

Abstract
Elimination of malignant cells is an unmet challenge for most human cancer types even with therapies targeting specific driver mutations. Therefore, a multi-pronged strategy to alter cancer cell biology on multiple levels is increasingly recognized as essential for cancer cure. One such aspect of cancer cell biology is the relative apoptosis resistance of tumor-initiating cells. Here, we provide an overview of the mechanisms affecting the apoptotic process in tumor cells emphasizing the differences in the tumor-initiating or stem-like cells of cancer. Further, we summarize efforts to exploit these differences to design therapies targeting that important cancer cell population.
Keywords: apoptosis, cancer stem cells, cancer therapy
Introduction
Programmed cell death, or apoptosis, plays essential roles throughout the lifetime of multi-cellular organisms, from early embryonic development to tissue homeostasis during adulthood. This self-destruction program is tightly regulated and mediated by a group of proteases named caspases. Based on the initiation mechanism, apoptosis can be classified into two types of death pathways: the extrinsic or receptor-mediated pathway and the intrinsic or mitochondria-dependent pathway (Fig1). The extrinsic death pathway is initiated by engagement of membrane-bound death receptors (DRs). Upon engagement with their cognate ligands such as TNF-α, FAS ligand, TNF-related apoptosis-inducing ligand (TRAIL) etc., DRs aggregate and recruit other adaptor proteins that bear the death domain to form a death-inducing signaling complex (DISC), which further triggers the execution phase of apoptosis. Apoptosis can also be initiated by receptor-independent stimuli such as radiation, free radicals, viral infection, growth factor withdrawal, and chemotherapeutic drugs, which trigger mitochondrial outer membrane permeabilization (MOMP) by activation of Bcl-2 homologous proteins 1. The process of apoptosis is positively regulated by tumor-suppressor p53 and negatively regulated by the pro-survival PI3K/AKT pathway. As a transcription factor, p53 induces the expression of many pro-apoptotic genes, including DRs and multiple pro-apoptotic Bcl-2 family members 2. p53 also represses the expression of anti-apoptotic genes such as Bcl-2, Bcl-xL, and survivin 2. Activation of PI3K/AKT suppresses apoptosis either directly by phosphorylating and inactivating pro-apoptotic proteins such as Bad and Bax or indirectly by activating downstream pro-survival targets such as FOXO, NF-κB, and GSK signaling cascades 3.
Figure 1. Schematic overview of the intrinsic and extrinsic apoptotic pathways.
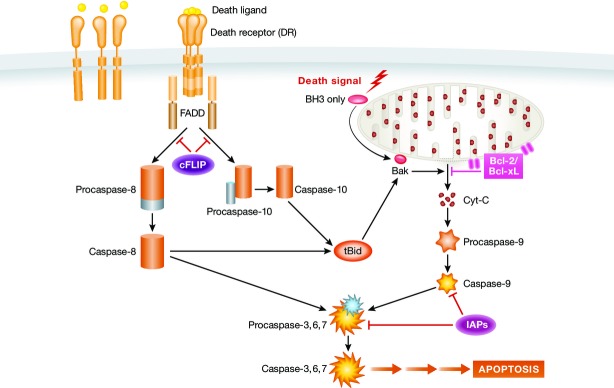
In the extrinsic pathway, the apoptotic program is initiated by engagement of membrane-bound DRs with their ligands. Ligand binding leads to aggregation of DRs and recruitment of adaptor proteins and initiator pro-caspases, such as pro-caspase-8 to form the DISC complex. Pro-caspase-8 is then converted to active caspase-8, which further activates the effector caspase-3. Active caspase-3 initiates the downstream caspase cascade leading to apoptosis. DR-initiated pathway is negatively regulated by c-FLIP, which inhibits DISC formation and activation of caspases 8 and 10 and prevents the downstream apoptotic signaling. The intrinsic pathway is initiated by receptor-independent stimuli, such as DNA damage, which triggers mitochondrial outer membrane permeabilization (MOMP) by activation of Bcl-2 homologous proteins. MOMP causes inner membrane permeability and initiates release of pro-apoptotic proteins such as cytochrome C. The proteins released from mitochondria activate caspase-9, which further activates downstream caspase cascade leading to programmed cell death.
A common hallmark of cancer is the dysregulated apoptosis machinery 4. Defects in both extrinsic and intrinsic apoptosis have been described in a variety of human cancers and are believed to contribute to tumor transformation and progression 5. It is frequently observed that tumor cells express higher levels of pro-survival genes and genes promoting cell proliferation and downregulate anti-apoptotic genes. Therefore, targeting the apoptotic pathway to induce cell death has been long considered a promising therapeutic strategy for cancer. In fact, for most anticancer therapies including chemotherapy, irradiation, and immunotherapy, induction of apoptosis represents the major mechanism by which cancer cells are eliminated. Many tumors are initially responsive to conventional therapy as reflected by tumor mass shrinkage and clinical remission. In most cases, however, development of secondary tumors often leads to disease relapse and increased resistance to therapy.
Cancer stem-like cells (CSCs) represent a population of cells within a tumor that are particularly endowed with the ability to initiate the tumor in transplantation models. Their ability to do so serially is regarded as a marker of self-renewal, and their ability to recapitulate phenotypic complexity of tumor cells upon transplant is regarded as evidence of a differentiation process. Self-renewal and differentiation being the cardinal features of stem cells has led to these cells being considered the stem-like cells of cancer. Compared to the bulk of tumor cells, CSCs have been reported to be less sensitive to apoptotic signals and more resistant to conventional therapy, though this remains controversial 6. This review will summarize work by others on the regulation of cell death and survival of CSCs, specifically focusing on modulation of pro- and anti-apoptotic signaling pathways to induce CSC death for cancer therapy.
Cancer stem-like cells
It has been recognized for many years that human cancers comprise a heterogeneous population of cells. Within the heterogeneous “swarm” of cells comprising a cancer, hierarchical relationships similar to cells in normal tissues have been hypothesized with stem cells serving as a self-renewing subset (Fig2). This population of cells is theorized to be capable of initiating new sites of tumor and so is also called “tumor-initiating cells”. They have been tested in a manner similar to normal stem cells using transplantation into irradiated, often immunodeficient murine hosts. The ability of a subset of cells to engraft an animal serially is an experimental definition of self-renewal and generation of cell populations bearing properties of the remaining tumor cells as evidence of differentiation.
Figure 2. Cancer stem cell model.

Based on the CSC model, cancers are composed of heterogeneous cell populations with a hierarchy similar to normal tissue. On top of the hierarchy are cancer stem cells (CSCs) that can self-renew and differentiate into more mature non-CSC cells. CSCs are usually rare and believed to be the driver of tumorigenesis and disease progression. Non-CSC cells do not self-renew, have limited proliferation potential and lifespan, and are not able to initiate the disease.
Because these tumor cells have features of tissue stem cells that are responsible for the maintenance of adult tissues, they are referred to as cancer stem cells or CSCs. They have been experimentally demonstrated in some but not all human tumors, and there is clear artificiality to the concept as it is based on engraftment in immunodeficient mice that may or may not reflect the context of the human body.
