

Abstract
Fe-S clusters are partners in the origin of life that predate cells, acetyl-CoA metabolism, DNA, and the RNA world. The double helix solved the mystery of DNA replication by base pairing for accurate copying. Yet, for genome stability necessary to life, the double helix has equally important implications for damage repair. Here we examine striking advances that uncover Fe-S cluster roles both in copying the genetic sequence by DNA polymerases and in crucial repair processes for genome maintenance, as mutational defects cause cancer and degenerative disease. Moreover, we examine an exciting, controversial role for Fe-S clusters in a third element required for life – the long-range coordination and regulation of replication and repair events. By their ability to delocalize electrons over both Fe and S centers, Fe-S clusters have unbeatable features for protein conformational control and charge transfer via double-stranded DNA that may fundamentally transform our understanding of life, replication, and repair.
Keywords: Fe-S cluster; DNA repair; DNA replication; DNA charge transfer; DNA charge transfer communication; conformational change, genome integrity; cancer; degenerative disease; DNA polymerase; DNA helicase; glycosylase; DNA nuclease
1. Introduction
Protein-bound iron-sulfur (Fe-S) clusters are among the most structurally and functionally versatile cofactors in biology [1]. Iron-sulfur clusters are extremely ancient components of modern protein chemistry, likely arising spontaneously on polypeptides when the Earth was anaerobic and iron and sulfur was abundant [2]. Indeed, Fe-S chemistry may have played a role in the origin of life itself [3]. For example, Fe-S clusters reacting with marine CO2 from undersea hydrothermal vents during the Hadean period (~4 × 109 years before present) are proposed to provide a primitive analogue of the acetyl-coenzyme-A (acetyl-CoA) pathway, in which hydrothermal H2 acted as an electron donor and marine CO2 as an electron acceptor effectively initiating a pathway for the transition from inorganic chemistry to biochemistry prior to cells [4]. More generally, the high levels of iron and sulfur on Earth, their ability to readily assemble into complexes with tunable charge transfer activity, and the pervasiveness of Fe-S as biological prosthetic groups across all domains of life support the hypothesis that Fe-S complexes were among life’s first catalysts [3].
As organisms evolved, they could employ Fe-S complexes for their metabolic pathways to generate organic molecules and to survive in different environments [4]. However, the rise of oxygen in the atmosphere and in the cell posed problems for Fe-S clusters as their biologically useful reactivity also makes them susceptible to inactivation through cluster oxidation [5]. Furthermore, iron mediated DNA damage via the Fenton reaction provided yet another mechanism for oxygen cytotoxicity that threatens genetic inheritance [6]. As a result, many Fe-S clusters in proteins were lost or replaced with other less oxygen sensitive metals like zinc unless there were specific selective advantages to the Fe-S cluster that outweighed its vulnerability to oxygen or they adapted to tolerate oxidation within the protein scaffold. In fact, redox inactive zinc is the metal of choice for nucleic acid binding proteins with zinc finger proteins being the largest family of regulatory proteins in mammals [7]. Zn also provides interactions among DNA-binding subunits as in the Rad50 Zn hook [8], and catalytic sites as in endonuclease IV, which cuts the DNA backbone at abasic sites [9].
Until recently, only a handful of DNA binding proteins—all glycosylases—were known to have Fe-S clusters, and it was generally assumed that most nucleic acid processing enzymes did not, and would not, have Fe-S clusters due to their oxygen sensitivity and possible toxicity. This view changed with the breakthrough discovery of an Fe-S cluster in the DNA helicase XPD and its associated family members that act in DNA repair [10]. In the last few years, all replicative DNA polymerases and the helicase-nuclease Dna2, which participates in Okazaki fragment processing during replication, were found to contain Fe-S clusters [11,12]. Researchers were slow to discover the Fe-S clusters in these enzymes due to the difficulty of purifying enzymes with intact clusters, and the lack of recognizable sequence motifs revealing the presence of an Fe-S cluster. Generally, protein metal sites are predicted using sequence-based strategy, however, many metal sites are missed before detailed structural and biochemical analyses, and many recombinant proteins can have an incorrect metal ion [13]. The features of Fe-S clusters make it difficult to replace them functionally with other metal ion cofactors. Among the activities under increasing interest and investigation is DNA-mediated charge transfer (DNA CT), an idea pioneered by Jacqueline K. Barton at the California Institute of Technology, that proposes Fe-S enzymes find DNA damage by probing DNA integrity electronically [14].
In general, charge transfer is actively investigated as a fascinating charge migration phenomena pertaining to novel aspects of molecular electronics in supramolecular-scale systems. These investigations are providing keystone information for relationships of structure, energetics, and electron transfer with potential for advances in biology, medicine and synthetic biology at the nano and mesoscales. For proteins, electron transfer is facilitated by electrostatic interactions that drive pre-collision orientation to promote transient complexes for direct electron transfer, as seen for plastocyanin-Cyctochrome C [15]. For enzymes, electron transfer is speeded by electrostatics that can drive substrate interactions to be faster than diffusion as computationally and experimentally shown for superoxide dismutase interactions with superoxide [16,17]. For DNA, charge transfer requires the intact DNA duplex and may be modified by bound proteins or other electrostatic modifiers. Understanding, predicting, and controlling DNA CT has implications for biology and nanomedicine as well as for the design of DNA-based sensors and single molecule devices [18-20].
For example, biological processes are typically considered in terms of interactomes, as lists of relevant direct macromolecular interactions. Molecular concentrations and binding affinity dictate which complexes are formed. The on and off rates determine the timescale, and their ratio determines the interaction affinity and thereby specificity. For chromosomes and DNA replication proteins, the concentrations in the cell are low; yet, the evolutionary selection for efficient and specific binding is extremely high. DNA CT provides a different way to consider possible interactomes and their timescale that merits attention.
As our knowledge of the roles of Fe-S clusters are emerging in critical DNA processing enzymes (Fig. 1), we are discovering that they are essential for their activities; yet the presence of Fe-S clusters remains puzzling. Why have Fe-S clusters been retained as key structural and functional components of DNA processing enzymes when iron mediated DNA damage poses such a threat to DNA integrity and genome maintenance? Replication and repair proteins having cofactors whose fundamental chemistry can endanger DNA is a paradox that has been largely ignored. We believe that there has been a huge hole in understanding DNA metabolism from the essential functions of Fe-S proteins in DNA replication and repair. This hole is being filled by solid biochemical, genetic and structural data that reveal Fe-S enzyme structures and roles in DNA replication and fidelity and how defects in Fe-S clusters can cause cancer and developmental diseases in humans.
Figure 1. The emerging roles of Fe-S cluster enzymes in DNA replication and repair.

Replication: Fe-S clusters are critical elements of DNA primase, all replicative DNA polymerases (DNA pols α and δ shown), and the nuclease/helicase Dna2 (shown on lagging strand 5′ flaps). Nucleotide Excision Repair (NER): the 5′->3′ Fe-S cluster helicase XPD opens a single stranded bubble around duplex distorting DNA damage allowing excision of the damaged strand by endonucleases and the gap filling by DNA polymerase (DNA pol ε shown). Base Excision Repair (BER): glycosylases Endo III / MutY and their role in the discovery and removal of damaged and mispaired bases. Telomere Maintenance: the helicase RTEL is involved in the unwinding of telomeric D-loops that affects telomere length maintenance and HR in the region.
Here we review the structural biochemistry of known classes of Fe-S cluster enzymes involved in nucleic acid processing, their shared Fe-S cluster maturation machinery, connections to disease, and unique features that suggest why these ancient co-factors have survived billions of years of evolution in an oxygen-rich world. In fact, current results suggest that Fe-S clusters are central to all life as key functional components of DNA replication and repair as well as of electron transfer and biochemical metabolism.
2. The nature and structure of Fe-S clusters
Why are potentially toxic and mutagenic Fe-S clusters conserved in proteins that directly interact with DNA? We know that reactive iron is used in DNA binding proteins when the iron is catalytically active, as shown for the oxidative demethylases, such as human ABH3 [21]. However, there are no cases, other than Fe-S clusters, where reactive iron has been proposed for structural roles in DNA binding. Two of the simplest and most common Fe-S clusters found in nature are [2Fe-2S] and [4Fe-4S], which can be spontaneously assembled by mixing Fe2+or Fe3+ and S2- in a reducing solution. Two [2Fe-2S] clusters can reductively couple to form one [4Fe-4S] cluster. And [4Fe-4S] clusters also can be oxidatively decoupled to form two 2Fe-2S clusters [22]. Fe-S clusters in proteins typically possess Fe2+/3+ and S2- and are ligated by cysteine residues. The main function of Fe-S clusters has generally been thought to be electron transfer and storage. The redox potential of Fe-S clusters can range from over -600 mV to over +400 mV [23]. Although the abundance of Fe-S cluster proteins varies from different organisms, [4Fe-4S] clusters are still nature’s favorite Fe-S clusters. In E. coli, an estimated 90% of Fe-S cluster proteins are [4Fe-4S] cluster proteins; the other 10% of Fe-S cluster proteins have [2Fe-2S] and [3Fe-4S] clusters [1].
The cubane-type [4Fe-4S] clusters (Fig. 2A) have four oxidation states: [4Fe-4S]0, [4Fe-4S]1+, [4Fe-4S]2+, and [4Fe-4S]3+. Even though they all have the same cluster arrangement, the electronic structure and redox properties of these oxidation states are different. Most [4Fe-4S] cluster proteins transfer one electron in each redox cycle using either [4Fe-4S]1+/2+ or [4Fe-4S]2+/3+, but in some particular cases, like nitrogenase, the Fe protein can have two redox cycles [4Fe-4S]2+/1+/0 [24]. A low redox potential identified as a [4Fe-4S]1+/[4Fe-4S]2+ redox couple can range from -300 mV to -700 mV; for high potential iron sulfur proteins (HIPIP) with a [4Fe-4S]2+/[4Fe-4S]3+ redox couple can have redox potential from +100 mV to +450 mV [23,25,26].
Figure 2. Structure of [4Fe-4S] clusters and their placement in DNA processing enzymes.

A) [4Fe-4S] cluster with 2Fo-Fc map in contour 4ζ (PDB: 1WEI [251]). Fe and S are shown as brown and goldenrod spheres, respectively. B) Distinct schematic sequence architecture for Fe-S clusters in DNA replication and repair proteins. The distinct patterns of placement for the Fe-S clusters relative to catalytic domains suggest their sequence location along with their three-dimensional topology provide a potential means for differential DNA CT activities suitable to coordinate replication and repair pathways. In one of the simplest models, the C-terminal placement of the Fe-S clusters in glycosylases and polymerases might impact the DNA affinity and hence exchange rate versus processivity. The Dna2 helicase/nuclease and the XPD family helicases have Fe-S clusters inserted into catalytic domains suggesting a tight linkage between cluster and catalytic activities. The unique placement of the XPD family Fe-S cluster within the HD1 catalytic domain supports its role as a sensor for double helix disruption. These and other testable roles emerge from the sequence architectures and structures analyzed here.
The inequivalence of Fe states (Fe3+/2.5+/2+) can be sensitive to the protein environment and electron properties of the cluster ligands. For example, nucleotide (ATP or ADP) binding in nitrogenase can shift the redox potential of the [4Fe-4S] cluster from -120 mV to -160 mV [27]. In E. coli nitrate reductase A, one of the [4Fe-4S] clusters is ligated with 3 cysteines and 1 histidine, and has a midpoint potential of -55 mV, which is higher than the 4 cysteine-ligated [4Fe-4S] clusters found in other subunits of this complex [28]. Interestingly, the cluster ligand Histidine to Cysteine substitution results in the loss of enzyme activity possibly due to the significant decrease of the midpoint potential to below -550 mV [28]. Besides electron transfer and storage roles, Fe-S clusters can function in many diverse roles including structural, substrate binding and activation, regulation of gene expression and enzyme activity, iron or cluster storage, and sulfur donor [22,29]. Other alternative cluster ligands such as histidine, arginine, aspartate, glutamate, tyrosine, threonine, enzyme substrates, glutathione, or S-adenosylmethionine (SAM) have been found in the increasing examples of proteins [23].
Fe-S clusters are best known for their activities in oxidation-reduction reactions of mitochondrial electron transport, catalysis by aconitase, generation of radicals by SAM-dependent enzymes, and sulfur donors in biosynthesis [22]. These functions are important, and mutations impacting such Fe-S cluster activities cause multiple human diseases [30]. Yet, these Fe-S proteins are vulnerable to attack by reactive oxygen species, which are regulated by enzymes such as superoxide dismutase [31], and by nitric oxide, which is regulated by its synthesis from arginine by nitric oxide synthases [32]. Yet, despite their inherent susceptibility to oxidation and degradation, Fe-S clusters have crucial advantages for some functions as they can bind or interact with electron-rich enzymatic substrates, accept or donate electrons and stabilize specific protein conformations.
3. Methods for Fe-S cluster determination and characterization
The first mammalian DNA polymerase was purified in 1965 [33], yet the discovery that DNA primase and replicative polymerases contain Fe-S clusters waited for over 40 years until 2007 [34] and 2011 [12] respectively. This discovery lag was likely due to the instability of Fe-S clusters during the multi-step purification schemes needed to isolate such enzymes and the lack of an easily recognizable conserved Fe-S cluster sequence motif. Given the importance of Fe-S clusters to biology and to aid more rapid discovery, we consider several methods that can be used to identify the possible existence of Fe-S clusters in proteins.
The first indication that a purified enzyme may contain an Fe-S cluster is the appearance of the protein solution. Fe-S cluster-containing proteins usually exhibit a brownish color due to ligand to metal charge transfer (LMCT). Such charge-transfer interactions are weak compared to covalent bonds, and the energy of their transition into an excited electronic state (charge-transfer or CT bands) occurs frequently in the visible region of the electro-magnetic spectrum, resulting in intense color for these complexes. The color is so striking that the papers describing the discovery of Fe-S clusters in the XPD helicase and the yeast replicative DNA polymerases showed photos of tubes or bottles filled with brown liquid [10,12]. The color of Fe-S cluster-containing protein solution varies depending on cluster ligands and Fe oxidation states. For example, a [2Fe-2S] cluster coordinated with two histidine and two cysteine ligands in Rieske protein shows a pinkish color. For [4Fe-4S] cluster proteins, a color change from brown to yellow or to loss of color during purification or storage in the presence of oxygen can signal oxidation of the Fe-S cluster. Smell, as well as sight, can be useful in suspecting the presence of an Fe-S cluster. Release of H2S gas upon acidification was the first indication that endonuclease III contained an Fe-S cluster [35]. If an enzyme is suspected to contain an Fe-S cluster, then it becomes important to consider anaerobic purification and storage to avoid damaging the cluster until tests show otherwise.
Sequence alignments of homologous proteins can help identify conserved cysteine residues. Three or more conserved cysteine residues might indicate that these cysteines participate in metal binding whether it is Fe or other metals such as Zn, Cu or Ni. Sequence alignment of eukaryotic and archaeal XPD homologs identified a conserved domain between the Walker A and B ATPase motifs that had four conserved cysteines [10]. Moreover, metallomics and metalloproteomics may increasingly find their place with genomics and transcriptomics as key approaches to understanding complex biological systems, as only half the existing metalloproteins are predicted to be known even in microorganisms [36]. Except for selenocysteine, which is incorporated by repurposing the UGA nonsense codon [37], metalloproteins depend upon the cell for proper metal site incorporation [38]. Misincorporated metal ions can be toxic and mutagenic as seen for cadmium, which can become incorporated into the MutS mismatch repair dimer resulting in a highly mutagenic phenotype [39]. Heterologously expressed proteins may have the incorrect metal ion or no metal ion inserted [36,40], so the absence of an Fe-S cluster in a recombinant protein does not rule out the cluster. Furthermore the use of a His-tagged, construct and its purification over metal affinity columns can remove metal ions from cysteine ligands [41]. If there is a question regarding the presence of an Fe-S cluster, native biomass is the best source for metal ion analyses [13]. During x-ray data collection for structural analyses, cysteine and metal ions are electrophilic targets for electrons ejected by synchrotron radiation, and ascorbate may protect against metal ion reduction and loss [42].
The iron content of purified protein can be quantified by colorimetric assay1, inductively coupled plasma mass spectrometry (ICP-MS)1, or atomic absorption spectroscopy (AAS). Colorimetric chelation assays can use several different reagents including ferrozine, bathophenanthroline, or phenanthroline [10,43,44]. A phenanthroline iron chelation assay was used to determine the iron content of Thermoplasma volcanium XPD mutants [10]. ICP-MS is a more sensitive method, and was used for determining the iron content of Ferroplasma acidarmanus Rad3 (XPD) [45]. AAS was used to measure the iron content of FancJ wild-type and mutant proteins [46]. A colorimetric assay is the most efficient method: it can detect the sample limit up to ppb 10−9 (nM) range as well as the AAS method [43]. While ICP-MS has a high running cost, it can detect sample limit to ppt 10−12 (pM) range.
Fe-S clusters have spectroscopic properties that can be measured using standard absorption methods or using electron paramagnetic resonance (EPR) or Mössbauer spectroscopy. [2Fe-2S]2+ clusters have an absorption band at 330 nm with broad shoulders at 460 nm and 550 nm [47], and [4Fe-4S]2+ clusters have a broad absorption centered at 390 nm [48] by UV-Vis spectroscopy. Furthermore, EPR and Mössbauer spectroscopy can provide the information of cluster types and their oxidation states. The [4Fe-4S]2+ clusters are diamagnetic (S = 0) at ground state and are EPR-silent, but can be reduced to an [4Fe-4S]1+ state to become EPR-active with a strong reducing agent such as sodium dithionite, which has redox potential of -660 mV [49], or Cr(II) EDTA with a redox potential of -1000 mV [50]. The [4Fe-4S]1+ state is paramagnetic, which has S = ½ ground state, and gives g = 1.94 EPR signal. The oxidized [4Fe-4S]3+ state, typically seen in HIPIP protein, exhibits S = ½ and a g = 2.01 EPR signal [51]. By Mössbauer spectroscopy, [2Fe-2S] clusters typically have quadrupole splitting δEQ = 0.4-0.8 mm/s and a chemical shift of δ = 0.25-0.30 mm/s [51], whereas [4Fe-4S]2+ and [4Fe-4S]1+ have δEQ = 1.22-1.6 and 0.83-0.98 mm/s and δ = 0.44-0.59 mm/s at 4.2K [52,53]. The above characteristics can support the existence of Fe-S clusters in a protein. However, EPR requires [Fe-S] cluster proteins to be EPR-active (i.e. electronic spin S>0, and non-integer) and usually needs 100 uM to 1 mM protein concentration, while Mössbauer experiment usually requires at least 0.5 mM of protein concentration [52,54]. Even though Mössbauer experiment needs high protein concentration, the Mössbauer spectroscopy has the capability to show electronic properties of each Fe site of the cluster, the coupling nature between Fe atoms, and oxidation state determination of the cluster. Circular Dichroism (CD) spectroscopy can detect unique spectroscopic characteristics of Fe-S clusters in visible wavelength region. It has been used to characterize the existence of Fe-S clusters and monitor the Fe-S cluster formation [55]. Magnetic Circular Dichroism (MCD) provides useful information of Fe-S cluster types and spin states, which can be a complementary method to EPR, especially for the EPR-silent Fe-S clusters [56]. Resonance Raman (RR) has been used to study the function and properties of Fe-S clusters. The useful Fe-S stretching region and vibrational frequencies were used to study the role of [4Fe-4S] cluster in Endonuclease III [57]. However, the fluorescence induced from the sample can overwhelm the Raman signals. X-ray absorption (XAS) spectroscopy has been used to investigate the detailed local geometry and electronic structure of Fe-S clusters and is never silent to x-ray absorption spectra [58]. The XAS provides the distances of Fe-Fe and Fe-S in oxidized and reduced states of [4Fe-4S] clusters, which can be used to study the impact on Fe-S clusters upon substrate binding or protein conformational change. The XAS also requires at least 1mM high protein concentration to generate good spectra.
