

Abstract
Obesity and metabolic syndrome diseases have exploded into a global epidemic. Consumption of calorie-dense food and diminished physical activity are the generally accepted causes for obesity. But, could environmental factors expose preexisting genetic differences or exacerbate the root causes of diet and exercise? The environmental obesogen model proposes that chemical exposure during critical developmental stages influences subsequent adipogenesis, lipid balance and obesity. Obesogens are chemicals that stimulate adipogenesis and fat storage or alter the control of metabolism, appetite and satiety to promote weight gain. Tributyltin (TBT) is a high-affinity agonistic ligand for the retinoid X receptor (RXR) and peroxisome proliferator activated receptor gamma (PPARγ). RXR-PPARγ signaling is a key component in adipogenesis and the function of adipocytes; activation of this heterodimer increases adipose mass in rodents and humans. Thus, inappropriate activation of RXR-PPARγ can directly alter adipose tissue homeostasis. TBT exposure promoted adipocyte differentiation, modulated adipogenic genes and increased adiposity in mice after in utero exposure. These results suggest that organotin exposure is a previously unappreciated risk factor for the development of obesity and related disorders. Based on the observed effects of TBT on adipogenesis, we hypothesized that organotin exposure during prenatal adipose tissue development would create an environment that led to more adipocytes. We observed that the multipotent stromal cell compartment was altered by prenatal TBT exposure leading to an increased number of preadipocytes. This increase in the number of preadipocytes could correspondingly increase the steady state number of adipocytes in the adult, which could favor the development of obesity over time.
Keywords: adipocyte, endocrine disrupter, multipotent stromal cell, obesity, obesogen, organotin, PPARγ
It is well known that there is an obesity epidemic in recent years and we commonly talk about this as a worldwide obesity epidemic. While it is true that obesity is on the rise around the world, the epidemic is probably most pronounced in the United States among developed nations. Current statistics show that 34% of US adults are clinically obese (i.e. a body mass index, BMI > 30), about double the worldwide average, and 68% are overweight (BMI > 25).1 The WHO estimates that 700 million adults, about 10% of the world’s population will be obese by 2015.2 The main problem with obesity is that it costs a lot of money in added healthcare costs, primarily due to its relationship with metabolic syndrome, diabetes and cardiovascular disease. In 2005, the additional costs ascribed to obesity were estimated at $75 billion; the amount for 2009 was 147 billion. This number is fairly close to the annual cost of implementing health care for all Americans, so obesity is obviously a very significant problem.
If obesity is a significant problem, one might reasonably ask how we get fat? Most doctors adopt the simplistic view that obesity is the result of too many calories in v. too few calories expended – what I call the ‘couch potato syndrome’. More formally, this is called positive energy balance. One classic example of positive energy balance would be sumo wrestlers who are very large men that perform at a high athletic level for a few years but who essentially eat their way into retirement when they get too large to fight competitively any longer.
While it is self-evident that one must consume more calories than are burned to gain weight, the situation is not as simple as the checkbook analogy. Many other factors in addition to positive energy balance have been associated with obesity (reviewed in3,4). We know that elevated glucocorticoid levels are linked with obesity and metabolic syndrome.5,6 Inadequate sleep, thrifty genes,7 viruses, gut microbes and single nucleotide polymorphisms in a variety of genes can all contribute to obesity in subsets of the population. There are a lot of data regarding prenatal nutrition, the in utero experience and adult disease from the work of Barker and colleagues.8–10 Maternal smoking is clearly associated with obesity in the exposed offspring.11 Overall, many factors in addition to excess calorie consumption contribute to obesity.
Interestingly, there was a paper published a few years ago by Paula Baillie Hamilton where she postulated a role for chemical toxins in obesity.12 She noted that the obesity epidemic roughly correlates with the increasing use of industrial chemicals after World War II. Sure enough, there is such a correlation. However, it is important to keep in mind that correlation is not causation and that many factors correlate with obesity, for example, global warming, and the number of dermatologists, climate scientists and SUVs on the road in the United States, among others. It is hard to imagine how any of these correlated factors might be involved in the obesity epidemic.
