

Abstract
This double-blind, placebo-controlled study evaluated the efficacy and safety of hydrocodone extended release (ER) developed with abuse-deterrence technology to provide sustained pain relief and limit effects of alcohol and tablet manipulation on drug release. Eligible patients with chronic moderate-to-severe low back or osteoarthritis pain were titrated to an analgesic dose of hydrocodone ER (15–90 mg) and randomized to placebo or hydrocodone ER every 12 hours. The primary efficacy measure was change from baseline to week 12 in weekly average pain intensity (API; 0=no pain, 10=worst pain imaginable). Secondary measures included percentage of patients with >33% and >50% increases from baseline in weekly API, change from baseline in weekly worst pain intensity, supplemental opioid usage, aberrant drug-use behaviors, and adverse events. Overall, 294 patients were randomized and received ≥1 dose of placebo (n=148) or hydrocodone ER (n=146). Weekly API did not differ significantly between hydrocodone ER and placebo at week 12 (P=0.134); although, in post hoc analyses, the change in weekly API was significantly lower with hydrocodone ER when excluding the lowest dose (15 mg; least squares mean, −0.20 vs 0.40; P=0.032). Significantly more patients had >33% and >50% increase in weekly API with placebo (P<0.05), and mean weekly worst pain intensity was significantly lower with hydrocodone ER at week 12 (P=0.026). Supplemental medication usage was higher with placebo (86%) than hydrocodone ER (79%). Incidence of aberrant drug-use behaviors was low, and adverse events were similar between groups. This study did not meet the primary endpoint, although results support the effectiveness of this hydrocodone ER formulation in managing chronic low back or osteoarthritis pain. Use of the hydrocodone ER 15-mg dose, a robust placebo response, and use of supplemental analgesics, particularly in the placebo group, may have limited detection of a statistically significant treatment effect, and additional research is needed to clarify these findings.
Keywords: clinical trial, aberrant behaviors, opioid analgesics, opioid diversion, opioid loss, abuse deterrent
Introduction
Chronic pain affects an estimated 100 million adults in the US and costs $560–$635 billion each year, exceeding annual cost estimates for heart disease, cancer, and diabetes.1,2 The conditions most frequently associated with chronic pain include low back pain and osteoarthritis.3,4 Chronic pain associated with these conditions can cause significant impairments in overall quality of life, mental health, employment status, sleep, and personal relationships.5–7
Clinical guidelines support the use of opioids as part of a comprehensive pain management program for patients with chronic, moderate-to-severe pain.8–11 Short-acting opioids (eg, immediate-release [IR] hydromorphone, IR hydrocodone, IR oxycodone) typically provide 4–6 hours of pain relief and have relatively short plasma half-lives that require frequent dosing, potentially reducing medication compliance.12,13 Extended-release (ER) opioid formulations may offer a longer duration of effect and less frequent dosing13; however, they require an increase in total medication load, which introduces a need to protect against the potential for abuse when tablets are manipulated for alternative routes of administration (eg, insufflation, injection) or dose dumping when mixed with alcohol.14 As a result, the US Food and Drug Administration has emphasized that the development of abuse-deterrent opioid formulations is a high public health priority.15
Until recently, hydrocodone bitartrate was available in the US only in IR formulations in combination with other medications such as acetaminophen and ibuprofen.16 Acetaminophen-induced hepatic toxicity and nonsteroidal anti-inflammatory drug (NSAID)-related gastrointestinal/renal/cardiovascular effects are important safety concerns for patients taking these hydrocodone combination products.17–19 Acetaminophen overdose is a leading cause of acute liver failure in the US, prompting the US Food and Drug Administration to reduce the maximum dosage strength of acetaminophen in prescription combination products.20,21 The non-opioid components of hydrocodone combination products can also limit successful opioid dose titration because they are associated with a dosage ceiling beyond which no further pain relief is generally seen, although additional adverse events (AEs) may occur.22
A new, single-agent (ie, acetaminophen and ibuprofen free) ER formulation of hydrocodone bitartrate (Teva Pharmaceuticals, Frazer, PA, USA) that employs the CIMA® Abuse-Deterrence Technology (ADT) platform (CIMA LABS, Inc., Brooklyn Park, MN, USA) was developed23 to provide sustained pain relief with twice-daily dosing. The physical properties of this formulation were designed to limit the effects of alcohol and tablet manipulation on drug release, thus reducing the potential for abuse and adverse consequences of overdose. In Phase I studies, this formulation of hydrocodone ER was well tolerated and shown to have a dose-proportional pharmacokinetic profile that was qualitatively similar after single- and multiple-dose administration.24,25 The objective of this study was to evaluate the efficacy and safety of this formulation of hydrocodone ER tablets at doses of 15–90 mg administered every 12 hours compared with placebo in alleviating moderate-to-severe chronic pain in patients with osteoarthritis or low back pain.
Methods
This Phase III, randomized, double-blind, placebo-controlled study was conducted at 71 sites in the US between November 2010 and August 2011 (http://ClinicalTrials.gov, NCT01240863). The study was conducted in full accordance with the International Conference on Harmonization Good Clinical Practice: Consolidated Guideline and any applicable national and local laws and regulations.26 Before the study was initiated, the protocol was reviewed and approved by each site’s independent ethics committee or institutional review board. Written informed consent was obtained from each patient before any study procedures or assessments were done.
Patients
Men and women of 18–80 years were eligible for inclusion in the study if they had pain of ≥3 months’ duration associated with osteoarthritis or low back pain and an average pain intensity (API) score ≥5 on an eleven-point numerical rating scale (0=no pain, 10=worst pain imaginable) during the previous 24 hours. For patients receiving physical therapy, biofeedback therapy, acupuncture therapy, or herbal remedies, these therapies had to remain unchanged during the study. Women of childbearing potential were required to use a medically accepted method of contraception and agree to continue its use for the duration of the study and for 30 days after participation in the study; patients had to have a negative pregnancy test at screening. Patients were categorized as either opioid naïve (taking <10 mg/day of oxycodone, or equivalent, for 14 days before screening) or opioid experienced (taking ≥10 mg/day but ≤135 mg/day of oxycodone, or equivalent, including around-the-clock medication and rescue medications, for 14 days before screening) at the time of enrollment.
Patients were excluded from the study if their primary painful condition was related to a source of chronic pain other than osteoarthritis or low back pain, if they had a recent history (≤5 years) or current evidence of alcohol or other substance abuse (except nicotine or caffeine), or were taking >135 mg/day of oxycodone, or equivalent, during the 14 days before screening. In addition, they were excluded if they had any medical or psychiatric disease that could compromise data collection or any cardiopulmonary disease that would significantly increase the risk of treatment with opioids; a positive urine drug screen that was not medically explainable; active malignancy, human immunodeficiency virus, or surgery planned during the study; treatment with a monoamine oxidase inhibitor within 14 days; any clinically significant deviation from normal in physical examination and/or clinical laboratory test values; and any hypersensitivities, allergies, or other contraindications to any ingredient in the study medication.
Study design
The study consisted of a screening period, an open-label titration period, and a double-blind treatment period (Figure 1). During the screening period, investigators screened patients per the inclusion/exclusion criteria, recorded patients’ medical and medication history, and conducted detailed pain assessments, clinical laboratory tests, electrocardiograms, urine drug screening, and a physical examination (including vital sign measurements).
