

Abstract
Clinical observations, as well as data obtained from the analysis of genetically engineered mouse models, firmly established the gain-of-function (GOF) properties of certain p53 mutations. However, little is known about the underlying mechanisms. We have used two independent microarray platforms to perform a comprehensive and global analysis of tumors arising in a model of metastatic skin cancer progression, which compares the consequences of a GOF p53R172H mutant vs. p53 deficiency. DNA profiling revealed a higher level of genomic instability in GOF vs. loss-of-function (LOF) p53 squamous cell carcinomas (SCCs). Moreover, GOF p53 SCCs showed preferential amplification of Myc with a corresponding increase in its expression and deregulation of Aurora Kinase-A. Fluorescent in situ hybridization confirmed amplification of Myc in primary GOF p53 SCCs and its retention in metastatic tumors. We also identified by RNA profiling distinct gene expression profiles in GOF p53 tumors, which included enriched integrin and Rho signaling, independent of tumor stage. Thus, the progression of GOF p53 papillomas to carcinoma was marked by the acquisition of epithelial to mesenchymal transition and metastatic signatures. In contrast, LOF p53 tumors showed enrichment of genes associated with cancer proliferation and chromosomal instability. Collectively, these observations suggest that genomic instability plays a prominent role in the early stages of GOF p53 tumor progression (i.e., papillomas), while it is implicated at a later stage in LOF p53 tumors (i.e., SCCs). This model will allow us to identify specific targets in mutant p53 SCCs, which may lead to the development of new therapeutic agents for the treatment of metastatic SCCs.
Keywords: Aurora Kinase A, genomic instability, Myc, p53, SCC, tumor progression
Introduction
Non-melanoma skin cancer (NMSC) is the most common neoplasm in the United States with a lifetime risk nearly equal to that of all other cancers combined (Jemal et al., 2009) and has been estimated to cost the healthcare systems over 1.4 billion dollars annually (Bickers et al., 2006). Squamous cell carcinoma (SCC) is the second most common skin cancer accounting for almost 200,000–300,000 new cases annually and the majority of deaths associated with NMSCs. Similar to other epithelial cancers, skin SCCs develop in a step-wise manner from pre-cursor lesions, to benign tumors (SCC in situ), to well differentiated SCCs, and lastly, to poorly differentiated spindle cell carcinomas that possess increased metastatic potential.
Activating RAS mutations occur in 5–40% of sporadic skin SCCs (Pierceall et al., 1991; Spencer et al., 1995). However, RAS mutations are found in ~62% of SCCs in individuals with the DNA repair deficiency syndrome, Xeroderma Pigmentosum (Daya-Grosjean and Sarasin, 2005) and in 46–76% of SCCs in psoriasis patients treated with PUVA (Psoralen with UVA treatment) (Kreimer-Erlacher et al., 2003; Wolf et al., 2004). In the absence of activating RAS mutations, elevated levels of active, GTP-bound RAS have been reported in a high number of SCCs (Dajee et al., 2003), as well as the overexpression of RAS family members (Lu et al., 2006; Paterson et al., 1996). Thus, activation of the RAS signaling pathway frequently occurs in cutaneous SCCs and contributes to malignant conversion of these tumors.
The p53 tumor suppressor gene is frequently mutated in skin cancers and over 73% of p53 mutations found in human SCCs are missense substitutions that result in the expression of mutant forms of p53, some of which abrogate the ability of p53 to turn on targets genes involved in cell cycle arrest, apoptosis, and other tumor suppression functions (Harris and Levine, 2005). Consequently, such mutations confer a loss-of-function (LOF) to p53. However, certain p53 mutants are capable of promoting tumorigenicity when introduced into p53 null cells, suggesting that they acquire gain-of-function (GOF) properties (Sun et al., 1993). One of the best characterized GOF mutations occurs at codon 175 (a human cancer “hot spot” in p53) and results in an arginine to histidine substitution (R175H or R172H in mice). Mice engineered to express the p53R172H mutant under the control of the endogenous p53 promoter recapitulate the spectrum of tumors observed in patients with Li-Fraumeni syndrome who carry the p53R175H mutation (Bougeard et al., 2008). It is unclear how various GOF p53 mutations contribute to the malignancy of skin SCCs or how these mutations co-operate with other oncogenic events such as the activation of RAS signaling during cancer progression.
We have recently generated an inducible mouse model that provides the strongest genetic evidence to date supporting GOF properties of mutant p53 in cutaneous SCCs (Caulin et al., 2007). The advantage of this system is that tumors are initiated by a common event, the deregulation of Ras signaling by the activation of an endogenously expressed KrasG12D allele in the skin, and allows a comparison of tumor promoting events such as the activation of the p53R172H mutant allele, or deletion of p53. The activation of the p53R172H mutant allele resulted in the increased frequency and earlier onset of tumor formation, accelerated cancer progression, and metastases relative to tumors lacking p53 (Caulin et al., 2007). SCCs from GOF p53 mice also showed hallmark features of genomic instability (Caulin et al., 2007) and are reminiscent of SCCs that develop in mice that overexpress the mitotic kinase, Aurora Kinase A (Aurora-A) (Torchia et al., 2009).
In this study, we analyzed GOF p53 tumors using whole genome approaches to understand how this p53 mutant promotes metastasis. We show that GOF p53 SCCs have distinct expression signatures and molecular alterations, including the gene amplification of Myc, deregulation of Aurora-A expression, and the upregulation of integrin and Rho gene signaling networks compared to LOF p53 SCCs.
Results
Expression profiling of papillomas and SCCs from GOF p53 mice
In the GOF p53 SCC model, activation of mutant p53 as compared to the loss of p53 resulted in the earlier emergence and greater numbers of precursor tumors (i.e., papillomas), and accelerated the malignant conversion of papillomas to SCCs with the induction of metastases (Caulin et al., 2007). This system allows the comparison of the GOF properties of mutant p53 in absence of wildtype (wt) p53 and thus eliminates the possibility that p53R172H mutant can act in a dominant negative fashion.
