

Abstract
Ubiquitylation is among the most prevalent post-translational modifications (PTMs) and regulates numerous cellular functions. Interestingly, ubiquitin (Ub) can be itself modified by other PTMs, including acetylation and phosphorylation. Acetylation of Ub on K6 and K48 represses the formation and elongation of Ub chains. Phosphorylation of Ub happens on multiple sites, S57 and S65 being the most frequently modified in yeast and mammalian cells, respectively. In mammals, the PINK1 kinase activates ubiquitin ligase Parkin by phosphorylating S65 of Ub and of the Parkin Ubl domain, which in turn promotes the amplification of autophagy signals necessary for the removal of damaged mitochondria. Similarly, TBK1 phosphorylates the autophagy receptors OPTN and p62 to initiate feedback and feedforward programs for Ub-dependent removal of protein aggregates, mitochondria and pathogens (such as Salmonella and Mycobacterium tuberculosis). The impact of PINK1-mediated phosphorylation of Ub and TBK1-dependent phosphorylation of autophagy receptors (OPTN and p62) has been recently linked to the development of Parkinson’s disease and amyotrophic lateral sclerosis, respectively. Hence, the post-translational modification of Ub and its receptors can efficiently expand the Ub code and modulate its functions in health and disease.
Keywords: mitophagy, phosphorylation, post-translational modification, ubiquitin
Ubiquitin—a versatile post-translational modifier
The cellular functions of most proteins are regulated by post-translational modifications (PTMs). As these PTMs are inducible and reversible, they allow eukaryotic cells to dynamically respond to external stimuli by modulating intracellular signal transduction pathways. Among the best-studied PTMs are ubiquitylation, phosphorylation and acetylation 1.
Ubiquitylation is a post-translational modification in which ubiquitin (Ub), a 76 amino acid protein, is attached to target proteins through the sequential actions of an E1 Ub-activating enzyme, an E2 Ub-conjugating enzyme and an E3 Ub ligase 2-4. This cascade requires the initial ATP-dependent activation of Ub by the E1, which links the C-terminal glycine residue of Ub via a thioester bond to a cysteine residue within the E1 active site 4. The activated Ub intermediate is then transferred to the catalytic cysteine residue of an E2 enzyme. The E3 Ub ligase conjugates the C-terminal glycine of Ub via an isopeptide bond to the ε-amino group of the target lysine (K) of the substrate 5. E3 ligases are divided into families, based on their catalytic domain structure and mode of catalysis. HECT E3 ligases temporarily accept activated Ub, whereas RING E3 ligases catalyse the direct transfer of Ub from the E2 to the substrate. Parkin belongs to the RBR family, which is formed by RING-HECT hybrid E3 ligases 6.
An important feature of ubiquitylation is that more Ub molecules can be added onto one of its own seven lysine residues or onto the α-amino terminus of the first Ub. Depending on which lysine residue within Ub is utilized to anchor the subsequent Ub molecule, chains of different types and lengths are formed. Linkages can occur on M1, K6, K11, K27, K29, K33, K48 or K63 of Ub 7, 8. Branched chains and mixed linkage types also occur 9, 10. These different linkage types function as signals that are specifically recognized by Ub-binding proteins, which relay the signal to ultimately determine the cellular response or regulate the modified protein by changing its enzymatic activity, localization or stability 11. The removal of Ub(s) attached to target proteins (known as deubiquitylation) is catalysed by deubiquitylating enzymes (DUBs) and represents an important regulatory mechanism of Ub signals in vivo 12.
Interplay between PTMs
Post-translational modifications can engage in extensive crosstalk, with the potential to either positively or negatively modulate signalling networks 13 (Fig1). For example, acetylation can compete with ubiquitylation at lysine residues and thus enhance the stability of target proteins by suppression of ubiquitylation 14, 15. On the other hand, acetylation of target proteins can trigger their subsequent Ub-mediated degradation by promoting ubiquitylation of another lysine within the substrate protein 16.
Figure 1. Generation and control of PTMs.
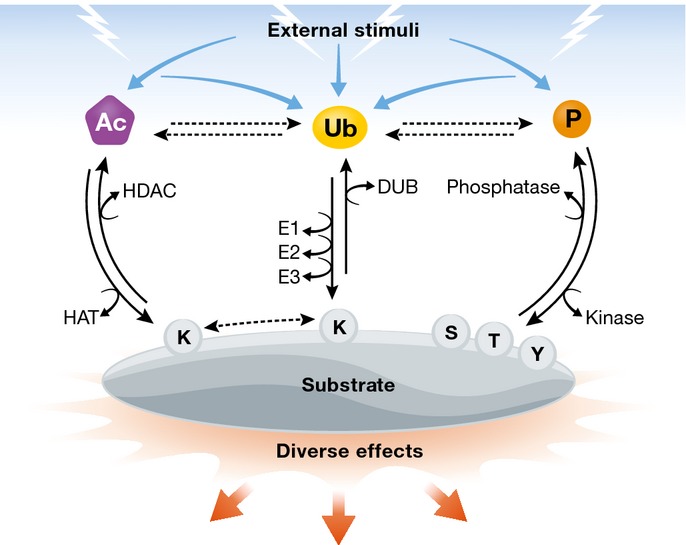
External stimuli result in the generation of different PTMs on target proteins. Phosphorylation, acetylation and ubiquitylation can influence each other and are attached to and removed from substrate proteins by different enzymes. Phosphorylation occurs mainly on serine, threonine and tyrosine residues, whereas acetylation and ubiquitylation target lysine residues. The attachment or removal of PTMs determines substrate fate.
In particular, ubiquitylation and phosphorylation are closely interlinked and mutually affect each other’s functions. For example, phosphorylation can stimulate or inhibit the activity of E3 Ub ligases 17, 18, 19 or DUBs 20. It can also promote or prevent interactions with their target proteins by controlling their intracellular localization 21. Additionally, phosphorylation can mark proteins for recognition by E3 Ub ligases 22 and DUBs 23, or can inhibit substrate recognition 24. Similarly, the phosphorylation of UBDs can alter the ability of a protein to recognize specific Ub modifications, thus affecting signal propagation or protein fate 25, 26, 27. Two prominent examples of the interplay between phosphorylation and ubiquitylation are cell cycle control and NF-κB activation. During cell cycle progression, the cyclin-dependent kinases (CDKs) and E3 ligases APC and SCF coordinate the controlled degradation of proteins 28. In the NF-κB pathway, multiple E3 ligases initiate the proximal ubiquitylation cascade—including TRAFs, IAPs, Pellino and LUBAC—that can be further regulated through phosphorylation events, modifying downstream substrate ubiquitylation. The ubiquitylation of substrates is recognized by the UBAN domain of IKK component NEMO, resulting in NF-κB activation 29, 30, 31.
This regulatory and functional interplay also holds true for the reverse situation, as ubiquitylation can activate or inactivate kinase activity by non-degradative 32, 33 and degradative mechanisms 34. For example, E3 ligases of the IAP family (inhibitor of apoptosis protein) have been recently identified as key regulators of ERK5 activation pathway, via physical and functional disassembly of the ERK5-MAPK module by non-degradative ubiquitylation 35.
Recent data indicate that the phosphorylation of Ub itself has an enormous impact on the ubiquitylation cascade, as it can promote polyUb chain formation, but also inhibit certain E2s, E3s and DUBs, as discussed in detail below. This review summarizes the current understanding of the regulation of Ub by the well-studied PTM phosphorylation and also by the less well-understood acetylation (see Box 1) and deamidation (see Box 2).
Box 1: Acetylation of ubiquitin.
Reversible lysine acetylation regulates several cellular processes—such as gene expression, cell cycle and cellular metabolism—and can impair phosphorylation-dependent protein–protein interactions 111, 112. The surface of Ub is theoretically amenable to most PTMs and acetylation of Ub was recently detected in cells 41.