Genetics and biochemistry may also provide clues as to the presence of an Fe-S cluster. A synthetic lethal screen with the pol3-13 allele, which is a mutation of a cysteine in the C-terminal domain of yeast DNA polymerase δ, identified several genes that are now known to be components of the cytosolic Fe-S protein assembly machinery (MMS19, NBP35, DRE2, and TAH18) [12,59]. Although this cysteine was widely believed to coordinate a Zn ion in DNA pol δ, this synthetic lethality data provided the first clue that this cysteine may actually coordinate an Fe-S cluster [12]. Yeast XPD had been genetically linked to MMS19 and was suspected to play a role in stabilizing XPD, but a clear role for MMS19 was confounded by proposed roles in diverse cellular pathways [60-62]. It was not until after XPD was known to contain an Fe-S cluster that the role for MMS19 in cluster assembly became clear. Co-immunoprecipitation experiments with MMS19 revealed 12 known Fe-S cluster proteins including the DNA processing enzymes XPD, FancJ, DNA pol δ, and Dna2 as well as members of the cytosolic Fe-S protein machinery [62]. Subsequent experiments confirmed a direct role for MMS19 in Fe-S cluster biogenesis and finally provided a molecular explanation as to why MMS19 had been implicated in many different cellular processes and protein complexes [62].
Lastly, determining the redox potential of Fe-S containing proteins both on and off the DNA is critical for gaining insights into protein functions. Redox potential can be measured using the electrode either by potentiometry [63,64] or voltammetry [65]. Redox potentiometry uses a redox mediator such as benzyl viologen (-360 mV), methylene blue (11 mV), and ferricyanide (360 mV) to titrate the solution and monitor the changes of the potential between two electrodes. Cyclic Voltammetry (CV) has been used extensively in Fe-S cluster containing proteins by applying constant or varying potential continuously or stepwise at an electrode and measuring the changes of potential in protein solution [66].
Additionally, Jacqueline K. Barton’s group developed a DNA modified electrode to specifically detect the charge transfer through DNA films on a gold surface [67] or DNA duplexes on an highly oriented pyrolytic graphite (HOPG) surface [68]. Electrons are transferred between protein (i.e. Fe-S clusters) and bound DNA through the DNA bases and conducted to modified gold or graphite surface. Several examples of Fe-S cluster containing DNA binding proteins have been reported using DNA modified electrodes to measure their redox potential [69,70], and these proteins have been proposed to transfer charge to one another through DNA in a process called DNA CT (discussed below). For DNA binding proteins, the redox potential of the Fe-S cluster is typically shifted by binding to polyanionic DNA. Notably, this shift will make the Fe-S cluster and also its protein more vulnerable to oxidative damage and degradation. Once an Fe-S cluster and probable cysteine ligands have been identified, protein data bank searches, as developed for metalloproteins, can give insights into possible Fe-S structures [71].
4. Unique role for Fe-S clusters in DNA processing enzymes
Charge transfer through DNA (DNA CT) occurs when electrons are transferred between redox partners, both of which are bound to DNA, in a path through the pi-stack of base pairs [72,73]. Intact double-stranded DNA is able to mediate DNA CT over long distances. Yet, DNA CT is extremely sensitive to perturbations in base pair stacking, such that damage to a DNA base or the presence of a base mismatch interrupts DNA CT. An intact DNA duplex is required for DNA CT: single-stranded DNA that is unstacked cannot transfer electrons. DNA CT thus provides an exquisitely sensitive mechanism to detect disrupted DNA structure over long distances. Proteins that contain redox centers such as [4Fe-4S] clusters can be both electron donors and acceptors when bound to DNA. However, in the absence of DNA binding, the potential of the 3+/2+ redox couple of the Fe-S cluster is significantly more positive and outside the range of physiological redox activity [68]. Upon DNA binding the redox potential shifts -200 mV into the physiological range, switching the cluster into a mode where it serves as a physiological redox switch. One of the many important insights that the Barton group made is that this switch is activated only in the DNA-bound form. DNA CT appears to be essentially distance-independent; the efficiency of DNA CT over 100 base pairs has been shown to be equal to that through a 17-mer [73]. The fact that DNA resembles a wire, mechanistically, means that both short and very long range signaling are equally achievable. Aspects of DNA CT seem to have potential differences from Marcus theory [74], including little distance dependence, making this type of charge migration of great interest for chemistry and physics.
DNA replication and repair processes require the orchestration of cooperative activities as the intermediates are toxic and mutagenic. Cooperative activities in general require efficient communication about the current state of the system. Cooperative activities in cellular replication and repair require communication via interactions by diffusing signaling molecules, post-translational modification machinery, or dynamic macromolecular interactions: all of these communication events involve relatively short-range interactions as intermolecular energies are extremely distance dependent. Such diffusion dependent interactions have apparent limitations for the communication among molecular machines engaged in replication and repair events across the genome. The DNA CT activity originally discovered as a possible means of damage transfer through the base stack [75,76] and extended into facilitating the damage search by altering DNA glycosylase binding [72] and to communication between glycosylases and helicases from distinct repair pathways [69,77] changed our view of DNA from insulator-like to wire-like for charge transfer [78]. Here we comprehensively examine Fe-S clusters structure-function in replication and repair. This broader analysis supports DNA CT concepts developed by the Barton group toward long-distance cooperative activities via DNA and extends them to propose a broader application of DNA CT that we term DNA CTC for DNA charge transfer communication (discussed in more detail in Section 10).
DNA CTC provides a potentially unifying mechanism of action for Fe-S proteins, with their specificity arising from differences in the rate of charge transport within individual proteins. The speed of moving an electron from the DNA through the protein depends upon the distance between the DNA and the Fe-S cluster as well as on the composition of the intervening side chains. Aromatic tyrosine and tryptophan residues can facilitate the transfer of electrons in proteins [79]. It follows that conformational changes within the protein that bring the Fe-S cluster closer to or further from the DNA contact point would change the efficiency and specificity of DNA CTC. Mutations or small-molecule effectors altering distance or side chain parameters would also affect the efficacy of DNA CTC. For example, the conserved Y82 residue in E. coli endonuclease III is positioned close to the DNA backbone [80] and an alanine substitution is defective in DNA CT [72].
The DNA processing enzymes we consider here have distinct sequence architectures regarding the placement of Fe-S clusters relative to catalytic domains (Fig. 2B), which may provide a means for different DNA CT activities and pathway coordination. The glycosylases and the polymerases all have Fe-S clusters that are separable from catalytic domains whereas the helicases and nuclease/helicase have insertions of Fe-S clusters into catalytic domains (Fig. 2B). In the simplest of models, the C-terminal placement of the Fe-S clusters in glycosylases and polymerases might impact the DNA affinity and hence exchange rate versus processivity. In contrast, the Fe-S cluster insertion into Dna2 and XPD family helicases catalytic domains suggests a tight linkage between cluster and catalytic activities. Furthermore, the unique placement of the XPD family Fe-S cluster within the HD1 catalytic domain supports its role as a sensor for double helix disruption. These sequence architectures are reflected in atomic resolution structures (Fig. 3).
Figure 3. Fe-S cluster domains and folds in DNA processing enzymes.

Top: Ribbon diagrams of overall protein architecture (orange ribbon with surface representation) and placement of Fe-S cluster domains (colored in blue). Bottom: close-up view of Fe-S cluster domains. [4Fe-4S] cluster is shown as brown (Fe) and yellow (S) spheres. (A) crystal structure of XPD helicase from S. acidocaldarius (PDB: 3CRV [112]); (B) crystal structure of C-terminal domain (CTD) of catalytic subunit (blue) and B subunit (orange) complex of yeast DNA polymerase α (PDB: 3FLO [133]). Two Zn metals (gray) were bound to CTD. Zn-2 (CysB) binding site was later experimentally shown to be a Fe-S cluster, but Zn is bound in this structure; (C) structure of C-terminal regulatory domain of human DNA primase (PDB: 3L9Q [150]).
The Fe-S cluster of the XPD helicase is more intimately connected to the globular structure of the catalytic core (Fig. 3A), whereas the Fe-S clusters of DNA polymerase α (solved with a Zn atom in place of the Fe-S cluster) and DNA primase are more separable from the cores (Fig. 3B-C). In keeping with a lack of a conserved Fe-S sequence motif, the folds around the Fe-S clusters are all different. The XPD Fe-S cluster is coordinated by mixed α-helices and loops, the DNA pol α Fe-S cluster will likely retain the mixed β-sheet and loop structure that coordinates the Zn atom seen in the structure, and the DNA primase Fe-S cluster is coordinated entirely by α-helices (Fig. 3A-C). As structures with DNA become available, more details about structural relationships between the Fe-S clusters and the DNA will inform our understanding of their activities and roles.
In particular, Fe-S clusters can greatly aid electron transfer by delocalization of electrons over both Fe and S centers. As a polyanion, DNA is resistant to nucleophilic attack, but not to Fe-mediated oxidation and radical damage. If Fe-S clusters are not acting in charge transfer in DNA replication and repair enzymes, but rather as structural co-factors, then there are other ways to accomplish similar structural roles for DNA binding proteins including replacement by metal ions such as Zn, which is not susceptible to oxidation and degradation. Indeed, both the Fe and the cysteine ligand sulfur are susceptible to oxidation, and removal of cysteine from enzymes can increase their stability, as seen for superoxide dismutase [81,82]. These collective observations suggest that Fe-S clusters in DNA metabolism result from genetic selection as biologically critical prosthetic groups with unusual chemical properties that enable Fe-S proteins to more effectively function than other structural elements and co-factors in pathways of DNA replication and repair as well as metabolism. To examine and integrate our knowledge of these Fe-S co-factor properties and functions for DNA metabolism, the structural biochemistry of these Fe-S cluster enzymes is analyzed below.
5. Glycosylases are on the front lines of finding and repairing frequent DNA damage
DNA glycosylases are a diverse family of enzymes that recognize and remove damaged bases in DNA. These enzymes are on the front lines of repairing damage due to spontaneous DNA decay from deamination, oxidation, or methylation [83-85] in the process that removes base lesions that do not generally distort the DNA helix called Base Excision Repair (BER). This damage is frequent, with an estimated 2,000-10,000 purine turnover events per cell per day just from hydrolytic depurination [86]. As seen from the first glycosylase-DNA structures, these enzymes bend double-stranded DNA and flip out the nucleotide containing the damaged base, which breaks base packing on the damaged strand [87]. Evolution has responded with many different DNA glycosylases, spanning six different structural superfamilies [83,88]. Two of these superfamilies contain members that have Fe-S clusters: 1) the helix-hairpin-helix (HhH) superfamily, named for a secondary structural element important for DNA binding, contains E. coli endonuclease III and MutY and their mammalian homologs, hNTH1 and MUTYH, also aided the discovery of Fe-S roles in DNA binding [89], and 2) the uracil DNA glycosylase family has thermophile-specific UDG enzymes that contain Fe-S clusters [90]. With or without Fe-S clusters, all DNA glycosylases have the remarkable ability to detect a single damaged base amongst a vast sea of normal bases [91]. Other DNA binding proteins that bend DNA and flip out nucleotides, such as ATL [92], have the potential to regulate DNA replication and repair processes by disrupting DNA CT. Despite forty years of research that followed the identification of the first DNA glycosylase [93], the basis of efficient and specific damage recognition is still controversial. Furthermore, the initiation of DNA repair by glycosylases and the subsequent abasic site cleavage by endonucleases creates intermediates that may be more toxic and mutagenic than the initial lesion. Product binding to control and coordinate a direct handoff of intermediates is likely essential for genome integrity, as proposed from the structural biochemistry of both the human uracil DNA glycosylase and the basic endonuclease APE1 [94,95]. From the analysis of structures, we are thus beginning to appreciate that processes such as DNA repair are choreographed by interrelated interactions [96], but how these processes are coordinated among pathways and with processes such as replication is poorly understood. Fe-S clusters may provide a means to control product binding and coordination with general implications for coordination within and among pathways [69].
5.1. Endonuclease III was the first Fe-S DNA repair enzyme discovered and DNA binding shifts the redox potential
The first DNA repair enzyme discovered to contain an [4Fe-4S] cluster was endonuclease III (EndoIII) from E. coli [35,89]. EndoIII is a bifunctional DNA glycosylase for oxidized pyrimidines that can cleave both the N-glycosidic bond between a damaged base and the deoxyribose sugar, and nick the DNA backbone [97]. Initial characterization of the purified enzyme showed that it contained a single [4Fe-4S] cluster in the 2+ oxidation state and that the cluster was not easily oxidized or reduced under physiological conditions [35], so a redox role for the cluster was not readily apparent. The crystal structure later revealed that the cluster positions conserved basic residues for interaction with the DNA phosphate backbone [98], suggesting that the cluster played an important structural role for DNA binding (Fig. 4A-B). A redox role for the cluster was not demonstrated until electrochemical experiments on DNA-modified electrodes revealed that DNA binding shifts the redox potential of the 3+/2+ couple into the physiological range [99]. In EndoIII, the Fe-S cluster, the protein structure, and the DNA all conspire to throw a redox active switch—the cluster is needed to position the protein structure such that the protein can bind DNA while DNA binding activates the cluster toward oxidation, ensuring that cluster oxidation is DNA-mediated (Fig 4B).
Figure 4. Important structure elements of EndoIII DNA interaction and MUTYH MAP mutations in the Fe-S domain.
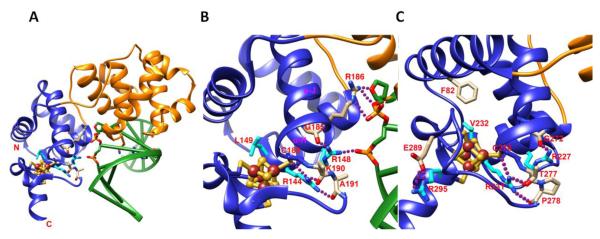
A) The overall structure of G. stearothermophilus Endonuclease III-DNA complex with [4Fe-4S] cluster (PDB:1P59 [80]). Fe-S cluster domain and DNA are shown in blue and green, respectively. B) Zoom in view of A show the interactions between Fe-S cluster domain and DNA form tightly H-bonding networks on Endo III-DNA complex (PDB:1P59 [80]). Conserved residue R144 forms H-bonding with residues C189, A191, and K190 that tightly hold Fe-S cluster domain with α-H helix together. That interaction positions conserved residue R148 to form H-bonding with DNA phosphate group and carbonyl group of G185 on α-J helix. And that positions residue R186 to form H-bonding with DNA phosphate and ribose groups. Interacting residues are shown in stick, H-bonding is shown in purple dot line, and Fe and S are shown in brown and yellow spheres, respectively. C) The interactions of human MUTYH associated polyposis (MAP) mutations with Fe-S cluster domain. MAP mutations R227W, R231C/H, V232F, and R195C (shown in cyan stick) form H-bonding network with Fe-S cluster domain. The similar interactions have been observed in EndoIII structure (Fig. 4B). Additionally, residue R295 forms H-bonding with E289 to fix the helix conformation for Fe-S cluster binding. Mutation of V232F can form steric clash with F82 that will destabilize the Fe-S cluster domain. (PDB:3N5N [108])
5.2. The MutY Fe-S cluster is important for enzymatic activity and organizes a hydrogen bond network important in cancer predisposition
MutY from E. coli was first discovered as an adenine glycosylase that removes adenine from G-A mispairs, but was later shown to be part of the E. coli GO system that removes oxidatively damaged guanine from DNA [100-103]. MutY was the second example of a DNA repair enzyme with an Fe-S cluster as it was cloned shortly after the discovery of the Fe-S cluster in EndoIII. Sequence alignment between the two enzymes revealed significant similarity in their N-terminal domains and a shared set of four identically spaced cysteines, suggesting these cysteines coordinate an [4Fe-4S] cluster in MutY as they do in EndoIII [102]. The crystal structure of MutY revealed that it conserves the overall bi-lobal architecture of EndoIII, with the buried [4Fe-4S] cluster organizing enzyme loops and alpha helices at the DNA binding surface [104]. The structure of MutY with DNA showed that the strand that contains the substrate adenine, which is flipped out from the DNA helix, runs through a deep cleft between the catalytic six-helix barrel domain and the [4Fe-4S] cluster domain [105]. Replacing the Fe-S cysteine ligands with serine, histidine, or alanine either dramatically effected solubility of MutY or decreased DNA substrate binding affinity, indicating the structural importance of the cluster in protein stability and activity [90,106]. Interestingly, the Fe-S cluster was not found to affect protein folding as MutY can be denatured and refolded in the absence of ferrous and sulfide ions without a change in thermal stability [107]. This refolded apo enzyme does not have adenine glycosylase or DNA binding activity, but these activities can be restored by the addition of ferrous and sulfide ions [107], providing further evidence of the critical role the Fe-S cluster plays in MutY activity.