However, we also know that many chemicals in the environment have effects on the endocrine system, so perhaps there is a kernel of truth to Dr Bailee Hamilton’s argument.13 We know that weight is under hormonal control.14 Appetite and satiety are controlled by a set of peptide hormones that control when we feel hungry, when we feel like we have had enough to eat, etc. The thyroid hormone receptor controls the basal metabolic rate, that is, how many calories it takes to maintain bodily functions at rest. The development of fat cells is also under hormonal control.15–17 The two key nuclear hormone receptors involved in adipogenesis are RXR and peroxisome proliferator activated receptor gamma (PPARγ). PPARγ is considered the master regulator of fat cell development. If you activate this receptor, cells that are capable of becoming fat cells will differentiate into fat cells and existing fat cells will store more fat reviewed in Tontonoz and Spiegelman.18
Therefore, one of the questions that we have been studying in my laboratory in recent years is whether disturbances in endocrine signaling pathways are somehow involved in the development of fat cells and in obesity. On the basis of the data described below, we proposed the ‘obesogen hypothesis’. 19,20 That is, that there are molecules called obesogens that inappropriately stimulate the development of fat cells, the storage of fat into existing fat cells, alter metabolic set points or the hormonal control of appetite and satiety leading to increased body fat mass. Are there any existing data that would support such a hypothesis?
We know from the work of Newbold et al.21 that exposure to diethylstilbesterol in the perinatal period increases weight at adulthood. Newbold has gone on to show that genistein has a similar effect,22 Beverly Rubin and Ana Soto23 have shown that bisphenol-A has a similar effect. We know that there are pharmaceutical obesogens. Thiazolidinediones such as Actos and Avandia that are used to treat type II diabetes increase the number of fat cells at all ages in humans.24,25 We know from Rick Stahlhut and Shanna Swann that urinary phthalate levels in men correlate with waist diameter and insulin resistance.26 Furthermore, there are a variety of cell culture-based studies which show that chemicals such as phthalates,27,28 bisphenol-A, alkyl phenols,13 and more recently PFOA29 and organotins20,30–32 can all cause the differentiation of fat cells. Taken together, these data indicate that obesogens exist and are worth studying.
We have been working on a compound called tributyltin (TBT), which is the most famous endocrine disrupter that most people have never heard of. I say this because TBT is the single example of an endocrine disrupter for which there is absolutely no controversy about dose and effect. It is found at parts per billion levels and exerts its effects at these levels. The classic effect of TBT is to cause ‘imposex’ the masculinization of female mollusks.33,34 TBT exposed mollusks grow an absolutely gigantic penis which creates a geometric problem for mating; therefore, the animals are sterile. We became interested in TBT when it was shown that TBT could masculinize genetically female Japanese flounder.35 We asked the question, ‘Which hormone receptor signaling pathway might be activated by TBT and therefore alter the sexual characteristics of both vertebrates and invertebrates?’ We expected to find an effect on one or more steroid signaling pathway. Instead, we found that TBT activated two other nuclear receptors: the 9-cis retinoic acid receptor, RXR and the peroxisome proliferator activated receptor gamma, PPARγ. We went on to show that TBT induced cultured 3T3-L1 cells to differentiate into fat, and TBT caused weight gain and the induction of adipogenic genes in mice.19 Some of these data are summarized below, followed by our new data on obesogens and stem cells.
Table 1 summarizes the effects of various organotins and other compounds on RXR and PPARγ in receptor activation assay. For RXR, the putative natural ligand 9-cis-retinoic acid and two very good synthetic rexinoids, AGN195203 and LG-100268, have EC50 values in the low nanomolar range. TBT and triphenyltin (TPT) are in the same range. On PPARγ, the thiazolidinedione, troglitazone, has an EC50 of about 1 μM and the organotins are actually much better. Clearly, these compounds are very potent activators for both PPARγ and RXR; do they bind to either receptor? That is important because the organotin structures do not look anything like the known ligands for these receptors. Figure 1 shows the structures of two rexinoids, 9-cRA and LGD100268 and rosiglitazone (ROSI), which is another of the thiazolidinediones, compared with TBT. It is obvious even to the untrained eye that these structures are not particularly similar. Therefore, we tested whether TBT binds to RXR and PPARγ using competitive ligand displacement studies. We found that TBT binds with low nM affinity to both receptors and this has been confirmed in other laboratories.20,30–32 It is clear that TBT binds very well to both receptors as an agonistic ligand. We next asked what effects it has in some standard adipogenic models.