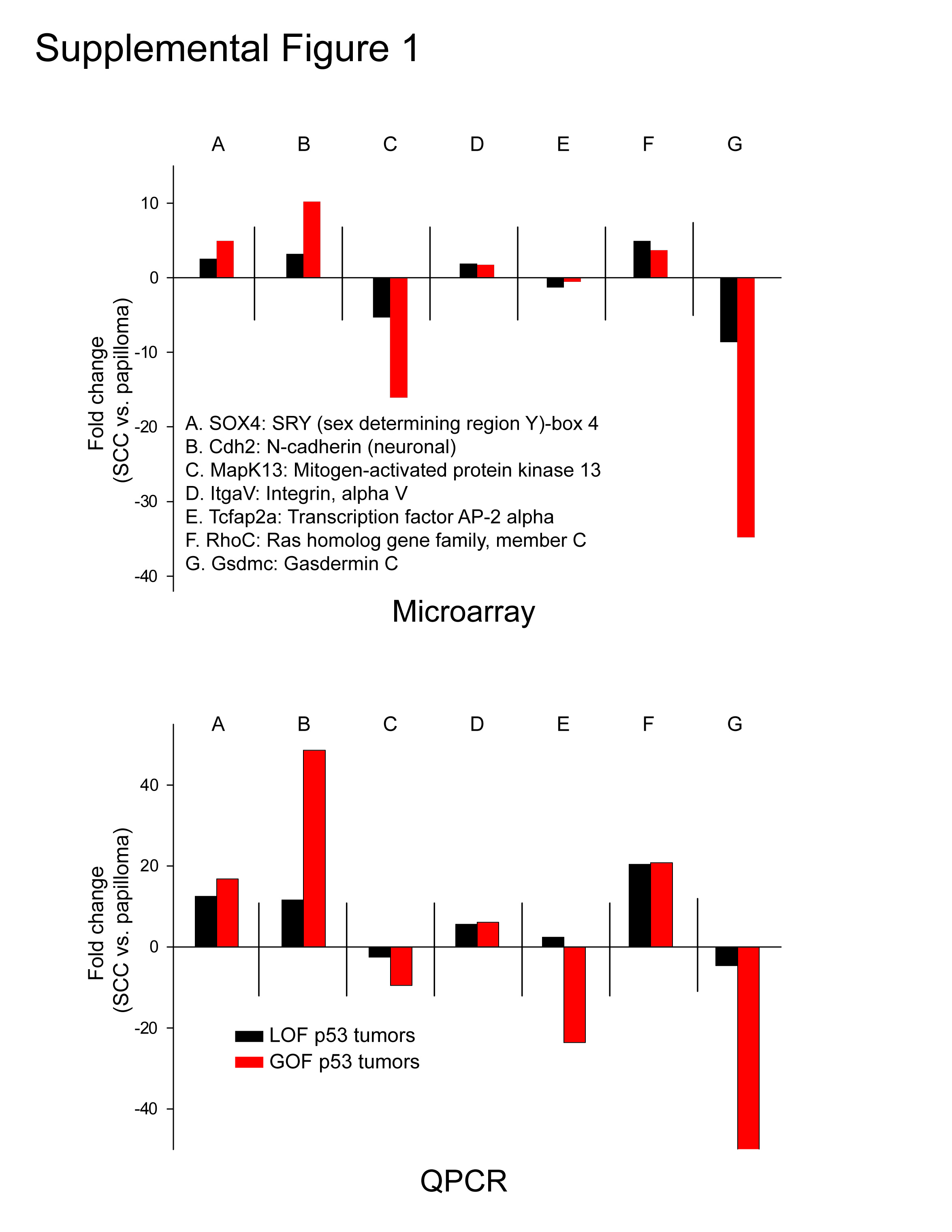
We analyzed all tumor groups by principal component analysis (PCA) as shown in Figure 1A using the most differentially regulated probesets identified by ANOVA (p<0.001). This experiment revealed that tumors expressing mutant p53 were distinct from p53 deficient tumors, regardless of tumor stage (Figure 1A). Next, 7 genes were selected at random for qPCR validation. As shown in Supplementary Figure 1, there were concordant fold changes in gene expression by qPCR as observed by microarray analysis. We then performed hierarchical clustering analysis using the probesets that were differentially regulated in papillomas (GOF vs. LOF p53 genotypes, Supplementary Dataset 1) and observed that GOF p53 papillomas clustered with both LOF and GOF p53 SCCs, indicating that GOF papillomas, the precursors to SCCs, had expression profiles more similar to advanced stages of tumor progression compared to the corresponding LOF p53 papillomas (Figure 1B). Clustering experiments using the probesets that were differentially regulated in SCCs (GOF vs. LOF p53 genotypes, Supplementary Dataset 2) showed that LOF p53 SCCs clustered together with all papillomas, while GOF p53 SCCs clustered in a separate branch (Figure 1C), suggesting that LOF p53 SCCs are at a less malignant stage of tumor progression compared to GOF p53 SCCs. Overall, these profiling and clustering analyses revealed four different stages of tumor progression as determined by the type of p53 mutation (Figure 1D) that correlate well with the kinetics of tumor development in p53R172H/− and p53−/− mice (Caulin et al., 2007).
Figure 1.

GOF p53 tumors are distinct and more malignant than LOF p53 tumors. (A) Principal component analysis using 1592 of the most differentially regulated probesets (Anova q<0.001) between all tumor groups (n=17). (B) Hierarchical clustering using 842 probesets differing between GOF vs. LOF p53 papillomas (paps) (t-test p<0.05) (n=7) or (C) 1485 probesets differing between GOF vs. LOF p53 SCCs (t-test, p<0.05) (n=10). (D) Profiling and clustering analyses revealed a hierarchy of cancer progression.
Effectors of Mutant p53 in SCCs
To identify potential targets of mutant p53 in SCCs, we determined the level of overlap between papillomas and SCCs, comparing GOF vs. LOF p53 tumors. We reasoned that any target genes of mutant p53 would most likely show concordant regulation in both precursor and advance tumors. First, we analyzed genes found altered in papillomas (GOF vs. LOF p53 genotypes, Supplementary Dataset 1) by gene ontology (GO) terms using the web-based functional and gene annotations tools, DAVID (Huang da et al., 2009) and GOTM (Dennis et al., 2003; Zhang et al., 2004). GO terms associated with Extracellular Matrix (ECM), cell adhesion, and cytoskeleton were enriched in upregulated genes of GOF p53 papillomas (q<0.05) (Supplementary Dataset 3). These categories included several matrix metalloproteinases, integrins, cytokines and ECM genes, which have been collectively implicated in ECM remodeling and cancer invasion. Moreover, no significant enrichment of GO terms was observed in downregulated genes. Analysis of SCCs (GOF vs. LOF p53 genotypes, Supplementary Dataset 4) did show the presence of blood vessel development, angiogenesis, cell adhesion, and ECM processes (q<0.05). GO terms associated with downregulated genes included desmosomes, cell to cell junctions, and cellular anchoring processes (q<0.05) (Supplementary Dataset 4). Thus, both GOF p53 papillomas and SCCs showed enhanced ECM interactions. We therefore explored these interactions using GSEA and genesets available from the Broad Institute (Mootha et al., 2003; Subramanian et al., 2005). GSEA revealed enrichment of genesets associated with integrin signaling and downstream Ras homolog gene family (Rho) GTPases (Figure 2A) in GOF vs. LOF p53 SCCs. Rho GTPases play a fundamental role in the regulation of cytoskeleton dynamics and other cellular function including cell cycle and cellular migration in normal and tumor cells (Karlsson et al., 2009). Enrichment of integrin complex genes was evident by clustering experiments showing the upregulation of these genes in both GOF p53 papillomas and SCCs (Figure 2B).
Figure 2.

