmonary antigen-presenting cells). Further studies are needed to test this specific hypothesis and clarify the importance of CCL5 and other chemokines in the development of LPS-induced lymphocytic lung inflammation.
In summary, our experiments suggest that lung rejection can be a local process, dependent upon innate immune activation of hematopoietic-derived cells in the lung. Our model provides a unique opportunity to further investigate mechanisms by which alloimmunity leads to bronchiolar injury and fibrosis. To our knowledge, there is no previous murine model of alloimmune orthotopic lung injury with predominant features of lymphocytic bronchiolitis and bronchiolar obliteration, a critical aspect of BOS in humans. Finally, our results suggest that inhibition of innate immune activation in hematopoietic cells represents a novel and potentially important therapeutic target to prevent post-transplant alloimmune lung injury after human lung or bone marrow transplant.
Supplementary Material
Acknowledgments
This study was supported by grants from the American Transplantation Society and the American Society of Transplantation (Partnership Grant T-05-004 to S.G.), National Heart Lung and Blood Institute (HL69978, P50HL-084917 to S.M.P.), and by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences (S.G., S.M.P., D.A.S.).
References
- 1.Dudek AZ, Mahaseth H. Hematopoietic stem cell transplant-related airflow obstruction. Curr Opin Oncol. 2006;18:115. doi: 10.1097/01.cco.0000208782.61452.08. [DOI] [PubMed] [Google Scholar]
- 2.Boucek MM, Faro A, Novick RJ, et al. The Registry of the International Society for Heart and Lung Transplantation: Fourth Official Pediatric Report–2000. J Heart Lung Transplant. 2001;20:39. doi: 10.1016/s1053-2498(00)00243-6. [DOI] [PubMed] [Google Scholar]
- 3.Chien JW, Martin PJ, Gooley TA, et al. Airflow obstruction after myeloablative allogeneic hematopoietic stem cell transplantation. Am J Respir Crit Care Med. 2003;168:208. doi: 10.1164/rccm.200212-1468OC. [DOI] [PubMed] [Google Scholar]
- 4.Cooke KR. Acute lung injury after allogeneic stem cell transplantation: From the clinic, to the bench and back again. Pediatr Transplant. 2005;9 (Suppl 7):25. doi: 10.1111/j.1399-3046.2005.00450.x. [DOI] [PubMed] [Google Scholar]
- 5.Cantu I, Appel EI, James Z, et al. Early fundoplication prevents chronic allograft dysfunction in patients with gastroesophageal reflux disease. Ann Thoracic Surg. 2004;78:1142. doi: 10.1016/j.athoracsur.2004.04.044. [DOI] [PubMed] [Google Scholar]
- 6.Palmer SM, Burch LH, Mir S, et al. Donor polymorphisms in Toll-like receptor-4 influence the development of rejection after renal transplantation. Clin Tranplant. 2006;20:30. doi: 10.1111/j.1399-0012.2005.00436.x. [DOI] [PubMed] [Google Scholar]
- 7.Savov JD, Brass DM, Lawson BL, et al. Toll-like receptor 4 antagonist (E5564) prevents the chronic airway response to inhaled lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol. 2005;289:L329. doi: 10.1152/ajplung.00014.2005. [DOI] [PubMed] [Google Scholar]
- 8.Iacono AT, Keenan RJ, Duncan SR, et al. Aerosolized cyclosporine in lung recipients with refractory chronic rejection. Am J Respir Crit Care Med. 1996;153:1451. doi: 10.1164/ajrccm.153.4.8616581. [DOI] [PubMed] [Google Scholar]
- 9.Belperio JA. Paying a toll for acute lung allograft rejection. Am J Respir Crit Care Med. 2003;168:623. doi: 10.1164/rccm.2306023. [DOI] [PubMed] [Google Scholar]
- 10.Belperio JA, Burdick MD, Keane MP, et al. The role of the CC chemokine, RANTES, in acute lung allograft rejection. J Immunol. 2000;165:461. doi: 10.4049/jimmunol.165.1.461. [DOI] [PubMed] [Google Scholar]