Table 1.
Activation of nuclear receptor ligand binding domain fusions by candidate ligands
| Ligand | hRXRα | hPPARγ |
|---|---|---|
| LG268 | 2–5 | NA |
| 9-cis RA | 15 | NA |
| All-trans RA | NA | NA |
| Monobutyltin chloride | NA | NA |
| Dibutyltin chloride | 3000 | NA |
| Tributyltin chloride | 3–8 | 20 |
| Tetrabutyltin chloride | 150 | ND |
| Di(triphenyltin) oxide | 2–10 | 20 |
| Butyltin-tris (2-ethylhexanoate) | NA | ND |
| Troglitazone | NA | 1000 |
NA, not active; ND, not determined.
The values are EC50 in nM.
Fig. 1.
Molecular structures of high-affinity nuclear receptor ligands. The structures of natural (9-cis RA) and synthetic (LGD100268) RXR ligands are given, as is the structure of a high-affinity synthetic ligand for PPARγ (rosiglitazone, ROSI). The structure of TBT is given for comparison.
One very standard adipogenic model utilizes 3T3-L1 cells. These are fibroblasts that can be induced to become adipocytes if treated in just the right way. We found that both TBT and RXR activators could induce 3T3-L1 cells to become adipocytes, as revealed by Oil Red O staining (stains triglycerides) and gene expression.20 This has also been shown by other investigators,30–32 although there is some controversy about the molecular mechanisms of TBT action.
Therefore, what happens when we expose animals to TBT? We treated pregnant mice with TBT at day 16 of gestation and then examined the effects of this exposure on fat accumulation at birth. We found that animals treated with vehicle control has no discernable triglyceride staining in fat depots, but that TBT treated animals were already storing fat in the liver, testis and mammary adipose depots at birth.20 If we instead allow the prenatally TBT-treated animals to develop, we find that by 10 weeks of age, the TBT-treated animals are smaller, but have stored more fat; therefore, TBT treatment causes increased fat deposition at the expense of overall body mass.20 By 6 months and 9 months, the TBT-treated animals are larger still, up to 15% larger than control animals. Therefore, a single prenatal exposure to TBT has altered the animals such that they are predisposed to weight gain later in life, despite normal diet and access to exercise.
The focus of our recent efforts has been to identify the molecular mechanisms underlying this observed predisposition to weight gain. One can imagine several possibilities. First, the prenatal TBT exposure has altered the hormonal control of appetite and satiety causing the animals to eat more. This is a plausible hypothesis, but we have not seen any evidence to support it. Second, perhaps the TBT treatment has altered the ability of fat cells to store and process lipids. Third, is it possible that TBT treatment has altered the number of preadipocytes or adipocytes? It turns out that this last model is the one for which we find the most support. There is a type of adult stem cell in the body that was previously called a mesenchymal stem cell and is now called a ‘multipotent stromal cell’ or MSC. MSCs can differentiate into multiple lineages in the body, including fat, bone, cartilage and muscle. We know from the literature that the PPARγ agonist ROSI can induce MSCs to differentiate into adipocytes.32 MSCs are found primarily in bone marrow and adipose tissue and it is currently controversial whether non-resident MSCs can home to adipose depots.