The concept of CSCs was first proposed and convincingly characterized in human acute myeloid leukemia (AML) 7, 8. Following these landmark studies, CSCs have been identified and isolated in a wide variety of solid tumors, including tumors of brain, breast, colon, lung, prostate, pancreas, liver, and ovary and other hematologic malignancy 9.
In many cancers, the CSC population can be identified using phenotypic surface markers (Table1).
Table 1.
Commonly used phenotypic markers for cancer stem cells
| Cancer types | Markers | References |
|---|---|---|
| Breast | CD44+CD24− | 10 |
| CD44+CD24−ESA+ | 10 | |
| ALDH+ | 11 | |
| Colorectal | CD133+ | 12 |
| EpCAM+ | 13 | |
| CD44+ | 14 | |
| CD166+ | 14 | |
| ALDH+ | 15 | |
| Brain | CD133+ | 16, 17 |
| CD15+ | 18 | |
| Ovarian | CD133+ | 19 |
| CD44+CD24− | 20 | |
| CD117+ | 21 | |
| Liver | CD133+ | 22, 23 |
| EpCAM+ | 24 | |
| CD90+ | 25 | |
| OV6+ | 26 | |
| Pancreatic | CD44+CD24+ESA+ | 27 |
| Lung | CD133+ and/or ALDHhi | 28 |
| AML | CD34+CD38− | 8 |
| CD34+CD38+ | 29 |
CSCs in most cancers are rare. Based on xenotransplantation using non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice as host, CSC frequency was estimated to be lower than 1 in 2,500 30. However, in certain tumor such as melanoma, up to 25% of tumor cells are CSCs when analyzed in more permissive host strain 31. Nonetheless, mounting evidence suggests that the CSC population is resistant to conventional therapies and may account for the relapse of the disease 32. Therefore, new strategies to target and eradicate CSCs have been regarded as essential to eradicate some cancers (Fig3). Various mechanisms are implicated in the evasion of CSCs from conventional therapy, including increased DNA damage repair, altered cell cycle checkpoint control, and overexpression of multi-drug-resistant proteins 33. Whereas CSCs generally display impaired apoptosis potential, recent studies suggest that such defects can be restored either by directly targeting components of the apoptotic machinery or by indirectly targeting other cellular pathways, such as bioenergetics.
Figure 3. Targeting CSCs for cancer therapy.

Conventional anticancer therapies kill fast cycling cancer cells and cause initial tumor regression. Slow cycling CSCs, however, are more resistant to conventional therapies. Spared CSCs self-renew and repopulate the entire tumor tissue, leading to disease relapse. Resistance to conventional therapy could be overcome by targeting CSCs. Studies based on preclinical models suggest that activation of apoptotic programs in CSCs alone or in combination with conventional therapies may eliminate the tumor-initiating population and achieve long-term tumor regression.
Activating death receptors to induce apoptosis in CSCs
Although CSCs are generally resistant to chemotherapy, there is vast diversity regarding the expression levels of DRs and sensitivity to DR-mediated apoptosis. In some cancers, the expression of DRs, such as FAS and TRAIL receptors, is lower in the CSC compartment than in other cells. This was first observed in human leukemia in which leukemia stem cells (LSCs, CD34+/CD38−) displayed lower expression of FAS and FAS ligand (FAS-L), decreased sensitivity to FAS-induced apoptosis, and increased resistance to chemotherapy compared to more differentiated cells (CD38+) 34. Human glioma cells were shown to be sensitive to apoptosis induction through the FAS/FAS-L pathway 35, 36. However, the CSC compartment of brain tumors expresses lower levels of FAS and FAS-L compared to cultured primary glioma cells, resulting in increased resistance to FAS-mediated cell death in glioma stem cells 37. The low expression levels of FAS on CSCs can be overcome by using novel synthetic FAS ligands such as Apo010 (a hexameric FAS-L) that display high avidity to FAS 38. Apo010 not only induces apoptosis in the bulk of glioma cells but also causes strong cytotoxicity in glioma CSC-like cells 39. Apo010 reduces viability of glioma cells and glioma stem cells in synergy with other cytotoxic drugs and prolongs survival of tumor-bearing mice 38, 39.
Normal and malignant cells appear to have different sensitivity to apoptosis induction by TRAIL, the natural ligand of DR4 and DR5, making TRAIL a promising anti-cancer agent. Recombinant soluble TRAIL was shown to induce apoptosis of primary human leukemia and myelodysplasia progenitors without affecting normal hematopoietic progenitors 40. The resistance of normal cells to TRAIL may be due to the expression of antagonistic decoy receptors that can compete with DR4 and DR5 for ligand binding 41. Glioblastoma cells treated with the proteasome inhibitor bortezomib increase surface expression of DR5 and are more sensitive to recombinant TRAIL-induced apoptosis. Bortezomib and TRAIL synergistically trigger cell death, reduce colony formation of glioblastoma stem cells, and suppress tumor growth in a mouse model of glioblastoma 42. In triple-negative breast cancer, TRAIL treatment increases the efficiency of cisplatin to induce cell death of CSCs and suppress mammosphere formation 43.
Despite potent anti-tumor effects in vitro, in vivo efficacy of soluble TRAIL is often limited by a short half-life in plasma due to a rapid clearance by the kidney. Such limitations can be overcome by engineering mesenchymal stromal cells (MSCs) to express TRAIL and provide continuous source of the protein. This was first shown in brain tumors 44, 45, where TRAIL-armed MSCs migrated to tumor sites following transplantation into mice bearing brainstem glioma xenografts and induced massive death of tumor cells, but not normal brain cells. Such treatment dramatically extended survival compared to groups treated with soluble TRAIL or MSC alone. Similar strategies have been applied to other types of cancers, including pancreatic cancer, breast cancer, melanoma and squamous lung cancers 46, 47, 48, 49. Importantly, engineered MSCs induce cell death not only in the bulk of tumor cells but also in the CSC population as assessed by decreased colony formation 49. These reports indicate that MSCs are promising vehicles for delivering the DR ligand TRAIL to tumor environment and may be used to eliminate CSCs.
In addition to their natural ligand, agonist antibodies against DRs have been shown to induce apoptosis in several tumor cell lines 50, 51. When treated alone or with other cytotoxic agents, anti-DR5 antibody displayed robust antitumor efficacy in mouse xenografts of tumor with minimum toxicity to normal cells 52, 53, 54. Importantly, in some cancers, agonist DR5 antibody also targets CSCs that are resistant to chemotherapy. In pancreatic ductal adenocarcinoma, for example, DR5 is enriched in CSCs 55. Treatment with the cytotoxic drug gemcitabine was effective in reducing tumor size but unable to eliminate the CSC pool. When gemcitabine was given in combination with a humanized DR5 agonist monoclonal antibody, both CSCs and the bulk of tumor cells were killed, resulting in marked tumor remission and delayed tumor progression 55. A similar effect was observed in breast cancer. While chemotherapy leads to enrichment of CSCs, anti-DR5 antibody treatment reduces the CSC pool and inhibits tumorigenicity 56. Notably, the efficiency of apoptotic induction in CSCs by DR5 agonist was fifty-fold higher than TRAIL or anti-DR4 antibody.