Figure 1.
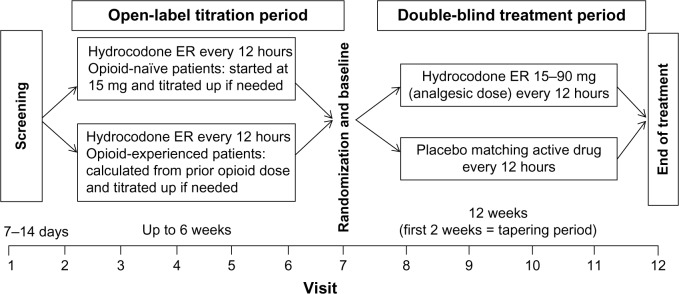
Phase III, placebo-controlled study of hydrocodone ER in treatment of chronic osteoarthritis or low back pain: study design.
Abbreviation: ER, extended release.
During the open-label titration period, eligible patients titrated to an analgesic dose (ie, stable pain relief without unacceptable AEs) of hydrocodone ER (15 mg, 30 mg, 45 mg, 60 mg, or 90 mg every 12 hours) (Teva Pharmaceuticals); patients were maintained on their analgesic dose for up to 7 days. Stable pain relief was defined as an API score of ≤4 within the past 24 hours for 3 consecutive days or for at least 3 of 5 consecutive days. Opioid-naïve patients initiated titration with hydrocodone ER 15 mg administered every 12 hours. Opioid-experienced patients switched from their previous opioid medications to a percentage of their calculated equianalgesic dose of hydrocodone ER (Figure S1). Follow-up visits (generally ~7 days apart) and daily telephone calls were scheduled to review pain intensity scores, medication usage, and other study assessments recorded in an electronic patient diary. If an analgesic dose was identified via telephone before a scheduled visit, patients were to continue taking hydrocodone ER at that dose and return to the clinic within 7 days for randomization into the double-blind treatment period. Patients who were unable to identify an analgesic dose were discontinued from the study. If needed, patients were allowed to administer NSAIDs (eg, ibuprofen at a maximum dosage of 2,400 mg/day) for conditions other than the primary pain condition for a maximum of 10 consecutive days; no additional analgesic medications were permitted during open-label titration. Patients completing the open-label titration period entered a double-blind treatment period.
During the double-blind treatment period, patients were randomized 1:1 to administer placebo or hydrocodone ER at the identified analgesic dose (15–90 mg) every 12 hours for 12 weeks. The randomization code was generated by the study sponsor and reviewed by a statistician not assigned to the study. Patients were randomly assigned with an interactive voice/web response system to ensure balance across treatment groups. Patients, investigators, and all study sponsor clinical personnel remained blinded throughout the study. Randomization was stratified by opioid treatment status at study enrollment (ie, opioid naïve or opioid experienced). To reduce the risk of opioid withdrawal in patients randomized to receive placebo, a step-wise, double-blind tapering schedule was implemented during the first 2 weeks of the 12-week, double-blind, treatment period. The tapering schedule varied according to the analgesic dose of hydrocodone ER identified in the titration period. During tapering, patients were permitted to administer hydrocodone/acetaminophen 5 mg/325 mg every 4–6 hours as needed, for a total daily hydrocodone dose of 30 mg.
Starting at week 3 and for the remainder of the double-blind treatment period until week 12 (or early termination), patients received placebo or the hydrocodone ER dose to which they were randomized. Patients were permitted to administer hydrocodone/acetaminophen tablets 5/325 mg as needed, up to a total daily hydrocodone dose of 10 mg. Those who increased the daily dose of supplemental medication above hydrocodone 10 mg/day on two occasions or who required 7 continuous days of rescue medication use were discontinued from the study. NSAIDs were also permitted on an as-needed basis (eg, ibuprofen at a maximum dosage of 2,400 mg/day). Daily telephone calls were scheduled for days 1–6, 8–13, and 15–20, and clinic visits were scheduled at weeks 1, 2, 4, 8, and 12 after randomization.
Study assessments
Efficacy
Patients recorded their API and worst pain intensity (WPI) scores daily in an electronic diary using an eleven-point numeric rating scale (0=no pain, 10=worst pain imaginable). The primary efficacy measure was the change from baseline (final visit of the open-label titration period) to week 12 in the mean weekly API. The baseline weekly API score was calculated by averaging API scores over the 3–12 days at the end of the open-label titration period, when the analgesic dose of hydrocodone ER was confirmed, before randomization. Decreases in mean weekly API score indicated that patients had less pain and showed improvement.
Secondary efficacy measures included the change from baseline in the weekly average of daily API scores at weeks 1, 2, 4, and 8; the percentage of patients with >33% and >50% increases from baseline in weekly API at weeks 1, 2, 4, 8, and 12; change from baseline in the weekly average of daily WPI scores at weeks 1, 2, 4, 8, and 12; and use of supplemental opioid medication during the double-blind treatment period.
Additional secondary measures included functional improvement and change in overall patient status, evaluated through the use of several questionnaires. The Patient Assessment of Function (PAF) and Clinician Assessment of Patient Function (CAPF) questionnaires, completed at weeks 4, 8, and 12, were used to measure a patient’s ability to function in normal activities. Answers on the PAF and CAPF were rated on a seven-point numeric scale (1=very much worsened, 7=very much improved). The Clinical Global Impression of Severity of Illness (CGI-S) questionnaire,27 completed prior to open-label titration, at baseline, and at weeks 1, 2, 4, 8, and 12, was used to assess the severity of the patient’s pain condition and response to treatment. Answers were rated based on seven categories: normal (shows no signs of illness), borderline ill, mildly (slightly) ill, moderately ill, markedly ill, severely ill, and most extremely ill. The Brief Pain Inventory-Short Form (BPI-SF),28 completed prior to open-label titration, at baseline, and at weeks 1, 2, 4, 8, and 12, was used to measure the severity of pain and impact of pain on daily functions. The 36-Item Short Form (SF-36) Health Survey,29,30 a clinician-rated scale completed prior to open-label titration, at baseline, and at week 12, was used to assess physical functioning, role limitations due to physical health problems, bodily pain, social functioning, general mental health, emotional problems, vitality, energy or fatigue, and general health perceptions.
A post hoc analysis was conducted to assess change in weekly API scores, which excluded patients administering the lowest dose of hydrocodone ER (15 mg, n=36) or matching placebo (n=43).
Safety
Safety and tolerability were assessed throughout the study by monitoring AEs and serious AEs, clinical laboratory test results (chemistry, hematology, and urinalysis), electrocardiogram findings, physical examination findings (including vital sign measurements), and concomitant medication use. Vital signs (pulse, systolic and diastolic blood pressure) were measured at screening and each study visit during the open-label titration and double-blind treatment periods. Clinical laboratory tests and 12-lead electrocardiograms were conducted at screening and the last visit during the double-blind treatment period (week 12 or early termination). Additional safety measures included pure tone audiometry, Subjective Opioid Withdrawal Scale (SOWS),31 Clinical Opiate Withdrawal Scale (COWS),32 Screener and Opioid Assessment for Patients with Pain–Revised (SOAPP-R),33 Addiction Behaviors Checklist (ABC),34 and the Current Opioid Misuse Measure (COMM).35 Pure tone audiometry was performed by the individual study centers before and during the open-label titration period (before study drug exposure at visit 2 and after 1 week of exposure to the study drug at visit 3), at baseline, and at the final visit of the double-blind treatment period (week 12 or early termination). Results of the SOWS were collected in the patient diary daily during the first 4 weeks of the double-blind treatment period, and then assessed at weeks 8 and 12. The COWS was administered at baseline and each visit of the double-blind treatment period. The SOAPP-R was administered at the beginning of the open-label titration period, and the ABC and COMM were administered at the beginning of the open-label titration period, at baseline, and at weeks 1, 4, 8, and 12 of the double-blind treatment period.