Integrin and Rho Signaling are enhanced in GOF p53 tumors. (A) GSEA comparing GOF vs. LOF p53 SCCs showed a positive correlation with integrin and Rho GTPases signaling associated genesets (n=10). (B) Clustering of integrin complex genes comparing GOF vs. LOF p53 tumors (n=17). Color bar represent normalized signal intensity. (C) Gene network involving ECM, integrin, and Rho/Rac signaling in GOF p53 tumors. Genes observed to be deregulated in both GOF p53 papillomas and SCCs (Supplementary Dataset 10) (n=10) were analyzed using Ingenuity gene network algorithm. One top rated network is shown for genes that interact (compiled from published literature). Green and red symbols represent downregulated and upregulated genes, respectively. Empty nodes depict genes that are not present in the dataset, but implied from literature. Solid and hatched lines correspond to direct and indirect interactions, respectively.
The overlap between genes differentially regulated in GOF p53 papillomas and SCCs, relative to the respective LOF p53 tumors, revealed 69 genes which showed concordant up or downregulation (53 upregulated and 16 downregulated) (Supplementary Dataset 5). In this list, we observed the previously identified targets of mutant p53, Rho/Rac guanine nucleotide exchange factor (GEF) 2 (Arghef2) and matrix metallopeptidase 3 (Mmp3) (Brosh and Rotter, 2009; Mizuarai et al., 2006). To further investigate the interconnection of these potential GOF p53 targets, we performed network analysis using the Ingenuity web-software. One such top-rated network represented the potential microenvironment and intracellular interactions, specifically those involving ECM components such as laminin, integrins, Rho GTPases, and GEFs signaling pathways, which help to regulate Rho signaling (Figure 2C) (van der Meel et al., 2011). Lastly, these genes may also account for more advanced stage of tumor progression observed in the gene expression profiles of GOF p53 papillomas relative to LOF p53 papillomas (Figure 1).
Analysis of the Molecular Events Associated with Progression in GOF and LOF p53 tumors
We compared the transition between papillomas to SCCs in both GOF (SCCs vs. papillomas, Supplementary Dataset 6) and LOF p53 tumors (SCCs vs. papillomas, Supplementary Dataset 7) and analyzed the genes found regulated in a similar manner between GOF and LOF p53 tumors. The overlap between Supplemental Datasets 7 and 8 represent molecular events previously described in cancers (e.g., loss of differentiation) as they evolve from pre-cancerous lesions (Hanahan and Weinberg, 2000) (See Supplementary Dataset 8 and Supplemental text for further discussion on commonly regulated genes and processes).
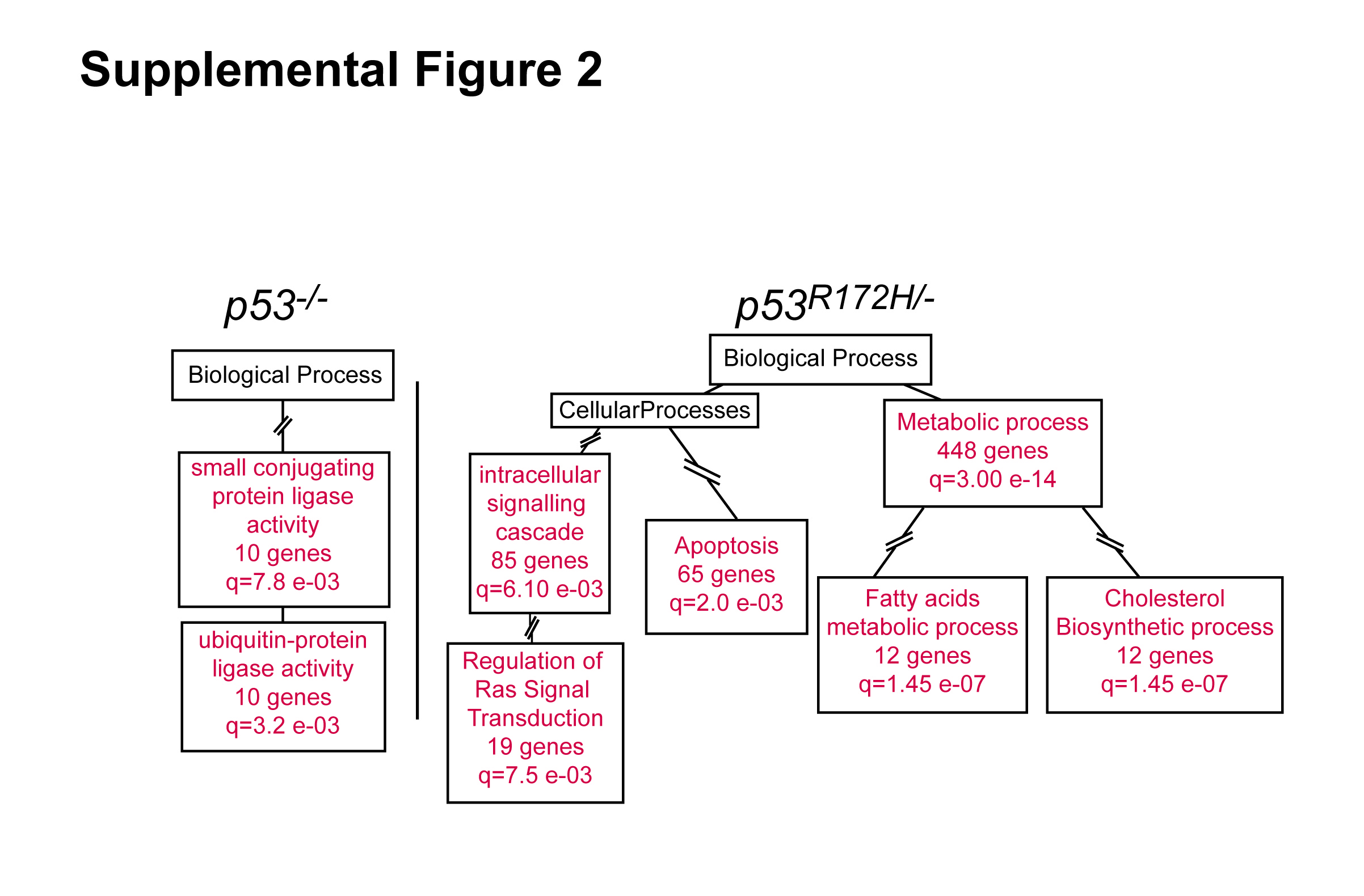
To understand the molecular events involved in the accelerated progression of GOF p53 skin cancers, we focused on non-overlapping genes which were deregulated in either LOF or GOF p53 tumors (SCCs vs. papillomas). Analysis of downregulated genes in LOF p53 tumors primarily revealed the enrichment of ubiquitin-protein ligase activity (Supplementary Figure 2 and Supplementary Dataset 9), while the upregulated genes in these tumors were enriched in processes associate with mitosis, including centrosome, spindle, and cell cycle checkpoint regulation events (Figure 3A and Supplementary Dataset 9). Thus, centrosome regulating genes such as NeK2, Aurora-A, Aurora-B, and Plk-1, and spindle and mitotic checkpoint genes such as Bub1, CenpE, Cdc6 and Cdc25C were present in these categories. Consistently, Ingenuity pathway analysis showed a significant overlap of canonical pathways involving the regulation of mitosis by Plk1 and G2/M checkpoints (Supplemental Table 1). In summary, these results indicate that deregulation of cell cycle checkpoints may be an important event in p53 deficient SCCs as they progress from papillomas. In GOF p53 tumors (SCCs vs. papillomas), GO terms associated with apoptosis, signal transduction, and lipid metabolism, were found in the downregulated genes (Supplementary Figure 2 and Supplementary Dataset 10). In contrast, terms associated with cellular movement, gene expression, and post translational protein modification were enriched in the upregulated genes (Figure 3A and Supplementary Dataset 10). These categories included genes implicated in cancer invasion and a small number of genes associated with centrosome function. However, there was a larger set of genes related to the modification of gene expression, including regulators of chromatin modification and numerous transcription factors and genes that regulate protein turnover, signal transduction, or cellular growth and survival. Collectively, these genes were part of intracellular signaling cascades associated with cellular migration and survival present in GOF p53 SCCs as revealed by Ingenuity pathway analysis (Supplemental Table 1).