In some cancers, the CSC population expresses higher levels of DRs, which provides a unique therapeutic opportunity to target this population. For example, the putative CSC compartment of human colon cancer cell line SW480, as defined by the dye-effluxing side population (SP), expresses ten-fold higher levels of DR4 than non-SP counterparts 57. Overexpression of DR4 in this model is driven by high cMyc activity through E-box DNA-response elements. As a result, the SP cells are more sensitive to TRAIL and other therapeutic agents than non-SP cells 57. In AT-3 mammary carcinoma cell line, the multi-potent, chemoresistant CSC-like population expresses higher level of FAS and DR5 than non-CSC-like cells and this correlates with increased sensitivity to apoptosis induced by FAS ligand and TRAIL 58.
Therefore, despite the refractory nature to conventional therapies, CSCs, at least in preclinical models, are sensitive to apoptosis induction by DR activation. Novel delivery approaches of DR ligands in combination with conventional therapies have shown potent anti-tumor effects, particularly in eradicating CSCs. The differential expression levels of DRs and/or sensitivity to DR ligands between normal and malignant cells further support the strategy of triggering the extrinsic apoptosis pathways for cancer therapy.
Antagonizing apoptosis inhibitory molecules in CSCs
In addition to reduced expression of DRs, CSCs also express higher levels of apoptosis inhibitory proteins, which further enhance resistance to cell death induction.
The DR-initiated apoptotic pathway is negatively regulated by cellular Fas-associated death domain-like IL-1β-converting enzyme (FLICE)-inhibitory protein (c-FLIP) 59. As a master anti-apoptotic regulator, cFLIP interacts with FADD, caspase-8 or 10 and DR5, prevents the formation of DISC and subsequent activation of the caspase cascade (Fig1) 60. cFLIP was found to be overexpressed in many cancers 59. In some tumors, such as leukemia, breast cancer, and glioblastoma, the expression of cFLIP is even higher in the CSC population than in non-CSC-like cancer cells 61, 62, 63. Consequently, CSCs from these tumors exhibit lower sensitivity to TRAIL-induced apoptosis compared to non-CSC-like counterparts. Knockdown of cFLIP by siRNA sensitizes CSCs to TRAIL-induced apoptosis, suggesting that death resistance of CSCs may be at least partially mediated by FLIP overexpression 61, 62. For breast tumors, TRAIL treatment in combination with cFLIP suppression inhibited CSC self-renewal and resulted in marked reduction of primary tumor growth and suppression of metastasis in a transplantation model 64. Importantly, inhibition of FLIP did not induce cell death of non-tumorigenic mammary cells 64.
Inhibition of FLIP also underlies the mechanism of action for several anti-cancer drugs. Sorafenib, a broad-spectrum kinase inhibitor targeting the RAF–MEK–ERK pathway, can induce apoptosis alone and enhances TRAIL- or FAS-mediated cell death in endometrial carcinoma cell lines that are otherwise resistant to TRAIL or FAS 65. The enhanced sensitization to DR-mediated apoptosis by sorafenib was attributed to downregulation of cFLIP. Similar phenomena were observed in colon cancer 66 and glioma cell lines 67 when treated with aspirin/sorafenib and cisplatin, respectively. Downregulation of cFLIP by anti-cancer agents is mediated through proteasome-dependent degradation of the protein.
The inhibitor of apoptosis (IAP) family proteins play important roles in promoting cell survival. IAPs can bind to caspases directly or indirectly and antagonize the apoptotic cascade (Fig1). Alternatively, some IAPs participate in signaling transduction, for instance, to activate the NF-κB pathway, and positively regulate cell survival. In many human cancers, the expression or function of IAP proteins is deregulated and the expression levels of IAP proteins and their antagonists have been correlated with clinical parameters and cancer prognosis 68, 69. Among the IAP family members, X chromosome-linked IAP (XIAP) has been a focus of investigation, in part because it is the most frequently overexpressed in human cancer 70. Small-molecule screening has identified a class of polyphenylureas that disrupt XIAP/caspase-3 interaction and reverse XIAP-mediated inhibition of caspase-3 71, 72. These XIAP inhibitors activate caspases and induce apoptosis alone and sensitize apoptosis induction by cytotoxic agents and TRAIL in a variety of cancers, including lung cancer, melanoma, leukemia, lymphoma, and pancreatic cancer 53, 73, 74, 75, 76, 77. While these XIAP antagonists suppress tumor cell growth both in vitro and in vivo, they appear to cause minimal toxicity to a variety of normal tissues 71, 72, 76, which makes them ideal candidates for anti-cancer drugs.
XIAP proteins have also been implicated in regulating apoptosis in CSCs. Similar to cFLIP, multiple XIAP proteins are expressed at higher levels in the glioblastoma stem-like cell compartment compared to non-CSC-like cells 78. In a radioresistant glioblastoma cell line, small-molecule XIAP inhibitors were shown to dramatically enhance mitochondria-dependent apoptosis induced by gamma-irradiation 79. Similarly, in TRAIL-resistant glioma cell lines, treatment with proteasome inhibitors sensitized TRAIL-induced apoptosis by downregulation of XIAP 80. Importantly, these studies demonstrated that inhibition of XIAP, either by small molecules or through protein degradation, induced apoptosis in both CSCs and non-CSC-like cells without impairing normal cells of the central nervous system.
In conclusion, apoptotic inhibitory proteins are frequently upregulated in cancer cells, particularly in CSCs. Despite increasing the threshold of apoptosis induction by conventional chemotherapeutic agents and DR ligands, elevated expression of these proteins also provides novel promising targets for anti-cancer therapy.
Targeting Bcl-2 family proteins in CSCs
The Bcl-2 family proteins play a pivotal role in mitochondrial-mediated apoptosis. Bcl-2, the founder member of the family, was initially identified in human follicular B-cell lymphoma at the chromosomal breakpoint of t(14:18) as a proto-oncogene 81, 82. Unlike many oncogenes, Bcl-2 does not promote proliferation, but instead blocks cell death and enhances tumor cell survival. During development, Bcl-2 expression generally correlates with the developmental state and lifespan of the cell. In the lymphoid lineage, for example, Bcl-2 is expressed at high levels in pro-B cells and naïve mature B cells and downregulated in pre-B cells, immature B cells, and germinal center B cells, stages where negative selection occurs 83. Within the myeloid compartment, Bcl-2 is expressed in early myeloid progenitors but downregulated during myeloid differentiation, correlating with the short lifespan of mature myeloid cells 84. Studies using Bcl-2 transgenic mice showed that overexpression of Bcl-2 protected hematopoietic stem cells (HSCs) from various apoptosis-inducing stimuli. Bcl-2 overexpression leads to increased HSC numbers in the bone marrow and enhances HSC colony formation in vitro and reconstitution capacity in vivo 85, 86. However, loss-of-function studies suggest that Bcl-2 is not required for HSC maintenance, but important for lymphoid cell survival 87, 88. Similarly, Bcl-xL, another member of the Bcl-2 family, is required for survival of immature lymphocytes and erythroid progenitors 89, 90, 91. In contrast, MCL-1 is indispensible for HSC survival 92. Mcl-1 is expressed at high levels in HSC, and its expression is regulated by growth factors such as stem cell factor 92, 93. Conditional deletion of Mcl-1 leads to complete ablation of bone marrow and premature lethality of mice due to massive loss of hematopoietic stem and progenitor cells 92.