Statistical analysis
An estimated 266 evaluable patients were required to be enrolled in the double-blind treatment period to achieve a 90% power to detect an effect size of ≥0.4 on the primary efficacy variable (two-sided t-test; significance level of 0.05). All randomized patients who received at least one dose of study medication during the double-blind treatment period were evaluable for primary and secondary efficacy analyses. The primary efficacy measure was analyzed using an analysis of covariance model, with treatment and previous opioid treatment status as fixed effects and screening and baseline pain intensities as covariates. Comparisons for secondary efficacy variables (weekly API, weekly WPI, CGI-S, BPI-SF, and SF-36) were assessed using the same analysis of covariance model. A multiple imputation method was used to manage missing API and WPI data. Comparisons between hydrocodone ER and placebo for percentage of patients with >33% and >50% increases in weekly API, CAPF, and PAF were assessed using a Cochran–Mantel–Haenszel test, controlling for the randomization stratus effect. All statistical tests were two sided. Nominal P-values were reported without adjusting for multiplicities. All statistical analyses were conducted using SAS version 9.1.3 (SAS Institute, Cary, NC, USA).
All patients who received at least one dose of study medication were included in the safety analyses. AEs were summarized by treatment group. All safety comparisons were summarized descriptively.
Post hoc analyses assessing the change in mean weekly API scores, excluding patients administering the lowest dose of hydrocodone ER (15 mg) or matching placebo, were also conducted.
Results
Patients
Patient disposition is summarized in Figure 2. A total of 519 patients were screened, 391 patients were enrolled, and 389 patients received at least one dose of hydrocodone ER in the titration period and were evaluable for safety. A total of 294 (75%) patients identified an analgesic dose of hydrocodone ER and were randomly assigned in the double-blind treatment period to receive hydrocodone ER (n=146) or placebo (n=148). Twenty-seven percent of patients were titrated to a hydrocodone ER analgesic dose of 30 mg/day, 26% were titrated to 60 mg/day, 22% were titrated to 90 mg/day, 15% were titrated to 120 mg, and 11% were titrated to 180 mg/day. In all, 293 patients were evaluable for efficacy (one patient randomized to receive placebo did not receive study medication) and 196 completed the double-blind treatment period, 94 (64%) in the hydrocodone ER group and 102 (69%) in the placebo group.
Figure 2.
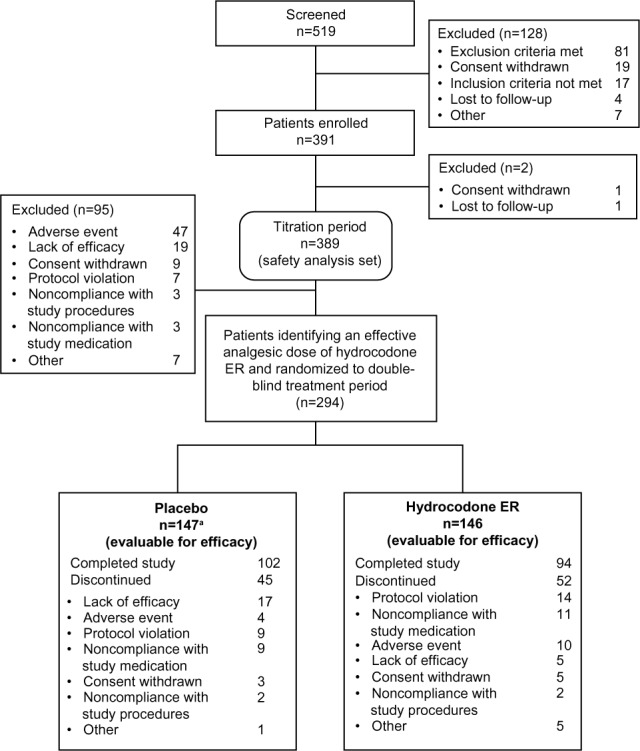
Patient disposition in Phase III study of hydrocodone ER in patients with chronic osteoarthritis or low back pain.
Note: aOne patient randomized to receive placebo did not receive study medication and was therefore not evaluable for efficacy.
Abbreviation: ER, extended release.
Patient demographics and other baseline characteristics were similar between treatment groups (Table 1). The mean (standard deviation) age of randomized patients was 53.1 (11.3) years, and the majority (72%) of patients had low back pain. Half of the patients were opioid naïve, and half were opioid experienced.
Table 1.
Baseline patient demographics and characteristics – full analysis set
| Placebo (n=148) | Hydrocodone ER (n=146) | Total (N=294) | P-value (placebo vs hydrocodone ER) | |
|---|---|---|---|---|
| Age, mean (SD), years | 52.7 (12.1) | 53.6 (10.4) | 53.1 (11.3) | 0.53a |
| Female, n (%) | 88 (59) | 87 (60) | 175 (60) | 0.98b |
| Race, n (%) | ||||
| White | 105 (71) | 115 (79) | 220 (75) | 0.21b |
| Black | 41 (28) | 28 (19) | 69 (23) | |
| Other | 2 (1) | 3 (2) | 5 (2) | |
| BMI, mean (SD), kg/m2 | 32.8 (7.3) | 33.0 (8.2) | 32.9 (7.7) | 0.87a |
| Type of pain, n (%) | ||||
| Low back pain | 107 (72) | 104 (71) | 211 (72) | –e |
| Osteoarthritis | 41 (28) | 42 (29) | 83 (28) | |
| Duration since diagnosis, mean (SD), years | 12.5 (9.1) | 12.1 (10.0) | 12.3 (9.5) | – |
| Opioid status, n (%) | ||||
| Opioid-naïve | 72 (49) | 75 (51) | 147 (50) | 0.64b |
| Not on opioid therapy | 45 (30) | 46 (32) | 91 (31) | |
| Opioid-experienced | 76 (51) | 71 (49) | 147 (50) | |
| Duration on opioid therapy, mean (SD), years | 4.1 (4.9) | 3.9 (4.8) | 4.0 (4.8) | – |
| Weekly average API, mean (SE)c,d | 6.6 (0.1) | 6.6 (0.1) | – | – |
Notes:
P-values based on ANOVA model with treatment as the factor for continuous variables;
P-values for categorical variables based on Pearson’s chi-square test;
values at screening prior to the open-label titration period;
n=147 for placebo group;
dashes indicate this value was not calculated.
Abbreviations: ANOVA, analysis of variance; API, average pain intensity; BMI, body mass index; ER, extended-release; SD, standard deviation; SE, standard error.