Our working hypothesis was that prenatal TBT treatment was altering the MSC compartment such that it produces more adipocytes. We first tested the ability of MSCs from either bone marrow or white adipose tissue (WAT) to differentiate into bone, cartilage or adipose tissue in culture. We found that it was possible to isolate relatively pure populations of mouse or human MSCs that expressed appropriate molecular markers and could be induced to differentiate into fat, bone or cartilage in vitro. We next asked the question whether MSCs derived from WAT could be induced to differentiate into adipocytes by TBT and whether TBT exposure altered the potential of MSCs to become bone or cartilage. We found that MSCs were quite responsive to adipogenic induction in vitro and that treatment of MSCs with TBT caused a substantial increase in the number of adipocytes.
We found that both mouse and human adipose derived MSCs (ADSCs) responded strongly to adipogenic induction and that this response was reflected in gene expression.36 TBT or ROSI treatment decreased expression of the adipogenesis inhibitor Pref-1 while increasing expression of both early (aP2/Fabp4, PPARγ) and late (leptin) adipocyte markers. Interestingly, TBT or ROSI treatment inhibits the ability of a bone differentiation cocktail to drive ADSCs toward the osteoblastic lineage.36
While it appears obvious that ROSI and TBT act through PPARγ to induce adipogenesis, this had never been formally tested. Therefore, we tested the ability of the PPARγ antagonists GW9662 and T0070907 to inhibit TBT or ROSI-induced adipogenesis. As expected, both compounds were able to inhibit the adipogenic effects of ROSI.36 In accord with our model that TBT acts through PPARγ, either antagonist was able to block the adipogenic effects of TBT. Addition of either ROSI or TBT to antagonist treated cultures was able to rescue the adipogenesis inducing effects of either compound, further confirming our results.36
Summarizing the in vitro effects, TBT induces adipogenesis in MSCs and inhibits the ability of the same cells to be induced into bone cells.36 The next question was whether prenatal exposure to TBT had any effect on the differentiation potential of MSCs. We treated pregnant dams at E16.5 with a single oral dose of TBT, ROSI or vehicle and either cross-fostered the pups to CD-1 foster mothers or left them with their dams (Fig. 2). ADSC were harvested at 8 weeks and tested for their ability to become adipocytes, osteocytes or chondrocytes after appropriate induction in vitro. We found that prenatal TBT or ROSI exposure increased the adipogenic potential of the ADSCs and that additional in vitro treatment with TBT or ROSI further induced adipogenesis and the expression of adipogenic genes such as Fabp4 and leptin (Fig. 2).36 There was no effect noted on chondrogenesis; however, prenatal TBT or ROSI exposure strongly inhibited calcium deposition and the expression of osteoblast markers (e.g. osteopontin and osteonectin) while simultaneously increasing adipogenesis and the expression of adipogenic markers (Fabp4, leptin; Fig. 2).36 Therefore, as with the in vitro experiments, prenatal TBT or ROSI exposure altered the fate of the ADSC compartment, biasing it to favor the adipogenic lineage at the expense of the osteogenic lineage.