Figure 3.

EMT and metastastic signatures are preferentially enriched in GOF p53 SCCs. (A) Graphic summary of GO term analysis using GOTM and adapted from GOTM output acyclic graphs. Genelists found in Supplementary Dataset 3 and 4 were compared and non overlapping list generated for LOF p53 SCCs vs. papillomas (1279 genes) (n=7) and GOF p53 SCCs vs. papillomas (2143 genes) (n=10). Panels show analysis of GO terms from non-overlapping upregulated genes in LOF and GOF p53 SCCs. ‘q’ denotes adjusted p values. Red boxes show significantly enriched GO categories and black boxes highlight non-enriched parent categories. Intermediate categories (broken lines) are omitted to simplify the presentation. (B) Lists of published cancer gene signatures were imported to Ingenuity web-software and used to determine the level of overlap with genes uniquely deregulated in LOF p53 and GOF p53 SCCs. Graphs shown represent the output from the ingenuity software. Blue bars denote` the q value for each analysis. The orange dots represent the ratio of overlap with each cancer signature geneset. The threshold line denotes p<0.05 significance.
To further characterize the dominant pathways driving cancer progression, we determined if previously identified cancer gene expression signatures were over-represented in either GOF or LOF p53 tumors (SCCs vs. papilloma). Specifically, we analyzed genes negatively regulated by wt p53 and whose expression is elevated in p53-null cells (Sur et al., 2009). Indeed, this gene signature was enriched in LOF p53 SCCs (Figure 3B). Furthermore, chromosomal instability and proliferation signatures identified in numerous cancers (Carter et al., 2006; Salvatore et al., 2007) were preferentially overrepresented in LOF p53 SCCs. However, these signatures were not found in either upregulated (Figure 3B) or downregulated genes of GOF p53 SCCs (not shown). Significant overlap was evident between upregulated genes in GOF p53 SCCs and genes associated with melanoma metastasis (Xu et al., 2008) and epithelial to mesenchymal transition (EMT) (Jechlinger et al., 2003) (Figure 3B) and between downregulated EMT genes and downregulated genes in GOF p53 SCCs (16% overlap; q=0.01). In summary, the transition from precursors to carcinomas in GOF p53 tumors is marked by the acquisition of cellular pathways favoring EMT and metastasis, while the transition in LOF p53 tumors is marked by the deregulation of cell cycle control leading to genomic instability at late stages of tumor evolution.
Analysis of Genomic Changes in GOF p53 SCCs
The induction of genomic instability is a common characteristic of the GOF properties of mutant p53 regardless of tumor type (Caulin et al., 2007; Lang et al., 2004; Olive et al., 2004). Furthermore, acquisition of key genomic alterations may further contribute to malignancy of GOF p53 tumors. To understand how expression of GOF p53 mutants can affect the genomic integrity of tumors and to identify genes that can co-operate with mutant p53 during carcinogenesis, we performed array comparative genome hybridization (aCGH) on SCCs that developed in p53+/+, p53+/−, p53R172H/+, p53−/−, or p53R172H/− mice. SCCs expressing the GOF p53 allele (p53R172H/+ or p53R172H/−) showed the highest copy number changes per tumor compared to p53+/+, p53+/−, or p53−/− SCCs (Table 1). To determine common regions of alterations, probe signals were averaged based on tumor genotype and alterations ascertained. Analyzed in this manner, SCCs expressing mutant p53 had more gains and deletions compared to tumors with deletion of one or two p53 wt alleles (Table 1). Thus, GOF p53 SCCs genomes appeared to be more unstable compared to tumors lacking p53, which is consistent with the aneuploidy of GOF p53 SCCs (Caulin et al., 2007).
Table 1. Summery of genomic alterations.
Genomic alterations in tumor DNA was examined by aCGH using the ADM-2 algorithm and the Agilent CGH Analytics V3.4 software. The range of alterations is shown for each tumor type. To ascertain common regions of genomic alterations, probe signals were averaged by genotype and deletions or gains determined as described above. The data are represented as % probes gained or lost.
| Tumor genotype |
Range | Averaged Signal | |||
|---|---|---|---|---|---|
| n | Gains | losses | Gains | losses | |
| p53+/+ | 5 | 0.1–10.5 | 0.1–0.3 | 0.2 | 0.4 |
| p53+/− | 6 | 0.1–21.0 | 0.2–1.4 | 0.2 | 0.7 |
| p53−/− | 7 | 0.1–12.0 | 0.1–13.0 | 5.7 | 0.5 |
| p53R172H/− | 10 | 5.8–30.1 | 0.4–6.2 | 11.2 | 1.7 |
| p53R172H/+ | 10 | 0.1–45.7 | 0.1–13.8 | 17 | 1.1 |
The majority of probes that showed alterations (Table 1) in LOF or GOF p53 SCCs were localized to chromosomes (Chrs) 3, 5, 6, 13, 15 and 18 (Figure 4). Regardless of p53 status, copy number gains (CNGs) were observed in chr 6 in all tumors (Figure 4). This accounted for a large proportion of probes showing amplification in p53−/− SCCs (Table 1). In subsets of GOF p53 SCCs, probes corresponding to chrs 3, 5, 13, 15, and 18 showed CNGs, with the most prevalent alterations occurring on chr 3 (25%), chr 5 (25%), and chr 15 (40%)(Figure 4). Furthermore, gains of whole regions on chrs 13 and 18 were evident in 30% of p53R172H/− tumors.
Figure 4.

Distribution of genomic alterations in LOF and GOF p53 SCCs. Graphical representation of the most commonly altered chromosomes in tumors (n=38) was generated using Agilent CGH Analytics software. Vertical bars represent individual tumors. Probes detecting gains or deletions are highlighted as red or green regions, respectively. Arrow represents the location of the Myc locus on chr 15qD1.