Efficacy
From baseline to week 12, the adjusted least squares mean (LSM) of weekly API decreased for patients in the hydrocodone ER group (−0.22), indicating the patients had less pain and showed improvement, but this value increased for patients in the placebo group (0.14), indicating patients had worsening pain. However, the primary efficacy measure of the change from baseline to week 12 in mean weekly API score was not significantly different between placebo and hydrocodone ER (P=0.134). Mean weekly API scores during the double-blind treatment period are shown in Figure 3. Significant differences between hydrocodone ER and placebo were observed at week 2 (LSM: −0.17 vs 0.14 [P=0.049]) and week 8 (LSM: −0.61 vs 0.02 [P=0.002]).
Figure 3.
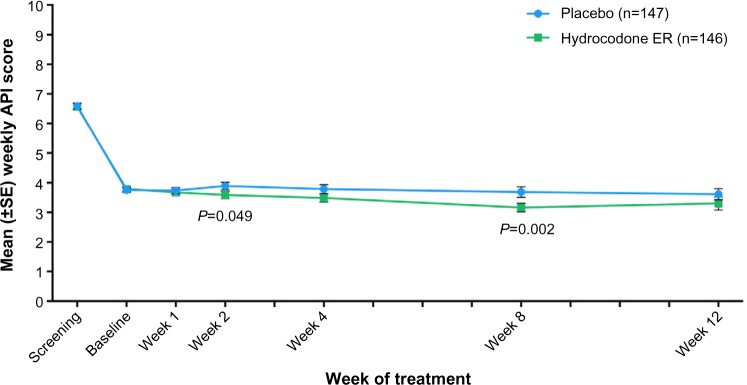
Mean (±SE) weekly average pain intensity (API) scores over time for patients in the placebo and hydrocodone extended release (ER) treatment groups.
Abbreviation: SE, standard error.
A significantly higher proportion of patients had >33% increases from baseline in mean weekly API with placebo vs hydrocodone ER starting at week 2 (22% vs 11% [P=0.016]) and persisting through week 12 (24% vs 11% [P=0.017]; Figure 4A). In addition, a significantly higher incidence of >50% increases from baseline were observed with placebo vs hydrocodone ER at week 8 (18% vs 6% [P=0.005]) and week 12 (15% vs 6% [P=0.041]; Figure 4B). The higher increases seen in the placebo group occurred irrespective of whether the patients were opioid naïve or opioid experienced.
Figure 4.

Percentage of patients with increases from baseline in the mean weekly average pain intensity (API) score exceeding (A) 33% and (B) 50%.
Abbreviation: ER, extended release.
Mean weekly WPI scores during the double-blind treatment period are shown in Figure 5. The change from baseline in the mean weekly WPI score was significantly different with placebo vs hydrocodone ER at week 2 (0.27 vs −0.15 [P=0.025]), week 8 (0.00 vs −0.68 [P=0.004]), and week 12 (−0.03 vs −0.60 [P=0.025]).
Figure 5.
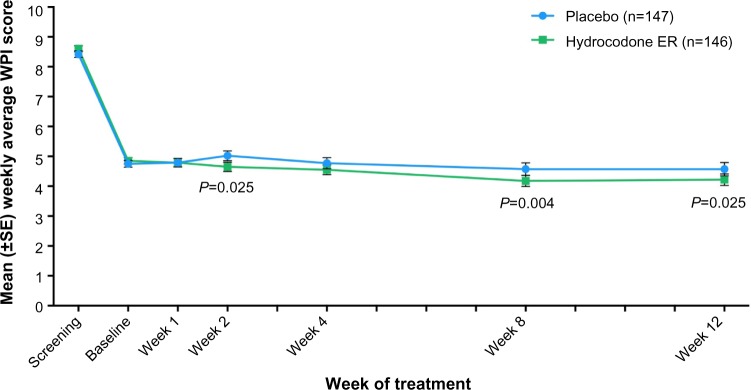
Mean (±SE) weekly average worst pain intensity (WPI) scores over time for patients in the placebo and hydrocodone extended-release (ER) treatment groups.
Abbreviation: SE, standard error.
A larger percentage of patients in the placebo group required supplemental opioid medication to control pain during the double-blind treatment period compared with patients in the hydrocodone ER group (86% vs 79%). During weeks 1–12, the mean daily dose of opioid rescue medication (hydrocodone equivalent dose) ranged from 2.2 mg to 5.4 mg in the placebo group and from 2.0 mg to 3.5 mg in the hydrocodone ER group (Figure 6). The mean daily dose of opioid rescue medication taken during the 2-week tapering schedule of the double-blind treatment period was higher among placebo-treated patients (4.4 mg) compared with hydrocodone ER-treated patients (2.7 mg). After the 2-week taper, during weeks 3–12 of the double-blind treatment period, the mean dose of rescue medication was 3.3 mg for the placebo-treated patients and 2.4 mg for the hydrocodone ER-treated patients.
Figure 6.
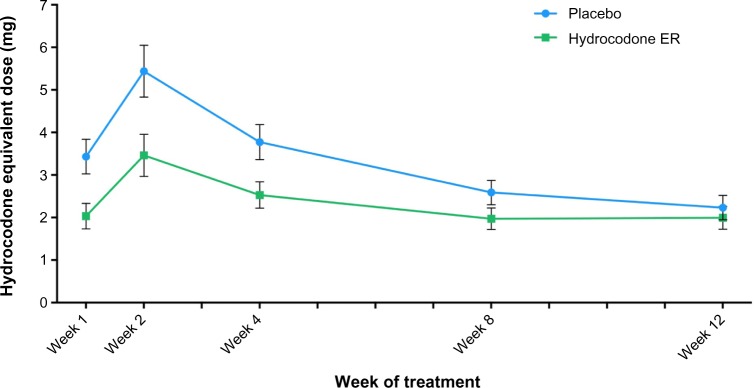
Mean (±SE) daily dosage of opioid supplemental medication (hydrocodone equivalent dose) in the placebo and hydrocodone extended-release (ER) treatment groups from week 1 to week 12 of the double-blind treatment period.
Abbreviation: SE, standard error.
No significant differences were observed between placebo and hydrocodone ER on the PAF, CAPF, CGI-S, or most subscales of the BPI-SF or SF-36.
Post hoc analyses
Post hoc analyses were conducted in an attempt to determine a possible reason that a statistically significant difference between the hydrocodone ER and placebo groups on the primary efficacy measure (API) was not attained. Patients receiving the lowest titration dose of hydrocodone ER (15 mg) or matching placebo composed 27% (79/293) of the patients randomized. Post hoc analyses that excluded these patients found that the change from baseline to week 12 in the mean weekly API score (primary efficacy measure) was significantly lower in the hydrocodone ER group vs the placebo group (LSM: −0.20 vs 0.40 [P=0.032]). Analyses of the change from baseline to week 12 in weekly API score by dose groups of hydrocodone ER did not show statistically significant differences between the hydrocodone ER and the placebo groups. Mean weekly API scores for patients in the hydrocodone ER 15 mg group and matching placebo group were lower at screening (5.86 and 6.07, respectively) compared with the higher dose hydrocodone ER and matching placebo groups (hydrocodone ER, 6.57–7.19; placebo, 6.63–7.19). However, at baseline of the double-blind treatment period, mean weekly API scores were similar between the hydrocodone ER 15 mg and matching placebo groups (3.69 and 3.70, respectively) and the higher dose hydrocodone ER and matching placebo groups (hydrocodone ER, 3.75–3.88; placebo, 3.67–4.01).