Members of the Bcl-2 pro-survival family are overexpressed in many cancers and believed to contribute to tumor initiation, maintenance, progression, and resistance to therapy 94. The Bcl-2 and Mcl-1 gene locus, for instance, are frequently amplified in multiple cancers 95. Knockdown of either Bcl-2 or Mcl-1 caused a more pronounced reduction in proliferation among Bcl-2- or Mcl-1-amplified cancer cells compared with non-amplified cancer cell lines. And the decreased proliferative rates were associated with increased apoptosis. Knockdown of Bcl-2 alone or in combination with other cytotoxic drugs has therapeutic effects in several cancer models, including leukemia, lung cancer, prostate cancer, melanoma, and brain tumors 96, 97, 98, 99, 100. The protein synthesis inhibitor omacetaxine exhibits moderate anti-cancer activity in various hematologic malignancies 101. In CML, omacetaxine induces apoptosis in LSCs via suppressing the expression of Mcl-1 and BCR-ABL 102, 103. As expected, however, because Mcl-1 is essential for the survival of normal HSC 92, omacetaxine also kills normal hematopoietic progenitor cells 103, underlying the side effects of the drug.
Several BH3-mimetic small molecules have been identified that either broadly inhibit multiple anti-apoptotic Bcl-2 proteins or selectively inhibit individual members. Among these, ABT-737 is the first that has been widely studied as a pan inhibitor binding to Bcl-2, Bcl-xL, and Bcl-w 104. ABT-737 exhibits single-agent-mechanism-based killing of lymphoma and small-cell lung carcinoma cell lines, as well as patient-derived primary tumor cells. In animal models of tumor xenograft, ABT-737 causes regression of established tumors and improves survival 104.
Overexpression of anti-apoptotic Bcl-2 family proteins is detected in the CSC pool of several tumors. The percentage of Bcl-2-positive cells is highest in the CD34+ LSCs, and Bcl-2 protein levels correlate with CD34 positivity 105. Moreover, high Bcl-2 expression is associated with poor response of leukemia to chemotherapy 105, 106, 107. In glioblastoma, Bcl-2 and Bcl-xL are expressed at higher levels in the CD133+ glioma stem cell population compared to the CD133− counterparts 78. Bcl-2 is also overexpressed in CD44+/CD24− subpopulation of breast cancer that is enriched with CSCs 108. Treatment with the pan Bcl-2 inhibitor ABT-737 induces apoptotic cell death in glioblastoma cells and enhances their sensitivity to chemotherapeutic drugs and death ligand TRAIL 109. However, apoptotic induction by ABT-737 is less efficient in glioma stem cells compared to non-CSC-like population. The death resistance is caused at least partially by overexpression of Mcl-1 in the CSC population, as knockdown of Mcl-1 increased the efficacy of ABT-737 to trigger cell death in these cells 109. The essential role of Mcl-1 in protecting glioma stem cells from cell death induction is further supported by another study using a natural BH3 mimetic gossypol. In contrast to ABT-737 that inhibits Bcl-2, Bcl-xl, and Bcl-m, gossypol also inhibits Mcl-1. Treatment with gossypol results in dramatically more cell death than other BH3 mimetics including ABT-737 110. Knockdown of Mcl-1 further enhances cell death induction by gossypol and ABT-737.
The therapeutic potential of targeting anti-apoptotic Bcl-2 family proteins has been extensively explored in hematopoietic malignancy. In a Myc-driven leukemia mouse model, all six members of the anti-apoptotic Bcl-2 family were shown to accelerate leukemia development 111. Expression of the anti-apoptotic Bcl-2 members did not increase cellular proliferation rate, suggesting that the effect was mainly attributed to pro-survival effects 111. The tyrosine kinase inhibitor (TKI) imatinib is effective in killing the majority of CML cells but unable to eradicate quiescent CML stem cells and has limited activity against blast crisis (BC) CML. BCR-ABL was shown to activate Mcl-1 in both primary CML and CML-derived cell lines. Knockdown of Mcl-1 synergizes with imatinib to induce cell death of CML cells 112. Although the levels of Bcl-2, Bcl-xL, and Mcl-1 are comparable between quiescent and proliferating CD34+ population of BC CML, inhibition of Bcl-2 and Bcl-xL by ABT-737 in TKI-resistant BC CML promotes apoptosis in quiescent CD34+ CML stem cells 113. Combination treatment with ABT-737 and imatinib synergistically induces death of both proliferative and quiescent CD34+ progenitor cells from TKI-resistant BC CML patients.
The feasibility of targeting Bcl-2 protein family in LSCs is further supported by two recent studies of human primary myeloid leukemia. In the first study, the expression of Bcl-2, Bcl-xL, and MCL-1 was found higher in BC LSCs (Lin−CD34+CD38+) than in chronic-phase CML cells and normal progenitor cells 114. When transplanted into immunodeficient mice, the BC LSCs engraft in bone marrow as well as other niches, but bone marrow-engrafted BC LSCs are more quiescent and resistant to TKI than cells engrafted in other sites. Moreover, marrow-engrafted BC LSCs express a pro-survival gene signature. Treatment with TKI increased the expression of Bcl-2 and MCL-1 and the proportion of quiescent BC LSCs, suggesting that upregulation of these pro-survival factors contributed to TKI resistance. In line with this, a pan Bcl-2 inhibitor, sabutoclax, was able to reduce LSC survival and disease burden in transplanted mice without impairing normal progenitor cells. In addition, sabutoclax markedly increased the sensitivity of BC LSCs to TKI treatment 114. In the second study, a functional approach was applied to identify LSCs from primary AML 115. AML stem cells exhibited low levels of reactive oxygen species (ROS), were metabolically inactive, and rely on oxidative respiration for energy biogenesis. Compared to ROS-high AML cells, the ROS-low cells were more quiescent and have higher engraftment efficiency when transplanted into immunodeficient mice. In addition, the expression of Bcl-2, but not other Bcl-2 family members, was overexpressed in ROS-low AML cells. Interestingly, when ROS was used to distinguish normal CD34+ progenitors, there was no difference in Bcl-2 expression, suggesting that upregulation of Bcl-2 in ROS-low AML cells correlates with malignant transformation. The study further demonstrated that inhibition of Bcl-2 with small molecules severely impairs oxidative phosphorylation in AML cells 115. In response to Bcl-2 inhibition, ROS-high leukemic cells and normal CD34+ progenitor cells robustly upregulate glycolytic metabolism to maintain energy production. In contrast, ROS-low AML cells are unable to induce glycolysis and maintain ATP generation, resulting in elevation of mitochondrial ROS. Consequently, inhibition of Bcl-2 disrupts mitochondrial energy production, induces apoptosis selectively in ROS-low LSCs, and impairs AML engraftment in NSG mice without impacting normal hematopoietic cells 115. Similar results were observed in a transgenic mouse model of AML, where Bcl-2 inhibition with ABT-737 targeted primitive LSK and progenitor cells and prolonged survival of diseased animals 116. The results are consistent with prior studies showing that bulk and stem-like AML cells are more dependent on BCL-2 than normal HSCs, and are therefore more sensitive to BCL-2 inhibition 87, 88, 117. Therefore, these reports provide a strong rationale for inhibition of BCL-2 as a therapeutic approach to myeloid malignancies.