A crystal structure of more than half of the human homolog of MutY (MUTYH) shows a hydrogen bond network around the Fe-S cluster [108] (Fig. 4C). Several of these residues are associated with MUTYH-associated polyposis, an inherited disorder that predisposes patients to colorectal tumors [109]. This H-bond network plus one of the cluster-coordinating cysteine residues are critical for orienting a helix of the interdomain connector (IDC) [108]. The IDC connects the N- and C-terminal domains that make up the bi-lobal architecture of MutY, but differs significantly in sequence and length in eukaryotes [108]. Mammalian IDCs have three additional conserved cysteine residues that were recently shown to coordinate a zinc ion [110]. Serine substitution mutants of the coordinating cysteines were found to have low iron content suggesting that a coordinated zinc ion in the IDC may be important for Fe-S cluster insertion [110]. The eukaryotic IDC is important for the interaction of MUTYH with the 9-1-1 complex, DNA damage selection, and robust enzymatic activity [108]. Therefore, the Fe-S cluster and the structural elements surrounding the cluster play key roles in MUTYH biology and cancer prevention. Like EndoIII, MutY becomes redox active when bound to DNA [99]. A model for how redox active glycosylases can increase the efficiency of damage detection is discussed below.
5.3. DNA charge transfer model of damage detection by DNA glycosylases
As DNA CT occurs only through an intact DNA duplex, DNA CT has been proposed by Jacqueline K. Barton and her laboratory, to be an efficient means for BER proteins that contain [4Fe-4S] clusters to redistribute in the vicinity of DNA damage and hence efficiently detect base damage in the vast sea of normal DNA [70] (Fig. 7A). This has been shown for MutY, which repairs oxodG-A mismatches, and EndoIII, which repairs hydroxylated pyrimidines [72,99]. In this model, DNA CT is a first step in lesion detection by localizing proteins near the damage. DNA CT is initiated by a guanine cation radical oxidizing a nearby MutY Fe-S cluster to a more tight-binding oxidized 3+ state. If the DNA is undamaged, the binding of a second repair enzyme with similar redox potential, e.g. another MutY molecule or EndoIII, reduces the first cluster to 2+ state, which decreases its affinity for DNA and the protein dissociates. This long range DNA CT to reduce EndoIII can only occur if the intervening DNA is intact and undamaged, effectively scanning this region of the genome. This cycle of binding, DNA CT, and release causes the local concentration of MutY (or EndoIII) to remain low on undamaged DNA. In the presence of damage, however, the second MutY (or EndoIII) cannot reduce the Fe-S cluster of the first MutY, so both molecules stay bound in the vicinity of damage to promote a higher local concentration of glycosylase around the damaged base for subsequent damage detection (Fig. 7A). This redistribution has been monitored in vitro by atomic force microscopy (AFM) where mutants unable to carry out DNA CT do not preferentially bind to damaged DNA [69,72]. In fact, in a series of mutants, a direct correlation was evident between the ability of EndoIII mutants to localize near a mismatch (which also inhibits DNA CT but is not a substrate for EndoIII) and their ability to carry out DNA CT [70].
Figure 7. The DNA mediated charge transport communication (DNA CTC) hypothesis as a method for protein-protein communication.

A) DNA CT provides a means for Endo III and MutY to preferentially localize to damaged DNA due to a disruption in the DNA pi stack B) Proposed DNA CTC mechanism controls rapid switching between replicative polymerases during lagging strand synthesis C) Nucleotide Excision Repair (NER) DNA CTC between DNA polymerase and XPD to coordinate DNA synthesis, incision, and release of the damaged strand. D) DNA CTC communication between replicative polymerases at adjacent origins to control origin firing.
By using DNA CT as a first step to localize near damage, the BER enzymes are hypothesized to effectively help one another search for damage within the cell. The more proteins involved with similar redox potentials that can exchange electrons, the more efficient the process, even if some repair enzymes are in low copy number. Using measured copy numbers, genome sizes, diffusion constants, etc., calculations show that a search of the E. coli genome by facilitated diffusive hopping of repair proteins (and even assuming no other protein traffic) is too slow to account for the efficiency of repair within the cell; yet, incorporating DNA CT over even a few hundred base pairs (distances for DNA CT that have been documented [73]) significantly improves the search time [72]. Consistent with this model, the MutY activity in E. coli is reduced in EndoIII deletion mutants, therefore EndoIII appears to aid MutY in repair. Furthermore, mutations that affect EndoIII CT activity that are otherwise active glycosylases also show attenuated cellular MutY activity [72], establishing a link in vivo between helping MutY and ability to perform DNA CT.
6. The XPD family of 5′-3′ helicases have diverse functions in DNA repair
The discovery that the XPD and FancJ helicases have Fe-S clusters [10] was truly a breakthrough in understanding the molecular mechanisms of this family of helicases. First, it allowed crystal structures of XPD to be solved [111-113], especially in our case as crystals of Sulfolobus acidocaldarius XPD could not be grown in the presence of oxygen [112]. Second, it suggested that Fe-S clusters may be a more general feature of DNA processing enzymes and not restricted to a few classes of glycosylases. Here, we consider four members of this unique family of Fe-S helicases and discuss their roles in DNA repair and related roles in DNA replication. Although the importance of these Fe-S helicases is underscored by their tight linkage to human disease and to cancer predisposition, the precise roles of their Fe-S clusters remain enigmatic.
6.1. XPD has a redox active Fe-S cluster with tight structural connections to catalytic domains
XPD is a SF2 DNA helicase with 5′-3′ polarity that serves as the primary helicase responsible for opening a DNA bubble during nucleotide excision repair (NER) and as an ATPase aiding transcription. In eukaryotes, XPD is part of the TFIIH machinery that participates in both transcription and DNA repair. Point mutations in XPD cause human diseases with increased cancer risk or premature aging: Xeroderma pigmentosum (XP), Cockayne syndrome (CS), trichothiodystrophy (TTD), or combinations including XP/CS or XP/TTD [114]. Chemical analyses and crystal structures of archaeal homologs of XPD revealed the presence of a [4Fe-4S] cluster in a domain that interrupts one of the two helicase domains [10,111-113]. In the absence of the Fe-S cluster, the Fe-S cluster domain becomes highly disordered, disrupting the structure of the nearby arch domain (Fig. 5A). One TTD point mutation K84H (R112H in humans) is located near the [4Fe-4S] cluster and forms hydrogen-bonding with the one of the cysteine ligands. The mutation of lysine to histidine can disrupt this H-bonding interaction due to the short length of the histidine residue (Fig. 5B), perturbing the protein environment around the Fe-S cluster. More importantly, this mutation could change the redox potential of Fe-S cluster, as hydrogen bonding interactions with Fe-S clusters play a key role in modulating the accessible redox couple [115]. The tight structural connection between the Fe-S cluster domain and the helicase domains provides mechanical coupling of the cluster domain motions to those in the ATP site. Mutations in Fe-S cluster cysteines or chemical oxidation of the cluster abolishes helicase activity and severely affects ATPase activity [112]. The Sulfolobus acidocaldarius XPD (SaXPD) Fe-S cluster has a DNA-bound redox potential of ~80 mV on DNA modified gold electrodes (versus a normal hydrogen electrode (NHE) reference), which is similar to the physiologically active redox potentials of the DNA glycosylases [116]. ATP hydrolysis increases DNA CT activity, suggesting that motions of the helicase domains are coupled to the Fe-S domain, resulting in increased DNA CT activity [116]. Surprisingly, SaXPD participates in damage detection with E. coli EndoIII even though a mismatch is not a substrate for XPD, suggesting that Fe-S repair proteins from different repair pathways and in this case, different species, can coordinate in the search for damage using DNA CT in vitro [69]. In vivo, a similar coordination between the DNA damage response helicase, DinG, and EndoIII has been observed genetically, suggesting that DNA CT signaling occurs within the cell [77].
Figure 5. The XPD Fe-S cluster coordinates key structural elements and is important in human disease.

A) Fe-S cluster stabilizes the local structure folding of Fe-S cluster domain in SaXPD (blue) (PDB:3CRV [112]). 55 residues in the Fe-S cluster domain becomes completely disordered without Fe-S cluster (cyan) (PDB:3CRW [112]). The Arch domain (orange) is also affected in the absence of Fe-S cluster (red). The disorder region is shown as green dot line. B) The impact of TTD mutation K84H on Fe-S cluster domain in SaXPD. Residue K84 forms hydrogen bonding (purple dot line) with one of the cysteine ligands of [4Fe-4S] cluster. [4Fe-4S] cluster was surrounded by many hydrophobic residues that control the solvent accessibility. The mutation of lysine to histidine can disrupt hydrogen-bonding interaction and impact the protein environment and redox potential of Fe-S cluster. The Fe-S cluster domain is shown in blue; residues K84 and hydrophobic residues are shown in stick.
6.2. FancJ is important in cancer predisposition and interstrand crosslink repair
Like XPD, mutations in FancJ are associated with predisposition to cancer. FancJ was first discovered as a protein that interacts with the BRCT motifs of BRCA1 and strong homology to the XPD family of DEAH helicases, so it was named BACH1 for Brca1-Associated C-terminal Helicase [10,117,118]. BACH1 was later found to be the product of the FANCJ gene that is deficient in Fanconi anemia (FA), a rare recessive disease with a high risk of developing leukemias and solid tumors [118-121]. The FA pathway is now known to include 16 gene products that repair DNA interstrand crosslinks [122]. FancJ is one of several factors referred to as downstream components of the FA pathway that link the FA pathway to homologous recombination (HR) including BRCA2 (FANCD1), PALB2 (FANCN), and RAD51C (FANCO) [122]. In vitro, FancJ can resolve G4 DNA structures, displaces protein bound to DNA, and forms a functional dimer [123]. A mutation in the Fe-S cluster domain (M299I) of the human protein, leads to early onset breast cancer and the enzyme showed increased in vitro ATPase activity without a corresponding increase in helicase activity, highlighting the biochemical and physiological importance of the Fe-S cluster domain in regulating the helicase activity of FancJ [124].
6.3. The Fe-S helicase RTEL1 is important for homologous recombination and telomere maintenance
RTEL1 was first identified as a factor that regulates telomere length in mice, but was later found to regulate homologous recombination in mitotic and meiotic cells [125,126]. RTEL1 is able to unwind displacement (D)-loop intermediates during HR and T-loops at telomeres [126]. Like XPD and FancJ, mutations in RTEL1 have been linked to cancer predisposition and human disease. Genome-wide association studies (GWAS) have linked single nucleotide polymorphisms (SNPs) in RTEL1 with brain tumors [126,127] and several nonsense and missense mutations in RTEL1 have been linked to the rare, bone-marrow condition, Hoyeraal-Hreidarsson syndrome (HH) [126]. Like FancJ, RTEL1 is much larger than XPD and is predicted to have additional domains in the C-terminal half. A Harmonin-N-like domain likely plays a role in protein-protein interactions and a cysteine rich C4C4 RING-finger domain may coordinate metal ions at the C-terminus [126,128]. The precise functions of these domains are unknown, but are of great interest as many patient mutations have been mapped to these domains. A recent study has suggested the N-terminal domain, containing 4Fe-4S cluster with a redox midpoint potential of −248 ± 10 mV, of human RTEL1 is not directly involved in DNA binding [129]. This could suggest that the Fe-S cluster domain of RTEL1 might be essential for interacting with other domains of RTEL1 during helicase activity.
6.4. Chlr1 is a Fe-S helicase with proposed roles in DNA replication and sister chromatid cohesion
Mutations in ChlR1 (DDX11) cause the recently identified Warsaw Breakage Syndrome that was named for an individual from Warsaw, Poland who was thought to have a chromosome-instability syndrome due to severe microcephaly, growth retardation and abnormal skin pigmentation [130,131]. Cytogenetic analysis of patient cells treated with mitomycin C (MMC) suggested a diagnosis of FA due to MMC sensitivity, but further analysis showed an increase in chromosome separation that is not observed in FA [130,131]. Sequence analysis revealed mutations in the ChlR1 (DDX11) gene in both alleles, and this patient remains the only known example of a genetic defect in ChlR1. Like other members of this helicase family, ChlR1 is a 5′-3′ SF2 family helicase with a [4Fe-4S] cluster between helicase motifs 1A and II [10,131]. Due to its unwinding activity on diverse DNA substrates and its interaction with several DNA replication factors such as FEN1 and PCNA, ChlR1 was proposed to play a role in processing lagging strand replication intermediates that affect sister chromatid cohesion [131], however the role of the Fe-S cluster in Chlr1 is unknown.
7. All replicative DNA polymerases have Fe-S clusters with unknown roles
The catalytic subunits of all eukaryotic B-family DNA polymerases can be separated into two domains—an N-terminal catalytic domain that contains conserved polymerase motifs and a C-terminal domain (CTD) that contains eight conserved cysteines (Fig. 2B). It was thought that these cysteines formed two zinc finger motifs [132] until the observation that a mutation in one of the cysteines of the second motif (CysB) of yeast Pol3 (a pol δ homolog) was synthetically lethal with essential Fe-S protein assembly machinery genes [12]. Radiolabeling experiments with 55Fe and cysteine mutants revealed that CysB in Pol1, Pol2, and Pol3 coordinates an Fe-S cluster [12]. Further analysis established that the [4Fe-4S] cluster in CysB is important for complex formation with accessory subunits while the CysA Zn-binding motif is crucial for PCNA binding to pol δ [12]. Therefore, one function of the Fe-S domain is structural, in mediating interactions with the accessory subunits of multi-subunit polymerase complexes [12]. However, the choice of iron is likely more than just structural, since the Fe-S cluster can be replaced by zinc, at least in pol α, without disrupting subunit-subunit interactions [133]. Furthermore, only the CTD of pol ε is essential for life, not the N-terminal catalytic domain [134], suggesting that the CTD domain and perhaps the Fe-S cluster plays essential roles in DNA replication and metabolism. The Fe-S cluster is thus associated with an essential function, but why has this potentially toxic Fe-S cluster not been replaced by other metals? We know from other types of metalloenzymes, such as the abasic site endonucleases, that replacement of one metal ion by another such as zinc can be accomplished while maintaining activity [9]. Indeed, different superoxide dismutases can use Cu, Mn, Fe, or Ni ions to accomplish the same reaction [31,135-137]. Evidently, there are regulatory functions that have strong and specific evolutionary selection for Fe-S clusters over other metal ions and structural elements.
7.1. DNA polymerases delta, epsilon, and zeta have Fe-S clusters in flexible CTD domains
DNA polymerase delta (Pol δ) and epsilon (Pol ε) are essential in lagging and leading strands DNA replication process, respectively. Pol δ consists of a catalytic subunit p125 and three accessory subunits p66, p50, and p12 that form a heterotetramer and interacts with proliferating cell nuclear antigen (PCNA) for processivity [138]. During Okazaki fragment maturation, pol δ coordinates with either flap endonuclease 1 (FEN1) or the Dna2 nuclease-helicase to remove priming RNA [139]. Pol δ has been shown to participate in PCNA-dependent base-excision repair, mismatch repair, and redundantly with pol ε in NER [140].
Pol ε is also a heterotetrameric protein that contains a catalytic subunit p261 and accessory subunits p59, 17, and p12 [141]. Pol ε is responsible for DNA leading strand replication and has more processive activity than pol δ with PCNA-dependent DNA elongation [142]. Pols δ and ε are extremely accurate with an estimated mutation rate of less than 1× 10−9 per base pair. Both pol ε and δ achieve high fidelity by high nucleotide selectivity, proofreading with their exonuclease activity, and the post-replication DNA mismatch repair [143]. The high nucleotide selectivity may result from the tight steric fit in the base-pair binding pocket that only fits the nascent Watson-Crick base pair [144]. Crystal structure of yeast pol δ catalytic domain with DNA complex shows the hydrogen-bonding networks between DNA and binding pocket that can potentially sense template-primer mismatch and switch to exonuclease editing mode [145].
Besides their high fidelity, pols ε and δ replicate DNA at a rate of 50 nucleotides per second. Yet, the [4Fe-4S] cluster was the chosen cofactor to be put in such a high speed and high accuracy process. What is the uniqueness or role of Fe-S cluster in these polymerases? Fe-S clusters possess sensitivity to protein environment (i.e. polarity) and delicate redox properties. The replication polymerase Fe-S clusters are all found in the polymerase C-terminal domains [12] of catalytic subunit. Yeast genetics revealed the puzzling result that the N-terminal catalytic domain of pol ∈ is dispensable; only the C-terminal domain is essential for life [134]. One clue comes from the cryo-electron microscopy (cryo-EM) structure of yeast DNA pol ∈ suggests that while the globular N-terminal catalytic domain is stable, the C-terminal domain containing the Fe-S cluster is both flexible and positioned to make extensive contact with the duplexed primer:template DNA [146]. The positioning of the Fe-S cluster near the DNA polyanion may shift its redox potential.
Pol ζ, another B-family DNA polymerase, is responsible for translesion DNA synthesis. Pol ζ is consists of a catalytic domain Rev3, an accessory domain Rev7, and shared accessory domains p50 and p66 of pol δ. The C-terminal domain of pol ζ, which contains Fe-S cluster, communicates with pol δ and switches with the catalytic domain of pol δ during translesion DNA synthesis [147]. It is possible that all the B-family DNA polymerases communicate each other in a similar manner by their essential Fe-S cluster domain, and this merits investigation.
7.2. The Fe-S cluster is substituted with zinc in the DNA polymerase alpha crystal structure
In eukaryotes, DNA polymerase alpha (pol α) works with primase to generate the primers necessary for DNA synthesis. DNA pol α consists of a p180 catalytic domain and a p70 accessory subunit B. It has been shown that C-terminal domain (CTD) of p180 forms complexes with p70 subunit B and primase p58 subunit that together stimulate primase activity [148,149]. Like pols δ, ε, and ζ, the pol α CTD contains two motifs with conserved cysteine residues.
Crystal structures of the heterodimeric complex of yeast pol α CTD and subunit B has been determined with Zn bound to both cysteine motifs (Fig. 3B) [133]. The structure reveals an intriguing fold with two Zn binding modules (Zn-1 and Zn-2) connected by a three-helix bundle. This helix bundle and the Zn-2 binding module are essential for interaction with subunit B based on the structure. Surprisingly, it was later demonstrated that the pol α CysB (Zn-2 binding module), and the CysB motifs of pols δ, ε, and ζ, all coordinate a [4Fe-4S] cluster, not zinc, in vivo [12]. Replacement of an Fe-S cluster with zinc is common when the protein is heterologous expressed [40]. The EM 3D-reconstruction of yeast pol α catalytic domain with subunit B has shown an elongated shape with distinct lobes. The CTD and subunit B complex was fitted to the smaller lobe, which is connected to the catalytic domain through a flexible linker [133]. A conformational change upon binding to primase may occur for RNA primer synthesis. Collective results show that the Fe-S cluster in the conserved CysB motif of pol α is essential for the binding with its accessory subunit B that regulates primase activity.