Fig. 2.
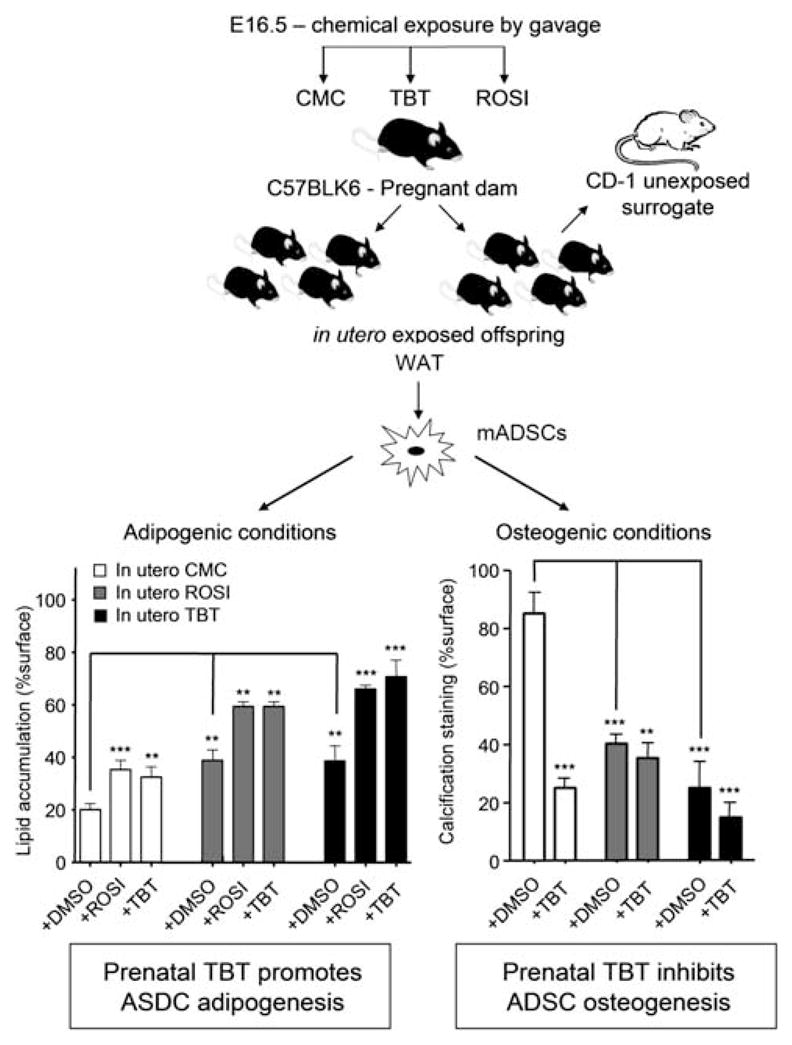
Prenatal TBT exposure predisposes ASDCs to become adipocytes at the expense of osteocytes. The top of the figure shows the experimental design for prenatal exposure. At 8 weeks of age, control or prenatally exposed animals were euthanized and MSCs were harvested from white adipose tissue. These cells were grown in culture and then induced to develop along the adipose or bone differentiation pathways. In each case, further exposure to TBT or ROSI or solvent controls was shown to increase (adipogenesis) or inhibit (bone) differentiation. The degree of adipogenic differentiation was assessed by Oil Red O staining, and the degree of calcium deposition assayed by Alizarin Red staining. Adapted from Kirchner et al.36
Of course, the next question is how has the MSC compartment been altered by prenatal TBT or ROSI exposure to favor the production of adipocytes? Our hypothesis was that prenatal obesogen exposure increased the number of preadipocytes; therefore, we analyzed the ADSC population for molecular markers of preadipocytes. Fabp4 is known to be expressed on preadipocytes and macrophages and CD-68 is expressed on macrophages but not preadipocytes. We asked how many cells are Fabp4+/CD-68− (i.e. preadipocytes) in the ADSC population from animals treated prenatally with vehicle. We found a 7–15% increase in the number of preadipocytes from obesogen exposed mice compared with controls and concluded that prenatal obesogen exposure increases the number of preadipocytes in the MSC population, an effect that could easily increase the number of fat cells in an adult.36 This observation also explains why ASDCs are inhibited from becoming bone – they are already enriched in preadipocytes.
We next asked whether the expression of genes known to be involved in specifying the adipocyte lineage was increased in untreated ADSCs harvested from obesogen treated v. control animals. We showed that Fabp4 and leptin mRNA levels were increased significantly whereas PPARγ mRNA was increased but the increase did not reach statistical significance. 36 Using methylation sensitive restriction enzyme digestion, we found that CpG islands in the Fabp4 promoter were undermethylated in the prenatally TBT treated animals. This suggests that prenatal exposure has caused an epigenetic alteration in the MSC compartment that increases the fraction of MSCs committed to the adipocyte lineage. Fabp4 is a downstream gene in the hierarchy of adipogenesis; hence, we are currently testing all of the genes known to be involved in adipogenesis to determine which others may be epigenetically altered in MSCs from animals treated prenatally with TBT.