Targeting energy metabolism in CSCs
In addition to altered expression of apoptotic genes, transformed cells also display metabolic abnormalities (Fig4). Cancer cells preferentially rely on a high rate of glycolysis followed by lactate fermentation even in the presence of oxygen, a phenomenon known as aerobic glycolysis or the Warburg effect 118. Targeting the glycolytic pathway as a cancer therapeutic strategy has been actively investigated. The exact metabolic pathways in CSCs, however, have not been fully characterized. Current literature suggests that CSCs of different cancer types may have distinct metabolic requirements.
Figure 4. Overview of central cellular metabolism.
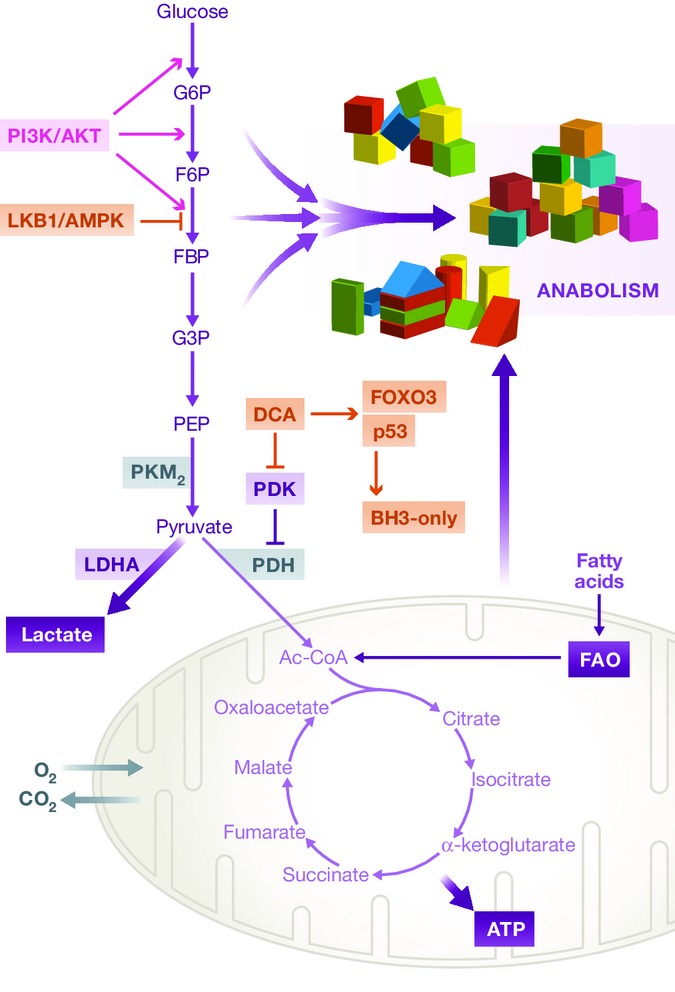
As the major fuel for mammalian cells, glucose is first metabolized to pyruvate through anaerobic glycolysis. In the bulk of cancer cells, the majority of pyruvate is further fermented to lactate, even in the presence of oxygen (the Warburg effect). Only a minor portion of pyruvate is catabolized via the TCA cycle in the mitochondria. Although inefficient in ATP generation, the rapid flux of glycolytic pathway is advantageous to provide metabolic intermediates as biosynthetic substrates to support daughter cell production (anabolism). The glycolytic pathway is regulated by signaling pathways such as the PI3K/AKT and LKB1/AMPK pathways. The metabolic programs in CSCs are more divergent than the bulk of tumor cells. For some cancer types, CSCs also preferentially uses aerobic glycolysis. In other cancers, CSCs appear to rely on oxidative metabolism. Targeting metabolic programs in CSCs represents a promising strategy for cancer therapy. In glioma CSCs, for instance, DCA drives a metabolic shift from glycolysis to oxidative phosphorylation. This is associated with increased expression of FOXO3 and p53, which further upregulates pro-apoptotic BH3-only proteins. As a consequence, DCA treatment sensitizes glioma CSCs to Bax-dependent apoptosis in response to chemotherapy or irradiation. In the hematopoietic tissue, leukemia stem cells depend on FAO. Inhibition of FAO sensitizes apoptosis induction by Bcl-2 antagonist. G6P, glucose 6-phosphate; F6P, fructose 6-phosphate; FBP, fructose 1,6-bisphosphate; G3P, glyceraldehyde 3-phosphate; PEP, phosphoenolpyruvate; PDH, pyruvate dehydrogenase; PDK, PDH kinase; FAO, fatty acid oxidation; DCA, dichloroacetate.
For some tumors, the CSCs population shares similar metabolic modes as the bulk of tumor cells. CSCs from human osteosarcoma cells, for instance, express higher levels of LDHA, produce more lactate, and consume less oxygen compared to parental cells, suggesting that osteosarcoma CSCs more preferentially rely on glycolytic metabolism 119. Accordingly, glucose withdrawal or inhibition of glycolysis leads to ATP deprivation and cell death induction in osteosarcoma CSCs, while oxidative phosphorylation inhibitors do not cause the same effect. 119. CSCs of glioma derived from rat also display a glycolytic phenotype 120. Treatment with the glycolysis inhibitor dichloroacetate (DCA) drives metabolic shifting from glycolysis to glucose oxidation in glioma CSCs but not neural stem cells 120. In this model, DCA induces FOXO3 and p53 expression, which further upregulates pro-apoptotic BH3-only proteins. Although unable to trigger apoptosis alone, DCA increases the sensitivity of glioma CSCs to etoposide or irradiation and leads to Bax-dependent apoptosis. Interestingly, in vivo treatment with DCA alone moderately inhibits tumor growth in a mouse xenograft model. While etoposide alone has no effect, the combination treatment of DCA and etoposide dramatically suppressed tumor formation 120.