7.3. The Fe-S cluster in primase is buried among α-helices
The heterotetrameric complex of DNA pol α and primase (pol-prim) is responsible for generating primers on both the leading and lagging strands of replication. Primase, the only known eukaryotic polymerase capable of initiating DNA synthesis de novo, is the first to engage the DNA template and synthesizes an 8-12 nucleotide RNA primer. Remarkably, primase is able to count the length of this initial primer and when the threshold is reached, it hands off the DNA substrate to pol α. Human primase is a heterodimer of the catalytic 48 kDa (p48) and regulatory 58 kDa (p58) subunits. Despite its unique biochemistry and fundamental importance in replication, the structural information available from any higher eukaryote is limited to the structures of the human p58 C-terminal [4Fe-4S] cluster domain (p58C) [150] and human p58 N-terminal domain (p58N)-p48 heterodimeric subunits [150,151]. The crystal structure of primase Fe-S cluster domain (p58C) reveals a fold unique from any Fe-S proteins (Fig. 3C). The [4Fe-4S] cluster is well buried in hydrophobic core of three α-helices. It will be interesting to see how the Fe-S cluster domain interacts with p48 subunit with RNA-DNA bound structure and even more exciting to see the interaction with the CTD of DNA pol α.
7.4. The Dna2 Fe-S cluster plays a critical role in its nuclease and helicase activities
Dna2 is a multifunctional enzyme with both nuclease and helicase domains fused in a single polypeptide. As Dna2 is required in vivo for Okazaki fragment maturation and resection of double-strand breaks in a complex with Sgs1/BLM [152-154], Dna2 links replication fork collapse and replication fork restart through recombinational repair mechanisms. Dna2, bacterial AddAB, and the CRISPR associated protein Cas4 nuclease are in the conserved RecB nuclease family but with an added Fe-S cluster [155-157]. Mutations in the Fe-S cysteines do not affect DNA binding activity, but change the way in which the protein binds DNA and abolish both DNA-dependent ATPase and helicase activity [11]. The Fe-S cluster mutants have defects in DNA replication and repair in vivo that correlate in intensity with their effect on the catalytic activities in vitro, confirming a critical role for the Fe-S cluster in Dna2 activities [11]. Dna2 knockdown makes normal cells sensitive to cisplatin but rescues the sensitivity of FANCD2−/− cells to cisplatin and formaldehyde [158]. In the absence of the FA pathway, Dna2 is deleterious to crosslink repair by causing excess resection [158].
We suggest that DNA CT from the polymerase to Dna2 may help coordinate synthesis events with FEN1, which bends and opens DNA to remove the final RNA base [159], and is associated with Dna2. Furthermore the break repair and replication fork nuclease Mre11, which must open double-stranded DNA to have it reach the active site metal ions [160], also interacts with Dna2 suggesting its Fe-S cluster could act in local regulation of partner enzymes as well as at a distance [152]. As in human cells, different MRE11 nuclease endonuclease and exonuclease activities control pathway choice [161], Dna2 and its Fe-S cluster are positioned to help determine pathway choice at breaks in concert with Mre11 and as a possible complement to direct conformational connections to Mre11 via phosphoprotein partner Nbs1 [162]. In general, we know from analyzing other DNA repair nucleases that they typically reshape the DNA and sometimes themselves to achieve extraordinary specificity and efficiency [163], suggesting that strict regulatory processes have evolved to inhibit resection nucleases, and Fe-S clusters may play a role in this regulation.
8. DNA processing enzymes with Fe-S clusters have critical roles in cancer and other diseases
Defects in Fe-S cluster-containing DNA processing enzymes are implicated in human diseases and cancer predisposition. Mutations in the human MutY cause MUTYH-associated polyposis that is an inherited autosomal recessive disease with a high predisposition to colorectal tumors [109]. The predominance of colorectal tumors is thought to be due to the high level of oxidative damage in the colon and the role of MUTYH in repairing oxidative damage [109]. Several of the residues implicated in this disease are important for coordinating a hydrogen bond network around the Fe-S cluster (Fig. 4C) [108].
Mutations in XPD helicase are linked to Xeroderma pigmentosum (XP), trichothiodystrophy (TTD), Cockayne syndrome (CS), and cerebral-ocular-facial syndrome (COFS) [164]. XP is a rare autosomal recessive disorder with sun sensitivity and UV radiation-induced skin cancer. XP, TTD, and CS patient mutations have been mapped on human XPD homolog crystal structure that show different impacts on structure [112,114]. XP mutants impact DNA and ATP-binding, XP/CS mutants impact helicase domain 1 and 2 conformational change, and TTD mutants impact the overall structural framework. One of the most frequent mutations found in TTD patients is the R112H substitution that is in the XPD Fe-S domain [165]. R112 (K84 in SaXPD) forms hydrogen bonds with one of the cluster-coordinating cysteines (Fig. 5B). Patients homozygous for this mutation have a moderate phenotype despite having a severe cellular defect in DNA repair [165]. Mutations of FancJ are linked to Fanconi anemia (FA), a rare genetic disorder characterized by bone marrow failure and retarded growth and high risk of ovarian cancer [119,166]. A M299I substitution that was detected in a case of early onset breast cancer [117] is directly adjacent to one of the cluster-coordinating cysteines in FancJ. And mutations of RTEL1 cause telomere instability and are linked to Dyskeratosis congenital (DC) and Hoyeraal-Hreidarsson syndrome (HHS) [167,168]. DC is a rare inherited disorder characterized by bone marrow failure and cancer predisposition syndrome. HHS is a severe variant of DC that characterized by bone marrow failure, cerebellar hypoplasia, immunodeficiency, and developmental defects. ChlR1 has been suggested as a tumor suppressor because deficiency of ChlR1 leads to high risk of cancer development [169]. Mutations in ChlR1 cause genome instability and are linked to Warsaw breakage syndrome (WABS), which shows a combination of features of Fanconi anemia and Roberts syndrome [130], characterized by drug-induced chromosomal breakage and sister chromatid cohesion defects.
Based on yeast studies, Dna2 helicase-endonuclease has been suggested to be associated with mitochondrial DNA deletion syndrome with progressive myopathy and Werner syndrome, which is a premature aging disorder [170,171]. Mutations of Dna2 in adult onset show features of mitochondrial myopathy with muscle mitochondrial DNA instability. Based on the genetic complementation studies in yeast, WRN (Werner syndrome) gene has been shown to rescue the Dna2 mutant phenotype cell growth and DNA replication and has interactions with human flap endonuclease 1 (FEN1), which physically and genetically interacts with Dna2 [172].
Due to the critical role that the replicative DNA polymerases play in the essential process of DNA replication, these enzymes are rarely associated with human disease. However, the extremely rare N syndrome, a multiple congenital anomaly mental retardation syndrome, was suggested to be caused by defect in pol α [173]. More recently, large-scale integrated genomic characterization of specific cancers have found a strong link between mutations in pol ε and sporadic colorectal cancers and endometrial carcinomas [174-177].
Defects of mitochondrial Fe-S cluster biosynthesis impair Fe-S cluster maturation and iron homeostasis and cause several human diseases [30,178], including one of the most frequent inherited ataxias, Friedreich ataxia (FRDA). FRDA is an autosomal recessive neurodegenerative disorder caused by the deficiency of frataxin protein [179] with a prevalence estimated at 1:50,000 individuals or 1:20,000–125,000 in the Caucasian population [180]. Several point mutations found from FRDA patients cause either instability of frataxin folding [181] or incapability of functioning in Fe-S cluster biogenesis [182]. The roles of frataxin in Fe-S cluster biogenesis have been proposed as an iron donor [183,184] and as an allosteric regulator [185-188] for sulfur transfer chemistry in cysteine desulfurase, which serve as a sulfur donor in Fe-S cluster biogenesis. However, increasing frataxin level or iron chelator strategies still cannot cure FRDA. Fe-S clusters are assembled in a scaffold protein ISCU2. Hereditary myopathy with lactic acidosis (HML) has been found to be associated with the deficiency of ISCU2, which lead to the impaired mitochondrial Fe-S cluster maturation. HML is a rare inherited disease with the deficiency of succinate dehydrogenase and aconitase, which both need Fe-S clusters for their function [189,190]. Another human disease sideroblastic anemia has been linked to the deficiency of glutaredoxin 5 (GLRX5), which is implicated for the maintenance of mitochondrial and cytosol iron homeostasis and the Fe-S cluster transfer [191,192]. The deficiency of GLRX5 results the impairment of heme biosynthesis, Fe-S cluster biosynthesis, and iron depletion in cytosol [193]. Other human diseases associated with defects in mitochondrial Fe-S cluster biogenesis have been discussed in recent reviews [30,178]. In the next section, we consider the cytosolic Fe-S cluster maturation machinery that is shared by DNA processing enzymes.
9. DNA processing enzymes share common Fe-S cluster maturation machinery
All of the DNA processing enzymes discussed above share common Fe-S biogenesis systems that assemble and deliver Fe-S clusters to target cytosolic and nuclear apoproteins. The Cytosolic Iron-Sulfur Protein Assembly (CIA) machinery includes a growing list of proteins responsible for Fe-S cluster maturation and targeting. Many of these factors were previously thought to be co-factors in divergent cellular pathways but have been uncovered as CIA operatives. Despite enormous advances and discoveries over the last decade, some factors are still being identified and precise molecular mechanisms for others have yet to be revealed.
The CIA process (Fig. 6) requires the function of the mitochondrial Iron Sulfur Cluster (ISC) assembly machinery that assembles its own Fe-S cluster proteins de novo [194]. An unknown product (S-X) of this pathway is exported by mitochondrial Atm1 and is essential for CIA [195,196]. There are indications Atm1 may export a glutathione (GSH)-coordinated 2Fe2S cluster that is ferried to the cytoplasm by Grx3/Grx4, though S-X remains unidentified [197-199]. It is also possible Atm1 exports the sulfur moiety via a GSH carrier that assembles with labile cytosolic iron on the scaffold of heterotetramer Nbp35/Cfd1 (Fig. 6), the first cytosolic component of CIA [200-203]. Cfd1 and Nbp35 are P-loop NTPases that hold up to four [4Fe-4S] clusters, a pair bridging homo and/or heterodimers and one on each N-termini of Nbp35 [202]. Binding and/or hydrolysis of nucleotide as well as an electron transfer from NADPH/Tah18/Dre2 to the Nbp35/Cfd1 complex is required for the Fe-S assembly in vivo [204,205]. The electron from Dre2 could be used to reduce the GSH carrier to its free form to facilitate delivery of S-X to Nbp35/Cfd1. The structural mechanism of this process remains unclear as in vitro experiments show that both Cdf1 and Nbp35 can individually coordinate and transfer clusters to apoproteins in the presence of free iron and sulfur without nucleotide binding or complex formation [203,206]. Plants and bacteria do not have Cfd1 but instead bind four [4Fe-4S] clusters as a homodimer, while Cdf1 in non-photosynthetic eukaryotes has been shown in vivo and in vitro to increase liability and transfer of Fe-S clusters to target apoproteins [206,207].
Figure 6. DNA processing enzymes share common Fe-S cluster assembly machinery.

A model for the Cytosolic Iron-Sulfur Cluster Assembly (CIA) in three steps. 1) The early steps of the mitochondrial Iron-Sulfur Cluster (ISC) machinery are essential for CIA. Many required proteins such as the cysteine desulfurase complex Nfs1-Isd11 and the Isu1 scaffold are not shown. An unknown sulfur-containing compound (X-S) is exported to the cytosol via ATM1, S-X potentially being a glutathione (GSH) coordinated [2Fe-2S] cluster. 2) X-S is transported to the Cfd1-Nbp35 scaffold complex that assembles the cytosolic [4Fe-4S] clusters. Monothiol glutaredoxins Grx3-Grx4 can transiently bind a [2Fe-2S] cluster and may help shuttle S-X to Cfd1-Nbp35. NTPase activity and electron transfer from Tah18-Dre2 is required for assembly in vivo. 3) IOP1 serves as a bridge between the scaffold and the CIA targeting complex of CIA1, CIA2B and MMS19. The targeting complex has been shown to have a long list of interactions with proteins known to have Fe-S clusters. The complex has not yet been shown to hold an Fe-S cluster, but may instead facilitate cluster handoff between IOP1 and target apoproteins.
The recipient for Nbp35/Cfd1 Fe-S cluster delivery is Nar1 in yeast and IOP1 in human [208-210] (Fig. 6). Nar1-IOP1 may serve as an adapter, transiently associating with Nbp35/Cfd1, binding a [4Fe-4S] cluster and passing it onto the ‘CIA targeting complex’ consisting of CIA1, CIA2 and MMS19 [208]. CIA2B (FAM96B, MIP18) is the human homolog of yeast CIA2, with CIA2A (FAM96A) being a human paralog absent in yeast yet involved in cellular iron regulation via its interaction with Iron Response Protein 2 (IRP2) [211]. CIA1, CIA2-CIA2B and MMS19 have not yet been shown to hold a Fe-S cluster; it’s possible that this heterotrimer serves as a scaffold for apoproteins that in turn associates with Nar1-IOP1 to deliver one of its two Fe-S clusters [212].
The core targeting complex consists of three proteins, CIA1, CIA2B and MMS19, that interacts with upstream Nar1-IOP1 adapter and downstream end targets [211,213]. The structure of CIA1 solved to 1.7 A resolution is a seven bladed WD40 repeat that forms a circular platform with ample space for protein partner docking, which would be consistent with the WD40 protein family [214]. The paralog of CIA2B, CIA2A, has three high-resolution structures in addition to three structures of related DUF59 family proteins [213,215,216]. CIA2A forms two crystallographic domain-swapped dimers that share characteristics with amyloidogenic proteins [213]. MMS19 is a large HEAT-repeat protein that is evolutionarily variable in length, sequence and number of HEAT-repeats [62,208,217,218]. Knocking down CIA1 or CIA2B results in reduced levels of the other, however MMS19 levels are not dependent on the other two, and MMS19 is the only CIA member that is not required for viability in yeast [62,208]. MMS19 deletion mutants do result in a variety of phenotypes that are likely to relate to a Fe-S cluster deficiency in a key protein that requires MMS19 for Fe-S delivery.
Precise molecular mechanisms and functions of these proteins and how together they facilitate delivery of [4Fe-4S] clusters to target apoproteins have yet to be revealed. Structures of these proteins together or in complex with upstream partner Nar1-IOP1 or downstream targets like XPD or Dna2 would shed needed light on the mechanism of the CIA targeting complex. Interaction mapping of CIA components would also be useful to determine what mutations maintain complex stability but inhibit interactions with target proteins. Identification of additional Fe-S biogenesis factors and further characterization of targeting complexes may aid the production of recombinant Fe-S proteins in heterologous expression systems. Overexpression of Fe-S proteins may overwhelm endogenous Fe-S biogenesis systems, requiring the co-expression of Fe-S assembly machinery components, deletion of ISC regulatory genes, or iron and cysteine supplementation. For example, deletion of an ISC regulator, IscR, in E. coli, enhanced the activities of heterologous [FeFe] hydrogenases up to 100-fold when combined with both iron and cysteine supplementation [219].
10. Hypothesis for communication between Fe-S cluster enzymes on DNA
If we step back from the protein level and look at these Fe-S cluster enzymes that act on DNA as a whole, a picture emerges of these enzymes all working to maintain or faithfully replicate the genome (Fig. 1). Disrupted DNA duplex structure is the substrate or product for all of these enzymes—from glycosylases that act on damaged bases to helicases that separate the DNA helix to polymerases that act at the interface of single and double stranded DNA. Furthermore, the DNA metabolic pathways in which these enzymes function are highly coordinated to prevent the formation of toxic intermediates. Since Fe-S clusters are the common structural feature among these enzymes that act on disrupted DNA substrates, we propose that Fe-S redox clusters provide a unique mechanism for key enzymes to rapidly interrogate DNA integrity and coordinate their activities by sending and receiving electrons that travel through the pi-stack of DNA [69]; a process we call DNA charge transfer communication (DNA CTC). Our DNA CTC hypothesis builds on the insightful DNA CT model proposed by Barton and her laboratory for DNA damage detection for glycosylases (Fig. 7A) and extends it to propose that DNA CT changes Fe-S cluster oxidation states in order to alter the conformations, interactions, and biochemical activities of their respective DNA-bound enzymes in ways that may orchestrate replication and repair steps.
Protein-protein interaction, fluorescently labeled protein cellular dynamics, and post-translational modification studies have shown that genome maintenance and propagation require the choreography of a complex and dynamic dance of multiple machineries. At present there are no known mechanisms that can fully explain how the action of different proteins in multi-protein genome maintenance machines, and the communication between these machines, is coordinated. The traditional mechanisms for protein communication, via direct protein-protein contacts and post-translational modifications, seem slow compared to the microsecond time scale required for ongoing processing of DNA. Moreover, these mechanisms do not provide a means for long-range communication, which appears to be required for the correct progression of biochemical activities provided by the participating proteins. Our DNA CTC hypothesis aligns the unknown role of Fe-S clusters with the unknown orchestration mechanism to explain communication between key DNA processing proteins at very high speeds and over long distances. DNA CTC provides an overarching mechanism for DNA-mediated communication between different proteins in multi-protein machines and across different machines in multiple pathways. The key feature is that communication through DNA can occur across long and short distances, independently of direct protein-protein interaction and chromosome structure. So the DNA CTC model provides a possible paradigm shift from traditional ideas about interactomes as having direct or linked protein interfaces rather than being potentially linked by communication through DNA.
10.1. Coordination of DNA synthesis during DNA replication and repair
DNA synthesis requires the interplay of several different polymerases, yet the mechanism for coordinating polymerase exchange even at a single replication fork remains unknown, particularly in eukaryotic cells. The pol α – primase complex (pol-prim) initiates synthesis by laying down an RNA-DNA primer. Two replicative polymerases, DNA pols ε and δ, synthesize the leading and lagging strands respectively. Specialized polymerases, such as DNA pol ζ, allow replication past DNA damage by translesion synthesis (TLS). DNA replication therefore requires efficient polymerase switching at the primer terminus. Current models of polymerase switching rely on post-translational modifications, but these likely act only in the subset of slow polymerase switching events that occur after DNA damage [220-222]. Such post-translational modifications result in altered conformation and interaction sites on protein partners, such as seen in the conformations of covalently bound ubiquitin on PCNA [223]. Most switching occurs during genomic replication, when primase hands off to pol α, and pol α in turn to pol δ or ∈. This switching occurs at each origin to initiate leading strand synthesis and millions of times during lagging strand synthesis. With the discoveries of Fe-S clusters in primase and pols α, δ, ∈, and ζ [12,150], DNA CTC provides an efficient signaling mechanism for polymerase switching during DNA replication (Fig. 7B) that does not require the steps of protein interaction and covalent modification. A primer of 40 nucleotides is required for pol ε to commit to its DNA substrate [146], and the newly synthesized primer may act as a conduit for DNA CTC between pols α and ε, allowing for efficient polymerase switching. Alternatively, positioning the Fe-S cluster in the flexible pol ε CTD [146] near the DNA polyanion may shift its redox potential and cause a conformational change that positions the globular catalytic domain to activate processive DNA synthesis. We know for example that ATP binding and hydrolysis can control the conformation of complexes and thereby biological outcomes, as seen for the RAD50 ABC ATPase [224,225]. In comparison, we have relatively little appreciation of the great ability of Fe-S clusters to effect such regulation of complexes as mediated by DNA.