Conclusions and prospects
In summary, our work and that in other laboratories has shown that organotins such as TBT and TPT are very potent activators and ligands for both RXR and PPARγ. Organotins bind and activate the receptors at low nanomolar levels – the levels at which they are found in humans and wildlife. Organotins drive adipocyte differentiation in mouse and human cell cultures, both in the mouse 3T3-L1 cell line and in human and mouse mesenchymal stem cells. Organotin exposure during development induces adipogenesis in two animal models: mouse and frog (Xenopus laevis). TBT induces the expression of target genes that would be expected if PPARγ were being activated and we have shown in MSC cultures that PPARγ activation is required for induction of adipogenesis by TBT or ROSI. Therefore, we conclude that induction of adipogenesis is a known, and previously unsuspected endocrine disrupting the effect of organotins such as TBT. While the bulk of current data suggest that TBT is acting through PPARγ, TBT is know to affect the expression of 11β-hydroxysteroid dehydrogenases, which in turn, alters glucocorticoid levels. TBT inhibits the expression of CYP19, which will alter estrogen levels and TBT is also a mitochondrial stressor. Future studies will reveal to what extent these other pathways may contribute to the adipogenic effects of TBT.
The main question arising from our work is whether obesogen exposure is a contributing factor in human obesity and, if so, how large is the contribution. Published studies from our lab and from other laboratories shows that organotin exposure increases weight and the expression of adipogenic genes. Importantly, we know that pharmaceutical obesogens that act through PPARγ can increase the number of fat cells and body weight in humans, therefore, it is highly likely that chemicals targeting PPARγ will have similar effects. We know that prenatal obesogen exposure permanently alters the adult phenotype such that exposed animals become fat on normal chow and we have shown that prenatal obesogen exposure recruits MSCs in WAT (and likely elsewhere) to become adipocytes. Therefore, obesogen exposure has the potential to increase adipogenesis in humans.
One very important question is whether and to what extent humans are exposed to obesogens such as TBT. Unhappily, we have very few data relating to organotin exposure. PVC plastic are 3% by weight of organotins, most of which are mono and dibutyltin which are inactive, or weak activators of PPARγ, respectively. TBT and TPT are still found as contaminants in seafood, particularly in cultured fish and shrimp, in the low nanomolar range. Fenbutatin is still used in agriculture on a variety of fruits and nuts. There is a single study in the United States that measured TBT in a small group of people at an average of 27 nM,37 TPT was measured in Europeans at an average of 0.5–2nM.38 These are well within the range required to activate PPARγ.20 Therefore, it appears reasonable to hypothesize that at least some of the population is exposed to organotins at levels sufficient to activate high affinity receptors. We need much more data on human exposures to make any reasonable inferences about the degree to which organotin exposure influences human obesity. This work is really in its infancy and future studies will be very informative. The list of chemicals known to act as obesogens is large and growing. Credible data exist for bisphenol A, phthalates, perfluorooctanoates, nicotine, fructose and organophosphate pesticides to name a few. We really need much more data about how these chemicals act and the extent to which humans are exposed and at what times during development in order to fully understand how obesogens affect body weight and obesity later in life.
In conclusion, it is becoming increasingly clear from our work and from studies in other laboratories that the traditional explanation for obesity – an imbalance in the caloric checkbook – is inadequate and overly simplistic to explain the epidemic of obesity, particularly in the very young (reviewed in3,4). Pharmaceutical obesogens, obesogenic chemicals and obesogenic foods exist and these can inappropriately alter development, physiology, metabolism and behavior to favor the increased storage of excess calories as fat. We know that prenatal exposure to one class of obesogen, organotins, reprograms the exposed animals to be fat. At least part of this process is the result of epigenetic changes that alter the expression of adipogenic genes.36 The overall conclusion is that it is high time that the focus on treating obesity is shifted toward prevention from the current focus of (typically unsuccessful) treatment. Once a person becomes obese, it is extraordinarily difficult to make them lean again. If it were a simple matter of balancing the caloric checkbook, only few people would be obese.