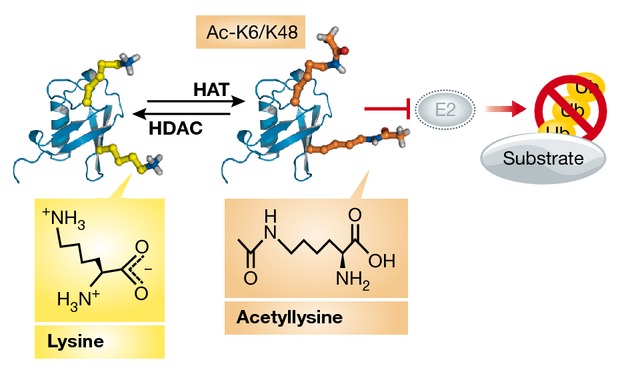
The acetylation of Ub at K6 and K48 is dynamically regulated by all three classes of histone deacetylases (HDACs) and blocks the synthesis of Ub chains. K6- or K48-acetylated Ub can be activated by the E1, transferred to the E2, but cannot be used for E2-mediated K11-, K48-, or K63-linked Ub chain assembly (see schematic illustration). Ub acetylation, which neutralizes the positive charges of lysine residues, was proposed to affect the non-covalent interaction of Ub with E2 enzymes 41. Through overexpression of acetyl-mimetic Ub (K6Q), K6 acetylation was shown to repress Ub chain elongation on substrates. Ideally, the conclusions of this study would need to be confirmed with K6-acetylated Ub rather than acetylmimetic Ub, as acetyl-lysine and glutamine are chemically different. In addition, only a small proportion of Ub is acetylated in vivo. By using mass spectrometry, acetylation on K6 and K48 was found on only 0.03 and 0.01% of total Ub in cells 41.
Interestingly, the acetylation of Ub occurs on two lysine residues that are frequently used for chain elongation. How the competition of ubiquitylation and acetylation is regulated on K6 and K48 residues of Ub is not yet known, but it might have a significant impact on the regulation of mono- versus polyubiquitylation of target proteins, and hence drastically impact the fate of these proteins. However, histone 2B is the only known endogenous substrate of acetylated Ub in cells 41.
UBDs recognize different Ub chain linkage types 11, and there is evidence that S65-phosphorylation of Ub may modulate the binding affinity to certain Ub-binding domains present in autophagic adaptor proteins 85. As K6 and K48 of Ub are close to the hydrophobic patch, which is the major binding site for Ub-binding motifs, acetylation or phosphorylation might have a direct impact on the interaction of Ub receptors with Ub. The identification of Ub-binding motifs specific for acetyl- or phospho-Ub would allow the identification of additional pathways regulated by Ub PTMs.
Box 2: Deamidation of ubiquitin, NEDD8 and Pup.
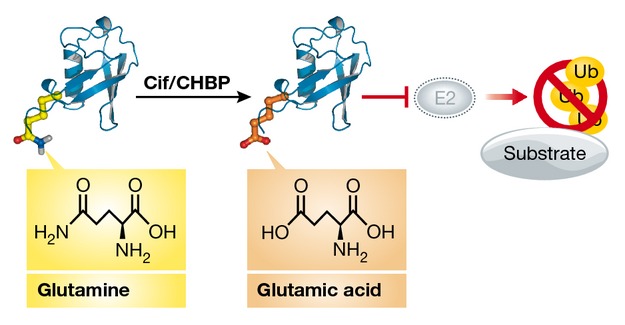
Deamidation is a ubiquitous protein modification in which an amide is converted into an acid. This irreversible conversion of glutamine and asparagine into glutamic acid and aspartic acid, respectively, can occur non-enzymatically without any reactant species or catalyst. Enzymatic in vivo deamidation of glutamine, in contrast to asparagine, has also been reported. Deamidation generally regulates protein turnover in vivo; however, several bacterial virulence factors have also evolved to use enzymatic glutamine deamidation to modify the functions of host proteins. At physiological pH, deamidation introduces a negative charge at the deamidation site and can thus alter the protein structure 113, 114. Deamidation can be detected in most proteins, including Ub and the Ub-like modifiers NEDD8 and Pup (see schematic illustration).
The bacterial effectors CHBP from Burkholderia pseudomallei and Cif from Enteropathogenic E. coli (EPEC) use a novel mechanism for the inhibition of Ub chain elongation in host cells 115. Cif/CHBP harbour a papain-like hydrolytic fold and deamidate Ub at Q40 to generate E40 in vitro and in infected cells. Ub chain synthesis catalysed in vitro by several different E2/E3s is abolished in the presence of catalytically active CHBP. Covalently modified Ub E40 does not affect the formation of E1 or E2 Ub thioester intermediates, but blocks the discharge of Ub from E2 to E3. How a single charge substitution in Ub (Q40E) could have such a dramatic effect on Ub chain elongation remains unclear. Crystallographic analysis of Ub Q40E could determine whether Ub deamidation significantly influences its 3D structure, thereby impeding chain formation, or whether the E2-binding motif is negatively affected.
In infected cells, the production of Ub E40 impairs TNFα-induced signalling, thereby preventing downstream NF-κB-dependent transcription. Hence, CHBP-mediated deamidation of Ub in Burkholderia-infected cells is an effective virulence mechanism that undermines the host Ub system 115. Notably, CHBP and Cif also target the Ub-like protein NEDD8, in which Q40 is conserved 115, 116. NEDD8 is conjugated to cullins—which are part of cullin-RING Ub E3 ligases (CRLs)—and stimulates E3 ligase activity 117, 118. However, this effect is abolished if deamidated NEDD8 is attached to cullins, resulting in impaired degradation of CRL substrates, such as p27, Nrf2, HIF-1α and RhoA 116. CHBP-mediated NEDD8 deamidation kills macrophages (while other cells remain viable), thus constituting an additional virulence mechanism for Burkholderia and EPEC to counteract macrophage-mediated host defence.
CHBP/Cif-mediated NEDD8 deamidation is obviously detrimental during infection, but could be exploited to serve therapeutic purposes, as the neddylation-mediated activation of CRL has a well-established role in cell proliferation, cancer progression and several other diseases 119, 120.
Modification by deamidation is not only relevant for Ub and NEDD8, but also for Pup, a bacterial Ub-like modifier. In analogy to Ub in eukaryotes, Pup is attached to lysine residues of proteins destined for proteasomal degradation. However, unlike Ub, Pup requires deamidation of its C-terminal glutamine to glutamate before its attachment 121.
Localization and stoichiometry of Ub phosphorylation
Ub can be phosphorylated at multiple sites in yeast and mammalian cells (Fig2). Phosphoproteomic analyses have shown that Ub may be phosphorylated at T7 36, 37, 38, T12 36, 39, T14 39, 40, 41, S20 42, 43, 44, S57 38, 39, 40, 45, 46, 47, 48, 49, 50, Y59 51, 52, 53, 54, S65 49, 55, 56, 57, 58, 59 and T66 39 (Fig2). However, little is known about the responsible kinases/phosphatases and stoichiometries of these Ub phosphorylation sites, and the functional relevance of most of them remains to be identified.
Figure 2. Structure of wild-type human and yeast ubiquitin.

The phosphorylation sites of human (T7, T12, T14, S20, S57, Y59, S65 and T66) and yeast (T7, T12, T14, S19, T22, S28, S57, Y59, S65 and T66) Ub are indicated, as well as the residues lining the ubiquitin hydrophobic patch (L8, I44, V70).
Multiple factors likely impact on the function of Ub phosphorylation within cells, including its intracellular localization and the stoichiometry of Ub or substrate phosphorylation. For example, a subpopulation of Ub can be modified on a specific site by a locally restricted kinase, indicating that only a small but locally enriched population of modified Ub is functionally relevant and thus detected by proteomic analyses. For instance, the stoichiometry of p-S65-Ub is below 0.5% of total Ub in exponentially growing yeast and ∼1.5% in mammalian cells 49, 55. Upon mitochondrial depolarization, the level of p-S65-Ub rises to ∼2.5% of total Ub in yeast, while in mammalian cells, ∼20% of Ub is phosphorylated at S65 on mitochondria, indicating that Ub p-S65 is locally concentrated there 55. In yeast cells, Ub S65 phosphorylation seems to be functionally relevant and dynamically regulated, as its concentration increases by 14-fold upon oxidative stress, whereas the concentration of Ub p-S57, which is the most abundant site, is not altered significantly 49.