Closer examination of chr 6 alterations showed Kras CNGs in 55% of GOF p53 tumors and 53% of p53+/+, p53+/− and p53−/− SCCs combined. Myc amplification (15qD1) was exclusively observed in 40% of both p53R172H/+ and p53R172H/− SCCs (Figure 4 Arrow, and Figure 5A Inset). We validated Myc CNGs by qPCR on SCC DNA and observed a high level increase in gene copy number compared to non-tumor tissue or LOF p53 SCCs (Figure 5A). Additionally, immunostaining confirmed high levels of Myc protein in GOF p53 SCCs harboring Myc amplification (Figure 5B). Using Fluorescent in situ Hybridization (FISH), we further characterized the CNGs present in the Kras and Myc loci in GOF p53 SCCs. Of the 8 tumors analyzed, most tumors showed gains in both Kras (2.7–9.5 mean gene copies (MGCs)) and Myc (2.1–15.7 MGCs) (Figure 5C). In 50% of tumors, Kras or Myc CNGs were detected as double minute or in clusters. Interestingly, one tumor showed a heterogeneous pattern of Kras and Myc amplification with regions of cells showing over 10 copies of each gene. No CNGs in the Kras and Myc loci were detected in four GOF p53 papillomas analyzed, suggesting that gene duplication of these loci occurred at a later stage of tumor development. In five different tumor biopsies from either lung or lymph nodes of GOF p53 mice, CNGs in Myc (2.7–4.5 MGCs) and Kras (2.8–8.5 MGCs) were evident (Figure 5C). GSEA analysis of our RNA microarray data revealed that, tumors harboring Myc CNGs showed a correlation with previously published Myc signatures (Kim et al., 2006; Lee et al., 2004) and Myc associated gene lists from the Broad Institute (Supplemental Table 2).
Figure 5.

GOF p53 SCCs show preferential deregulation of Myc. (A) Inset, probes corresponding to the Myc locus at 15qD1 show amplification in GOF p53 tumors (n=20). Red dots represent individual probes showing gains while green dots represent probes showing losses. Graph shows qPCR analysis of Myc gene copy number in GOF p53 SCCs. Representative tumors are shown for each tumor genotype. Myc copy number was normalized to Pgam1, which was not found altered by array CGH in the tumors analyzed. (B) Immunohistochemical detection of Myc protein levels in GOF p53 tumors with and without Myc amplification. Bar = 50 µm. Representative images shown from the staining of 10 different tumors. (C) Detection of Kras and Myc copy number gains by FISH in primary (right panels) or metastatic tumor cells (left panel) from GOF p53 mice. FISH probes in green detect Kras and those in red, Myc. Nuclei were stained with Dapi. Note the presence of greater than 2 green or red dots in each nuclei. Bar = 10 µm.
Further analysis of array CGH data revealed the presence of CNGs of the Aurora-A (AurkA) locus in 3/20 GOF p53 SCCs and the deletion of its negative regulator, AurkAIP1 (Lim and Gopalan, 2007) in 2/20 separate GOF p53 tumors. Moreover, CNGs in the Aurora-A activator, Nedd9 (Karthigeyan et al., 2010), were also preferentially found in 4/20 GOF p53 SCCs. Of these, 2/20 did not overlap with tumors harboring Aurora-A or AurkAIP1 alterations. Furthermore, examination of our expression profiling data for known Aurora-A regulators revealed downregulation of the negative regulator Chfr (Rao et al., 2009; Yu et al., 2005) in GOF vs. LOF p53 SCCs (p<0.05). Based on these observations, we examined the expression pattern Aurora-A in GOF p53 SCCs by immunostaning. Aurora-A was readily detectable in dividing cells from p53+/+ tumors, localizing to spindle poles in metaphase tumor cells (Figure 6 and inset). This pattern of expression was preserved in p53+/− and p53−/− tumors. In contrast, we observed a higher level of Aurora-A staining in p53R172H/+ SCC cells (See graph in Figure 6), but a more diffused pattern in p53R172H/− tumor cells, reminiscent of a staining pattern observed in poorly differentiated human SCCs (Torchia et al., 2009).
Figure 6.

Aurora-A is aberrantly expressed in GOF p53 SCCs. Representative detection of Aurora-A by immunohistochemistry (IHC) in tumor sections of p53+/+ (n=4), p53+/− (n=5), p53R172H/+ (n=5), p53−/− (n=5), and p53R172H/− (n=6). Inset in the right hand panel (p53+/+) illustrates the typical localization of Aurora-A at spindle poles of cells in metaphase. Scale=50 µm. Graph on the left depicts quantification of Aurora-A staining. X-axis shows log transformation of the raw IHC score. A simple student t-test was used compare groups.
DISCUSSION
We present in this study a comprehensive and global molecular analysis of a well characterized progression model of metastatic skin SCCs. We used two independent microarray platforms to correlate the in vivo phenotypic presentation of GOF and LOF p53 tumors (Caulin et al., 2007) with the molecular alterations at the RNA and DNA level that promote metastasis. This unique approach revealed the pathways involved in mutant p53 driven tumorigenesis such as genomic stability, ECM interactions and cytoskeletal signaling. The enhanced genomic instability in GOF p53 SCCs led to the retention of specific genomic alterations targeting powerful oncogenes as Myc and Aurora-A. Furthermore, the presence of metastatic and EMT signatures coupled with the absence of genomic instability signatures suggests that GOF p53 SCCs acquired genomic instability at an early stage of tumor evolution, consistent with the presence of centrosome amplification, a hallmark feature of genomic instability in cancer (Fukasawa, 2005), previously observed in papillomas that expressed the GOF p53 mutant (Caulin et al., 2007; Wang et al., 1998). Taken together our results indicate that LOF p53 SCCs acquire genomic instability at a late stage in their tumor development, thereby delaying the emergence of metastases which is consistent with previous reports for p53-null skin tumors (Caulin et al., 2007; Kemp et al., 1993), whereas GOF p53 tumors acquire genomic instability at an early stage of cancer progression.
It is well known that mutant p53 can induce defective cell cycle checkpoint regulation, which combined with the deregulation of Aurora-A activity or its expression may further promote genomic instability and drive the selection of other oncogenes (e.g., Myc) which enhance tumor invasion and metastasis. We have shown that Aurora-A overexpression can lead to genomic instability in tumors and promote skin SCC metastasis, with the concomitant loss of p53 expression (Torchia et al., 2009). Interestingly, amplification of Aurora-A was only observed in a small number of GOF p53 SCCs, while a much higher frequency of tumors showed altered protein expression. Thus, the regulation of Aurora-A function in GOF p53 tumors may be complex and involve post-translation mechanisms that control its overall protein level and/or activity.
The preferential amplification of Myc seen in GOF p53 primary SCCs and the presence of Myc CNGs in metastases suggest a dominant role for Myc signaling in highly malignant and metastatic SCCs. Overexpression of Myc in skin has been shown to enhance SCC formation (Rounbehler et al., 2001) and to promote genomic instability in tumor cells (Prochownik and Li, 2007). Alone, overexpression of Myc can enhance invasiveness of breast carcinoma cells (Cho et al., 2010) and the amplification of Myc has been associated with poor patient prognosis and more aggressive tumors (Boelens et al., 2009; Haughey et al., 1992; Kozma et al., 1994; Ozakyol et al., 2006; Yakut et al., 2003). Moreover, MYC amplification was reported in over 50% of SCCs found in organ transplant recipients, which are 65–250 times more likely to develop highly malignant and metastatic SCCs (Boukamp, 2005; Euvrard et al., 2003).