Lastly, it is the environment that is making us fat? It is probably premature to make such a provocative statement, but the weight of the evidence suggests that environmental exposures to obesogenic drugs and chemicals is a contributing factor to obesity that has been largely ignored in the past. Identifying the range of foods, drugs and chemicals that act as obesogens and understanding their mode of action will greatly illuminate our understanding of obesity. Establishing safer replacements for obesogens to which we are currently exposed will ultimately be required. In this respect, the burgeoning field of green chemistry offers great potential.39
Acknowledgments
Work in the author’s laboratory was supported by grants from the National Institutes of Health (ES015849) and U.S. Environmental Protection Agency (STAR-R830686).
Footnotes
Statement of Interest
Dr Blumberg is a named inventor on several US patents related to PPARγ.
References
- 1.Flegal KM, Carroll MD, Ogden CL, et al. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Global Strategy on Diet, Physical Activity and Health: Obesity and Overweight. 2009 Retrieved 13 May 2010 from http://www.who.int/dietphysicalactivity/publications/facts/obesity/en/
- 3.Grun F, Blumberg B. Endocrine disrupters as obesogens. Mol Cell Endocrinol. 2009;304:19–29. doi: 10.1016/j.mce.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grun F, Blumberg B. Minireview: the case for obesogens. Mol Endocrinol. 2009;23:1127–1134. doi: 10.1210/me.2008-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nyirenda MJ, Lindsay RS, Kenyon CJ, et al. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J Clin Invest. 1998;101:2174–2181. doi: 10.1172/JCI1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Achard V, Boullu-Ciocca S, Desbriere R, et al. Perinatal programming of central obesity and the metabolic syndrome: role of glucocorticoids. Metab Syndr Relat Disord. 2006;4:129–137. doi: 10.1089/met.2006.4.129. [DOI] [PubMed] [Google Scholar]
- 7.Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5–20. doi: 10.1093/bmb/60.1.5. [DOI] [PubMed] [Google Scholar]
- 8.Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077–1081. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- 9.Barker DJ, Osmond C. Low birth weight and hypertension. BMJ. 1988;297:134–135. doi: 10.1136/bmj.297.6641.134-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barker DJ, Osmond C, Golding J, et al. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–567. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Power C, Jefferis BJ. Fetal environment and subsequent obesity: a study of maternal smoking. Int J Epidemiol. 2002;31:413–419. [PubMed] [Google Scholar]
- 12.Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med. 2002;8:185–192. doi: 10.1089/107555302317371479. [DOI] [PubMed] [Google Scholar]
- 13.Heindel JJ. Endocrine disruptors and the obesity epidemic. Toxicol Sci. 2003;76:247–249. doi: 10.1093/toxsci/kfg255. [DOI] [PubMed] [Google Scholar]
- 14.Badman MK, Flier JS. The gut and energy balance: visceral allies in the obesity wars. Science. 2005;307:1909–1914. doi: 10.1126/science.1109951. [DOI] [PubMed] [Google Scholar]
- 15.Rangwala SM, Lazar MA. Transcriptional control of adipogenesis. Annu Rev Nutr. 2000;20:535–559. doi: 10.1146/annurev.nutr.20.1.535. [DOI] [PubMed] [Google Scholar]
- 16.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 17.Lazar MA. How obesity causes diabetes: not a tall tale. Science. 2005;307:373–375. doi: 10.1126/science.1104342. [DOI] [PubMed] [Google Scholar]
- 18.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 19.Grun F, Blumberg B. Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology. 2006;147:S50–S55. doi: 10.1210/en.2005-1129. [DOI] [PubMed] [Google Scholar]
- 20.Grun F, Watanabe H, Zamanian Z, et al. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. 2006;20:2141–2155. doi: 10.1210/me.2005-0367. [DOI] [PubMed] [Google Scholar]
- 21.Newbold RR, Padilla-Banks E, Snyder RJ, et al. Developmental exposure to estrogenic compounds and obesity. Birth Defects Res A Clin Mol Teratol. 2005;73:478–480. doi: 10.1002/bdra.20147. [DOI] [PubMed] [Google Scholar]