More broadly, DNA CTC has the potential to explain the coordination of origin firing across the genome. For example, both the spatial and temporal orders of replication origin firing seem to correspond to the linear genome order. Current proposals suggest that this “replication wave” may propagate through changes in chromatin structure, i.e. replication causes a local chromatin destabilization, so neighboring regions become more accessible for initiation [226]. In our DNA CTC hypothesis, key replication components (e.g. replicative polymerases and the Dna2 helicase) contain redox centers, which may allow one replication fork complex to probe DNA integrity and communicate to the next fork complex along the linear order of the genome (Fig. 7C). Notably, DNA CTC proceeds effectively through duplex DNA with and without bound histones [75]. Thus, the DNA CTC mechanism is rapid, independent of nucleosomes [75], provides a mechanism for the observed linear order, and may explain observed changes in replication dynamics during development or cell differentiation.
Protein communication through DNA CTC also provides an additional level of regulation during excision repair to prevent the release of toxic intermediates. Bulky DNA lesions are repaired by NER, in which ~30 nucleotides containing the damage is excised and new DNA is synthesized. Although defects in the NER machinery are directly association with human disease, including Xeroderma pigmentosum (XP) and other disorders, the precise mechanism for coordinating excision and synthesis are not known. The textbook model of NER depicts that excision of the damaged strand occurs before DNA synthesis. However, the observation that incision on only one side of the lesion (5′ incision by ERCC1-XPF) is sufficient to initiate DNA synthesis suggests a new NER model in which DNA synthesis is initiated before excision is complete [227]. Since the XPG endonuclease that makes the second incision is closely associated with the XPD helicase [228] that contains a Fe-S cluster, we propose that DNA CTC from the polymerase to XPD (Fig. 7D) may help coordinate synthesis with XPG incision. Furthermore, the completion of DNA synthesis may signal for the release of XPD/TFIIH through the reduction of the XPD Fe-S cluster by the DNA polymerase. In yeast XPD, the rad3-102 mutant blocks post-incision events due to defective release of TFIIH at the sites of damage and leads to replication fork breakage [229]. Therefore, controlling the efficient release of XPD and TFIIH may well be as important as coordinating incision events during NER.
11. Synopsis and perspectives
Building upon the results discussed here and aided by ongoing structural, biochemical, and genetic studies, we can expect major new emerging insights on Fe-S clusters and their activities in cell biology of DNA replication, repair and transcription. In particular, as DNA CT provides an exquisitely sensitive mechanism to detect disrupted double-stranded DNA structure over long distances, such as occurs in most replication and repair intermediates, it is likely that these DNA CT mechanisms will be under increasing study both in vitro and in vivo. Cas9 and other tools for gene targeting make the testing of Fe-S roles in human cells practical [230-232]. Furthermore, biophysical methods such as SAXS, atomic force microscopy, fluorescent energy transfer, and electron microscopy are enabling the characterization of solution architectures, assemblies and conformations [233,234] that will help address the roles of Fe-S in conformational states and charge transfer in cells. SAXS can be powerfully combined with x-ray crystallography [235] and advanced SAXS methods, which define shape, flexibility, and agreement to atomic models [236-238] have, for example, allowed the characterization of multicomponent Fe-S proteins and membrane complexes [239], as well as conformation change for proteins acting in DNA complexes [224,240]. Collective X-ray scattering results suggest conformational variation is a general functional feature of macromolecules [241], but we know too little about the roles of Fe-S clusters in conformational changes of DNA bound complexes.
As Fe-S clusters occur across the domains of life, comparative structures and biochemistry from archaeal hyperthermophiles will likely continue to be useful for the general understanding of Fe-S functions in DNA replication and repair proteins [242]. Furthermore, as proteins can mimic specific target DNA structures [243], another interesting issue will be whether protein mimics of DNA may help regulate the activities of Fe-S cluster enzymes in DNA replication and repair. The determination of metalloprotein structures can provide an informed basis for their design [244] and transfer to other frameworks to test an understanding of their functions [245,246], and this would provide a possible means to test the role of Fe-S in DNA CTC in vitro and in vivo. Possible chemical inhibitors of DNA CTC might include redox active cage metal complexes, but they would have to bind DNA and act in the low nanomolar range to avoid non-specific pleiotropic effects. Yet, such complexes could potentially form potent metal-based inhibitors for DNA CTC functions. Gold nanoparticles, such as used in ultrasensitive SAXS studies on protein-DNA complexes [247] and in cancer medicine [248], might be harnessed to alter change transfer in DNA while providing a markers for visualization. Furthermore, conducting atomic force microscopy coupled with G-quadruplex DNA wires [249,250] could aid study of Fe-S cluster proteins in DNA replication and repair.
Overall, Fe-S clusters in DNA replication and repair are not simply structural features and remnants from early evolution. Fe-S clusters seem likely to act in the coordination of replication and repair events either by local conformational changes and direct interactions or by longer-range and DNA CTC or both. Whatever the case, ongoing structural and biophysical elucidations will take our understanding of nanoscale DNA assemblies and their control of the energetics of charge transfer and conformational chemistry to the next level of the molecular circuitry coordinating DNA replication and repair. This knowledge may directly integrate our understanding of interaction and signaling networks for DNA replication and repair events. More generally, a deeper knowledge of the critical roles for Fe-S clusters in DNA replication and repair enzymes is fundamental to cell biology and medicine in solving a great mystery of the DNA enzymes critical to life.
Supplementary Material
Highlights.
A growing list of DNA processing enzymes contain Fe-S clusters despite risks to DNA
Fe-S prediction remains elusive and Fe-S confirmation relies on empirical methods
Fe-S cluster enzymes have the unique ability to transfer electrons even via DNA
That Fe-S clusters play key roles in DNA processing enzymes is of growing interest
DNA Charge Transfer between Fe-S enzymes merits study of its biological importance
Acknowledgements
We thank our pioneering colleagues in the area of Fe-S cluster functions in DNA replication and repair for many discussions and insights, especially Jacqueline Barton, Peter Burgers, Judith Campbell, Walter Chazin, and Stephen Kowalczykowski along with Dale Wigley, Sheila David, and Richard Cunningham. We also thank Fe-S experts Brian Crane, Douglas Rees, Lou Noodleman, David Case, David Barondeau, Harry Gray, Elizabeth Getzoff, David Stout, and Michael Adams for sharing their thoughts over the years. Stuart Linn and Irwin Fridovich contributed to our thoughts on iron-mediated Fenton reactions in the presence of DNA. Steven Yannone provided critical edits of this manuscript and contributed to many rich discussions about Fe-S proteins over the years. Analyses of Fe-S clusters relevant to XPD in the authors’ laboratory are funded by the National Institutes of Health (R01 CA112093).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Fontecave M. Iron-sulfur clusters: ever-expanding roles. Nat Chem Biol. 2006;2:171–174. doi: 10.1038/nchembio0406-171. doi:10.1038/nchembio0406-171. [DOI] [PubMed] [Google Scholar]
- [2].Imlay JA. Iron-sulphur clusters and the problem with oxygen. Mol Microbiol. 2006;59:1073–1082. doi: 10.1111/j.1365-2958.2006.05028.x. doi:10.1111/j.1365-2958.2006.05028.x. [DOI] [PubMed] [Google Scholar]
- [3].Koonin EV, Martin W. On the origin of genomes and cells within inorganic compartments. Trends in Genetics. 2005;21:647–654. doi: 10.1016/j.tig.2005.09.006. doi:10.1016/j.tig.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Russell MJ, Martin W. The rocky roots of the acetyl-CoA pathway. Trends Biochem Sci. 2004;29:358–363. doi: 10.1016/j.tibs.2004.05.007. doi:10.1016/j.tibs.2004.05.007. [DOI] [PubMed] [Google Scholar]
- [5].Imlay JA. Pathways of oxidative damage. Annu. Rev. Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. doi:10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- [6].Imlay JA, Linn S. DNA damage and oxygen radical toxicity. Science. 1988;240:1302–1309. doi: 10.1126/science.3287616. [DOI] [PubMed] [Google Scholar]
- [7].Iuchi S. Three classes of C2H2 zinc finger proteins. Cell Mol Life Sci. 2001;58:625–635. doi: 10.1007/PL00000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen B, et al. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002;418:562–566. doi: 10.1038/nature00922. doi:10.1038/nature00922. [DOI] [PubMed] [Google Scholar]
- [9].Tsutakawa SE, Shin DS, Mol CD, Izumi T, Arvai AS, Mantha AK, et al. Conserved Structural Chemistry for Incision Activity in Structurally Non-homologous Apurinic/Apyrimidinic Endonuclease APE1 and Endonuclease IV DNA Repair Enzymes. Journal of Biological Chemistry. 2013;288:8445–8455. doi: 10.1074/jbc.M112.422774. doi:10.1074/jbc.M112.422774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rudolf J, Makrantoni V, Ingledew WJ, Stark MJR, White MF. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. 2006. doi:10.1016/j.molcel.2006.07.019. [DOI] [PubMed]
- [11].Pokharel S, Campbell JL. Cross talk between the nuclease and helicase activities of Dna2: role of an essential iron-sulfur cluster domain. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks534. doi:10.1093/nar/gks534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Netz DJA, Stith CM, Stümpfig M, Köpf G, Vogel D, Genau HM, et al. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat Chem Biol. 2011;8:125–132. doi: 10.1038/nchembio.721. doi:10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cvetkovic A, Menon AL, Thorgersen MP, Scott JW, Poole FL, II, Jenney FE, Jr, et al. Microbial metalloproteomes are largely uncharacterized. Nature. 2010;466:779–782. doi: 10.1038/nature09265. doi:10.1038/nature09265. [DOI] [PubMed] [Google Scholar]
- [14].Service RF. Live wire. Science. 2014;346:1284–1287. doi: 10.1126/science.346.6215.1284. doi:10.1126/science.346.6215.1284. [DOI] [PubMed] [Google Scholar]
- [15].Roberts VA, Freeman HC, Olson AJ, Tainer JA, Getzoff ED. Electrostatic orientation of the electron-transfer complex between plastocyanin and cytochrome c. J Biol Chem. 1991;266:13431–13441. [PubMed] [Google Scholar]
- [16].Getzoff ED, Tainer JA, Weiner PK, Kollman PA, Richardson JS, Richardson DC. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature. 1983;306:287–290. doi: 10.1038/306287a0. [DOI] [PubMed] [Google Scholar]
- [17].Getzoff ED, Cabelli DE, Fisher CL, Parge HE, Viezzoli MS, Banci L, et al. Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature. 1992;358:347–351. doi: 10.1038/358347a0. doi:10.1038/358347a0. [DOI] [PubMed] [Google Scholar]
- [18].Jortner J, Bixon M, Heitele H, Michelbeyerle ME. Long-Range Electron-Transfer in Solvent-Free Supermolecules. Chemical Physics Letters. 1992;197:131–135. [Google Scholar]
- [19].Jortner J, Bixon M, Langenbacher T, Michel-Beyerle ME. Charge transfer and transport in DNA. Proc Natl Acad Sci USA. 1998;95:12759–12765. doi: 10.1073/pnas.95.22.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fujitsuka M, Majima T. Hole and excess electron transfer dynamics in DNA. Phys. Chem. Chem. Phys. 2012;14:11234. doi: 10.1039/c2cp41576c. doi:10.1039/c2cp41576c. [DOI] [PubMed] [Google Scholar]
- [21].Sundheim O, Vagbo CB, Bjørås M, Sousa MML, Talstad V, Aas PA, et al. Human ABH3 structure and key residues for oxidative demethylation to reverse DNA/RNA damage. Embo J. 2006;25:3389–3397. doi: 10.1038/sj.emboj.7601219. doi:10.1038/sj.emboj.7601219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Johnson DC, Dean DR, Smith AD, Johnson MK. Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. doi:10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- [23].Bak DW, Elliott SJ. Alternative FeS cluster ligands: tuning redox potentials and chemistry. Current Opinion in Chemical Biology. 2014;19:50–58. doi: 10.1016/j.cbpa.2013.12.015. doi:10.1016/j.cbpa.2013.12.015. [DOI] [PubMed] [Google Scholar]
- [24].Yoo SJ, Angove HC, Burgess BK, Hendrich MP, Münck E. Mössbauer and Integer-Spin EPR Studies and Spin-Coupling Analysis of the [4Fe-4S] Cluster of the Fe Protein from Azotobacter vinelandii Nitrogenase. J Am Chem Soc. 1999;121:2534–2545. doi:10.1021/ja9837405. [Google Scholar]
- [25].Meyer TE, Przysiecki CT, Watkins JA, Bhattacharyya A, Simondsen RP, Cusanovich MA, et al. Correlation between rate constant for reduction and redox potential as a basis for systematic investigation of reaction mechanisms of electron transfer proteins. Proc Natl Acad Sci USA. 1983;80:6740–6744. doi: 10.1073/pnas.80.22.6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dey A, Jenney FE, Adams MWW, Babini E, Takahashi Y, Fukuyama K, et al. Solvent tuning of electrochemical potentials in the active sites of HiPIP versus ferredoxin. Science. 2007;318:1464–1468. doi: 10.1126/science.1147753. doi:10.1126/science.1147753. [DOI] [PubMed] [Google Scholar]
- [27].Ryle MJ, Lanzilotta WN, Seefeldt LC. Elucidating the mechanism of nucleotide-dependent changes in the redox potential of the [4Fe-4S] cluster in nitrogenase iron protein: the role of phenylalanine 135. Biochemistry. 1996;35:9424–9434. doi: 10.1021/bi9608572. doi:10.1021/bi9608572. [DOI] [PubMed] [Google Scholar]
- [28].Rothery RA, Bertero MG, Spreter T, Bouromand N, Strynadka NCJ, Weiner JH. Protein Crystallography Reveals a Role for the FS0 Cluster of Escherichia coli Nitrate Reductase A (NarGHI) in Enzyme Maturation. Journal of Biological Chemistry. 2010;285:8801–8807. doi: 10.1074/jbc.M109.066027. doi:10.1074/jbc.M109.066027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bandyopadhyay S, Chandramouli K, Johnson MK. Iron–sulfur cluster biosynthesis. Biochem. Soc. Trans. 2008;36:1112. doi: 10.1042/BST0361112. doi:10.1042/BST0361112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rouault TA, Tong WH. Iron–sulfur cluster biogenesis and human disease. Trends in Genetics. 2008;24:398–407. doi: 10.1016/j.tig.2008.05.008. doi:10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Perry JJP, Shin DS, Getzoff ED, Tainer JA. The structural biochemistry of the superoxide dismutases. BBA - Proteins and Proteomics. 2010;1804:245–262. doi: 10.1016/j.bbapap.2009.11.004. doi:10.1016/j.bbapap.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, et al. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- [33].Yoneda M, Bollum FJ. Deoxynucleotide-polymerizing enzymes of calf thymus gland. I. Large scale purification of terminal and replicative deoxynucleotidyl transferases. J Biol Chem. 1965;240:3385–3391. [PubMed] [Google Scholar]
- [34].Weiner BE, Huang H, Dattilo BM, Nilges MJ, Fanning E, Chazin WJ. An iron-sulfur cluster in the C-terminal domain of the p58 subunit of human DNA primase. J Biol Chem. 2007;282:33444–33451. doi: 10.1074/jbc.M705826200. doi:10.1074/jbc.M705826200. [DOI] [PubMed] [Google Scholar]
- [35].Cunningham RP, Asahara H, Bank JF, Scholes CP, Salerno JC, Surerus K, et al. Endonuclease III is an iron-sulfur protein. Biochemistry. 1989;28:4450–4455. doi: 10.1021/bi00436a049. [DOI] [PubMed] [Google Scholar]
- [36].Yannone SM, Hartung S, Menon AL, Adams MW, Tainer JA. Metals in biology: defining metalloproteomes. Current Opinion in Biotechnology. 2012;23:88–94. doi: 10.1016/j.copbio.2011.11.005. doi:10.1016/j.copbio.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mullenbach GT, Tabrizi A, Irvine BD, Bell GI, Tainer JA, Hallewell RA. Selenocysteine’s mechanism of incorporation and evolution revealed in cDNAs of three glutathione peroxidases. Protein Eng. 1988;2:239–246. doi: 10.1093/protein/2.3.239. [DOI] [PubMed] [Google Scholar]
- [38].O’Halloran TV, Culotta VC. Metallochaperones, an Intracellular Shuttle Service for Metal Ions. Journal of Biological Chemistry. 2000;275:25057–25060. doi: 10.1074/jbc.R000006200. doi:10.1074/jbc.R000006200. [DOI] [PubMed] [Google Scholar]
- [39].McMurray CT, Tainer JA. Cancer, cadmium and genome integrity. Nat Genet. 2003;34:239–241. doi: 10.1038/ng0703-239. doi:10.1038/ng0703-239. [DOI] [PubMed] [Google Scholar]
- [40].Kocabas E, Hernick M. Metalloenzymes: Use of Recombinant Protein Expression and Affinity Tags to Aid Identification of Native Metal Ion Cofactors. Biochem Anal Biochem. 2013;2:2161–1009. 1000132. [Google Scholar]