Notably, the same phosphorylation site (such as p-S65-Ub) might be modulated by different kinases in other cellular processes. For example, although the mitochondrial kinase PINK1 is the only one demonstrated to catalyse Ser65 phosphorylation of Ub to date, another kinase that can phosphorylate this site must exist, as yeast cells lack a PINK1 orthologue 49. To identify proteins that are preferentially modified by Ub p-S65, Villen and colleagues performed quantitative proteomics in yeast using His-tagged Ub mutants (wt Ub, phosphomimetic UbS65E and non-phosphoryatable UbS65A). Purification and MS analyses of the proteins conjugated to His-Ub variants revealed that the main targets of UbS65E ubiquitylation were histone H2B (HTB2), histone H2B nuclear import protein Kap95 and the histone deacetylase Rpd3, as well as SNARE proteins, which are involved in vesicle trafficking 49. This indicates a pleiotropic role for the phospho-Ub and likely implicates distinct kinases in modifying multiple pathways in yeast. Nevertheless, the observed S65-Ub-modified proteome probably varies from pS65-Ub effects under physiological conditions, as phosphomimetic mutants of S65-Ub do not faithfully reproduce the behaviour of natively phosphorylated S65-Ub 59, 60. Moreover, the stoichiometry of Ub phosphorylation varies depending on the cellular context and can play a critical role in triggering signalling pathways within the cell (see below).
Phosphorylation of ubiquitin modulates the ubiquitylation cycle
Since the ubiquitylation cascade involves a variety of different players 5, it can be assumed that the phosphorylation of Ub influences components of the ubiquitylation machinery as well as Ub-binding proteins. Current studies point to significant phosphorylation-mediated structural and biophysical changes of Ub and Ub chains, which might have a wide spectrum of consequences on Ub biology 49, 59. In yeast, Ub S65E is preferentially incorporated into K6- and K11-chains, while K27-linkages are disfavoured (K63 could not be assessed by MS due to the S/E mutation) 49. Moreover, proteins modified by S65E-Ub are more ubiquitylated, suggesting that either the disassembly of chains containing Ub S65E is impeded 49, 59, or Ub S65E increases the conjugation activity of certain E2-E3 pairs, or both 49. Indeed, Ub chain generation in vitro is hindered through the phosphorylation of S65 on Ub, as it impairs the activity of several E2 and E3 enzymes in mammalian cells 59, 61 and yeast 49. E1-mediated E2 charging with p-S65-Ub is not affected, whereas a subset of E2-conjugating enzymes (mammalian: UBE2R1, UBE3E1, UBE2T, UBE2K, UBE2N/UBE2V1, and yeast: Ubc13/Mms2, Ubc1) were unable to efficiently discharge phosphorylated Ub to form Ub chains, even though S65 does not contact the active site or interact with any other part of the enzyme (with the exception of Ubc13/Mms2) 49, 59, 61. Although one could speculate that the close proximity of p-S65 to K63 might disturb K63-linked chain formation, several of the tested K63-specific E2/E3 pairs were not inhibited in chain formation 55, 59 and K63-linked p-S65-Ub chains could be detected by mass spectrometry 55, 62. Also the mammalian E3 ligases TRAF6 and HOIP (RNF31) were unable to use phosphorylated Ub for Ub chain synthesis, whereas phosphorylated Ub had no negative impact on mammalian Nedd4L, cIAP1- or yeast Rsp5-mediated chain assembly 49, 59. Nevertheless, there is currently no evidence that p-S65 exists as monoUb in cells or that it is used for chain synthesis. Interestingly, Parkin E3 ligase activity is activated by p-S65-Ub, but also hindered by concentrations exceeding 20% of total Ub in in vitro ubiquitylation assays 55, 59. Taken together, S65 phosphorylation can impact the activity of certain E2-E3 combinations to assemble polyUb chains, although the structural and mechanistic basis for this phenomenon remains unclear.
Phosphorylation of Ub chains also affects the activity of DUBs 49, 59, 61. The phosphorylation of S65 on Ub impedes efficient deubiquitylation by mammalian USP8, USP15, USP30, ataxin-3, USP2, AMSH and USP5 49, 59. Interestingly, the site of phosphorylation in diUb (whether distal or proximal) is highly relevant: USP2 (a promiscuous DUB) and AMSH (a K63-linkage-specific DUB) are inactive against the doubly phosphorylated diUb 61, whereas AMSH is inhibited by phosphorylation of the proximal Ub and USP2 is inhibited by phosphorylation at the distal site but not affected by the modification of the proximal Ub 61.
S65 phosphorylation of ubiquitin activates Parkin
The functional interplay between phosphorylation and ubiquitylation has been extensively studied in the context of mitochondrial autophagy (mitophagy), mediated by PINK1 and the E3 Ub ligase Parkin 63, 64 (Fig3A). This process involves the selective engulfment of damaged mitochondrial material by a specialized double membrane that forms the autophagosome and delivers its cargo to the lysosome for degradation 63, 64. Disturbances in this pathway, for example by mutations affecting the activities of PINK1 or Parkin, are associated with neurodegenerative diseases such as Parkinson’s disease 65, 66, 67.
Figure 3. PINK1-mediated phosphorylation of ubiquitin and Parkin.

(A) Upon mitochondrial damage, PINK1 is recruited and activated at the mitochondrial surface resulting in the sequential phosphorylation of both Ub and the Parkin Ubl domain on their respective Ser65 sites. This leads to Parkin activation and ubiquitylation of multiple substrates on the mitochondrial outer membrane (MOM), which in turn become favourable substrates for PINK1. In such a way, PINK1 and Parkin generate high-density p-S65-Ub chains on MOM proteins. This increases the binding and retention time of Parkin on mitochondria, leading to the amplification of Ub signals. Phosphorylation of Ub chains also serves as a commitment step, as p-Ub chains are resistant to deubiquitylation by the majority of DUBs. The multiple ubiquitylation signals then commit mitochondria for degradation by attracting multiple autophagy receptors, such as OPTN and NDP52. (B) Conformational domain rearrangements for Parkin activation. (a) Inactive PARKIN: Closed auto-inhibited structure of full-length PARKIN (PDB: 4k95). The catalytic cysteine 431 of the RING2 domain (in red) is blocked by the UPD. The I44 patch of the Ubl domain interacts with the RING1 helix and the IBR interacts with Ubl, thereby covering the S65 phosphorylation site of the Ubl domain. Additionally, the C-terminal Ubl and REP block the E2 access to the RING1 domain. (b) Dynamic intermediate Parkin (PDB: 5caw; 143–461 a.a.) in complex with p-Ub. The interaction between p-Ub and Parkin straightens the kinked pUBH helix, opens the IBR and releases the Ubl and its phosphorylation site from the Parkin RBR core. (c) Model of the active form of Parkin. The RING domain of a Cbl-UBCH7 crystal structure (PDB: 1fbv; magenta) conjugated to ubiquitin (PDB: 4q5e; yellow) is superposed to PARKIN RING1 (PDB: 5caw). The Ubl and REP domains are released, making the RING1 domain accessible to the E2∼Ub. The interaction of Parkin with the E2∼Ub could induce conformational changes in that active site of the RING2 domain and fully activate Parkin.
Mechanistically, mitophagy is induced through stabilization and autophosphorylation of PINK1 at the mitochondrial outer membrane (MOM) upon mitochondrial depolarization 68, 69. PINK1 activity is required to recruit Parkin to mitochondria and to activate its E3 ligase activity (Fig3A and B). Parkin then promotes the ubiquitylation of a variety of MOM proteins 70, 71, thereby priming the organelle for recognition by autophagic adaptor proteins that link it to the autophagosomal membrane 63. However, how PINK1 activates and recruits Parkin to mitochondria has been unclear until recently. Crucial insights into this process were gained through quantitative proteomic approaches and novel enrichment strategies for ubiquitylated and phosphorylated proteins. PINK1 was shown to have two critical phosphorylation targets required for optimal activation of Parkin, mitochondrial ubiquitylation and mitophagy: serine 65 of the Ubl domain of Parkin 72, 73, and serine 65 of either Ub 55, 56, 57, 58 or Ub chains assembled on mitochondria 55, 62 (Fig3A).