- 22.Newbold RR, Padilla-Banks E, Jefferson WN. Environmental estrogens and obesity. Mol Cell Endocrinol. 2009;304:84–89. doi: 10.1016/j.mce.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubin BS, Murray MK, Damassa DA, et al. Perinatal exposure to low doses of bisphenol A affects body weight, patterns of estrous cyclicity, and plasma LH levels. Environ Health Perspect. 2001;109:675–680. doi: 10.1289/ehp.01109675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larsen TM, Toubro S, Astrup A. PPARgamma agonists in the treatment of type II diabetes: is increased fatness commensurate with long-term efficacy? Int J Obes Relat Metab Disord. 2003;27:147–161. doi: 10.1038/sj.ijo.802223. [DOI] [PubMed] [Google Scholar]
- 25.Rubenstrunk A, Hanf R, Hum DW, et al. Safety issues and prospects for future generations of PPAR modulators. Biochim Biophys Acta. 2007;1771:1065–1081. doi: 10.1016/j.bbalip.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Stahlhut RW, van Wijgaarden E, Dye TD, et al. Concentrations of urinary phthalate metabolites are associated with increased waist circumferece and insulin resistance in adult U.S. males. Environ Health Perspect. 2007;115:876–882. doi: 10.1289/ehp.9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hurst CH, Waxman DJ. Activation of PPARalpha and PPARgamma by environmental phthalate monoesters. Toxicol Sci. 2003;74:297–308. doi: 10.1093/toxsci/kfg145. [DOI] [PubMed] [Google Scholar]
- 28.Feige JN, Gelman L, Rossi D, et al. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor gamma modulator that promotes adipogenesis. J Biol Chem. 2007;282:19152–19166. doi: 10.1074/jbc.M702724200. [DOI] [PubMed] [Google Scholar]
- 29.Hines EP, White SS, Stanko JP, et al. Phenotypic dichotomy following developmental exposure to perfluorooctanoic acid (PFOA) in female CD-1 mice: low doses induce elevated serum leptin and insulin, and overweight in mid-life. Mol Cell Endocrinol. 2009;304:97–105. doi: 10.1016/j.mce.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 30.Inadera H, Shimomura A. Environmental chemical tributyltin augments adipocyte differentiation. Toxicol Lett. 2005;159:226–234. doi: 10.1016/j.toxlet.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 31.Kanayama T, Kobayashi N, Mamiya S, et al. Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoid X receptor pathway. Mol Pharmacol. 2005;67:766–774. doi: 10.1124/mol.104.008409. [DOI] [PubMed] [Google Scholar]
- 32.Carfi M, Croera C, Ferrario D, et al. TBTC induces adipocyte differentiation in human bone marrow long term culture. Toxicology. 2008;249:11–18. doi: 10.1016/j.tox.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 33.Blaber SJM. The occurrence of a penis-like outgrowth behind the right tentacle in spent females of Nucella lapillus. Proc Malacol Soc Lon. 1970;39:231–233. [Google Scholar]
- 34.Gibbs P, Bryan G. Reproductive failure in populations of the dog-whelk, Nucella lapillus, caused by imposex induced by tributyltin from antifouling paints. J Mar Biol Assoc UK. 1986;66:767–777. [Google Scholar]
- 35.Shimasaki Y, Kitano T, Oshima Y, et al. Tributyltin causes masculinization in fish. Environ Toxicol Chem. 2003;22:141–144. [PubMed] [Google Scholar]
- 36.Kirchner S, Kieu T, Chow C, et al. Prenatal exposure to the environmental obesogen tributyltin predisposes multipotent stem cells to become adipocytes. Mol Endocrinol. 2010;24:526–539. doi: 10.1210/me.2009-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kannan K, Senthilkumar K, Giesy J. Occurrence of butyltin compounds in human blood. Environ Sci Technol. 1999;33:1776–1779. [Google Scholar]
- 38.Rantakokko P, Turunen A, Verkasalo PK, et al. Blood levels of organotin compounds and their relation to fish consumption in Finland. Sci Total Environ. 2008;399:90–95. doi: 10.1016/j.scitotenv.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 39.Anastas P, Eghbali N. Green chemistry: principles and practice. Chem Soc Rev. 2010;39:301–312. doi: 10.1039/b918763b. [DOI] [PubMed] [Google Scholar]