Safety and tolerability
AEs occurring in ≥5% of patients in any treatment group are summarized by study period in Table 2. A total of 111 (59%) opioid-naïve and 116 (58%) opioid-experienced patients reported at least one AE during the open-label titration period. The most frequent AE was nausea, occurring in 39 (21%) opioid-naïve and 31 (16%) opioid-experienced patients. During the double-blind treatment period, 93 (64%) patients in the hydrocodone ER group and 91 (62%) patients in the placebo group reported at least one AE. The most frequently occurring AEs were constipation (13% vs 5%, respectively) and nausea (13% vs 6%, respectively).
Table 2.
Adverse events occurring in ≥5% of patients
| Open-label titration period – safety analysis set
|
Double-blind treatment period – full analysis set
|
|||
|---|---|---|---|---|
| Opioid-naïve (n=189) | Opioid-experienced (n=200) | Placebo (n=147) | HydrocodoneER (n=146) | |
| Patients with ≥1 AE, n (%) | 111 (59) | 116 (58) | 91 (62) | 93 (64) |
| AEs with incidence ≥5%, n (%) | ||||
| Nausea | 39 (21) | 31 (16) | 9 (6) | 19 (13) |
| Headache | 17 (9) | 26 (13) | 8 (5) | 10 (7) |
| Back pain | 0 | 3 (2) | 7 (5) | 1 (<1) |
| Constipation | 28 (15) | 27 (14) | 7 (5) | 19 (13) |
| URTI | 3 (2) | 1 (<1) | 7 (5) | 7 (5) |
| Vomiting | 15 (8) | 10 (5) | 5 (3) | 9 (6) |
| Dry mouth | 9 (5) | 6 (3) | 2 (1) | 2 (1) |
| Fatigue | 8 (4) | 10 (5) | 2 (1) | 1 (<1) |
| Dizziness | 14 (7) | 8 (4) | 1 (<1) | 3 (2) |
| Pruritus | 16 (8) | 4 (2) | 1 (<1) | 3 (2) |
| Somnolence | 23 (12) | 22 (11) | 1 (<1) | 3 (2) |
Note: All values are number of patients (percentage of patients).
Abbreviations: AE, adverse event; ER, extended release; URTI, upper respiratory tract infection.
Sixty patients were discontinued from the study because of AEs; 48 during the open-label titration period (opioid naïve, n=33; opioid experienced, n=15) and 12 during the double-blind treatment period (hydrocodone ER, n=9; placebo, n=3). AEs leading to discontinuation in at least two patients included nausea (n=17), somnolence (n=14), vomiting (n=6), dizziness (n=6), constipation (n=5), headache (n=3), and fatigue, decreased appetite, sedation, insomnia, nervousness, and hyperhidrosis (n=2 each) in the open-label titration period and somnolence (n=2) in the double-blind treatment period.
No deaths or AEs of respiratory depression were reported during the study. Eight patients had at least one serious AEs during the study; two (1%) opioid-experienced patients during the open-label titration period and six patients during the double-blind treatment period (hydrocodone ER, n=3 [2%]; placebo, n=3 [2%]). The only serious AEs considered by the investigator to be treatment related were two episodes of acute pancreatitis, which occurred in one patient with a history of cholecystectomy in the hydrocodone ER group. This patient subsequently developed chronic pancreatitis.
There were no clinically meaningful changes from baseline to endpoint in pulse or systolic and diastolic blood pressure values after treatment with hydrocodone ER. Small mean changes from baseline to endpoint in electrocardiogram parameters were seen among patients receiving hydrocodone ER compared with patients receiving placebo. The changes were consistent across time and were not considered to be clinically meaningful. There were also no clinically meaningful trends in mean changes from baseline to endpoint in pure tone audiometry results for patients treated with hydrocodone ER. Mean changes from baseline to final values were minimal at all hearing thresholds (range −2.6 to 1.3 decibel levels).
Mean SOWS scores and the proportion of patients in each category of withdrawal symptoms (normal, mild, moderate, moderately severe, or severe withdrawal) were similar for patients in the placebo and hydrocodone ER treatment groups at each visit. At endpoint (last observation carried forward), the majority of patients classified their opioid withdrawal symptoms as normal (hydrocodone ER, 74%; placebo, 79%). No patients in either treatment group classified themselves as having severe withdrawal symptoms at any time point during the study. Mean COWS scores and the proportions of patients in each category of withdrawal symptoms were also similar between treatment groups at each visit. At endpoint, the majority of patients in both treatment groups were categorized by their physicians as having normal opioid withdrawal symptoms (hydrocodone ER, 95%; placebo, 97%). No patient in either treatment group was categorized as having moderately severe or severe withdrawal symptoms at any time point.
The majority (~75%) of patients randomized to hydrocodone ER (n=104; 71%) and placebo (n=116; 78%) had SOAPP-R total scores <18, which was predictive of a low risk for aberrant drug-related behaviors. At the beginning of the open-label titration period (visit 2), 22 patients (hydrocodone ER, n=15; placebo, n=7) had positive results for aberrant drug-use behaviors, as indicated by SOAPP-R total score ≥18 and COMM total score ≥9, and three patients (hydrocodone ER, n=2; placebo, n=1) had positive results for inappropriate opioid use behaviors, as indicated by SOAPP-R total score ≥18 and ABC total score ≥3. At endpoint, the majority (72%) of patients had no indication of aberrant drug-use behavior or inappropriate opioid use; 210 patients (hydrocodone ER, n=100; placebo, n=110) had SOAPP-R total score <18 and COMM total score <9, and 212 patients (hydrocodone ER, n=101; placebo, n=111) had SOAPP-R total score <18 and ABC total score <3.
Four patients (1%) reported study drug diversion during the study; three reported theft of rescue medication (hydrocodone/acetaminophen; two of these patients were withdrawn from the study) and the fourth patient returned unknown medication instead of hydrocodone ER. Thirty-five (<9%) patients enrolled in the study reported loss of study drug (hydrocodone ER, rescue medication, and/or placebo tablets). The most frequent reason for loss of study drug was the patient dropping the tablets and not being able to retrieve them (eg, in the sink, in the toilet, behind the refrigerator, or on the floor). Twenty nine of these 35 patients reported loss of at least one tablet of hydrocodone ER, four reported loss of at least one tablet of rescue medication, and eight reported loss of at least one tablet of placebo.
Discussion
In this randomized, double-blind, placebo-controlled study of patients with osteoarthritis or low back pain, the difference between the hydrocodone ER and placebo groups was not statistically significant for the primary efficacy measure of change from baseline to week 12 in mean weekly API score. A post hoc analysis of the primary efficacy measure showed that the change from baseline to week 12 in mean weekly API was significantly lower in the hydrocodone ER group vs the placebo group when the lowest hydrocodone ER dose group (15 mg) and matching placebo group were excluded from the analysis. Significant differences were observed on several secondary measures, including the proportion of patients with >33% and >50% increases in weekly API and mean WPI scores. For the other secondary efficacy measures (PAF, CAPF, CGI-S, BPI-SF, and SF-36 scores), there were generally no statistically significant differences (P<0.05) between the hydrocodone ER and placebo groups.