Before mitochondrial depolarization, interactions between the Parkin N-terminal Ubl domain, its C-terminus 74 and an inhibitory interface involving its catalytically important RBR domains 75, 76, 77 render cytoplasmic Parkin inactive. This native inactive state is partially released upon phosphorylation of serine 65 in the Ubl domain; however, to be fully active p-S65-Parkin also requires the presence of S65-phosphorylated Ub generated by PINK1. Phosphorylated Ub is hardly used by Parkin for conjugation in vitro, but it non-covalently binds to Parkin’s RBR region 57, 58 and accelerates the discharge of Ub loaded on the E2 UBCH7 57, 58, 60. p-S65-Ub (and not p-S20-Ub or p-S57-Ub) binds Parkin to allosterically trigger its E3 ligase activity 58, 60, 78.
Recent structural studies have provided crucial atomic insights into the role of p-S65-Ub binding to Parkin, which initiates the multistep process required for its activation (Fig3B) 78, 79, 80, 81. Parkin is inhibited at multiple levels and disruption of the autoinhibited conformation primes Parkin for activation. Binding of the Ubl domain to the RING1 interface keeps the molecule in a closed conformation 74, the REP element blocks access for the E2-Ub loading 75 and the catalytic cysteine—which is C431 in the RING2—is occluded by the RING0 domain, thus blocking catalysis 75, 76, 77. Binding of p-S65-Ub to Parkin leads to a conformational reorganization in its RING1 domain, and displacement of the Ubl domain and REP element, opening a binding surface to engage Ub conjugated with E2 78, 79, 80, 81. Minimal conformational changes in the RING2 domain upon binding of p-Ub reveal that the thiol group of the active site C431 is capable of accepting ubiquitin from the E2∼Ub conjugate 80. Thus, the conformational change that enables access of the E2-Ub conjugate in the proximity of the active site seems to be an essential step for the catalytic activation of Parkin 80. The p-Ub-binding site is located on the opposite side of the Ubl-binding patch, on a hinge region at RING0/RING1, indicating that antagonistic binding of the Ubl and pUb is mediated through negative allostery 80, 81. Mutations in the p-S65-Ub-binding interface of Parkin (K151, H302, R305, A320 and G284) impede Parkin activation and its recruitment to depolarized mitochondria 78, 79, 80, 81. Interestingly, the G284R mutation in Parkin is associated with AR-JP and the AR-JP mutation L283P and cancer-associated H279P mutations might also interfere with p-Ub binding 79. Parkinson’s disease missense mutations in the Parkin IBR-RING1 interface (R275W, Q311H, G328E) could interfere with sequential p-Ub binding and Ubl release 80. Further AR-JP mutations that occur in the Ubl domain (R42P, A46P and R33Q) 74 could result in loosening of the Ubl from the RING1 and thus in enhanced E3 ligase activity 81. The structural model nicely accounts for the two required steps in Parkin activation: binding of p-S65-Ub to Parkin and PINK1-mediated phosphorylation of S65-Ubl of Parkin (Fig3A and B). Yet, the situation in vivo might be more complex, as Ub-replacement experiments in cells indicate that Parkin can efficiently ubiquitylate a subset of mitochondrial substrates in cells expressing non-phosphorylatable Ub S65A (approximately 90% of total Ub) 60.
The stoichiometry of non-phosphorylated vs. phosphorylated Ub on different substrates, or within Ub chains, might be critical in controlling Parkin activity and in the recruitment of specific effector proteins to p-Ub-decorated mitochondria. For example, whether Parkin can be activated by a single p-Ub on a substrate or a p-Ub moiety within a larger non-phosphorylated Ub chain is still unclear. Notably, unphosphorylated Ub does not induce Parkin activation 57, but has to be present in the reaction for chain building 55, 59.
The stoichiometry of Ub phosphorylation will also depend on phosphatases that can reverse the process. The PINK1-mediated phosphorylation of Ub is reversible and declines with cessation of mitochondrial stress 82. A mitochondrial phosphatase PGAM5 has been shown to interact with PINK1 83, 84 and could be a potential candidate to dephosphorylate p-S65 on Ub. It will be intriguing to identify phosphatases that specifically remove the phosphate group from free Ub or from Ub chains attached to substrates, thereby regulating not only Parkin activation but more broadly the biological responses mediated by binding of UBD-containing effector proteins.
Phosphorylation of Ub expands the repertoire of Ub-binding proteins
The unique conjugation machinery of ubiquitin enables the assembly of diverse and complex Ub modifications on substrate proteins. More than 20 different types of UBDs implicated in decoding the multiple functions of specific Ub modifications have been characterized to date 11. The large majority of them bind with a relatively low affinity to a hydrophobic patch centred on isoleucine 44 of Ub. Phosphorylation of Ub changes its surface properties, inducing a modest conformational change adjacent to the hydrophobic patch 59. Therefore, the recognition of phosphorylated monoUb and polyUb chains by Ub-binding proteins is likely altered upon phosphorylation. Recent work has indicated that the Ub-binding proteins NDP52 and OPTN are the primary autophagic receptors for PINK1/Parkin-mediated mitophagy 85. The UBAN domain of OPTN and the UBZ domain of NDP52 are essential domains required for the removal of both cytosolic Salmonella 86, 87 and mitochondria 85, 88. In the case of mitophagy, Youle and colleagues have shown that PINK1-mediated phosphorylation of Ub generates a specific signal required for recruiting OPTN and NDP52 to damaged mitochondria thereby activating mitophagy 85. Interestingly, OPTN and NDP52 were not only shown to act as receptors for LC3-positive autophagosomal membranes, but also to recruit additional key components of the autophagy machinery including ULK1, DFCP1 and WIPI1 in order to promote in situ growth of the autophagosome and efficient mitophagy 85. The kinase activity of PINK1 and the ability of OPTN and NDP52 to bind to Ub are essential for this process, whereas Parkin-mediated ubiquitylation amplifies it (Fig3A). Yet, the basis for the specific interaction of OPTN and NDP52 with pS65-Ub is not understood. Both autophagy receptors preferentially bind to p-S65-Ub and to phosphomimetic S65D-Ub in vivo 85. However, in vitro OPTN, NDP52 and p62 interacted less well with phosphorylated K63-Ub chains 60. Similarly, none of the tested UBD-containing proteins that bind to the conserved hydrophobic patch around I44 on Ub showed increased affinities to phosphomimetic monoUbS65E 49 or phosphorylated Ub chains 60 in vitro. Thus, the cellular milieu favours the binding of autophagy receptors to phosphorylated Ub through a mechanism that cannot be recapitulated in vitro using purified proteins. Harper and colleagues showed that around 20% of mitochondrial Ub is phosphorylated 55. Ub chains consisting of both phosphorylated and non-phosphorylated Ub molecules could control the recruitment of autophagic receptors and LC3 in vivo. In addition, phosphorylation of Ub at multiple sites may generate a new set of signalling cues that are recognized by autophagy receptor complexes (harbouring known or novel UBDs).