In contrast to osteosarcoma, an accumulating body of evidence suggests that CSCs of many tumors appear to depend on oxidative metabolism. As discussed above, inhibition of anti-apoptotic Bcl-2 proteins in human AML stem cells disrupts oxidative phosphorylation and induces apoptosis of these cells 115, suggesting that intact mitochondrial respiration is required for the maintenance of LSCs. Similarly, CSCs of the human glioma cell line U87 appear to be less glycolytic than differentiated glioma cells 121. Glioma CSCs consumed less glucose and produced less lactate while maintaining higher ATP levels. Interestingly, glioma CSCs express both PKM1 and PKM2 isoforms of pyruvate kinase. Inhibition of either glycolysis or oxidative phosphorylation, however, has minimal effect on energy production in glioma CSCs, suggesting that U87 CSCs are able to use multiple pathways to produce energy and targeting individual metabolic pathway in glioblastoma may spare CSCs 121. The insulin-like growth factor 2 mRNA binding protein 2 (IMP2) plays an essential role for the maintenance of CSCs in primary glioblastoma 122. IMP2 binds several mRNAs that encode mitochondrial respiratory chain complex subunits and interacts with complex I proteins. Depletion of IMP2 in gliomaspheres impairs oxidative phosphorylation and energy production without affecting glycolysis, resulting in compromised gliomasphere formation and tumorigenicity. Inhibition of mitochondrial respiration by rotenone mimics the effects of IMP2 depletion. However, inhibition of anaerobic glycolysis does not impair clonogenic potential of glioma CSCs 122. Disrupting mitochondrial energy metabolism was also shown to impact ovarian CSCs that are resistant to conventional chemotherapy. Treatment of ovarian CSCs with an isoflavone derivative depresses mitochondrial function leading to reduction of ATP, decreased expression of Cox-1 and Cox-IV, loss of mitochondrial membrane potential, and increased ROS levels 123. The mitochondrial dysfunction activates AMPKα1–mTOR pathway and ERK–Bax pathway, resulting in caspase-independent cell death 123.
Recently, several studies have identified an important role of fatty acid oxidation (FAO) in the maintenance and function of normal and cancer stem cells, particularly in the hematopoietic tissue. For example, Samudia et al 124 reported that pharmacological inhibition of FAO inhibits proliferation and sensitizes human leukemia-initiating cells to apoptosis induction by the Bcl-2 antagonist ABT-737. FAO was shown to regulate the activity of Bak-dependent mitochondrial permeability transition 124. In a more recent study, Ito et al 125 demonstrated that normal HSC function was dependent on FAO downstream of promyelocytic leukemia (PML)–peroxisome proliferator-activated receptor δ (PPAR-δ) pathway. Genetic depletion of this pathway or pharmacological interference of FAO impaired HSC asymmetric division and resulted in exhaustion of stem cell pool 125. As a tumor suppressor, PML was shown to play an essential role in the maintenance of LSCs in CML 126. Whether the PML–PPAR-δ–FAO pathway is essential for the maintenance of LSCs has remained to be addressed. However, these studies suggest that targeting FAO represents a promising therapeutic strategy for leukemia by eradicating LSCs.
Therefore, in contrast to proliferating cancer cells that exclusively depend on glycolytic metabolism for energy and biomass generation, CSCs appear to be more versatile in terms of metabolic dependency. While CSCs of certain cancers preferentially rely on aerobic glycolysis, other CSCs employ mitochondrial respiration for energy production. Targeting metabolism in CSCs has to be designed based on the specific metabolic needs of individual cancer.
Restoring the p53 pathway in CSCs
p53 is a tumor suppressor that has been dubbed “the guardian of the genome”. In response to cellular stresses, such as hypoxia and DNA damage, p53 protein is stabilized and induces cell cycle arrest, senescence, or apoptosis 127. Therefore, activation of the p53 pathway prevents the accumulation of genetic mutations and maintains the genomic stability of the cell. The tumor-suppressor function of p53 has been supported by both human and animal studies. Deletion of P53 in mice leads to the development of spontaneous tumors 128, 129. Mutation of the P53 gene has been detected in the majority of human cancers 130, 131, and restoration of p53 activity retards cancer cell growth 132. p53 also regulates stem cell self-renewal and differentiation. For instance, upon DNA damage, p53 activation induces differentiation of mouse embryonic stem cells (ESCs) by suppressing Nanog expression 133. Similarly, p53 suppresses proliferation and self-renewal of neural stem cells 133, 134. In the hematopoietic system, p53 regulates HSC quiescence during steady-state hematopoiesis 135.
Given the role of p53 in regulating normal stem cell survival, self-renewal, and proliferation, it has been speculated that p53 may also regulate CSC function. It has been shown that inactivation of p53 promotes self-renewal and transformation of myeloid progenitor cells expressing oncogenic Kras into LSCs in mice 136. p53 loss has been shown to inhibit differentiation of multiple cancers, including lung, skin, mammary, and colorectal cancers 137. In ErbB2-driven breast tumor, CSCs undergo higher frequency of self-renewal divisions than normal mammary stem cells 138. In this model, p53 is not mutated, but its activity is attenuated 138. Interestingly, targeted mutation of p53 in mammary tissue also drives the symmetric self-renewal division of mammary stem cells and increases stem cell frequency in the premalignant mammary gland 138. Pharmacological restoration of p53 promotes asymmetric division in breast CSCs and inhibits tumor growth 138. The deacetylase SIRT1, known to down-modulate p53, is overexpressed in CD34+ human CML stem cells that are resistant to TKI treatment 139. Pharmacological inhibition of SIRT1 or SIRT1 knockdown increases apoptosis selectively in CML stem cells and inhibits their growth in vitro and in vivo. This effect is mediated through increased acetylation and activation of p53 139. In colon cancer cell line, deletion of p53 leads to enrichment of dye-effluxing CSCs and restoration of p53 enhances the cytotoxicity of chemotherapeutic drugs by depletion of the CSC population 140.
In addition to loss of function mutations, some cancer cells express a mutant p53 that has oncogenic activities (gain-of-function). In breast cancer, for instance, depletion of a mutant p53 gene reverses the oncogenic potential of cancer cells by inducing a normal cell-like phenotype characterized by the acini-like morphology 141. The oncogenic effect of p53 mutants appears to be mediated, at least partially, through activation of the mevalonate metabolic pathway, as deletion of mutant p53 dramatically downregulates genes of mevalonate pathway and inhibition of these genes also reverses the breast cancer phenotype 141. Interestingly, mevalonate metabolism has been identified as a key regulator of breast CSCs. Treatment with small-molecule inhibitors of key enzymes in this pathway decreases breast CSC population both in vitro and in primary breast cancer xenografts 142. The mevalonate pathway also appears to have a role in supporting LSCs as inhibition of this pathway by lovastatin selectively kills LSCs over normal stem and progenitor cells in the setting of MLL-AF9-induced AML 143. The effects on LSCs by lovastatin were through inhibition of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase 143.
These studies suggest that p53 mutation contributes to CSC maintenance and cancer drug resistance and targeting p53 pathway might be a beneficial adjuvant to conventional therapies in eliminating CSCs.
Targeting the PI3K pathway in CSCs
The phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway plays important roles in multiple cellular processes, including proliferation, differentiation, metabolism, apoptosis, and migration. Aberrantly upregulated PI3K/Akt/mTOR signaling has been observed in many cancers and is implicated in tumor resistance to conventional therapies 144. A number of chemical compounds have been identified that inhibit either individual or multiple components of this pathway and display anticancer activity in various neoplasms 144. Recent studies indicate that this signaling pathway also plays a ke y role in CSC biology.