- [41].Daniels DS, Mol CD, Arvai AS, Kanugula S, Pegg AE, Tainer JA. Active and alkylated human AGT structures: a novel zinc site, inhibitor and extrahelical base binding. Embo J. 2000;19:1719–1730. doi: 10.1093/emboj/19.7.1719. doi:10.1093/emboj/19.7.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Crane BR, Rosenfeld RJ, Arvai AS, Ghosh DK, Ghosh S, Tainer JA, et al. N-terminal domain swapping and metal ion binding in nitric oxide synthase dimerization. Embo J. 1999;18:6271–6281. doi: 10.1093/emboj/18.22.6271. doi:10.1093/emboj/18.22.6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Analytical Biochemistry. 2004;331:370–375. doi: 10.1016/j.ab.2004.03.049. doi:10.1016/j.ab.2004.03.049. [DOI] [PubMed] [Google Scholar]
- [44].Paraskevopoulou C, Fairhurst SA, Lowe DJ, Brick P, Onesti S. The Elongator subunit Elp3 contains a Fe4S4 cluster and binds S-adenosylmethionine. Mol Microbiol. 2006;59:795–806. doi: 10.1111/j.1365-2958.2005.04989.x. doi:10.1111/j.1365-2958.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- [45].Pugh RA, Honda M, Leesley H, Thomas A, Lin Y, Nilges MJ, et al. The iron-containing domain is essential in Rad3 helicases for coupling of ATP hydrolysis to DNA translocation and for targeting the helicase to the single-stranded DNA-double-stranded DNA junction. J Biol Chem. 2008;283:1732–1743. doi: 10.1074/jbc.M707064200. doi:10.1074/jbc.M707064200. [DOI] [PubMed] [Google Scholar]
- [46].Wu Y, Sommers JA, Suhasini AN, Leonard T, Deakyne JS, Mazin AV, et al. Fanconi anemia group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood. 2010;116:3780–3791. doi: 10.1182/blood-2009-11-256016. doi:10.1182/blood-2009-11-256016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dailey HA, Finnegan MG, Johnson MK. Human ferrochelatase is an iron-sulfur protein. Biochemistry. 1994;33:403–407. doi: 10.1021/bi00168a003. [DOI] [PubMed] [Google Scholar]
- [48].Aono S, Bryant FO, Adams MW. A novel and remarkably thermostable ferredoxin from the hyperthermophilic archaebacterium Pyrococcus furiosus. J. Bacteriol. 1989;171:3433–3439. doi: 10.1128/jb.171.6.3433-3439.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mayhew SG. The redox potential of dithionite and SO-2 from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur. J. Biochem. 1978;85:535–547. doi: 10.1111/j.1432-1033.1978.tb12269.x. [DOI] [PubMed] [Google Scholar]
- [50].Crane BR, Siegel LM, Getzoff ED. Structures of the siroheme- and Fe4S4-containing active center of sulfite reductase in different states of oxidation: heme activation via reduction-gated exogenous ligand exchange. Biochemistry. 1997;36:12101–12119. doi: 10.1021/bi971065q. doi:10.1021/bi971065q. [DOI] [PubMed] [Google Scholar]
- [51].Munck E, Kent TA. Structure and Magnetism of Iron-Sulfur Clusters in Proteins. Hyperfine Interactions. 1986;27:161–172. [Google Scholar]
- [52].Angove HC, Yoo SJ, Burgess BK, Munck E. Mossbauer and EPR evidence for an all-ferrous Fe4S4 cluster with S = 4 in the Fe protein of nitrogenase. J Am Chem Soc. 1997;119:8730–8731. [Google Scholar]
- [53].Middleton P, Dickson DPE, Johnson CE, Rush JD. Interpretation of Mossbauer-Spectra of 4-Iron Ferredoxin From Bacillus-Stearothermophilus. Eur. J. Biochem. 1978;88:135–141. doi: 10.1111/j.1432-1033.1978.tb12430.x. [DOI] [PubMed] [Google Scholar]
- [54].Berto P, Di Valentin M, Cendron L, Vallese F, Albertini M, Salvadori E, et al. Biochimica et Biophysica Acta. BBA - Bioenergetics. 2012;1817:2149–2157. doi: 10.1016/j.bbabio.2012.09.004. doi:10.1016/j.bbabio.2012.09.004. [DOI] [PubMed] [Google Scholar]
- [55].Bridwell-Rabb J, Fox NG, Tsai C-L, Winn AM, Barondeau DP. Human Frataxin Activates Fe–S Cluster Biosynthesis by Facilitating Sulfur Transfer Chemistry. Biochemistry. 2014;53:4904–4913. doi: 10.1021/bi500532e. doi:10.1021/bi500532e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Johnson MK, Morningstar JE, Bennett DE, Ackrell BAC, Kearney EB. Magnetic Circular-Dichroism Studies of Succinate-Dehydrogenase - Evidence for [2fe-2s], [3fe-Xs], and [4fe-4s] Centers in Reconstitutively Active Enzyme. J Biol Chem. 1985;260:7368–7378. [PubMed] [Google Scholar]
- [57].Fu W, O’Handley S, Cunningham RP, Johnson MK. The role of the iron-sulfur cluster in Escherichia coli endonuclease III. A resonance Raman study. J Biol Chem. 1992;267:16135–16137. [PubMed] [Google Scholar]
- [58].Mitra D, George SJ, Guo Y, Kamali S, Keable S, Peters JW, et al. Characterization of [4Fe-4S] Cluster Vibrations and Structure in Nitrogenase Fe Protein at Three Oxidation Levels via Combined NRVS, EXAFS, and DFT Analyses. J Am Chem Soc. 2013;135:2530–2543. doi: 10.1021/ja307027n. doi:10.1021/ja307027n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chanet R, Heude M. Characterization of mutations that are synthetic lethal with pol3-13 , a mutated allele of DNA polymerase delta in Saccharomyces cerevisiae. Current Genetics. 2003;43:337–350. doi: 10.1007/s00294-003-0407-2. doi:10.1007/s00294-003-0407-2. [DOI] [PubMed] [Google Scholar]
- [60].Lauder S, Bankmann M, Guzder SN, Sung P, Prakash L, Prakash S. Dual requirement for the yeast MMS19 gene in DNA repair and RNA polymerase II transcription. Mol Cell Biol. 1996;16:6783–6793. doi: 10.1128/mcb.16.12.6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kou H, Zhou Y, Gorospe RMC, Wang Z. Mms19 protein functions in nucleotide excision repair by sustaining an adequate cellular concentration of the TFIIH component Rad3. Proc Natl Acad Sci USA. 2008;105:15714–15719. doi: 10.1073/pnas.0710736105. doi:10.1073/pnas.0710736105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gari K, Leon Ortiz AM, Borel V, Flynn H, Skehel JM, Boulton SJ. MMS19 Links Cytoplasmic Iron-Sulfur Cluster Assembly to DNA Metabolism. Science. 2012;337:243–245. doi: 10.1126/science.1219664. doi:10.1126/science.1219664. [DOI] [PubMed] [Google Scholar]
- [63].O’Reilly JE. Oxidation-reduction potential of the ferro-ferricyanide system in buffer solutions. Biochim. Biophys. Acta. 1973;292:509–515. doi: 10.1016/0005-2728(73)90001-7. [DOI] [PubMed] [Google Scholar]
- [64].Dutton PL. Redox potentiometry: determination of midpoint potentials of oxidation-reduction components of biological electron-transfer systems. Meth. Enzymol. 1978;54:411–435. doi: 10.1016/s0076-6879(78)54026-3. [DOI] [PubMed] [Google Scholar]
- [65].Heinze J. Cyclic Voltammetry—“Electrochemical Spectroscopy.” New Analytical Methods (25) Angewandte Chemie International Edition in English. 1984;23:831–847. [Google Scholar]
- [66].Hudson JM, Heffron K, Kotlyar V, Sher Y, Maklashina E, Cecchini G, et al. Electron Transfer and Catalytic Control by the Iron–Sulfur Clusters in a Respiratory Enzyme, E.coliFumarate Reductase. J Am Chem Soc. 2005;127:6977–6989. doi: 10.1021/ja043404q. doi:10.1021/ja043404q. [DOI] [PubMed] [Google Scholar]
- [67].Kelley SO, Boon EM, Barton JK, Jackson NM, Hill MG. Single-base mismatch detection based on charge transduction through DNA. Nucleic Acids Res. 1999;27:4830–4837. doi: 10.1093/nar/27.24.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gorodetsky AA, Barton JK. Electrochemistry Using Self-Assembled DNA Monolayers on Highly Oriented Pyrolytic Graphite. Langmuir. 2006;22:7917–7922. doi: 10.1021/la0611054. doi:10.1021/la0611054. [DOI] [PubMed] [Google Scholar]
- [69].Sontz PA, Mui TP, Fuss JO, Tainer JA, Barton JK. DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proc Natl Acad Sci USA. 2012;109:1856–1861. doi: 10.1073/pnas.1120063109. doi:10.1073/pnas.1120063109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sontz PA, Muren NB, Barton JK. DNA Charge Transport for Sensing and Signaling. Acc. Chem. Res. 2012;45:1792–1800. doi: 10.1021/ar3001298. doi:10.1021/ar3001298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Castagnetto JM, Hennessy SW, Roberts VA, Getzoff ED, Tainer JA, Pique ME. MDB: The metalloprotein database and browser at the scripps research institute. Nucleic Acids Res. 2002;30:379–382. doi: 10.1093/nar/30.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Boal AK, Genereux JC, Sontz PA, Gralnick JA, Newman DK, Barton JK. Redox signaling between DNA repair proteins for efficient lesion detection. Proc Natl Acad Sci USA. 2009;106:15237–15242. doi: 10.1073/pnas.0908059106. doi:10.1073/pnas.0908059106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Slinker JD, Muren NB, Renfrew SE, Barton JK. DNA charge transport over 34 nm. Nature Chemistry. 2011;3:230–235. doi: 10.1038/nchem.982. doi:10.1038/nchem.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Winkler JR, Gray HB. Long-Range Electron Tunneling. J Am Chem Soc. 2014;136:2930–2939. doi: 10.1021/ja500215j. doi:10.1021/ja500215j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Núñez ME, Noyes KT, Barton JK. Oxidative charge transport through DNA in nucleosome core particles. Chem. Biol. 2002;9:403–415. doi: 10.1016/s1074-5521(02)00121-7. [DOI] [PubMed] [Google Scholar]
- [76].Hall DB, Holmlin RE, Barton JK. Oxidative DNA damage through long-range electron transfer. Nature. 1996;382:731–735. doi: 10.1038/382731a0. doi:10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- [77].Grodick MA, Segal HM, Zwang TJ, Barton JK. DNA-Mediated Signaling by Proteins with 4Fe–4S Clusters Is Necessary for Genomic Integrity. J Am Chem Soc. 2014;136:6470–6478. doi: 10.1021/ja501973c. doi:10.1021/ja501973c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kelley SO, Barton JK. Electron transfer between bases in double helical DNA. Science. 1999;283:375–381. doi: 10.1126/science.283.5400.375. [DOI] [PubMed] [Google Scholar]
- [79].Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Bilio AJ, Sudhamsu J, et al. Tryptophan-Accelerated Electron Flow Through Proteins. Science. 2008;320:1760–1762. doi: 10.1126/science.1158241. doi:10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- [80].Fromme JC, Verdine GL. Structure of a trapped endonuclease III-DNA covalent intermediate. Embo J. 2003;22:3461–3471. doi: 10.1093/emboj/cdg311. doi:10.1093/emboj/cdg311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].McRee DE, Redford SM, Getzoff ED, Lepock JR, Hallewell RA, Tainer JA. Changes in crystallographic structure and thermostability of a Cu,Zn superoxide dismutase mutant resulting from the removal of a buried cysteine. J Biol Chem. 1990;265:14234–14241. doi: 10.2210/pdb3sod/pdb. [DOI] [PubMed] [Google Scholar]
- [82].Parge HE, Hallewell RA, Tainer JA. Atomic structures of wild-type and thermostable mutant recombinant human Cu,Zn superoxide dismutase. Proc Natl Acad Sci USA. 1992;89:6109–6113. doi: 10.1073/pnas.89.13.6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Krokan HE, Bjoras M. Base Excision Repair. Cold Spring Harbor Perspectives in Biology. 2013;5:a012583–a012583. doi: 10.1101/cshperspect.a012583. doi:10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: Implications for DNA damage recognition, removal, and repair. DNA Repair (Amst) 2007;6:410–428. doi: 10.1016/j.dnarep.2006.10.004. doi:10.1016/j.dnarep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- [85].Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: New twists and grooves. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. doi:10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- [86].Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. doi:10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- [87].Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. doi:10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- [88].Dalhus BR, Laerdahl JK, Backe PH, Bjà rà s M. DNA base repair â❟❟ recognition and initiation of catalysis. FEMS Microbiology Reviews. 2009;33:1044–1078. doi: 10.1111/j.1574-6976.2009.00188.x. doi:10.1111/j.1574-6976.2009.00188.x. [DOI] [PubMed] [Google Scholar]
- [89].Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. Embo J. 1995;14:4108–4120. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lukianova OA, David SS. A role for iron–sulfur clusters in DNA repair. Current Opinion in Chemical Biology. 2005;9:145–151. doi: 10.1016/j.cbpa.2005.02.006. doi:10.1016/j.cbpa.2005.02.006. [DOI] [PubMed] [Google Scholar]
- [91].Mol CD, Parikh SS, Putnam CD, Lo TP, Tainer JA. DNA repair mechanisms for the recognition and removal of damaged DNA bases. Annu Rev Biophys Biomol Struct. 1999;28:101–128. doi: 10.1146/annurev.biophys.28.1.101. doi:10.1146/annurev.biophys.28.1.101. [DOI] [PubMed] [Google Scholar]
- [92].Tubbs JL, Latypov V, Kanugula S, Butt A, Melikishvili M, Kraehenbuehl R, et al. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature. 2009;459:808–813. doi: 10.1038/nature08076. doi:10.1038/nature08076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Lindahl T. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc Natl Acad Sci USA. 1974;71:3649–3653. doi: 10.1073/pnas.71.9.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected] Nature. 2000;403:451–456. doi: 10.1038/35000249. doi:10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- [95].Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. Embo J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. doi:10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Parikh SS, Mol CD, Hosfield DJ, Tainer JA. Envisioning the molecular choreography of DNA base excision repair. Curr Opin Struct Biol. 1999;9:37–47. doi: 10.1016/s0959-440x(99)80006-2. [DOI] [PubMed] [Google Scholar]
- [97].Demple B, Linn S. DNA N-glycosylases and UV repair. Nature. 1980;287:203–208. doi: 10.1038/287203a0. [DOI] [PubMed] [Google Scholar]
- [98].Kuo CF, McRee DE, Fisher CL, O’Handley SF, Cunningham RP, Tainer JA. Atomic structure of the DNA repair [4Fe-4S] enzyme endonuclease III. Science. 1992;258:434–440. doi: 10.1126/science.1411536. [DOI] [PubMed] [Google Scholar]
- [99].Boal AK, Yavin E, Lukianova OA, O’Shea VL, David SS, Barton JK. DNA-Bound Redox Activity of DNA Repair Glycosylases Containing [4Fe-4S] Clusters †. Biochemistry. 2005;44:8397–8407. doi: 10.1021/bi047494n. doi:10.1021/bi047494n. [DOI] [PubMed] [Google Scholar]
- [100].Michaels ML, Tchou J, Grollman AP, Miller JH. A repair system for 8-oxo-7,8-dihydrodeoxyguanine. Biochemistry. 1992;31:10964–10968. doi: 10.1021/bi00160a004. [DOI] [PubMed] [Google Scholar]
- [101].Michaels ML, Cruz C, Grollman AP, Miller JH. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc Natl Acad Sci USA. 1992;89:7022–7025. doi: 10.1073/pnas.89.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Michaels ML, Pham L, Nghiem Y, Cruz C, Miller JH. MutY, an adenine glycosylase active on G-A mispairs, has homology to endonuclease III. Nucleic Acids Res. 1990;18:3841–3845. doi: 10.1093/nar/18.13.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Michaels ML, Miller JH. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) J. Bacteriol. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, et al. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat. Struct. Biol. 1998;5:1058–1064. doi: 10.1038/4168. doi:10.1038/4168. [DOI] [PubMed] [Google Scholar]
- [105].Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004;427:652–656. doi: 10.1038/nature02306. doi:10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- [106].Golinelli M-P, Chmiel NH, David SS. Site-Directed Mutagenesis of the Cysteine Ligands to the [4Fe–4S] Cluster of Escherichia coli MutY. Biochemistry. 1999;38:6997–7007. doi: 10.1021/bi982300n. doi:10.1021/bi982300n. [DOI] [PubMed] [Google Scholar]
- [107].Porello SL, Cannon MJ, David SS. A substrate recognition role for the [4Fe-4S]2+ cluster of the DNA repair glycosylase MutY. Biochemistry. 1998;37:6465–6475. doi: 10.1021/bi972433t. doi:10.1021/bi972433t. [DOI] [PubMed] [Google Scholar]
- [108].Luncsford PJ, Chang D-Y, Shi G, Bernstein J, Madabushi A, Patterson DN, et al. A Structural Hinge in Eukaryotic MutY Homologues Mediates Catalytic Activity and Rad9–Rad1–Hus1 Checkpoint Complex Interactions. J Mol Biol. 2010;403:351–370. doi: 10.1016/j.jmb.2010.08.045. doi:10.1016/j.jmb.2010.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Cheadle JP, Sampson JR. MUTYH-associated polyposis—From defect in base excision repair to clinical genetic testing. DNA Repair (Amst) 2007;6:274–279. doi: 10.1016/j.dnarep.2006.11.001. doi:10.1016/j.dnarep.2006.11.001. [DOI] [PubMed] [Google Scholar]
- [110].Engstrom LM, Brinkmeyer MK, Ha Y, Raetz AG, Hedman B, Hodgson KO, et al. A Zinc Linchpin Motif in the MUTYH Glycosylase Interdomain Connector Is Required for Efficient Repair of DNA Damage. J Am Chem Soc. 2014;136:7829–7832. doi: 10.1021/ja502942d. doi:10.1021/ja502942d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Liu H, Rudolf J, Johnson K, Mcmahon S, Oke M, Carter L, et al. Structure of the DNA Repair Helicase XPD. Cell. 2008;133:801–812. doi: 10.1016/j.cell.2008.04.029. doi:10.1016/j.cell.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, et al. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. doi:10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Wolski SC, Kuper J, Hänzelmann P, Truglio JJ, Croteau DL, Van Houten B, et al. Crystal structure of the FeS cluster-containing nucleotide excision repair helicase XPD. PLoS Biol. 2008;6:e149. doi: 10.1371/journal.pbio.0060149. doi:10.1371/journal.pbio.0060149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Fuss JO, Tainer JA. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair (Amst) 2011;10:697–713. doi: 10.1016/j.dnarep.2011.04.028. doi:10.1016/j.dnarep.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Torres RA, Lovell T, Noodleman L, Case DA. Density Functional and Reduction Potential Calculations of Fe 4S 4Clusters. J Am Chem Soc. 2003;125:1923–1936. doi: 10.1021/ja0211104. doi:10.1021/ja0211104. [DOI] [PubMed] [Google Scholar]