Several potential reasons may have contributed to the lack of a statistically significant difference in the primary efficacy measure between hydrocodone ER and placebo. Patients who were randomized to receive placebo did not show a worsening in pain scores as would be expected. Only 24 of 98 (24%) patients in the placebo group showed a >33% increase in weekly API and only 15 of 98 (15%) showed a >50% increase; the vast majority of patients in the placebo group showed decreases in weekly API. In part, these results may be explained by a high placebo response rate, a phenomenon commonly observed in chronic pain studies.36 Placebo response has been cited as a potential cause of study failure in several previous pain studies in which the medications being evaluated did not significantly separate from placebo on the primary efficacy analyses.37–39
In the current study, more patients in the placebo group compared with the hydrocodone ER group took supplemental opioid medication (86% vs 79%, respectively) and had a higher mean dose of opioid medication (2.2–5.4 mg vs 2.0–3.5 mg, respectively). Patients were also permitted to administer NSAIDs on an as-needed basis. This may have provided adequate pain relief in patients receiving placebo and prevented the demonstration of worsening pain as was predicted. Additionally, it is possible that at the time of randomization, patients may not have appropriately titrated to an analgesic dose of hydrocodone ER. The weekly API scores reported in patients at the end of the open-label titration period (3.75–3.79 on an eleven-point numeric rating scale) were relatively high compared with post-titration pain scores observed in previous similarly designed studies of oxymorphone ER in opioid-naïve patients (API, 18.5–19.3 on a 100-point visual analog scale)40 and opioid-experienced patients (API, 22.2–23.9 on a 100-point visual analog scale).41
Another possible reason why a statistically significant difference between treatment groups was not demonstrated in this study was the inclusion of the hydrocodone ER 15 mg dose as an analgesic dose. Hydrocodone ER 15 mg every 12 hours is equivalent to approximately 10–15 mg of oxycodone controlled release (CR) tablets taken every 12 hours or 5–10 mg of oxymorphone ER tablets taken every 12 hours. In clinical practice, 10 mg of oxycodone CR or 5 mg of oxymorphone ER is a starting dose in opioid-naïve patients.42,43 While a starting dose may be efficacious in clinical practice, it is unlikely to statistically separate from placebo in randomized, placebo-controlled clinical studies. This was shown in a study conducted by Roth et al in which oxycodone CR 20 mg significantly reduced pain intensity vs placebo but 10 mg did not.44 In other randomized placebo-controlled studies that assessed the efficacy of oxymorphone ER in patients with chronic low back pain, opioid-naïve patients were stabilized on a mean daily dose of approximately 40 mg/day and opioid-experienced patients were stabilized on a mean daily dose of approximately 90 mg/day prior to randomization, which is equal to approximately 80 mg/day and 180 mg/day of hydrocodone ER, respectively.40,41 In the current study, approximately half of the patients were treated with doses of hydrocodone ER that were lower than 80–180 mg/day. Thus, it is likely that doses higher than 15 mg of hydrocodone ER are needed to show differences from placebo, as was demonstrated in the post hoc analysis of the primary efficacy measure conducted without the approximately 27% of patients in the hydrocodone ER 15 mg dose group. This post hoc analysis showed a statistically significant difference in weekly mean API from baseline to week 12 between the hydrocodone ER and placebo treatment groups (P=0.032). Thus, the 15 mg dose of hydrocodone ER should have been considered to be a titration dose rather than an analgesic dose. Patients who responded to hydrocodone ER 15 mg had a slightly lower baseline level of pain (API score of ~6) compared with the higher dose hydrocodone ER groups (API score of ~6.5–7). These patients may have also had increased pain tolerance, which may have made it more difficult to demonstrate a between-group difference in the double-blind treatment period of the study.
Hydrocodone ER was generally well tolerated in this study. The majority of AEs were opioid-related and consistent with the known safety profile of hydrocodone.19,45 The incidences of AEs were similar between treatment groups during the open-label and double-blind periods; the most common AEs that occurred during both the open-label titration period and double-blind treatment period with hydrocodone ER were nausea and constipation. Eight patients had serious AEs; only for one of these patients was the serious AE of pancreatitis determined by the investigator to be related to study medication (ie, hydrocodone ER). All opioids have the potential to cause spasm of the sphincter of Oddi, causing subsequent pancreatitis, particularly in patients with histories of cholecystectomy,46 as was the case for this patient. Thus, the occurrence of pancreatitis in a single hydrocodone ER-treated patient in this study does not affect the overall safety profile of hydrocodone ER.
There are limited data on rates of abuse, addiction, or other aberrant drug-related behaviors from randomized controlled trials of chronic opioid therapy in patients with chronic noncancer pain. A systematic review and meta-analysis of primarily open-label, uncontrolled, observational studies in patients receiving opioids for chronic noncancer pain reported opioid addiction in 0.05% (1/2,042) and abuse in 0.43% (3/685) of patients.47 A separate systematic review of opioids for low back pain reported aberrant drug-related behaviors or substance use disorders in 5%–24% of patients.48 The majority of studies included in both reviews did not report rates of opioid addiction, abuse, or aberrant drug-related behaviors; among those that did, differences in study methodologies and diagnostic criteria used to assess addiction/abuse likely contribute to the varying rates reported. Overall in this study, there was a relatively low occurrence of inappropriate opioid use and aberrant drug-use behavior, as predicted by SOAPP-R total scores at visit 2 and confirmed by the results of the ABC questionnaire, which showed a low occurrence of inappropriate opioid use, and the COMM questionnaire, which showed that most patients had no aberrant drug-use behaviors. Incidence of study drug diversion (ie, theft; 1%) and loss (<9%) was also low.
ER opioid formulations offer several advantages over short-acting formulations. The release of opioid medication over time in a controlled manner provides therapeutic levels of analgesia with minimal fluctuations in the degree of exposure,49 and the convenience of once- or twice-daily dosing may improve patient compliance.50,51 Furthermore, no hydrocodone ER formulations with potentially abuse-deterrent properties are currently available. Thus, the safety and efficacy of this hydrocodone ER formulation developed with the CIMA ADT platform merits further study, and the findings from this study provide several important considerations for the design of future analgesic trials. Careful attention should be given to the types of rescue and concomitant analgesics permitted during the study. In the current study, patients could administer NSAIDs on an as-needed basis, as well as hydrocodone/acetaminophen tablets (5 mg/325 mg) as needed, to a maximum total daily hydrocodone dose of 10 mg, which may have contributed to the improvements in pain intensity observed in patients administered placebo. Additionally, although low doses of opioids may be efficacious in clinical practice, they may be unlikely to statistically separate from placebo in randomized controlled trials. As such, future trials should consider limiting low doses of opioids to titration doses but not as an option for an analgesic dose during the actual treatment phase. A Phase III study that does not include the 15 mg dose of hydrocodone ER is currently ongoing to further evaluate the efficacy and safety of this hydrocodone ER tablet formulated with the CIMA ADT platform in patients with chronic pain.
Conclusion
Hydrocodone ER tablets, at doses of 15–90 mg administered every 12 hours, were generally safe and well tolerated. Although this study did not meet its primary endpoint, hydrocodone ER was associated with a statistically significant improvement in patient’s pain vs placebo as measured by the change from baseline to week 12 in mean weekly WPI score (secondary endpoint). These results combined with the established efficacy of hydrocodone as an analgesic generally supports the effectiveness of this hydrocodone ER formulation for treating chronic pain. The AEs observed in this study were consistent with those observed with other opioid products, and there was a low occurrence of inappropriate opioid use and aberrant drug-use behaviors.