TBK1-mediated phosphorylation controls ubiquitin-dependent autophagy
In addition to PINK1, TBK1 was found to play a key role in engaging selective autophagy pathways for efficient removal of pathogens, protein aggregates and mitochondria 25, 27, 87, 89. OPTN, as well as TANK, Sintbad and Nap1 act as adaptor proteins that direct TBK1 to distinct complexes within cells 90. For example in the case of bacterial invaders, TBK1 is activated and recruited via OPTN and NDP52 to Ub-decorated cytosolic bacteria 86, 87. Analogously, TBK1 acts in concert with OPTN to mediate mitophagy of depolarized mitochondria 85. Notably, direct binding of OPTN to Ub chains on cargoes has been proposed to promote TBK1 oligomerization and activation 91. Locally accumulated TBK1 in turn phosphorylates the autophagy receptors OPTN and p62 on multiple sites, including the UBA domain of p62 and the UBAN domain of OPTN 25, 27, 87 (Richter and Dikic, unpublished observation) (Fig4). This feedback phosphorylation increases the binding affinity of the autophagy receptors to Ub chains, thus strengthening their commitment to selected cargoes. This occurs, for example, on TBK1-mediated phosphorylation of S403 in the UBA domain of p62, which strongly enhances its binding to K63- and K48-linked polyUb chains 25 and has been implicated in the autophagic elimination of Mycobacterium tuberculosis 27 and polyubiquitylated mitochondria during PINK1/Parkin-mediated mitophagy 25. Phosphorylation of S403-p62 by CK2 can also promote targeting of polyubiquitylated proteins into sequestosomes and their subsequent clearance by aggrephagy 26. One interesting observation is that TBK1 may phosphorylate OPTN and p62 that are localized to different mitochondrial surface microdomains, with likely distinct functional consequences in the mitophagy pathway 85, 88. This is reminiscent of microdomain structures found on the Salmonella surface in the early phase of autophagy 87, 92.
Figure 4. TBK1 in control of ubiquitin-dependent autophagy.
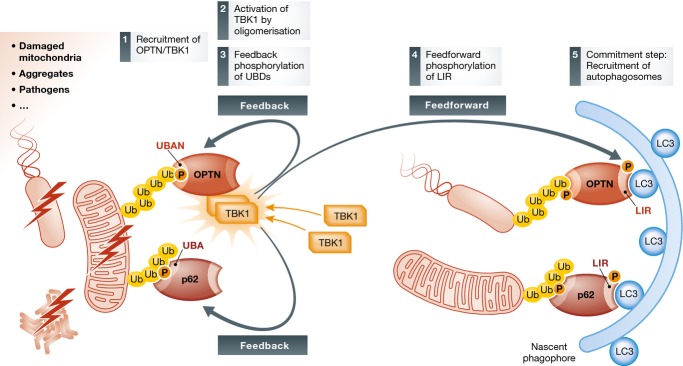
TBK1 mediates feedback and feedforward regulation of the two autophagic adaptor proteins p62 and OPTN. Phosphorylation on their UBDs promotes binding to ubiquitylated cargo, whereas phosphorylation of the LIR motifs promotes the recruitment of autophagosomal membranes. Thus, TBK1 amplifies cargo recognition and commits it to degradation.
TBK1 can also phosphorylate S177 adjacent to the LIR motif of OPTN, resulting in an increase in LC3 binding that provides a feedforward mechanism for the clearance of cytosolic Salmonella and restriction of intracellular bacterial proliferation 87. Moreover, S332 in the LIR motif of p62 is also subjected to phosphorylation 26, indicating that, in analogy to OPTN, the LIR/LC3 interactions of p62 might be regulated through phosphorylation. However, further studies are required to assign a functional role for S332 phosphorylation of p62.
Thus, both the Ub/UBD interface that couples autophagy receptors to cargo, as well as the LIR/LC3 interface that links cargo to autophagic membranes, undergo phosphorylation (Fig4). In this context, TBK1-mediated phosphorylation of autophagy receptors effectively prolongs their residence time on the surface of cargoes and also promotes directionality and the delivery of cargoes for autophagosomal degradation.
Integrative model for phosphorylation-based regulation of mitophagy
The modulation of critical binding interfaces by phosphorylation is emerging as a general regulatory concept in selective autophagy pathways as nicely demonstrated for the PINK1- and TBK1-dependent amplification loop in the control of mitophagy (Fig5). PINK1-mediated phosphorylation of Ub and Parkin Ubl on their conserved serine 65 residues acts as an early event leading to the recruitment and activation of Parkin on mitochondria. Parkin ensures its own continuance on mitochondria through assembling Ub chains on MOM proteins. Simultaneously, autophagy receptors (such as OPTN and p62) are recruited to the sites of mitochondrial damage. Through TBK1-mediated phosphorylation on their UBDs and LIRs, autophagy receptors facilitate the removal of damaged mitochondria by bridging the p-ubiquitylated cargo to LC3-coated phagophores (Fig5). Taken together, PINK1-mediated phosphorylation of Ub and Parkin and TBK1-mediated phosphorylation of autophagy receptors establish a robust mechanism that drives the selective labelling of damaged mitochondria to ensure their delivery for lysosomal degradation.
Figure 5. Integrative model for PINK1/Parkin- and TBK1/OPTN-dependent mitophagy.
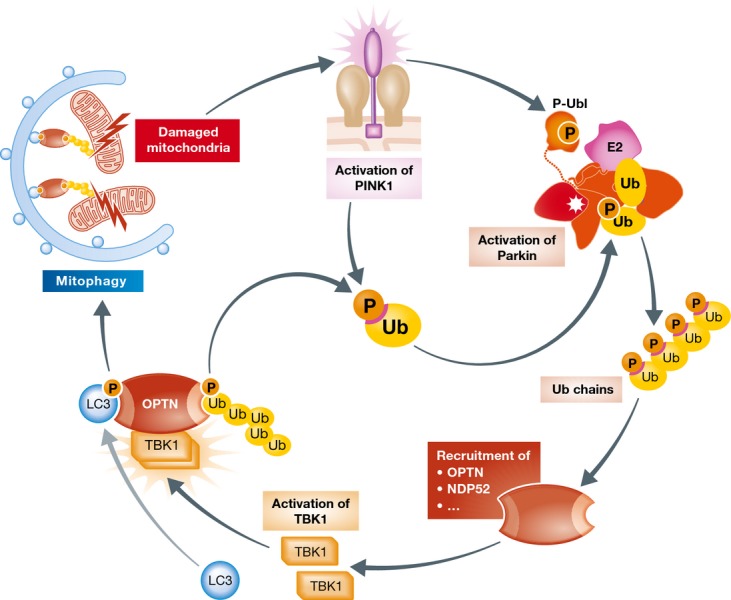
Mitochondrial damage leads to the activation of the kinase PINK1, which phosphorylates Ub and Parkin on S65. The phosphorylation-dependent activation of Parkin results in the generation of phosphorylated polyUb chains on MOM proteins. Autophagic receptors such as OPTN are recruited to the site of mitochondrial damage, together with the kinase TBK1, which becomes activated through dimerization and autophosphorylation. Active TBK1 phosphorylates the Ub-binding regions within OPTN. The p-Ub MOM substrates are subsequently bound by autophagic receptors, which link damaged mitochondria to autophagosomal membranes through binding to LC3s thereby resulting in mitophagy.
Phosphorylation of Ub and UBDs in neurodegenerative disease pathogenesis
Deregulation of the Ub system is often associated with severe phenotypes and diseases, including a variety of neurodegenerative disorders 93. The potential physiological relevance of the PINK1- and TBK1-mediated phosphorylation loops is emphasized by genetic and biochemical studies showing that mutations in autophagy receptors that affect either their Ub-binding or their LC3-binding ability are linked to neurodegenerative disorders, in particular frontotemporal lobar degeneration (FTLD), amyotrophic lateral sclerosis (ALS) and Parkinson’s disease (PD) 88, 94, 95, 96. Most recently, genetic alterations impairing TBK1 activity or its binding to OPTN have been linked to FTLD and ALS 97, 98, 99. Likewise, defects in the PINK1/Parkin pathway that disrupt signal amplification cause Parkinson’s disease 67, 100, 101. Given the crucial role of p-S65-Ub in mitophagy, anti-p-S65-Ub antibodies may prove to be a useful tool for future studies of Parkinson’s disease. Indeed, p-S65-Ub can be detected under endogenous conditions in stressed neurons and accumulates in human brain during ageing and Parkinson’s disease, whereas it is largely absent in material obtained from Parkinson patients carrying PINK1 mutations 82. Moreover, proteomics could potentially be developed as a diagnostic tool, as its quantitative nature would be crucial for the discrimination between basal mitophagy and pathological conditions.