- [116].Mui TP, Fuss JO, Ishida JP, Tainer JA, Barton JK. ATP-Stimulated, DNA-Mediated Redox Signaling by XPD, a DNA Repair and Transcription Helicase. J Am Chem Soc. 2011 doi: 10.1021/ja207222t. doi:10.1021/ja207222t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- [118].Suhasini AN, Brosh RM., Jr Mutation Research/Reviews in Mutation Research. Mutation Research-Reviews in Mutation Research. 2013;752:138–152. doi: 10.1016/j.mrrev.2012.12.004. doi:10.1016/j.mrrev.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet. 2005;37:934–935. doi: 10.1038/ng1625. doi:10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- [120].Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005;37:931–933. doi: 10.1038/ng1624. doi:10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- [121].Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, et al. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. doi:10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- [122].Walden H, Deans AJ. The Fanconi Anemia DNA Repair Pathway: Structural and Functional Insights into a Complex Disorder. Annu Rev Biophys. 2014;43:257–278. doi: 10.1146/annurev-biophys-051013-022737. doi:10.1146/annurev-biophys-051013-022737. [DOI] [PubMed] [Google Scholar]
- [123].Wu Y, Sommers JA, Loiland JA, Kitao H, Kuper J, Kisker C, et al. The Q Motif of Fanconi Anemia Group J Protein (FANCJ) DNA Helicase Regulates Its Dimerization, DNA Binding, and DNA Repair Function. Journal of Biological Chemistry. 2012;287:21699–21716. doi: 10.1074/jbc.M112.351338. doi:10.1074/jbc.M112.351338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, et al. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci USA. 2004;101:2357–2362. doi: 10.1073/pnas.0308717101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Zhu L, Hathcock KS, Hande P, Lansdorp PM, Seldin MF, Hodes RJ. Telomere length regulation in mice is linked to a novel chromosome locus. Proc Natl Acad Sci USA. 1998;95:8648–8653. doi: 10.1073/pnas.95.15.8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Vannier J-B, Sarek G, Boulton SJ. RTEL1: functions of a disease-associated helicase. Trends in Cell Biology. 2014;24:416–425. doi: 10.1016/j.tcb.2014.01.004. doi:10.1016/j.tcb.2014.01.004. [DOI] [PubMed] [Google Scholar]
- [127].Wrensch M, Jenkins RB, Chang JS, Yeh R-F, Xiao Y, Decker PA, et al. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet. 2009;41:905–908. doi: 10.1038/ng.408. doi:10.1038/ng.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Faure G, Revy P, Schertzer M, Londono-Vallejo A, Callebaut I. The C-terminal extension of human RTEL1, mutated in Hoyeraal-Hreidarsson syndrome, contains harmonin-N-like domains. Proteins. 2014;82:897–903. doi: 10.1002/prot.24438. doi:10.1002/prot.24438. [DOI] [PubMed] [Google Scholar]
- [129].Landry AP, Ding H. The N-Terminal Domain of Human DNA Helicase Rtel1 Contains a Redox Active Iron-Sulfur Cluster. BioMed Research International. 2014;2014:1–8. doi: 10.1155/2014/285791. doi:10.1016/j.freeradbiomed.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].van der Lelij P, Chrzanowska KH, Godthelp BC, Rooimans MA, Oostra AB, Stumm M, et al. Warsaw Breakage Syndrome,a Cohesinopathy Associated with Mutationsin the XPD Helicase Family Member DDX11/ChlR1. Am. J. Hum. Genet. 2010;86:262–266. doi: 10.1016/j.ajhg.2010.01.008. doi:10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Bharti SK, Khan I, Banerjee T, Sommers JA, Wu Y, Brosh RM. Molecular functions and cellular roles of the ChlR1 (DDX11) helicase defective in the rare cohesinopathy Warsaw breakage syndrome. Cell Mol Life Sci. 2014 doi: 10.1007/s00018-014-1569-4. doi:10.1007/s00018-014-1569-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Sanchez Garcia J. The C-terminal zinc finger of the catalytic subunit of DNA polymerase is responsible for direct interaction with the B-subunit. Nucleic Acids Res. 2004;32:3005–3016. doi: 10.1093/nar/gkh623. doi:10.1093/nar/gkh623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Klinge S, Nunez-Ramirez R, Llorca O, Pellegrini L. 3D architecture of DNA Pol alpha reveals the functional core of multi-subunit replicative polymerases. Embo J. 2009;28:1978–1987. doi: 10.1038/emboj.2009.150. doi:10.1038/emboj.2009.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Dua R, Levy DL, Campbell JL. Analysis of the Essential Functions of the C-terminal Protein/Protein Interaction Domain of Saccharomyces cerevisiae pol and Its Unexpected Ability to Support Growth in the Absence of the DNA Polymerase Domain. Journal of Biological Chemistry. 1999;274:22283–22288. doi: 10.1074/jbc.274.32.22283. doi:10.1074/jbc.274.32.22283. [DOI] [PubMed] [Google Scholar]
- [135].Barondeau DP, Kassmann CJ, Bruns CK, Tainer JA, Getzoff ED. Nickel Superoxide Dismutase Structure and Mechanism †. Biochemistry. 2004;43:8038–8047. doi: 10.1021/bi0496081. doi:10.1021/bi0496081. [DOI] [PubMed] [Google Scholar]
- [136].Tainer JA, Getzoff ED, Richardson JS, Richardson DC. Structure and mechanism of copper, zinc superoxide dismutase. Nature. 1983;306:284–287. doi: 10.1038/306284a0. [DOI] [PubMed] [Google Scholar]
- [137].Borgstahl GE, Parge HE, Hickey MJ, Beyer WF, Hallewell RA, Tainer JA. The structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundles. Cell. 1992;71:107–118. doi: 10.1016/0092-8674(92)90270-m. [DOI] [PubMed] [Google Scholar]
- [138].Xie B, Mazloum N, Liu L, Rahmeh A, Li H, Lee MYWT. Reconstitution and Characterization of the Human DNA Polymerase Delta Four-Subunit Holoenzyme †. Biochemistry. 2002;41:13133–13142. doi: 10.1021/bi0262707. doi:10.1021/bi0262707. [DOI] [PubMed] [Google Scholar]
- [139].Burgers PMJ. Polymerase Dynamics at the Eukaryotic DNA Replication Fork. Journal of Biological Chemistry. 2009;284:4041–4045. doi: 10.1074/jbc.R800062200. doi:10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Iyama T, Wilson DM., III DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 2013;12:620–636. doi: 10.1016/j.dnarep.2013.04.015. doi:10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Henninger EE, Pursell ZF. DNA polymerase ε and its roles in genome stability. IUBMB Life. 2014;66:339–351. doi: 10.1002/iub.1276. doi:10.1002/iub.1276. [DOI] [PubMed] [Google Scholar]
- [142].Bermudez VP, Farina A, Raghavan V, Tappin I, Hurwitz J. Studies on Human DNA Polymerase and GINS Complex and Their Role in DNA Replication. Journal of Biological Chemistry. 2011;286:28963–28977. doi: 10.1074/jbc.M111.256289. doi:10.1074/jbc.M111.256289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kunkel TA. Evolving Views of DNA Replication (In)Fidelity. Cold Spring Harbor Symposia on Quantitative Biology. 2010;74:91–101. doi: 10.1101/sqb.2009.74.027. doi:10.1101/sqb.2009.74.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Kim TW, Delaney JC, Essigmann JM, Kool ET. Probing the active site tightness of DNA polymerase in subangstrom increments. Proc Natl Acad Sci USA. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. doi:10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase. Nat Struct Mol Biol. 2009;16:979–986. doi: 10.1038/nsmb.1663. doi:10.1038/nsmb.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Asturias FJ, Cheung IK, Sabouri N, Chilkova O, Wepplo D, Johansson E. Structure of Saccharomyces cerevisiae DNA polymerase epsilon by cryo–electron microscopy. Nat Struct Mol Biol. 2005;13:35–43. doi: 10.1038/nsmb1040. doi:10.1038/nsmb1040. [DOI] [PubMed] [Google Scholar]
- [147].Baranovskiy AG, Lada AG, Siebler HM, Zhang Y, Pavlov YI, Tahirov TH. DNA Polymerase and Switch by Sharing Accessory Subunits of DNA Polymerase. Journal of Biological Chemistry. 2012;287:17281–17287. doi: 10.1074/jbc.M112.351122. doi:10.1074/jbc.M112.351122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Copeland WC, Wang TSF. Enzymatic Characterization of the Individual Mammalian Primase Subunits Reveals a Biphasic Mechanism for Initiation of Dna-Replication. J Biol Chem. 1993;268:26179–26189. [PubMed] [Google Scholar]
- [149].Zhang Y, Baranovskiy AG, Tahirov TH, Pavlov YI. The C-terminal Domain of the DNA Polymerase Catalytic Subunit Regulates the Primase and Polymerase Activities of the Human DNA Polymerase -Primase Complex. Journal of Biological Chemistry. 2014;289:22021–22034. doi: 10.1074/jbc.M114.570333. doi:10.1074/jbc.M114.570333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Vaithiyalingam S, Warren EM, Eichman BF, Chazin WJ. Insights into eukaryotic DNA priming from the structure and functional interactions of the 4Fe-4S cluster domain of human DNA primase. Proc Natl Acad Sci USA. 2010;107:13684–13689. doi: 10.1073/pnas.1002009107. doi:10.1073/pnas.1002009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Kilkenny ML, Longo MA, Perera RL, Pellegrini L. Structures of human primase reveal design of nucleotide elongation site and mode of Pol α tethering. Proc Natl Acad Sci USA. 2013;110:15961–15966. doi: 10.1073/pnas.1311185110. doi:10.1073/pnas.1311185110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Budd ME, Campbell JL. Interplay of Mre11 Nuclease with Dna2 plus Sgs1 in Rad51-Dependent Recombinational Repair. PLoS ONE. 2009;4:e4267. doi: 10.1371/journal.pone.0004267. doi:10.1371/journal.pone.0004267.t001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, et al. DNA end resection by Dna2. Nature. 2010;467:112–116. doi: 10.1038/nature09355. doi:10.1038/nature09355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes & Development. 2011;25:350–362. doi: 10.1101/gad.2003811. doi:10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Saikrishnan K, Yeeles JT, Gilhooly NS, Krajewski WW, Dillingham MS, Wigley DB. Insights into Chi recognition from the structure of an AddAB-type helicase–nuclease complex. Embo J. 2012;31:1568–1578. doi: 10.1038/emboj.2012.9. doi:10.1038/emboj.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Krajewski WW, Fu X, Wilkinson M, Cronin NB, Dillingham MS, Wigley DB. Structural basis for translocation by AddAB helicase– nuclease and its arrest at x sites. Nature. 2014;508:416–419. doi: 10.1038/nature13037. doi:10.1038/nature13037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Zhang J, Kasciukovic T, White MF. The CRISPR Associated Protein Cas4 Is a 5′ to 3′ DNA Exonuclease with an Iron-Sulfur Cluster. PLoS ONE. 2012;7:e47232. doi: 10.1371/journal.pone.0047232. doi:10.1371/journal.pone.0047232.g007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Karanja KK, Lee EH, Hendrickson EA, Campbell JL. Preventing over-resection by DNA2 helicase/nuclease suppresses repair defects in Fanconi anemia cells. Cell Cycle. 2014;13:1540–1550. doi: 10.4161/cc.28476. doi:10.4161/cc.28476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, Guenther G, et al. Human Flap Endonuclease Structures, DNA Double-Base Flipping, and a Unified Understanding of the FEN1 Superfamily. Cell. 2011;145:198–211. doi: 10.1016/j.cell.2011.03.004. doi:10.1016/j.cell.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, et al. Mre11 Dimers Coordinate DNA End Bridging and Nuclease Processing in Double-Strand-Break Repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. doi:10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois M-M, et al. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol Cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. doi:10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, et al. Nbs1 Flexibly Tethers Ctp1 and Mre11- Rad50 to Coordinate DNA Double-Strand Break Processing and Repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. doi:10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Tsutakawa SE, Lafrance-Vanasse J, Tainer JA. The cutting edges in DNA repair, licensing, and fidelity: DNA and RNA repair nucleases sculpt DNA to measure twice, cut once. DNA Repair (Amst) 2014;19:95–107. doi: 10.1016/j.dnarep.2014.03.022. doi:10.1016/j.dnarep.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].DiGiovanna JJ, Kraemer KH. Shining a Light on Xeroderma Pigmentosum. Journal of Investigative Dermatology. 2012;132:785–796. doi: 10.1038/jid.2011.426. doi:10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Botta E, Nardo T, Broughton BC, Marinoni S, Lehmann AR, Stefanini M. Analysis of mutations in the XPD gene in Italian patients with trichothiodystrophy: site of mutation correlates with repair deficiency, but gene dosage appears to determine clinical severity. Am. J. Hum. Genet. 1998;63:1036–1048. doi: 10.1086/302063. doi:10.1086/302063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Rafnar T, Gudbjartsson DF, Sulem P, Jonasdottir A, Sigurdsson A, Jonasdottir A, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011;43:1104–1107. doi: 10.1038/ng.955. doi:10.1038/ng.955. [DOI] [PubMed] [Google Scholar]
- [167].Ballew BJ, Yeager M, Jacobs K, Giri N, Boland J, Burdett L, et al. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum Genet. 2013;132:473–480. doi: 10.1007/s00439-013-1265-8. doi:10.1007/s00439-013-1265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Le Guen T, Jullien L, Touzot F, Schertzer M, Gaillard L, Perderiset M, et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum. Mol. Genet. 2013;22:3239–3249. doi: 10.1093/hmg/ddt178. doi:10.1093/hmg/ddt178. [DOI] [PubMed] [Google Scholar]
- [169].Parish JL, Bean AM, Park RB, Androphy EJ. ChlR1 Is Required for Loading Papillomavirus E2 onto Mitotic Chromosomes and Viral Genome Maintenance. Mol Cell. 2006;24:867–876. doi: 10.1016/j.molcel.2006.11.005. doi:10.1016/j.molcel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- [170].Sharma S. In vivo function of the conserved non-catalytic domain of Werner syndrome helicase in DNA replication. Hum. Mol. Genet. 2004;13:2247–2261. doi: 10.1093/hmg/ddh234. doi:10.1093/hmg/ddh234. [DOI] [PubMed] [Google Scholar]
- [171].Ronchi D, Di Fonzo A, Lin W, Bordoni A, Liu C, Fassone E, et al. Mutations in DNA2 Link Progressive Myopathy to Mitochondrial DNA Instability. Am. J. Hum. Genet. 2013;92:293–300. doi: 10.1016/j.ajhg.2012.12.014. doi:10.1016/j.ajhg.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Bae SH. Characterization of the Enzymatic Properties of the Yeast Dna2 Helicase/Endonuclease Suggests a New Model for Okazaki Fragment Processing. Journal of Biological Chemistry. 2000;275:38022–38031. doi: 10.1074/jbc.M006513200. doi:10.1074/jbc.M006513200. [DOI] [PubMed] [Google Scholar]
- [173].Floy KM, Hess RO, Meisner LF. DNA polymerase alpha defect in the N syndrome. Am. J. Med. Genet. 1990;35:301–305. doi: 10.1002/ajmg.1320350302. doi:10.1002/ajmg.1320350302. [DOI] [PubMed] [Google Scholar]
- [174].Kane DP, Shcherbakova PV. A Common Cancer-Associated DNA Polymerase Mutation Causes an Exceptionally Strong Mutator Phenotype, Indicating Fidelity Defects Distinct from Loss of Proofreading. Cancer Res. 2014;74:1895–1901. doi: 10.1158/0008-5472.CAN-13-2892. doi:10.1158/0008-5472.CAN-13-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Network TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2013;487:330–337. doi: 10.1038/nature11252. doi:10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Church DN, Briggs SEW, Palles C, Domingo E, Kearsey SJ, Grimes JM, et al. DNA polymerase and exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013;22:2820–2828. doi: 10.1093/hmg/ddt131. doi:10.1093/hmg/ddt131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Getz G, Gabriel SB, Cibulskis K, Lander E, Sivachenko A, Sougnez C, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. doi:10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Stehling O, Wilbrecht C, Lill R. Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie. 2014;100:61–77. doi: 10.1016/j.biochi.2014.01.010. doi:10.1016/j.biochi.2014.01.010. [DOI] [PubMed] [Google Scholar]
- [179].Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- [180].Cossée M, Schmitt M, Campuzano V, Reutenauer L, Moutou C, Mandel JL, et al. Evolution of the Friedreich’s ataxia trinucleotide repeat expansion: founder effect and premutations. Proc Natl Acad Sci USA. 1997;94:7452–7457. doi: 10.1073/pnas.94.14.7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Correia AR, Pastore C, Adinolfi S, Pastore A, Gomes CM. Dynamics, stability and iron-binding activity of frataxin clinical mutants. FEBS Journal. 2008;275:3680–3690. doi: 10.1111/j.1742-4658.2008.06512.x. doi:10.1111/j.1742-4658.2008.06512.x. [DOI] [PubMed] [Google Scholar]
- [182].Tsai C-L, Bridwell-Rabb J, Barondeau DP. Friedreich’s Ataxia Variants I154F and W155R Diminish Frataxin-Based Activation of the Iron–Sulfur Cluster Assembly Complex. Biochemistry. 2011;50:6478–6487. doi: 10.1021/bi200666h. doi:10.1021/bi200666h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Yoon T, Cowan JA. Iron–Sulfur Cluster Biosynthesis. Characterization of Frataxin as an Iron Donor for Assembly of [2Fe-2S] Clusters in ISU-Type Proteins. J Am Chem Soc. 2003;125:6078–6084. doi: 10.1021/ja027967i. doi:10.1021/ja027967i. [DOI] [PubMed] [Google Scholar]
- [184].Layer G, Ollagnier-de Choudens S, Sanakis Y, Fontecave M. Iron-Sulfur Cluster Biosynthesis: Characterization of Escherichia coli CyaY as an iron donor for the assembly of [2Fe-2S] clusters in the scaffold IscU. Journal of Biological Chemistry. 2006;281:16256–16263. doi: 10.1074/jbc.M513569200. doi:10.1074/jbc.M513569200. [DOI] [PubMed] [Google Scholar]
- [185].Tsai C-L, Barondeau DP. Human Frataxin Is an Allosteric Switch That Activates the Fe–S Cluster Biosynthetic Complex. Biochemistry. 2010;49:9132–9139. doi: 10.1021/bi1013062. doi:10.1021/bi1013062. [DOI] [PubMed] [Google Scholar]