To date, few universal mechanisms or effectors have been described to account for the highly metastatic tumors expressing the p53R172H mutant in various non-cutaneous tissues (Doyle et al., 2010; Hingorani et al., 2005; Liu et al., 2000; Zheng et al., 2007). This may reflect a propensity of mutant p53 to act in a tissue or tumor-stage dependent manner. Our studies identified numerous cellular processes that were preferentially deregulated in GOF p53 SCCs and two previously identified targets of mutant p53, Arhgef2 and Mmp3 (Brosh and Rotter, 2009; Mizuarai et al., 2006), both of which have been implicated in promoting invasive cancer phenotypes (Birkenfeld et al., 2008; Ramos et al., 2002). Further, it has been established that remodeling of the extracellular environment is crucial for the development of metastatic tumors (Denys et al., 2009). Hence, any alterations in the ECM coupled with specific intracellular signaling events will play a critical role in increasing the invasive potential of mutant p53 tumor cells. Based on our microarray analyses, both integrin signaling and its downstream mediators (e.g., Rho GTPase and GEFs) may contribute to the invasive phenotype of GOF p53 SCCs, which is consistent with previous studies (Muller et al., 2009; Sauer et al., 2010).
Currently, histopathological evaluation may be insufficient to determine if highly malignant skin tumors will recur or undergo metastasis. Our study shows that the mutational status of p53 in tumor cells dictates which molecular pathways or genetic alterations can predominate in highly malignant and metastasis prone skin tumors. Moreover, our data suggest that crosstalk between Aurora-A, Myc, and effectors of mutant p53 occur in GOF p53 SCCs. Thus, Aurora-A can upregulate Myc (Yang et al., 2010), which can upregulate Aurora-A protein levels (den Hollander et al., 2010). Myc also regulates RhoA expression (Chan et al., 2010) and Aurora-A can regulate Arhgef2 activity (Birkenfeld et al., 2007), thereby affecting tumor cell invasiveness. This potential oncogene crosstalk offers an opportunity for therapeutic intervention depending on the p53 mutational status of the patient’s tumor. For example, defective checkpoint regulation in cancer cells may be exploited to selectively kill tumor cells with wt or LOF mutant p53 (Cheok et al., 2011). However, unlike wt or LOF p53 tumors, the treatment GOF p53 tumors with p53 pathway activators such as nutlin-3 (Shen and Maki, 2011) may have devastating effects, since previous studies have shown that GOF mutant forms of p53 are also stabilized by nutlin-3 (Terzian et al., 2008). However, nutlin-3 in combination with an Aurora Kinase inhibitor such as VX680 may be very effective in selectively killing GOF p53 tumors (Cheok et al., 2010). Alternatively, drugs such as Prima-1 which restores wt activity to mutant p53 (Saha et al., 2010) in combination with small molecule inhibitors targeting Myc interactions with its binding partner Max (Shi et al., 2009), may be useful in treating SCC metastases with mutant p53 and MYC amplification.
In summary, we have compared skin SCCs by the type of p53 mutation (either LOF of GOF) present in these tumors and revealed the genetic and molecular alterations that are specific for GOF p53 tumors. This analysis further suggests that the pathways governed by Aurora-A, Myc and integrin/Rho signaling play an important role in mediating the oncogenic properties of GOF p53 in skin tumors and offer potential strategies for therapeutic intervention in aggressive and metastatic SCCs. Our GOF p53 model offers a unique tool to test p53 based therapeutic strategies in vivo and future studies will determine if skin SCCs harboring GOF p53 mutations can be selectively targeted by therapies against Aurora Kinase or Myc signaling pathways.
Materials and Methods
RNA and DNA microarray Profiling and qPCR analysis
RNA and DNA microarray profiling was performed at the Baylor College of Medicine Microarray Core Facility. Tumor RNA was isolated, processed for hybridization to Affymetrix Mouse 430 2.0 genechips. Expression microarray data was processed using dchip, Genespring GX v11, and Ingenuity (www.ingenuity.com) software. GO terms were analyzed using DAVID (david.abcc.ncifcrf.gov) and GOTM (bioinfo.vanderbilt.edu/gotm) web based software. Published gene lists were imported into Ingenuity software to determine the level overlap with gene lists generated from the analysis of expression microarray data. Gene Set Enrichment Analysis (GSEA) was performed in Genespring GX v11 software using imported gene sets from the Broad Institute (www.broadinstitute.org). Array CGH analysis was performed as previously described using Agilent CGH Analytics V3.4 software (Torchia et al., 2009). The ADM-2 aberration algorithm was applied with the threshold set to 6 in order to determine regions of amplification or deletion in tumors (Torchia et al., 2009). Tumor DNA or cDNA was used for qPCR analysis using a Roche Lighcycler 2.0 system. Myc gene copy number was normalized by detection of Phosphoglycerate mutase 1 (Pgam1). See Supplemental text for probe sequence and additional details on microarray experiments.
Immunohistochemical Analysis
Immunostaining was performed as previously described (Torchia et al., 2009). The antibodies used were against Myc (Sc-764, Santa Cruz Biotechnology) and Aurora-A (610938, BD Bioscience). Quantification of Aurora positive cells was performed using a three-point scale (1=low, 2=medium, and 3=high) for staining intensity multiplied by number of positive cells. Three separate fields were evaluated for each sample and final score averaged. See Supplemental text for additional details.
FISH Analyses
Detection of Kras and Myc copy number changes was conducted at the University of Colorado Cancer Center Cytogenetic Core using BACS encoding the murine Kras and Myc loci. Stained slides were analyzed by fluorescence microscopy. Mean copy numbers per cell were determined using at least 50 nuclei per specimen. See Supplemental text for additional details.
Statistical Analyses
Statistical tests were performed using dChip, Genespring, DAVID, GOTM, Graph Pad Prism v5.0, and Ingenuity software. A ‘q value’ denotes adjusted p-values derived from multiple testing corrections (Hochberg and Benjamini, 1990).
The tumors analyzed in this study were derived as described in (Caulin et al., 2007). For ease of reading, tumors from KrasG12D;p53+/+, KrasG12D;p53+/−, KrasG12D/p53R172H/+, KrasG12D;p53−/− or KrasG12D;p53R172H/− mice (Caulin et al., 2007) will be referred by the p53 genotype (e.g., p53R172H/−). p53−/− tumors will also be referred as LOF p53 SCCs and p53R172H/− tumors as GOF p53 tumors.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Grant Support: NIH grants: DE015344 (CC), CA52607 and CA105491 (D.R.R.). We acknowledge Dr. Lisa White, Laura Liles for their assistance with microarray experiments; Dr. Leila Garcia for her assistance with the FISH experiments and Dr. Ariefdjohan for the reading of the manuscript..