The PI3K pathway has been demonstrated to regulate survival of medulloblastoma CSCs residing in the perivascular niche following radiation 145. Inhibition of Akt signaling sensitizes CSCs in the perivascular region to radiation-induced apoptosis. The PI3K/Akt pathway is highly active in brain tumor stem cells. The anticancer drug sorafenib inhibits proliferation of glioblastoma cells in culture. This effect depends partially on the inhibition of PI3K/Akt and induction of apoptosis via downregulation of Mcl-1 146. Importantly, sorafenib appears to have a selective action on glioblastoma stem cells. Sorafenib treatment causes glioblastoma cell differentiation, resulting in reduced clonogenic ability in vitro and tumorigenic potential in vivo 146. In patient-derived glioma, inhibition of PI3K by Ly294002 or PI-103 reduces the number of clonogenic stem cells 147. But such an effect is due to inhibition of proliferation, but not apoptosis induction. Interestingly, PI3K inhibitors induce autophagy of CSCs in response to gamma-irradiation. Combination treatment with the autophagy inhibitor chloroquine and PI3K inhibitors significantly promotes cell death induction by irradiation 147. PI3K inhibitors also synergize with oncolytic herpes simplex virus to induce apoptosis of glioblastoma stem cells, but not human astrocytes in vitro, and significantly extend survival of mice of with brain tumor xenograft, as compared with either agent alone 148.
Constitutive activation of PI3K is required for survival of some AML blasts 149. Interestingly, mTOR activation is required for long-term, but not short-term survival of AML cells. Inhibition of mTOR with rapamycin synergizes with etoposide to decrease engraftment of primary AML cells in NOD/SCID animals, suggesting that mTOR regulates survival of AML stem cells and that targeting of mTOR may be combined with cytotoxic agents to eradicate the LSC compartment 150. Genetic ablation in mice of either mTOR complex 1 (mTORC1) or mTORC2 prevented T-cell leukemia initiated by loss of the upstream inhibitor of PI3K, Pten, likely through effects on leukemia-initiating cells 151, 152. In a mouse model of CML, Naka et al 153 revealed a critical role of TGF-β–FOXO3a in maintaining LSC survival. TGF-β regulates Akt activation in LSCs and controls FOXO3a localization. Depletion of FOXO3a induced apoptosis of LSCs, and combination of TGF-β inhibition, FOXO3a deficiency, and imatinib treatment led to efficient depletion of CML LSCs both in vitro and in vivo. It should be noted that in addition to the anti-apoptotic effects, FOXOs also act as differentiation blockade. In AML, for instance, low AKT and high FOXOs activities were detected in the LSC population 154. Activation of AKT or inhibition of FOXOs drove LSC myeloid differentiation and impaired leukemogenic activity 154.
In most tumor types, however, AKT activation drives cancer cell proliferation. Multi-kinase inhibitor EI210 inhibits phosphorylation of AKT, mTOR, and several downstream mediators of the mTOR pathway and induces both apoptosis and autophagy in several cancer cell lines 155. Importantly, EI201 suppresses self-renewal and tumorigenic capacity of CSCs, as revealed by decreased tumorsphere formation in vitro and reduced tumor burden in vivo 155. Plant-derived agents such as rottlerin and harmine hydrochloride were shown to induce apoptosis and autophagy in prostate and glioblastoma CSCs through inhibition of Akt and mTOR phosphorylation 156, 157. Treatment of glioblastoma with harmine hydrochloride inhibits self-renewal and promotes differentiation of glioblastoma stem cells, resulting in decreased neurosphere formation and tumorigenicity 157. In colorectal cancer, the PP2A/AKT/mTOR pathway is activated in the CSC population. Inhibition of AKT/mTOR by silibinin suppresses CSC maintenance and tumorsphere formation in vitro and impedes tumor growth in vivo 158. The AKT/cyclin D1/Cdk4 survival signaling pathway is activated in radioresistant CSCs of glioblastoma and liver cancer cell lines 159. Inhibition of AKT, cyclin D1, or Cdk4 was shown to suppress DNA damage repair in response to radiation and leads to reduction of CSCs, suggesting combination of radiation and targeting of the AKT/cyclin D1/Cdk4 pathway as an effective therapeutic strategy 159.
Conclusions and future directions
It has been experimentally demonstrated in many tumors that CSCs are on the top of a tumor cell hierarchy participating in tumor initiation and, perhaps, progression. Like normal tissue stem cells, CSCs have the capacity to self-renew and repopulate the entire tumor population. Accumulating evidence suggests that in some tumors, CSCs are more quiescent than the bulk of tumor cells 160 and less sensitive to therapies that are designed to target proliferating cells 32. The escape of cells from conventional therapy usually leads to disease relapse, characterized by decreased sensitivity to previous treatment and enrichment of the CSC pool. Thus, effective anticancer therapy not only requires elimination of the bulk of tumor mass but also depends on eradication of CSCs. Although induction of apoptosis is a common mechanism of anticancer drugs, most of these agents do not target the apoptotic machinery directly. Given that the apoptosis pathway is frequently impaired in cancer cells and CSCs, restoration or activation of cell death pathway via targeting components of the apoptotic machinery has been an attractive strategy for cancer therapy. CSCs exhibit increased resistance to apoptosis induction, either due to defects in the death receptor pathway or through an impaired mitochondrial-dependent pathway. Downregulation of death receptors, overexpression of anti-apoptotic Bcl-2 family members and apoptosis inhibitory proteins, increased PI3K signaling, and defective p53 function all contribute to increased resistance to cell death induction in CSCs. In recent pre-clinical studies, engagement of death receptors by recombinant ligands, inhibition of anti-apoptotic proteins by small-molecule antagonists or interfering RNAs, restoration of p53 pathway, inhibition of PI3K pathway, and disruption of energy metabolism have all been shown to induce cell death in CSCs that are resistant to conventional therapy. Although beyond the scope of the review, research over the past several years has begun to uncover roles of microRNAs in cancer and CSCs. Dysregulated microRNAs have been implicated in CSC function and tumorigenesis 161. Similar to protein-coding genes, two types of microRNAs have been described in terms of their function in cancer, tumor-suppressor microRNAs, and oncogenic microRNAs. Ectopic expression of tumor-suppressor microRNAs and inhibition of oncogenic microRNAs have been both reported to generate anti-tumor effects. For example, overexpression of let-7, a tumor-suppressor microRNA, was shown to inhibit cell proliferation and induce cell death in lung and breast cancers 162, 163, 164. In breast cancer, let-7 reduces self-renewal and promotes differentiation. In contrast, antagonizing miR21 and miR-182 using oligonucleotides induced apoptosis and suppressed tumorigenesis in breast cancer and melanoma, respectively 165, 166. Inhibition of miR-181 in hepatocellular carcinoma led to a reduction in the CSC compartment 167.
Therefore, activation of apoptosis programs in tumor cells, particularly in CSCs, appears to be a potent adjuvant to current anticancer regimens and may open new directions for effective treatment of cancer. For clinical application, however, many questions remain to be addressed before novel therapies can be developed to specifically targeting CSCs. For example, do CSCs from different tumor types share common mechanisms of apoptotic resistance? What is the cellular and molecular basis for abnormal apoptotic programs in CSCs? What is the role of tumor microenvironment in regulating CSCs survival and function? Can CSCs be eliminated by targeting the tumor microenvironment? If CSCs initially respond to apoptosis-targeted therapies, do they evolve and develop resistance, as seen in conventional chemotherapy? Do CSCs and their normal tissue counterparts share common apoptotic regulation? What are the molecular mechanisms that determine the differential sensitivities between CSCs and normal stem cells, as discussed above in multiple tissues? How to combine conventional chemotherapy with targeted therapy to elicit maximum cell death in CSCs without harming normal tissues? Answers to these questions will not only provide deeper insights into the pathophysiology of cancer, but reveal novel therapeutic opportunities to treat this devastating disease.