A caveat when studying the effects of Parkin mutations on the development of Parkinsonism in mouse models is that Parkin knockout mice do not display a neurodegenerative phenotype. In order to overcome this issue and study Parkin function in vivo, the Youle laboratory created a mouse model with accelerated accumulation of mitochondrial DNA mutations 102. The loss of Parkin paired with the accumulation of dysfunctional mitochondria caused a loss of dopaminergic neurons, but neurons from other neuroanatomical regions appeared unaffected. The levels of p-S65-Ub measured by quantitative proteomics increase in the brains of these mice, indicating that in vivo mitochondrial damage leads to the accumulation of a specific p-Ub autophagic signal 102.
Conclusions
Initial work on the crosstalk of ubiquitylation with other PTMs revealed interesting bidirectional regulatory mechanisms. PTMs on the Ub moiety fine-tune signalling and increase the repertoire and selectivity of downstream interacting proteins. Recent work on PINK1/Parkin and TBK1/OPTN, showing interdependency of ubiquitylation and phosphorylation in selective mitophagy pathways, has provided new mechanistic insight and generated great excitement in the field (Figs5).
There is likely a much broader spectrum of phosphorylation-dependent Ub functions in different pathways. Moreover, the identification of other mammalian kinases, as well as the yeast kinase(s) that can phosphorylate Ub on S65, S57 and other residues, is likely to reveal many of the pleiotropic effects of the expanded Ub code in cellular signalling. One interesting avenue for future research is to further explore the crosstalk between PINK1 and TBK1, which modify the Ub code and Ub receptors, respectively, thereby influencing their binding properties and their physiological roles. In addition, the role of other Ub ligases that act together or in parallel with Parkin should be further investigated. There is evidence that MUL1/MAPL/MULAN, another RING E3 ligase associated with mitochondria, can act as an alternative E3 ligase to Parkin to mediate mitophagy 103, 104. Furthermore, Parkin has been shown to maintain mitochondrial integrity by increasing linear ubiquitylation of NEMO, and this autophagy-independent pathway involves Parkin-mediated activation of LUBAC 105.
Phosphomimetic Ub mutants—which are commonly used in research—provide a pragmatic experimental tool as the entire population of the studied protein is changed. Nevertheless, caution should be exerted when using these approaches regarding the physiological relevance of the conclusions drawn—even though they could be correct. Diverting results may partly be ascribed to the varying stoichiometry of phosphorylated substrates, such as Ub or Ub receptors. Moreover, the effect of serine to glutamic/aspartic acid mutations only partially mimics the altered surface or the conformational change induced by phosphorylation and could also account for a non-physiological consequence of the mutation. For example, MEK that is phosphorylated at S217/S221 by Raf has a 7,000-fold higher activity than the dephosphorylated enzyme, but the phosphomimetic mutations S217E/S221E of MEK only induce a 180-fold higher activity 106. Similarly, due to the fast off-rates, a physiological binding partner of phosphorylated Ub is unlikely to be trapped in a complex with a Ub species that harbours a phosphomimetic residue (E/D). Likewise, phosphomimetic Ub or Ubl Parkin proteins do not mimic their stoichiometrically phosphorylated forms, as phosphomimetics lack activity during Ub chain synthesis, whereas phosphorylated proteins are active 60.
The tRNA-based method to directly incorporate O-phosphoserine at desired positions and to stoichiometrically generate milligram quantities of serine-phosphorylated proteins in E. coli has already been used for S65-phosphorylated Ub (as well as other serine sites) 60, 107, 108. Another recombinant approach to generate phosphorylated Ub molecules even included threonine- and tyrosine-phosphorylated forms of Ub 109. These new techniques could lead to improved analysis, aiding the definition of genuine interactors of phosphorylated Ub.
Future studies will reveal whether there are even more PTMs that control the function of Ub and it will be important to identify the modifying enzymes that catalyse these PTMs. Moreover, using powerful quantitative proteomics will enable the identification, quantification and tracking of new PTMs on Ub with great sensitivity. Advanced proteomic tools to identify the flux and stoichiometry of rate limiting intermediates for phosphorylation and ubiquitylation are already available 110; the technology would have to be further advanced for less studied PTMs.
Taken together, the modification of Ub and its receptors through phosphorylation, acetylation (Box 1) or deamidation (Box 2) can efficiently diversify the Ub code, thus adding another layer of complexity to Ub biology with important medical implications.
Acknowledgments
We thank Wade Harper, Richard Youle, Keiji Tanaka, Dario Alessi, David Komander, Helen Walden, Noriyuki Matsuda, Miratul Muqit, David McEwan, Alexandra Stolz, Sagar Bhogaraju, Daniela Hoeller and Alban Ordureau for insightful comments and critical reading of the manuscript. We are grateful to Masato Akutsu for technical help with PyMOLE and a model presented in Figs2 and 3B. L.H. is funded by the EMBO long-term postdoctoral fellowship. I.D. is supported by grants from Deutsche Forschungsgemeinschaft (DI 931/3-1), the Cluster of Excellence “Macromolecular Complexes” of the Goethe University Frankfurt (EXC115), LOEWE grant Ub-Net and LOEWE Centrum for Gene and Cell therapy Frankfurt and the European Research Council/ERC grant agreement n° 250241-LineUb to I.D.
Glossary
- AMSH
associated molecule with the SH3 domain of STAM
- APC
anaphase promoting complex
- AR-JP
autosomal recessive juvenile parkinsonism
- Cif
cycle inhibiting factor
- CHBP
cycle inhibiting factor homolog in Burkholderia pseudomallei
- CK2
casein kinase 2
- DUB
deubiquitylating enzyme
- ERK
extracellular-signal-regulated kinase
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- IAP
inhibitor of apoptosis
- IKK
IkappaB kinase
- LUBAC
linear ubiquitin assembly complex
- MAPK
mitogen activated protein kinase
- MS
mass spectrometry
- MUL1
mitochondrial E3 ubiquitin protein ligase 1
- MAPL
mitochondrial-anchored protein ligase
- MULAN
mitochondrial ubiquitin ligase activator of NF-κB
- NEDD8
neural precursor cell expressed, developmentally down-regulated 8
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PGAM5
phosphoglycerate mutase 5
- PINK1
PTEN-induced putative kinase 1
- RING
really interesting new gene
- RBR
RING in between RING
- SCF
Skp, Cullin, F-box containing complex
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment receptor
- TBK-1
TANK-binding kinase 1
- TRAF
TNF receptor-associated factor
- UBA
ubiquitin-associated domain
- UBAN
ubiquitin binding in Abin and NEMO
- UBD
ubiquitin-binding domain
- UBZ
ubiquitin-binding zinc finger domain
- Ubl
ubiquitin-like
- USP
ubiquitin-specific protease
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol. 2010;17:666–672. doi: 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- Varshavsky A. The early history of the ubiquitin field. Protein Sci. 2006;15:647–654. doi: 10.1110/ps.052012306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009;458:422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Schulman BA. Twists and turns in ubiquitin-like protein conjugation cascades. Protein Sci. 2011;20:1941–1954. doi: 10.1002/pro.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt DE, Walden H, Shaw GS. RBR E3 ubiquitin ligases: New structures, new insights, new questions. Biochem J. 2014;458:421–437. doi: 10.1042/BJ20140006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Meyer HJ, Rape M. Enhanced protein degradation by branched ubiquitin chains. Cell. 2014;157:910–921. doi: 10.1016/j.cell.2014.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, Cohen P. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci USA. 2013;110:15247–15252. doi: 10.1073/pnas.1314715110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem. 2012;81:291–322. doi: 10.1146/annurev-biochem-051810-094654. [DOI] [PubMed] [Google Scholar]
- Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ, Urbe S. Deubiquitylases from genes to organism. Physiol Rev. 2013;93:1289–1315. doi: 10.1152/physrev.00002.2013. [DOI] [PubMed] [Google Scholar]
- Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–6245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronroos E, Hellman U, Heldin CH, Ericsson J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell. 2002;10:483–493. doi: 10.1016/s1097-2765(02)00639-1. [DOI] [PubMed] [Google Scholar]