This work was funded by grants from the National Institute of Cancer, USA
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
References
- Bickers DR, Lim HW, Margolis D, Weinstock MA, Goodman C, Faulkner E, et al. The burden of skin diseases: 2004 a joint project of the American Academy of Dermatology Association and the Society for Investigative Dermatology. J Am Acad Dermatol. 2006;55:490–500. doi: 10.1016/j.jaad.2006.05.048. [DOI] [PubMed] [Google Scholar]
- Birkenfeld J, Nalbant P, Bohl BP, Pertz O, Hahn KM, Bokoch GM. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev Cell. 2007;12:699–712. doi: 10.1016/j.devcel.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenfeld J, Nalbant P, Yoon SH, Bokoch GM. Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: is altered GEF-H1 activity a crucial determinant of disease pathogenesis? Trends Cell Biol. 2008;18:210–219. doi: 10.1016/j.tcb.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Boelens MC, Kok K, van der Vlies P, van der Vries G, Sietsma H, Timens W, et al. Genomic aberrations in squamous cell lung carcinoma related to lymph node or distant metastasis. Lung Cancer. 2009;66:372–378. doi: 10.1016/j.lungcan.2009.02.017. [DOI] [PubMed] [Google Scholar]
- Bougeard G, Sesboue R, Baert-Desurmont S, Vasseur S, Martin C, Tinat J, et al. Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet. 2008;45:535–538. doi: 10.1136/jmg.2008.057570. [DOI] [PubMed] [Google Scholar]
- Boukamp P. Non-melanoma skin cancer: what drives tumor development and progression? Carcinogenesis. 2005;26:1657–1667. doi: 10.1093/carcin/bgi123. [DOI] [PubMed] [Google Scholar]
- Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043–1048. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- Caulin C, Nguyen T, Lang GA, Goepfert TM, Brinkley BR, Cai WW, et al. An inducible mouse model for skin cancer reveals distinct roles for gain- and loss-of-function p53 mutations. J Clin Invest. 2007;117:1893–1901. doi: 10.1172/JCI31721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Lee SW, Li CF, Wang J, Yang WL, Wu CY, et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat Cell Biol. 2010;12:457–467. doi: 10.1038/ncb2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheok CF, Kua N, Kaldis P, Lane DP. Combination of nutlin-3 and VX-680 selectively targets p53 mutant cells with reversible effects on cells expressing wild-type p53. Cell Death Differ. 2010;17:1486–1500. doi: 10.1038/cdd.2010.18. [DOI] [PubMed] [Google Scholar]
- Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nat Rev Clin Oncol. 2011;8:25–37. doi: 10.1038/nrclinonc.2010.174. [DOI] [PubMed] [Google Scholar]
- Cho KB, Cho MK, Lee WY, Kang KW. Overexpression of c-myc induces epithelial mesenchymal transition in mammary epithelial cells. Cancer Lett. 2010;293:230–239. doi: 10.1016/j.canlet.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- Daya-Grosjean L, Sarasin A. The role of UV induced lesions in skin carcinogenesis: an overview of oncogene and tumor suppressor gene modifications in xeroderma pigmentosum skin tumors. Mutat Res. 2005;571:43–56. doi: 10.1016/j.mrfmmm.2004.11.013. [DOI] [PubMed] [Google Scholar]
- den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood. 2010;116:1498–1505. doi: 10.1182/blood-2009-11-251074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Denys H, Braems G, Lambein K, Pauwels P, Hendrix A, De Boeck A, et al. The extracellular matrix regulates cancer progression and therapy response: implications for prognosis and treatment. Curr Pharm Des. 2009;15:1373–1384. doi: 10.2174/138161209787846711. [DOI] [PubMed] [Google Scholar]
- Doyle B, Morton JP, Delaney DW, Ridgway RA, Wilkins JA, Sansom OJ. p53 mutation and loss have different effects on tumourigenesis in a novel mouse model of pleomorphic rhabdomyosarcoma. J Pathol. 2010 doi: 10.1002/path.2748. [DOI] [PubMed] [Google Scholar]
- Euvrard S, Kanitakis J, Claudy A. Skin cancers after organ transplantation. N Engl J Med. 2003;348:1681–1691. doi: 10.1056/NEJMra022137. [DOI] [PubMed] [Google Scholar]
- Fukasawa K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005;230:6–19. doi: 10.1016/j.canlet.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- Haughey BH, von Hoff DD, Windle BE, Wahl GM, Mock PM. c-myc oncogene copy number in squamous carcinoma of the head and neck. Am J Otolaryngol. 1992;13:168–171. doi: 10.1016/0196-0709(92)90117-c. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9:811–818. doi: 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jechlinger M, Grunert S, Tamir IH, Janda E, Ludemann S, Waerner T, et al. Expression profiling of epithelial plasticity in tumor progression. Oncogene. 2003;22:7155–7169. doi: 10.1038/sj.onc.1206887. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- Karlsson R, Pedersen ED, Wang Z, Brakebusch C. Rho GTPase function in tumorigenesis. Biochim Biophys Acta. 2009;1796:91–98. doi: 10.1016/j.bbcan.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Karthigeyan D, Prasad SB, Shandilya J, Agrawal S, Kundu TK. Biology of Aurora A kinase: Implications in cancer manifestation and therapy. Med Res Rev. 2010 doi: 10.1002/med.20203. [DOI] [PubMed] [Google Scholar]
- Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993;74:813–822. doi: 10.1016/0092-8674(93)90461-x. [DOI] [PubMed] [Google Scholar]
- Kim YH, Girard L, Giacomini CP, Wang P, Hernandez-Boussard T, Tibshirani R, et al. Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene. 2006;25:130–138. doi: 10.1038/sj.onc.1208997. [DOI] [PubMed] [Google Scholar]
- Kozma L, Kiss I, Szakall S, Ember I. Investigation of c-myc oncogene amplification in colorectal cancer. Cancer Lett. 1994;81:165–169. doi: 10.1016/0304-3835(94)90198-8. [DOI] [PubMed] [Google Scholar]
- Kreimer-Erlacher H, Seidl H, Back B, Cerroni L, Kerl H, Wolf P. High frequency of ultraviolet mutations at the INK4a-ARF locus in squamous cell carcinomas from psoralenplus-ultraviolet-A-treated psoriasis patients. J Invest Dermatol. 2003;120:676–682. doi: 10.1046/j.1523-1747.2003.12085.x. [DOI] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Lee JS, Chu IS, Mikaelyan A, Calvisi DF, Heo J, Reddy JK, et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004;36:1306–1311. doi: 10.1038/ng1481. [DOI] [PubMed] [Google Scholar]