- 11.Sekine Y, Yasufuku K, Heidler KM, et al. Monocyte chemoattractant protein-1 and RANTES are chemotactic for graft infiltrating lymphocytes during acute lung allograft rejection. Am J Respir Cell Mol Biol. 2000;23:719. doi: 10.1165/ajrcmb.23.6.3825. [DOI] [PubMed] [Google Scholar]
- 12.Suga M, Maclean AA, Keshavjee S, et al. RANTES plays an important role in the evolution of allograft transplant-induced fibrous airway obliteration. Am J Respir Crit Care Med. 2000;162:1940. doi: 10.1164/ajrccm.162.5.9910082. [DOI] [PubMed] [Google Scholar]
- 13.Neuringer IP, Mannon RB, Coffman TM, et al. Immune cells in a mouse airway model of obliterative bronchiolitis. Am J Respir Cell Mol Biol. 1998;19:379. doi: 10.1165/ajrcmb.19.3.3023m. [DOI] [PubMed] [Google Scholar]
- 14.Heath DG, Hohneker K, Carriker C, et al. Six-year molecular analysis of Burkholderia cepacia complex isolates among cystic fibrosis patients at a referral center for lung transplantation. J Clin Microbiol. 2002;40:1188. doi: 10.1128/JCM.40.4.1188-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neuringer IP, Chalermskulrat W, Aris R. Obliterative bronchiolitis or chronic lung allograft rejection: A basic science review. J Heart Lung Transplant. 2005;24:3. doi: 10.1016/j.healun.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Hadjiliadis D, Howell DN, Davis RD, et al. Anastomotic infections in lung transplant recipients. Ann Transplant. 2000;5:13. [PubMed] [Google Scholar]
- 17.Fiser SM, Tribble CG, Long SM, et al. Ischemia-reperfusion injury after lung transplantation increases risk of late bronchiolitis obliterans syndrome. Ann Thorac Surg. 2002;73:1041. doi: 10.1016/s0003-4975(01)03606-2. [DOI] [PubMed] [Google Scholar]
- 18.Andrade CF, Kaneda H, Der S, et al. Toll-like receptor and cytokine gene expression in the early phase of human lung transplantation. J Heart Lung Transplant. 2006;25:1317. doi: 10.1016/j.healun.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 19.Rao PN, Zeevi A, Snyder J, et al. Monitoring of acute lung rejection and infection by bronchoalveolar lavage and plasma levels of hyaluronic acid in clinical lung transplantation. J Heart Lung Transplant. 1994;13:958. [PubMed] [Google Scholar]
- 20.Tesar BM, Jiang D, Liang J, et al. The role of hyaluronan degradation products as innate alloimmune agonists. Am J Transplant. 2006;6:2622. doi: 10.1111/j.1600-6143.2006.01537.x. [DOI] [PubMed] [Google Scholar]
- 21.Cooke KR, Kobzik L, Martin TR, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230. [PubMed] [Google Scholar]
- 22.Hildebrandt GC, Olkiewicz KM, Corrion LA, et al. Donor-derived TNF-alpha regulates pulmonary chemokine expression and the development of idiopathic pneumonia syndrome after allogeneic bone marrow transplantation. Blood. 2004;104:586. doi: 10.1182/blood-2003-12-4259. [DOI] [PubMed] [Google Scholar]
- 23.Cooke KR, Hill GR, Crawford JM, et al. Tumor necrosis factor-alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest. 1998;102:1882. doi: 10.1172/JCI4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by toll-like receptors and hyaluronan. Nature Med. 2005;11:1173. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 25.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 26.Lewkowicz P, Lewkowicz N, Sasiak A, Tchorzewski H. Lipopolysaccharide-activated CD4+CD25+ T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J Immunol. 2006;177:7155. doi: 10.4049/jimmunol.177.10.7155. [DOI] [PubMed] [Google Scholar]
- 27.Zhai Y, Meng L, Gao F, et al. CD4+ T regulatory cell induction and function in transplant recipients after CD154 blockade is TLR4 independent. J Immunol. 2006;176:5988. doi: 10.4049/jimmunol.176.10.5988. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.