- Du Z, Song J, Wang Y, Zhao Y, Guda K, Yang S, Kao HY, Xu Y, Willis J, Markowitz SD, et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci Signal. 2010;3:ra80. doi: 10.1126/scisignal.2001462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher E, Gao M, Liu YC, Karin M. Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change. Proc Natl Acad Sci USA. 2006;103:1717–1722. doi: 10.1073/pnas.0510664103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Zhou W, M-s J, Demydenko D, Harada Y, Zhou H, Liu Y-C. Negative regulation of the E3 ubiquitin ligase itch via Fyn-mediated tyrosine phosphorylation. Mol Cell. 2006;21:135–141. doi: 10.1016/j.molcel.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Persaud A, Alberts P, Mari S, Tong J, Murchie R, Maspero E, Safi F, Moran MF, Polo S, Rotin D. Tyrosine phosphorylation of NEDD4 activates its ubiquitin ligase activity. Sci Signal. 2014;7:ra95. doi: 10.1126/scisignal.2005290. [DOI] [PubMed] [Google Scholar]
- Huang OW, Ma X, Yin J, Flinders J, Maurer T, Kayagaki N, Phung Q, Bosanac I, Arnott D, Dixit VM, et al. Phosphorylation-dependent activity of the deubiquitinase DUBA. Nat Struct Mol Biol. 2012;19:171–175. doi: 10.1038/nsmb.2206. [DOI] [PubMed] [Google Scholar]
- Grabbe C, Husnjak K, Dikic I. The spatial and temporal organization of ubiquitin networks. Nat Rev Mol Cell Biol. 2011;12:295–307. doi: 10.1038/nrm3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell. 2003;11:1445–1456. doi: 10.1016/s1097-2765(03)00234-x. [DOI] [PubMed] [Google Scholar]
- Herhaus L, Al-Salihi M, Macartney T, Weidlich S, Sapkota GP. OTUB1 enhances TGFbeta signalling by inhibiting the ubiquitylation and degradation of active SMAD2/3. Nat Commun. 2013;4:2519. doi: 10.1038/ncomms3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet. 2015;24:4429–4442. doi: 10.1093/hmg/ddv179. [DOI] [PubMed] [Google Scholar]
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–289. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37:223–234. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ. Regulatory modules: coupling protein stability to phopshoregulation during cell division. FEBS Lett. 2012;586:2773–2777. doi: 10.1016/j.febslet.2012.05.045. [DOI] [PubMed] [Google Scholar]
- Ordureau A, Smith H, Windheim M, Peggie M, Carrick E, Morrice N, Cohen P. The IRAK-catalysed activation of the E3 ligase function of Pellino isoforms induces the Lys63-linked polyubiquitination of IRAK1. Biochem J. 2008;409:43–52. doi: 10.1042/BJ20071365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Wertz IE, Dixit VM. Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harb Perspect Biol. 2010;2:a003350. doi: 10.1101/cshperspect.a003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- Peschard P, Ishiyama N, Lin T, Lipkowitz S, Park M. A conserved DpYR motif in the juxtamembrane domain of the Met receptor family forms an atypical c-Cbl/Cbl-b tyrosine kinase binding domain binding site required for suppression of oncogenic activation. J Biol Chem. 2004;279:29565–29571. doi: 10.1074/jbc.M403954200. [DOI] [PubMed] [Google Scholar]
- Takeda AN, Oberoi-Khanuja TK, Glatz G, Schulenburg K, Scholz RP, Carpy A, Macek B, Remenyi A, Rajalingam K. Ubiquitin-dependent regulation of MEKK2/3-MEK5-ERK5 signaling module by XIAP and cIAP1. EMBO J. 2014;33:1784–1801. doi: 10.15252/embj.201487808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Na K, Kwon MS, Kim H, Kim KS, Paik YK. Quantitative analysis of phosphopeptides in search of the disease biomarker from the hepatocellular carcinoma specimen. Proteomics. 2009;9:3395–3408. doi: 10.1002/pmic.200800943. [DOI] [PubMed] [Google Scholar]
- Shiromizu T, Adachi J, Watanabe S, Murakami T, Kuga T, Muraoka S, Tomonaga T. Identification of missing proteins in the neXtProt database and unregistered phosphopeptides in the PhosphoSitePlus database as part of the Chromosome-centric Human Proteome Project. J Proteome Res. 2013;12:2414–2421. doi: 10.1021/pr300825v. [DOI] [PubMed] [Google Scholar]
- Bian Y, Song C, Cheng K, Dong M, Wang F, Huang J, Sun D, Wang L, Ye M, Zou H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J Proteomics. 2014;96:253–262. doi: 10.1016/j.jprot.2013.11.014. [DOI] [PubMed] [Google Scholar]
- Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJ, Mohammed S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res. 2013;12:260–271. doi: 10.1021/pr300630k. [DOI] [PubMed] [Google Scholar]
- Sharma K, D’Souza RC, Tyanova S, Schaab C, Wisniewski JR, Cox J, Mann M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8:1583–1594. doi: 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- Ohtake F, Saeki Y, Sakamoto K, Ohtake K, Nishikawa H, Tsuchiya H, Ohta T, Tanaka K, Kanno J. Ubiquitin acetylation inhibits polyubiquitin chain elongation. EMBO Rep. 2015;16:192–201. doi: 10.15252/embr.201439152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, Olsen JV. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun. 2012;3:876. doi: 10.1038/ncomms1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PN, Bohmer FD, Gerke V, Schmidt-Arras DE, Berdel WE, Muller-Tidow C, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009;36:326–339. doi: 10.1016/j.molcel.2009.09.019. [DOI] [PubMed] [Google Scholar]
- Manes NP, Dong L, Zhou W, Du X, Reghu N, Kool AC, Choi D, Bailey CL, Petricoin EF, 3rd, Liotta LA, et al. Discovery of mouse spleen signaling responses to anthrax using label-free quantitative phosphoproteomics via mass spectrometry. Mol Cell Proteomics. 2011;10:M110 000927. doi: 10.1074/mcp.M110.000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen MV, Larsen DH, Bunkenborg J, Bartek J, Lukas J, Andersen JS. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol Cell Proteomics. 2010;9:1314–1323. doi: 10.1074/mcp.M900616-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Brumbaugh J, Wenger CD, Tian S, Probasco MD, Bailey DJ, Swaney DL, Tervo MA, Bolin JM, Ruotti V, et al. Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat Methods. 2011;8:821–827. doi: 10.1038/nmeth.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Lenobel R, Santamaria A, Ries A, Nigg EA, Korner R. Quantitative analysis of the human spindle phosphoproteome at distinct mitotic stages. J Proteome Res. 2009;8:4553–4563. doi: 10.1021/pr9003773. [DOI] [PubMed] [Google Scholar]
- Villen J, Beausoleil SA, Gerber SA, Gygi SP. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci USA. 2007;104:1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney DL, Rodríguez-Mias RA, Villén J. Phosphorylation of ubiquitin at Ser65 affects its polymerization, targets and proteome-wide turnover. EMBO Rep. 2015;16:1131–1144. doi: 10.15252/embr.201540298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- Gu TL, Deng X, Huang F, Tucker M, Crosby K, Rimkunas V, Wang Y, Deng G, Zhu L, Tan Z, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One. 2011;6:e15640. doi: 10.1371/journal.pone.0015640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, Kornhauser J, Sprott K, Zhou J, Possemato A, et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal. 2010;3:ra64. doi: 10.1126/scisignal.2000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Bodenmiller B, Campbell D, Gerrits B, Lam H, Jovanovic M, Picotti P, Schlapbach R, Aebersold R. PhosphoPep–a database of protein phosphorylation sites in model organisms. Nat Biotechnol. 2008;26:1339–1340. doi: 10.1038/nbt1208-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56:360–375. doi: 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460:127–139. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- Wauer T, Swatek KN, Wagstaff JL, Gladkova C, Pruneda JN, Michel MA, Gersch M, Johnson CM, Freund SM, Komander D. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 2015;34:307–325. doi: 10.15252/embj.201489847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Heo J, Duda DM, Olszewski JL, Yanishevski D, Rinehart J, Schulman B, Harper JW. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci USA. 2015;112:6637–6642. doi: 10.1073/pnas.1506593112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondalapati S, Mansour W, Nakasone MA, Maity SK, Glickman MH, Brik A. Chemical synthesis of phosphorylated ubiquitin and diubiquitin exposes positional sensitivities of E1-E2 enzymes and deubiquitinases. Chem Eur J. 2015;21:7360–7364. doi: 10.1002/chem.201500540. [DOI] [PubMed] [Google Scholar]
- Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, Matsuda N. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol. 2015;209:111–128. doi: 10.1083/jcb.201410050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun. 2012;3:1016. doi: 10.1038/ncomms2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol. 2012;4:a011338. doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2:1002. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, Walden H. Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 2011;30:2853–2867. doi: 10.1038/emboj.2011.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013;340:1451–1455. doi: 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- Wauer T, Komander D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013;32:2099–2112. doi: 10.1038/emboj.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BE, Lougheed JC, Callaway K, Velasquez M, Brecht E, Nguyen L, Shaler T, Walker D, Yang Y, Regnstrom K, et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun. 2013;4:1982. doi: 10.1038/ncomms2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Martínez-Torres RJ, Wilkie S, Kumar A, Peltier J, Gonzalez A, Johnson C, Zhang J, Hope AG, Peggie M, et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep. 2015;16:939–954. doi: 10.15252/embr.201540352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Simicek M, Schubert A, Komander D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature. 2015 doi: 10.1038/nature14879. doi: 10.1038/nature14879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvé V, Lilov A, Seirafi M, Vranas M, Rasool S, Kozlov G, Sprules T, Wang J, Trempe JF, Gehring K. A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 2015 doi: 10.15252/embj.201592237. doi: 10.15252/embj.201592237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Aguirre JD, Condos TEC, Martinez-Torres RJ, Chaugule VK, Toth R, Sundaramoorthy R, Mercier P, Knebel A, Spratt DE, et al. Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J. 2015 doi: 10.15252/embj.201592337. doi: 10.15252/embj.201592337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiesel FC, Ando M, Hudec R, Hill AR, Castanedes-Casey M, Caulfield TR, Moussaud-Lamodière EL, Stankowski JN, Bauer PO, Lorenzo-Betancor O, et al. PINK1-Dependent Phosphorylation of Ubiquitin is (Patho-)Physiologically Relevant. EMBO Rep. 2015;16:1114–1130. doi: 10.15252/embr.201540514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kanao T, Sawada T, Kobayashi Y, Moriwaki Y, Ishida Y, Takeda K, Ichijo H, Lu B, Takahashi R. The loss of PGAM5 suppresses the mitochondrial degeneration caused by inactivation of PINK1 in Drosophila. PLoS Genet. 2010;6:e1001229. doi: 10.1371/journal.pgen.1001229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Karuppagounder SS, Springer DA, Allen MD, Zheng L, Chao B, Zhang Y, Dawson VL, Dawson TM, Lenardo M. Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson’s-like movement disorder. Nat Commun. 2014;5:4930. doi: 10.1038/ncomms5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. PINK1 phosphorylation of ubiquitin recruits autophagy receptors to mitochondria and upstream autophagy machinery for mitophagy. Nature. 2015 doi: 10.1038/nature14893. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci USA. 2014;111:E4439–E4448. doi: 10.1073/pnas.1405752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, Terzic J, Dikic I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013;126:580–592. doi: 10.1242/jcs.114926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves A, Burckstummer T, Dixit E, Scheicher R, Gorna MW, Karayel E, Sugar C, Stukalov A, Berg T, Kralovics R, et al. Functional dissection of the TBK1 molecular network. PLoS One. 2011;6:e23971. doi: 10.1371/journal.pone.0023971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason CE, Ordureau A, Gourlay R, Arthur JS, Cohen P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon beta. J Biol Chem. 2011;286:35663–35674. doi: 10.1074/jbc.M111.267567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy. 2011;7:341–345. doi: 10.4161/auto.7.3.14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. 2014;20:1242–1253. doi: 10.1038/nm.3739. [DOI] [PubMed] [Google Scholar]
- Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, Fenoglio P, Grinberg Y, Isaia G, Calvo A, Gentile S, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79:1556–1562. doi: 10.1212/WNL.0b013e31826e25df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen WC, Li HY, Chen GC, Chern Y, Tu PH. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy. 2015;11:685–700. doi: 10.4161/auto.36098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, Brown P, Ravenscroft T, van Blitterswijk M, Nicholson AM, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015;130:77–92. doi: 10.1007/s00401-015-1436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, Marroquin N, Nordin F, Hubers A, Weydt P, et al. Haploin-sufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–636. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Muqit MM. PINK1 and Parkin - mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson’s disease. FEBS J. 2015;282:215–223. doi: 10.1111/febs.13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiesel FC, Caulfield TR, Moussaud-Lamodiere EL, Dourado KO, Flores SC, Ross OA, Springer W. Structural and functional impact of Parkinson disease-associated mutations in the E3 ubiquitin ligase Parkin. Hum Mutat. 2015;36:774–786. doi: 10.1002/humu.22808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ. Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron. 2015;87:371–381. doi: 10.1016/j.neuron.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Puri R, Yang H, Lizzio MA, Wu C, Sheng ZH, Guo M. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. eLife. 2014;3:e01958. doi: 10.7554/eLife.01958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambivero CT, Cilenti L, Main S, Zervos AS. Mulan E3 ubiquitin ligase interacts with multiple E2 conjugating enzymes and participates in mitophagy by recruiting GABARAP. Cell Signal. 2014;26:2921–2929. doi: 10.1016/j.cellsig.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Muller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, Peis R, Deinlein A, Schweimer C, Kuhn PH, et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell. 2013;49:908–921. doi: 10.1016/j.molcel.2013.01.036. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Rogerson DT, Sachdeva A, Wang K, Haq T, Kazlauskaite A, Hancock SM, Huguenin-Dezot N, Muqit MM, Fry AM, Bayliss R, et al. Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nat Chem Biol. 2015;11:496–503. doi: 10.1038/nchembio.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, Soll D. Expanding the genetic code of Escherichia coli with phosphoserine. Science. 2011;333:1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Pao KC, Kazlauskaite A, Muqit MM, Virdee S. A versatile strategy for the semisynthetic production of Ser65 phosphorylated ubiquitin and its biochemical and structural characterization. ChemBioChem. 2015;16:1574–1579. doi: 10.1002/cbic.201500185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Munch C, Harper JW. Quantifying ubiquitin signaling. Mol Cell. 2015;58:660–676. doi: 10.1016/j.molcel.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Robinson NE, Robinson AB. Use of Merrifield solid phase peptide synthesis in investigations of biological deamidation of peptides and proteins. Biopolymers. 2008;90:297–306. doi: 10.1002/bip.20852. [DOI] [PubMed] [Google Scholar]
- Washington EJ, Banfield MJ, Dangl JL. What a difference a Dalton makes: bacterial virulence factors modulate eukaryotic host cell signaling systems via deamidation. Microbiol Mol Biol Rev. 2013;77:527–539. doi: 10.1128/MMBR.00013-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Yao Q, Li S, Ding X, Lu Q, Mao H, Liu L, Zheng N, Chen S, Shao F. Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science. 2010;329:1215–1218. doi: 10.1126/science.1193844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q, Cui J, Wang J, Li T, Wan X, Luo T, Gong YN, Xu Y, Huang N, Shao F. Structural mechanism of ubiquitin and NEDD8 deamidation catalyzed by bacterial effectors that induce macrophage-specific apoptosis. Proc Natl Acad Sci USA. 2012;109:20395–20400. doi: 10.1073/pnas.1210831109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14:1050–1061. doi: 10.1038/embor.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nat Rev Drug Discovery. 2014;13:889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Nakatani T, Kamitani T. Inhibition of NEDD8-conjugation pathway by novel molecules: potential approaches to anticancer therapy. Mol Oncol. 2012;6:267–275. doi: 10.1016/j.molonc.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striebel F, Imkamp F, Sutter M, Steiner M, Mamedov A, Weber-Ban E. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol. 2009;16:647–651. doi: 10.1038/nsmb.1597. [DOI] [PubMed] [Google Scholar]