- Lim SK, Gopalan G. Aurora-A kinase interacting protein 1 (AURKAIP1) promotes Aurora-A degradation through an alternative ubiquitin-independent pathway. Biochem J. 2007;403:119–127. doi: 10.1042/BJ20061272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, McDonnell TJ, Montes de Oca Luna R, Kapoor M, Mims B, El-Naggar AK, et al. High metastatic potential in mice inheriting a targeted p53 missense mutation. Proc Natl Acad Sci U S A. 2000;97:4174–4179. doi: 10.1073/pnas.97.8.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SL, Herrington H, Reh D, Weber S, Bornstein S, Wang D, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20:1331–1342. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuarai S, Yamanaka K, Kotani H. Mutant p53 induces the GEF-H1 oncogene, a guanine nucleotide exchange factor-H1 for RhoA, resulting in accelerated cell proliferation in tumor cells. Cancer Res. 2006;66:6319–6326. doi: 10.1158/0008-5472.CAN-05-4629. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–1341. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Ozakyol A, Ozdemir M, Artan S. Fish detected p53 deletion and N-MYC amplification in colorectal cancer. Hepatogastroenterology. 2006;53:192–195. [PubMed] [Google Scholar]
- Paterson IC, Eveson JW, Prime SS. Molecular changes in oral cancer may reflect aetiology and ethnic origin. Eur J Cancer B Oral Oncol. 1996;32B:150–153. doi: 10.1016/0964-1955(95)00065-8. [DOI] [PubMed] [Google Scholar]
- Pierceall WE, Goldberg LH, Tainsky MA, Mukhopadhyay T, Ananthaswamy HN. Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol Carcinog. 1991;4:196–202. doi: 10.1002/mc.2940040306. [DOI] [PubMed] [Google Scholar]
- Prochownik EV, Li Y. The ever expanding role for c-Myc in promoting genomic instability. Cell Cycle. 2007;6:1024–1029. doi: 10.4161/cc.6.9.4161. [DOI] [PubMed] [Google Scholar]
- Ramos DM, But M, Regezi J, Schmidt BL, Atakilit A, Dang D, et al. Expression of integrin beta 6 enhances invasive behavior in oral squamous cell carcinoma. Matrix Biol. 2002;21:297–307. doi: 10.1016/s0945-053x(02)00002-1. [DOI] [PubMed] [Google Scholar]
- Rao CV, Yamada HY, Yao Y, Dai W. Enhanced genomic instabilities caused by deregulated microtubule dynamics and chromosome segregation: a perspective from genetic studies in mice. Carcinogenesis. 2009;30:1469–1474. doi: 10.1093/carcin/bgp081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rounbehler RJ, Schneider-Broussard R, Conti CJ, Johnson DG. Myc lacks E2F1's ability to suppress skin carcinogenesis. Oncogene. 2001;20:5341–5349. doi: 10.1038/sj.onc.1204691. [DOI] [PubMed] [Google Scholar]
- Saha MN, Micallef J, Qiu L, Chang H. Pharmacological activation of the p53 pathway in haematological malignancies. J Clin Pathol. 2010;63:204–209. doi: 10.1136/jcp.2009.070961. [DOI] [PubMed] [Google Scholar]
- Salvatore G, Nappi TC, Salerno P, Jiang Y, Garbi C, Ugolini C, et al. A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res. 2007;67:10148–10158. doi: 10.1158/0008-5472.CAN-07-1887. [DOI] [PubMed] [Google Scholar]
- Sauer L, Gitenay D, Vo C, Baron VT. Mutant p53 initiates a feedback loop that involves Egr-1/EGF receptor/ERK in prostate cancer cells. Oncogene. 2010;29:2628–2637. doi: 10.1038/onc.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Maki CG. Pharmacologic activation of p53 by small-molecule MDM2 antagonists. Curr Pharm Des. 2011;17:560–568. doi: 10.2174/138161211795222603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Stover JS, Whitby LR, Vogt PK, Boger DL. Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Bioorg Med Chem Lett. 2009;19:6038–6041. doi: 10.1016/j.bmcl.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JM, Kahn SM, Jiang W, DeLeo VA, Weinstein IB. Activated ras genes occur in human actinic keratoses, premalignant precursors to squamous cell carcinomas. Arch Dermatol. 1995;131:796–800. [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Nakamura K, Wendel E, Colburn N. Progression toward tumor cell phenotype is enhanced by overexpression of a mutant p53 tumor-suppressor gene isolated from nasopharyngeal carcinoma. Proc Natl Acad Sci U S A. 1993;90:2827–2831. doi: 10.1073/pnas.90.7.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Jr, Kinzler KW, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A. 2009;106:3964–3969. doi: 10.1073/pnas.0813333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchia EC, Chen Y, Sheng H, Katayama H, Fitzpatrick J, Brinkley WR, et al. A genetic variant of Aurora kinase A promotes genomic instability leading to highly malignant skin tumors. Cancer Res. 2009;69:7207–7215. doi: 10.1158/0008-5472.CAN-09-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meel R, Symons MH, Kudernatsch R, Kok RJ, Schiffelers RM, Storm G, et al. The VEGF/Rho GTPase signalling pathway: A promising target for anti-angiogenic/antiinvasion therapy. Drug Discov Today. 2011 doi: 10.1016/j.drudis.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Greenhalgh DA, Jiang A, He D, Zhong L, Brinkley BR, et al. Analysis of centrosome abnormalities and angiogenesis in epidermal-targeted p53172H mutant and p53-knockout mice after chemical carcinogenesis: evidence for a gain of function. Mol Carcinog. 1998;23:185–192. doi: 10.1002/(sici)1098-2744(199811)23:3<185::aid-mc7>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Wolf P, Kreimer-Erlacher H, Seidl H, Back B, Soyer HP, Kerl H. The ultraviolet fingerprint dominates the mutational spectrum of the p53 and Ha-ras genes in psoralen + ultraviolet A keratoses from psoriasis patients. J Invest Dermatol. 2004;122:190–200. doi: 10.1046/j.0022-202X.2004.22118.x. [DOI] [PubMed] [Google Scholar]
- Xu L, Shen SS, Hoshida Y, Subramanian A, Ross K, Brunet JP, et al. Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol Cancer Res. 2008;6:760–769. doi: 10.1158/1541-7786.MCR-07-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakut T, Egeli U, Gebitekin C. Investigation of c-myc and p53 gene alterations in the tumor and surgical borderline tissues of NSCLC and effects on clinicopathologic behavior: by the FISH technique. Lung. 2003;181:245–258. doi: 10.1007/s00408-003-1026-x. [DOI] [PubMed] [Google Scholar]
- Yang S, He S, Zhou X, Liu M, Zhu H, Wang Y, et al. Suppression of Aurora-A oncogenic potential by c-Myc downregulation. Exp Mol Med. 2010;42:759–767. doi: 10.3858/emm.2010.42.11.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Minter-Dykhouse K, Malureanu L, Zhao WM, Zhang D, Merkle CJ, et al. Chfr is required for tumor suppression and Aurora A regulation. Nat Genet. 2005;37:401–406. doi: 10.1038/ng1538. [DOI] [PubMed] [Google Scholar]
- Zhang B, Schmoyer D, Kirov S, Snoddy J. GOTree Machine (GOTM): a web-based platform for interpreting sets of interesting genes using Gene Ontology hierarchies. BMC Bioinformatics. 2004;5:16. doi: 10.1186/1471-2105-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, El-Naggar AK, Kim ES, Kurie JM, Lozano G. A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene. 2007;26:6896–6904. doi: 10.1038/sj.onc.1210493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.