Supplementary material
Selection of hydrocodone ER starting dose for opioid-experienced patients.
Note: aIf the patient reported a range of daily doses of opioid medication, the highest dose reported was taken as the base to calculate patient’s total daily dose of opioid treatment.
Abbreviation: ER, extended release.
Acknowledgments
Medical writing assistance was provided by Bina J Patel, PharmD, CMPP, of Peloton Advantage, LLC, and was funded by Teva Branded Pharmaceutical Products R & D, Inc. (Frazer, PA). Teva provided a full review of the article. This study was sponsored by Cephalon, Inc. (Frazer, PA), now a wholly owned subsidiary of Teva Branded Pharmaceutical Products R & D, Inc. (Frazer, PA).
Footnotes
Author contributions
All authors participated in the research, acquisition of data, analysis and interpretation of data and/or the development and critical review of the paper. All authors approved the final article for submission and agree to be accountable for all aspects of the work.
Disclosure
MEH conducts research for Teva Pharmaceuticals; RY and RM are employees of Teva Pharmaceuticals; CL was a consultant of Teva Pharmaceuticals at the time of the study; AN was an employee of Cephalon, now a wholly owned subsidiary of Teva Branded Pharmaceutical Products R & D, Inc., at the time of the study. The authors report no other conflicts of interest in this work.
References
- 1.Gaskin DJ, Richard P. The economic costs of pain in the United States. J Pain. 2012;13(8):715–724. doi: 10.1016/j.jpain.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 2.National Heart Lung, and Blood Institute . NHLBI Factbook, Fiscal Year 2012. Bethesda: National Institutes of Health; 2013. [Google Scholar]
- 3.Deyo RA, Mirza SK, Martin BI. Back pain prevalence and visit rates: estimates from US national surveys, 2002. Spine. 2006;31(23):2724–2727. doi: 10.1097/01.brs.0000244618.06877.cd. [DOI] [PubMed] [Google Scholar]
- 4.Johannes CB, Le TK, Zhou X, Johnston JA, Dworkin RH. The prevalence of chronic pain in United States adults: results of an internet-based survey. J Pain. 2010;11(11):1230–1239. doi: 10.1016/j.jpain.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 5.McCarberg BH, Nicholson BD, Todd KH, Palmer T, Penles L. The impact of pain on quality of life and the unmet needs of pain management: results from pain sufferers and physicians participating in an Internet survey. Am J Ther. 2008;15(4):312–320. doi: 10.1097/MJT.0b013e31818164f2. [DOI] [PubMed] [Google Scholar]
- 6.Dominick KL, Ahern FM, Gold CH, Heller DA. Health-related quality of life among older adults with arthritis. Health Qual Life Outcomes. 2004;2:5. doi: 10.1186/1477-7525-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salaffi F, Carotti M, Stancati A, Grassi W. Health-related quality of life in older adults with symptomatic hip and knee osteoarthritis: a comparison with matched healthy controls. Aging Clin Exp Res. 2005;17(4):255–263. doi: 10.1007/BF03324607. [DOI] [PubMed] [Google Scholar]
- 8.Trescot AM, Helm S, Hansen H, et al. Opioids in the management of chronic non-cancer pain: an update of American Society of the Interventional Pain Physicians’ (ASIPP) Guidelines. Pain Physician. 2008;11(2 Suppl):S5–S62. [PubMed] [Google Scholar]
- 9.Trescot AM, Boswell MV, Atluri SL, et al. Opioid guidelines in the management of chronic non-cancer pain. Pain Physician. 2006;9(1):1–39. [PubMed] [Google Scholar]
- 10.Chou R, Fanciullo GJ, Fine PG, et al. American Pain Society-American Academy of Pain Medicine Opioids Guidelines Panel Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain. 2009;10(2):113–130. doi: 10.1016/j.jpain.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chou R, Qaseem A, Snow V, et al. Clinical Efficacy Assessment Subcommittee of the American College of Physicians. American College of Physicians. American Pain Society Low Back Pain Guidelines Panel Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147(7):478–491. doi: 10.7326/0003-4819-147-7-200710020-00006. [DOI] [PubMed] [Google Scholar]
- 12.Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296–1310. doi: 10.1016/s0149-2918(01)80109-0. [DOI] [PubMed] [Google Scholar]
- 13.Argoff CE, Silvershein DI. A comparison of long- and short-acting opioids for the treatment of chronic noncancer pain: tailoring therapy to meet patient needs. Mayo Clin Proc. 2009;84(7):602–612. doi: 10.1016/S0025-6196(11)60749-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walden M, Nicholls FA, Smith KJ, Tucker GT. The effect of ethanol on the release of opioids from oral prolonged-release preparations. Drug Dev Ind Pharm. 2007;33(10):1101–1111. doi: 10.1080/03639040701377292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.US Food Drug Administration Guidance for Industry. Abuse-deterrent opioids – evaluation and labeling. 2013. [Accessed November 21, 2013]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM334743.pdf.
- 16.The American Society of Health-System Pharmacists Hydrocodone. 2011. [Accessed: February 24, 2015]. Available from: http://www.nlm.nih.gov/medlineplus/druginfo/meds/a601006.html.
- 17.Co-Gesic – hydrocodone bitartrate and acetaminophen tablet [package insert] Smyrna, GA: UCB, Inc.; 2011. [Google Scholar]
- 18.Barkin RL. Acetaminophen, aspirin, or ibuprofen in combination analgesic products. Am J Ther. 2001;8(6):433–442. doi: 10.1097/00045391-200111000-00008. [DOI] [PubMed] [Google Scholar]
- 19.Vicodin® (hydrocodone bitartrate and acetaminophen tablets, USP) 5 mg/500 mg [package insert] North Chicago, IL: AbbVie Inc.; 2014. [Google Scholar]
- 20.US Food and Drug Administration Prescription drug products containing acetaminophen; actions to reduce liver injury from unintentional overdose. Fed Register. 2011;76(10):2691–2697. [Google Scholar]
- 21.Bower WA, Johns M, Margolis HS, Williams IT, Bell BP. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007;102(11):2459–2463. doi: 10.1111/j.1572-0241.2007.01388.x. [DOI] [PubMed] [Google Scholar]
- 22.American Pain Society. National Pharmaceutical Counsel Pain: Current Understanding of Assessment, Management, and Treatment. Section III: Types of treatments. 2012. [Accessed July 2, 2013]. Available from: http://pain-topics.org/pdf/Pain_APS_2006.pdf.
- 23.CIMA LABS Inc OraGuard™: tamper-deterrent, alcohol-resistant extended release technology. 2012. [Accessed January 12, 2015]. Available from: http://www.cimalabs.com/technology/oraguard.