- [186].Bridwell-Rabb J, Iannuzzi C, Pastore A, Barondeau DP. Effector Role Reversal during Evolution: The Case of Frataxin in Fe–S Cluster Biosynthesis. Biochemistry. 2012;51:2506–2514. doi: 10.1021/bi201628j. doi:10.1021/bi201628j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Pandey A, Gordon DM, Pain J, Stemmler TL, Dancis A, Pain D. Frataxin Directly Stimulates Mitochondrial Cysteine Desulfurase by Exposing Substrate-binding Sites, and a Mutant Fe-S Cluster Scaffold Protein with Frataxin-bypassing Ability Acts Similarly. Journal of Biological Chemistry. 2013;288:36773–36786. doi: 10.1074/jbc.M113.525857. doi:10.1074/jbc.M113.525857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Colin F, Martelli A, Clémancey M, Latour J-M, Gambarelli S, Zeppieri L, et al. Mammalian Frataxin Controls Sulfur Production and Iron Entry during de Novo Fe 4S 4Cluster Assembly. J Am Chem Soc. 2013;135:733–740. doi: 10.1021/ja308736e. doi:10.1021/ja308736e. [DOI] [PubMed] [Google Scholar]
- [189].Olsson A, Lind L, Thornell LE, Holmberg M. Myopathy with lactic acidosis is linked to chromosome 12q23.3-24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum. Mol. Genet. 2008;17:1666–1672. doi: 10.1093/hmg/ddn057. doi:10.1093/hmg/ddn057. [DOI] [PubMed] [Google Scholar]
- [190].Mochel F, Knight MA, Tong WH, Hernandez D, Ayyad K, Taivassalo T, et al. Splice Mutation in the Iron-Sulfur Cluster Scaffold Protein ISCU Causes Myopathy with Exercise Intolerance. The American Journal of Human Genetics. 2008;82:652–660. doi: 10.1016/j.ajhg.2007.12.012. doi:10.1016/j.ajhg.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110:1353–1358. doi: 10.1182/blood-2007-02-072520. doi:10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]
- [192].Mühlenhoff U, Gerber J, Richhardt N, Lill R. Components involved in assembly and dislocation of iron-sulfur clusters on the scaffold protein Isu1p. Embo J. 2003;22:4815–4825. doi: 10.1093/emboj/cdg446. doi:10.1093/emboj/cdg446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Ye H, Jeong SY, Ghosh MC, Kovtunovych G, Silvestri L, Ortillo D, et al. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Invest. 2010;120:1749–1761. doi: 10.1172/JCI40372. doi:10.1172/JCI40372DS1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O, et al. Biochimica et Biophysica Acta. BBA - Molecular Cell Research. 2012;1823:1491–1508. doi: 10.1016/j.bbamcr.2012.05.009. doi:10.1016/j.bbamcr.2012.05.009. [DOI] [PubMed] [Google Scholar]
- [195].Kispal G, Csere P, Prohl C, Lill R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. Embo J. 1999;18:3981–3989. doi: 10.1093/emboj/18.14.3981. doi:10.1093/emboj/18.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Gerber J, Neumann K, Prohl C, Muhlenhoff U, Lill R. The Yeast Scaffold Proteins Isu1p and Isu2p Are Required inside Mitochondria for Maturation of Cytosolic Fe/S Proteins. Mol Cell Biol. 2004;24:4848–4857. doi: 10.1128/MCB.24.11.4848-4857.2004. doi:10.1128/MCB.24.11.4848-4857.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Srinivasan V, Pierik AJ, Lill R. Crystal Structures of Nucleotide-Free and Glutathione-Bound Mitochondrial ABC Transporter Atm1. Science. 2014;343:1137–1140. doi: 10.1126/science.1246729. doi:10.1126/science.1246729. [DOI] [PubMed] [Google Scholar]
- [198].Pondarre C. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum. Mol. Genet. 2006;15:953–964. doi: 10.1093/hmg/ddl012. doi:10.1093/hmg/ddl012. [DOI] [PubMed] [Google Scholar]
- [199].Mühlenhoff U, Molik S, Godoy JR, Uzarska MA, Richter N, Seubert A, et al. Cytosolic Monothiol Glutaredoxins Function in Intracellular Iron Sensing and Trafficking via Their Bound Iron-Sulfur Cluster. Cell Metabolism. 2010;12:373–385. doi: 10.1016/j.cmet.2010.08.001. doi:10.1016/j.cmet.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Roy A, Solodovnikova N, Nicholson T, Antholine W, Walden WE. A novel eukaryotic factor for cytosolic Fe-S cluster assembly. Embo J. 2003;22:4826–4835. doi: 10.1093/emboj/cdg455. doi:10.1093/emboj/cdg455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Hausmann A, Aguilar Netz DJ, Balk J, Pierik AJ, Mühlenhoff U, Lill R. The eukaryotic P loop NTPase Nbp35: an essential component of the cytosolic and nuclear iron-sulfur protein assembly machinery. Proc Natl Acad Sci USA. 2005;102:3266–3271. doi: 10.1073/pnas.0406447102. doi:10.1073/pnas.0406447102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Netz DJA, Pierik AJ, Stumpfig M, Bill E, Sharma AK, Pallesen LJ, et al. A Bridging [4Fe-4S] Cluster and Nucleotide Binding Are Essential for Function of the Cfd1-Nbp35 Complex as a Scaffold in Iron-Sulfur Protein Maturation. Journal of Biological Chemistry. 2012;287:12365–12378. doi: 10.1074/jbc.M111.328914. doi:10.1074/jbc.M111.328914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Netz DJA, Pierik AJ, Stümpfig M, Mühlenhoff U, Lill R. The Cfd1-Nbp35 complex acts as a scaffold for iron-sulfur protein assembly in the yeast cytosol. Nat Chem Biol. 2007;3:278–286. doi: 10.1038/nchembio872. doi:10.1038/nchembio872. [DOI] [PubMed] [Google Scholar]
- [204].Zhang Y, Lyver ER, Nakamaru-Ogiso E, Yoon H, Amutha B, Lee DW, et al. Dre2, a Conserved Eukaryotic Fe/S Cluster Protein, Functions in Cytosolic Fe/S Protein Biogenesis. Mol Cell Biol. 2008;28:5569–5582. doi: 10.1128/MCB.00642-08. doi:10.1128/MCB.00642-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Netz DJA, Stümpfig M, Doré C, Mühlenhoff U, Pierik AJ, Lill R. tah18 transfers electrons to dre2 in cytosolic iron-sulfur protein biogenesis. Nat Chem Biol. 2010;6:758–765. doi: 10.1038/nchembio.432. doi:10.1038/nchembio.432. [DOI] [PubMed] [Google Scholar]
- [206].Pallesen LJ, Solodovnikova N, Sharma AK, Walden WE. Interaction with Cfd1 Increases the Kinetic Lability of FeS on the Nbp35 Scaffold. Journal of Biological Chemistry. 2013;288:23358–23367. doi: 10.1074/jbc.M113.486878. doi:10.1074/jbc.M113.486878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Balk J, Pilon M. Ancient and essential: the assembly of iron. Trends in Plant Science. 2011;16:218–226. doi: 10.1016/j.tplants.2010.12.006. doi:10.1016/j.tplants.2010.12.006. [DOI] [PubMed] [Google Scholar]
- [208].Netz DJA, Mascarenhas J, Stehling O, Pierik AJ, Lill R. Maturation of cytosolic and nuclear iron–sulfur proteins. Trends in Cell Biology. 2014;24:303–312. doi: 10.1016/j.tcb.2013.11.005. doi:10.1016/j.tcb.2013.11.005. [DOI] [PubMed] [Google Scholar]
- [209].Balk J, Pierik AJ, Netz DJA, Mühlenhoff U, Lill R. The hydrogenase-like Nar1p is essential for maturation of cytosolic and nuclear iron-sulphur proteins. Embo J. 2004;23:2105–2115. doi: 10.1038/sj.emboj.7600216. doi:10.1038/sj.emboj.7600216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Beinert H, Holm RH, Munck E. Iron-sulfur clusters: nature’s modular, multipurpose structures. Science. 1997;277:653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- [211].Stehling O, Mascarenhas J, Vashisht AA, Sheftel AD, Niggemeyer B, Rösser R, et al. Human CIA2A-FAM96A and CIA2B-FAM96B Integrate Iron Homeostasis and Maturation of Different Subsets of Cytosolic-Nuclear Iron-Sulfur Proteins. Cell Metabolism. 2013;18:187–198. doi: 10.1016/j.cmet.2013.06.015. doi:10.1016/j.cmet.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Seki M, Takeda Y, Iwai K, Tanaka K. IOP1 Protein Is an External Component of the Human Cytosolic Iron-Sulfur Cluster Assembly (CIA) Machinery and Functions in the MMS19 Protein-dependent CIA Pathway. Journal of Biological Chemistry. 2013;288:16680–16689. doi: 10.1074/jbc.M112.416602. doi:10.1074/jbc.M112.416602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Chen KE, Richards AA, Ariffin JK, Ross IL, Sweet MJ, Kellie S, et al. The mammalian DUF59 protein Fam96a forms two distinct types of domain-swapped dimer. Acta Cryst. D. 2012:637–648. doi: 10.1107/S0907444912006592. doi:10.1107/S0907444912006592. [DOI] [PubMed] [Google Scholar]
- [214].Srinivasan V, Netz DJA, Webert H, Mascarenhas J, Pierik AJ, Michel H, et al. Structure of the Yeast WD40 Domain Protein Cia1, a Component Acting Late in Iron-Sulfur Protein Biogenesis. Structure. 2007;15:1246–1257. doi: 10.1016/j.str.2007.08.009. doi:10.1016/j.str.2007.08.009. [DOI] [PubMed] [Google Scholar]
- [215].Almeida MS, Herrmann T, Peti W, Wilson IA, Wüthrich K. NMR structure of the conserved hypothetical protein TM0487 from Thermotoga maritima: Implications for 216 homologous DUF59 proteins. Protein Science. 2005;14:2880–2886. doi: 10.1110/ps.051755805. doi:10.1110/ps.051755805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Ouyang B, Wang L, Wan S, Luo Y, Wang L, Lin J, et al. Solution structure of monomeric human FAM96A. J Biomol NMR. 2013;56:387–392. doi: 10.1007/s10858-013-9746-6. doi:10.1007/s10858-013-9746-6. [DOI] [PubMed] [Google Scholar]
- [217].Queimado L, Rao M, Schultz RA, Koonin EV, Aravind L, Nardo T, et al. Cloning the human and mouse MMS19 genes and functional complementation of a yeast mms19 deletion mutant. Nucleic Acids Res. 2001;29:1884–1891. doi: 10.1093/nar/29.9.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Stehling O, Vashisht AA, Mascarenhas J, Jonsson ZO, Sharma T, Netz DJA, et al. MMS19 Assembles Iron-Sulfur Proteins Required for DNA Metabolism and Genomic Integrity. Science. 2012;337:195–199. doi: 10.1126/science.1219723. doi:10.1126/science.1219723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Kuchenreuther JM, Grady-Smith CS, Bingham AS, George SJ, Cramer SP, Swartz JR. High-Yield Expression of Heterologous [FeFe] Hydrogenases in Escherichia coli, PLoS. ONE. 2010;5:e15491. doi: 10.1371/journal.pone.0015491. doi:10.1371/journal.pone.0015491.t002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Friedberg E, Lehmann A, Fuchs R. Trading Places: How Do DNA Polymerases Switch during Translesion DNA Synthesis? Mol Cell. 2005;18:499–505. doi: 10.1016/j.molcel.2005.03.032. doi:10.1016/j.molcel.2005.03.032. [DOI] [PubMed] [Google Scholar]
- [221].Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2011;465:951–955. doi: 10.1038/nature09097. doi:10.1038/nature09097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Karras GI, Jentsch S. The RAD6 DNA Damage Tolerance Pathway Operates Uncoupled from the Replication Fork and Is Functional Beyond S Phase. Cell. 2010;141:255–267. doi: 10.1016/j.cell.2010.02.028. doi:10.1016/j.cell.2010.02.028. [DOI] [PubMed] [Google Scholar]
- [223].Tsutakawa SE, Van Wynsberghe AW, Freudenthal BD, Weinacht CP, Gakhar L, Washington MT, et al. Solution X-ray scattering combined with computational modeling reveals multiple conformations of covalently bound ubiquitin on PCNA. Proc Natl Acad Sci USA. 2011;108:17672–17677. doi: 10.1073/pnas.1110480108. doi:10.1073/pnas.1110480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Deshpande RA, Williams GJ, Limbo O, Williams RS, Kuhnlein J, Lee JH, et al. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. Embo J. 2014;33:482–500. doi: 10.1002/embj.201386100. doi:10.1002/embj.201386100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Williams GJ, Williams RS, Williams JS, Moncalian G, Arvai AS, Limbo O, et al. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat Struct Mol Biol. 2011;18:423–431. doi: 10.1038/nsmb.2038. doi:10.1038/nsmb.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Casas-Delucchi CS, Cardoso MC. Epigenetic control of DNA replication dynamics in mammals. Nucleus. 2011;2:370–382. doi: 10.4161/nucl.2.5.17861. doi:10.4161/nucl.2.5.17861. [DOI] [PubMed] [Google Scholar]
- [227].Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand-Sauthier I, et al. Coordination of dual incision and repair synthesis in human nucleotide excision repair. 2009. pp. 1–10. doi:10.1038/emboj.2009.49. [DOI] [PMC free article] [PubMed]
- [228].Iyer N, Reagan MS, Wu KJ, Canagarajah B, Friedberg EC. Interactions involving the human RNA polymerase II transcription/nucleotide excision repair complex TFIIH, the nucleotide excision repair protein XPG, and Cockayne syndrome group B (CSB) protein. Biochemistry. 1996;35:2157–2167. doi: 10.1021/bi9524124. doi:10.1021/bi9524124. [DOI] [PubMed] [Google Scholar]
- [229].Moriel-Carretero M, Aguilera A. A Postincision-Deficient TFIIH Causes Replication Fork Breakage and Uncovers Alternative Rad51- or Pol32-Mediated Restart Mechanisms. Mol Cell. 2010;37:690–701. doi: 10.1016/j.molcel.2010.02.008. doi:10.1016/j.molcel.2010.02.008. [DOI] [PubMed] [Google Scholar]
- [230].Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. doi:10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-Guided Human Genome Engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. doi:10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. doi:10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Uchihashi T, Kodera N, Ando T. Guide to video recording of structure dynamics and dynamic processes of proteins by high-speed atomic force microscopy. Nat Protoc. 2012;7:1193–1206. doi: 10.1038/nprot.2012.047. doi:10.1038/nprot.2012.047. [DOI] [PubMed] [Google Scholar]
- [234].Robinson CV, Sali A, Baumeister W. The molecular sociology of the cell. Nature. 2007;450:973–982. doi: 10.1038/nature06523. doi:10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- [235].Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quart. Rev. Biophys. 2007;40 doi: 10.1017/S0033583507004635. doi:10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- [236].Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, Tsutakawa SE, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. doi:10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Rambo RP, Tainer JA. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 2011;95:559–571. doi: 10.1002/bip.21638. doi:10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Rambo RP, Tainer JA. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2014;496:477–481. doi: 10.1038/nature12070. doi:10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].McTernan PM, Chandrayan SK, Wu CH, Vaccaro BJ, Lancaster WA, Yang Q, et al. Intact Functional Fourteen-subunit Respiratory Membrane-bound [NiFe]-Hydrogenase Complex of the Hyperthermophilic Archaeon Pyrococcus furiosus. Journal of Biological Chemistry. 2014;289:19364–19372. doi: 10.1074/jbc.M114.567255. doi:10.1074/jbc.M114.567255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Hura GL, Budworth H, Dyer KN, Rambo RP, Hammel M, McMurray CT, et al. Comprehensive macromolecular conformations mapped by quantitative SAXS analyses. Nat Methods. 2013;10:453–454. doi: 10.1038/nmeth.2453. doi:10.1038/nmeth.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Rambo RP, Tainer JA. Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies bysmall-angle X-ray scattering. Curr Opin Struct Biol. 2010;20:128–137. doi: 10.1016/j.sbi.2009.12.015. doi:10.1016/j.sbi.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Shin DS, Pratt AJ, Tainer JA. Review Article. Archaea. 2014:1–24. doi: 10.1155/2014/206735. doi:10.1155/2014/206735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Putnam CD, Shroyer MJ, Lundquist AJ, Mol CD, Arvai AS, Mosbaugh DW, et al. Protein mimicry of DNA from crystal structures of the uracil-DNA glycosylase inhibitor protein and its complex with Escherichia coli uracil-DNA glycosylase. J Mol Biol. 1999;287:331–346. doi: 10.1006/jmbi.1999.2605. doi:10.1006/jmbi.1999.2605. [DOI] [PubMed] [Google Scholar]
- [244].Tainer JA, Roberts VA, Getzoff ED. Metal-binding sites in proteins. Current Opinion in Biotechnology. 1991;2:582–591. doi: 10.1016/0958-1669(91)90084-i. [DOI] [PubMed] [Google Scholar]
- [245].Barondeau DP, Kassmann CJ, Tainer JA, Getzoff ED. Structural Chemistry of a Green Fluorescent Protein Zn Biosensor. J Am Chem Soc. 2002;124:3522–3524. doi: 10.1021/ja0176954. doi:10.1021/ja0176954. [DOI] [PubMed] [Google Scholar]
- [246].Roberts VA, Iverson BL, Iverson SA, Benkovic SJ, Lerner RA, Getzoff ED, et al. Antibody remodeling: a general solution to the design of a metal-coordination site in an antibody binding pocket. Proc Natl Acad Sci USA. 1990;87:6654–6658. doi: 10.1073/pnas.87.17.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Hura GL, Tsai C-L, Claridge SA, Mendillo ML, Smith JM, Williams GJ, et al. DNA conformations in mismatch repair probed in solution by X-ray scattering from gold nanocrystals. Proc Natl Acad Sci USA. 2013;110:17308–17313. doi: 10.1073/pnas.1308595110. doi:10.1073/pnas.1308595110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Yang C, Neshatian M, van Prooijen M. Cancer nanotechnology: enhanced therapeutic response using peptide-modified gold nanoparticles. J Nanosci Nanotechnol. 2014;14:4813–4819. doi: 10.1166/jnn.2014.9280. [DOI] [PubMed] [Google Scholar]
- [249].Livshits GI, Stern A, Rotem D, Borovok N, Eidelshtein G, Migliore A, et al. Long-range charge transport in singleG-quadruplex DNA molecules. Nature Nanotechnology. 2014;9:1040–1046. doi: 10.1038/nnano.2014.246. doi:10.1038/nnano.2014.246. [DOI] [PubMed] [Google Scholar]
- [250].Scheer E. A DNA that conducts. Nature Nanotechnology. 2014;9:960–961. doi: 10.1038/nnano.2014.293. doi:10.1038/nnano.2014.293. [DOI] [PubMed] [Google Scholar]
- [251].Manuel RC, Hitomi K, Arvai AS, House PG, Kurtz AJ, Dodson ML, et al. Reaction Intermediates in the Catalytic Mechanism of Escherichia coli MutY DNA Glycosylase. Journal of Biological Chemistry. 2004;279:46930–46939. doi: 10.1074/jbc.M403944200. doi:10.1074/jbc.M403944200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.