- 24.Darwish M, Yang R, Tracewell W, Robertson P, Jr, Bond M. Single- and multiple-dose pharmacokinetics of a hydrocodone bitartrate extended-release tablet formulated with abuse-deterrence technology in healthy, naltrexone-blocked volunteers. Clin Ther. 2015;37(2):390–401. doi: 10.1016/j.clinthera.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Darwish M, Yang R, Tracewell W, Robertson P, Bond M. Dose proportionality of a hydrocodone extended-release tablet formulated with abuse-deterrence technology. Clin Drug Investig. 2015;35(5):291–297. doi: 10.1007/s40261-015-0280-z. [DOI] [PubMed] [Google Scholar]
- 26.International Conference on Harmonisation Working Group ICH Harmonised Tripartite Guideline: Guideline for Good Clinical Practice E6 (R1); International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; June 10, 1996; Washington DC. [Accessed March 1, 2011]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. [Google Scholar]
- 27.Guy W. ECDEU Assessment Manual for Psychopharmacology (Revised) Washington, DC: National Institute of Mental Health, US Department of Health, Education and Welfare; 1976. [Google Scholar]
- 28.Cleeland CS, Ryan KM. Pain assessment: global use of the brief pain inventory. Ann Acad Med Singapore. 1994;23(2):129–138. [PubMed] [Google Scholar]
- 29.Ware JE, Jr, Kosinski M, Bayliss MS, McHorney CA, Rogers WH, Raczek A. Comparison of methods for the scoring and statistical analysis of SF-36 health profile and summary measures: summary of results from the Medical Outcomes Study. Med Care. 1995;33(4 Suppl):AS264–AS279. [PubMed] [Google Scholar]
- 30.Ware JE, Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care. 1992;30(6):473–483. [PubMed] [Google Scholar]
- 31.Handelsman L, Cochrane KJ, Aronson MJ, Ness R, Rubinstein KJ, Kanof PD. Two new rating scales for opiate withdrawal. Am J Drug Alcohol Abuse. 1987;13(3):293–308. doi: 10.3109/00952998709001515. [DOI] [PubMed] [Google Scholar]
- 32.Wesson DR, Ling W. The clinical opiate withdrawal scale (COWS) J Psychoactive Drugs. 2003;35(2):253–259. doi: 10.1080/02791072.2003.10400007. [DOI] [PubMed] [Google Scholar]
- 33.Butler SF, Fernandez K, Benoit C, Budman SH, Jamison RN. Validation of the revised screener and opioid assessment for patients with pain (SOAPP-R) J Pain. 2008;9(4):360–372. doi: 10.1016/j.jpain.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu SM, Compton P, Bolus R, et al. The Addiction Behaviors Checklist: validation of a new clinician-based measure of inappropriate opioid use in chronic pain. J Pain Symptom Manage. 2006;32(4):342–351. doi: 10.1016/j.jpainsymman.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 35.Butler SF, Budman SH, Fernandez KC, et al. Development and validation of the current opioid misuse measure. Pain. 2007;130(1–2):144–156. doi: 10.1016/j.pain.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dworkin RH, Turk DC, Peirce-Sandner S, et al. Research design considerations for confirmatory chronic pain clinical trials: IMMPACT recommendations. Pain. 2010;149(2):177–193. doi: 10.1016/j.pain.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 37.Vojtassak J, Vojtassak J, Jacobs A, Rynn L, Waechter S, Richarz U. A phase IIIb, multicentre, randomised, parallel-group, placebo-controlled, double-blind study to investigate the efficacy and safety of OROS hydromorphone in subjects with moderate-to-severe chronic pain induced by osteoarthritis of the hip or the knee. Pain Res Treat. 2011;2011:239501. doi: 10.1155/2011/239501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vinik AI, Tuchman M, Safirstein B, et al. Lamotrigine for treatment of pain associated with diabetic neuropathy: results of two randomized, double-blind, placebo-controlled studies. Pain. 2007;128(1–2):169–179. doi: 10.1016/j.pain.2006.09.040. [DOI] [PubMed] [Google Scholar]
- 39.Beydoun A, Shaibani A, Hopwood M, Wan Y. Oxcarbazepine in painful diabetic neuropathy: results of a dose-ranging study. Acta Neurol Scand. 2006;113(6):395–404. doi: 10.1111/j.1600-0404.2006.00631.x. [DOI] [PubMed] [Google Scholar]
- 40.Katz N, Rauck R, Ahdieh H, et al. A 12-week, randomized, placebo-controlled trial assessing the safety and efficacy of oxymorphone extended release for opioid-naive patients with chronic low back pain. Curr Med Res Opin. 2007;23(1):117–128. doi: 10.1185/030079906x162692. [DOI] [PubMed] [Google Scholar]
- 41.Hale ME, Ahdieh H, Ma T, Rauck R. Efficacy and safety of OPANA ER (oxymorphone extended release) for relief of moderate to severe chronic low back pain in opioid-experienced patients: a 12-week, randomized, double-blind, placebo-controlled study. J Pain. 2007;8(2):175–184. doi: 10.1016/j.jpain.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 42.Opana ER [package insert] Chadds Ford, PA: Endo Pharmaceuticals Inc.; 2013. [Google Scholar]
- 43.Oxycontin [package insert] Stamford, CT: Purdue Pharma LP; 2013. [Google Scholar]
- 44.Roth SH, Fleischmann RM, Burch FX, et al. Around-the-clock, controlled-release oxycodone therapy for osteoarthritis-related pain: placebo-controlled trial and long-term evaluation. Arch Intern Med. 2000;160(6):853–860. doi: 10.1001/archinte.160.6.853. [DOI] [PubMed] [Google Scholar]
- 45.Palangio M, Morris E, Doyle RT, Jr, Dornseif BE, Valente TJ. Combination hydrocodone and ibuprofen versus combination oxycodone and acetaminophen in the treatment of moderate or severe acute low back pain. Clin Ther. 2002;24(1):87–99. doi: 10.1016/s0149-2918(02)85007-x. [DOI] [PubMed] [Google Scholar]
- 46.Benyamin R, Trescot AM, Datta S, et al. Opioid complications and side effects. Pain Physician. 2008;11(2 Suppl):S105–S120. [PubMed] [Google Scholar]
- 47.Noble M, Tregear SJ, Treadwell JR, Schoelles K. Long-term opioid therapy for chronic noncancer pain: a systematic review and meta-analysis of efficacy and safety. J Pain Symptom Manage. 2008;35(2):214–228. doi: 10.1016/j.jpainsymman.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 48.Martell BA, O’Connor PG, Kerns RD, et al. Systematic review: opioid treatment for chronic back pain: prevalence, efficacy, and association with addiction. Ann Intern Med. 2007;146(2):116–127. doi: 10.7326/0003-4819-146-2-200701160-00006. [DOI] [PubMed] [Google Scholar]
- 49.Amabile CM, Bowman BJ. Overview of oral modified-release opioid products for the management of chronic pain. Ann Pharmacother. 2006;40(7–8):1327–1335. doi: 10.1345/aph.1G259. [DOI] [PubMed] [Google Scholar]
- 50.Cramer MP, Saks SR. Translating safety, efficacy and compliance into economic value for controlled release dosage forms. Pharmacoeconomics. 1994;5(6):482–504. doi: 10.2165/00019053-199405060-00005. [DOI] [PubMed] [Google Scholar]
- 51.Greenberg RN. Overview of patient compliance with medication dosing: a literature review. Clin Ther. 1984;6(5):592–599. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Selection of hydrocodone ER starting dose for opioid-experienced patients.
Note: aIf the patient reported a range of daily doses of opioid medication, the highest dose reported was taken as the base to calculate patient’s total daily dose of opioid treatment.
Abbreviation: ER, extended release.