Conflict of interest
The authors declare that they have no conflict of interest.
Glossary
- Bax
Bcl-2-associated X protein
- Bcl-2
B-cell CLL/lymphoma 2
- Bcl-xL
B-cell lymphoma-extra large or Bcl-2-related gene, long form
- BH3
Bcl-2 homology domain 3
- Cdk4
cyclin-dependent kinase 4
- cFLIP
cellular homologue of Fas-associated death domain-like IL-1-converting enzyme (FLICE)-inhibitory protein
- DCA
dichloroacetate
- DR
death receptor
- ESA
anti-epithelial-specific antigen
- FAO
fatty acid oxidation
- FAS
apoptosis-stimulating fragment
- FAS-L
FAS death receptor ligand
- FOXO
Forkhead box protein O
- GSK
glycogen synthase kinase
- IAP
inhibitor of apoptosis protein
- LDHA
lactate dehydrogenase A
- Mcl-1
myeloid cell leukemia-1
- mTOR
mammalian target of rapamycin
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PI3K
phosphoinositide 3-kinase
- PKM2
pyruvate kinase M2
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
- TNF-α
tumor necrosis factor alpha
- TRAIL
TNF-related apoptosis-inducing ligand
- XIAP
x-linked inhibitor of apoptosis protein
References
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Zhao J, Lu Y, Shen HM. Targeting p53 as a therapeutic strategy in sensitizing TRAIL-induced apoptosis in cancer cells. Cancer Lett. 2012;314:8–23. doi: 10.1016/j.canlet.2011.09.040. [DOI] [PubMed] [Google Scholar]
- Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008;415:333–344. doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer. 2009;9:501–507. doi: 10.1038/nrc2663. [DOI] [PubMed] [Google Scholar]
- Signore M, Ricci-Vitiani L, De Maria R. Targeting apoptosis pathways in cancer stem cells. Cancer Lett. 2013;332:374–382. doi: 10.1016/j.canlet.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, Fields JZ, Wicha MS, Boman BM. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009;69:3382–3389. doi: 10.1158/0008-5472.CAN-08-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Mao XG, Zhang X, Xue XY, Guo G, Wang P, Zhang W, Fei Z, Zhen HN, You SW, Yang H. Brain tumor stem-like cells identified by neural stem cell marker CD15. Transl Oncol. 2009;2:247–257. doi: 10.1593/tlo.09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandina G, Bonanno G, Pierelli L, Perillo A, Procoli A, Mariotti A, Corallo M, Martinelli E, Rutella S, Paglia A, et al. Expression of CD133-1 and CD133-2 in ovarian cancer. Int J Gynecol Cancer. 2008;18:506–514. doi: 10.1111/j.1525-1438.2007.01056.x. [DOI] [PubMed] [Google Scholar]
- Meng E, Long B, Sullivan P, McClellan S, Finan MA, Reed E, Shevde L, Rocconi RP. CD44+/CD24− ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin Exp Metastasis. 2012;29:939–948. doi: 10.1007/s10585-012-9482-4. [DOI] [PubMed] [Google Scholar]
- Luo L, Zeng J, Liang B, Zhao Z, Sun L, Cao D, Yang J, Shen K. Ovarian cancer cells with the CD117 phenotype are highly tumorigenic and are related to chemotherapy outcome. Exp Mol Pathol. 2011;91:596–602. doi: 10.1016/j.yexmp.2011.06.005. [DOI] [PubMed] [Google Scholar]
- Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, Guan XY. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542–2556. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- Yin S, Li J, Hu C, Chen X, Yao M, Yan M, Jiang G, Ge C, Xie H, Wan D, et al. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer. 2007;120:1444–1450. doi: 10.1002/ijc.22476. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, Jia H, Ye Q, Qin LX, Wauthier E, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, Chu PW, Lam CT, Poon RT, Fan ST. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Yang W, Yan HX, Chen L, Liu Q, He YQ, Yu LX, Zhang SH, Huang DD, Tang L, Kong XN, et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008;68:4287–4295. doi: 10.1158/0008-5472.CAN-07-6691. [DOI] [PubMed] [Google Scholar]
- Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- Akunuru S, James Zhai Q, Zheng Y. Non-small cell lung cancer stem/progenitor cells are enriched in multiple distinct phenotypic subpopulations and exhibit plasticity. Cell Death Dis. 2012;3:e352. doi: 10.1038/cddis.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, Lillington D, Oakervee H, Cavenagh J, Agrawal SG, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- Ishizawa K, Rasheed ZA, Karisch R, Wang Q, Kowalski J, Susky E, Pereira K, Karamboulas C, Moghal N, Rajeshkumar NV, et al. Tumor-initiating cells are rare in many human tumors. Cell Stem Cell. 2010;7:279–282. doi: 10.1016/j.stem.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727–738. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- Morrison R, Schleicher SM, Sun Y, Niermann KJ, Kim S, Spratt DE, Chung CH, Lu B. Targeting the mechanisms of resistance to chemotherapy and radiotherapy with the cancer stem cell hypothesis. J Oncol. 2011;2011:941876. doi: 10.1155/2011/941876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello RT, Mallet F, Gaugler B, Sainty D, Arnoulet C, Gastaut JA, Olive D. Human acute myeloid leukemia CD34+/CD38− progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000;60:4403–4411. [PubMed] [Google Scholar]
- Weller M, Frei K, Groscurth P, Krammer PH, Yonekawa Y, Fontana A. Anti-Fas/APO-1 antibody-mediated apoptosis of cultured human glioma cells. Induction and modulation of sensitivity by cytokines. J Clin Invest. 1994;94:954–964. doi: 10.1172/JCI117462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei K, Ambar B, Adachi N, Yonekawa Y, Fontana A. Ex vivo malignant glioma cells are sensitive to Fas (CD95/APO-1) ligand-mediated apoptosis. J Neuroimmunol. 1998;87:105–113. doi: 10.1016/s0165-5728(98)00065-4. [DOI] [PubMed] [Google Scholar]
- Tao J, Qiu B, Zhang D, Wang Y. Expression levels of Fas/Fas-L mRNA in human brain glioma stem cells. Mol Med Rep. 2012;5:1202–1206. doi: 10.3892/mmr.2012.791. [DOI] [PubMed] [Google Scholar]
- Eisele G, Roth P, Hasenbach K, Aulwurm S, Wolpert F, Tabatabai G, Wick W, Weller M. APO010, a synthetic hexameric CD95 ligand, induces human glioma cell death in vitro and in vivo. Neuro Oncol. 2011;13:155–164. doi: 10.1093/neuonc/noq176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele G, Wolpert F, Decrey G, Weller M. APO010, a synthetic hexameric CD95 ligand, induces death of human glioblastoma stem-like cells. Anticancer Res. 2013;33:3563–3571. [PubMed] [Google Scholar]
- Plasilova M, Zivny J, Jelinek J, Neuwirtova R, Cermak J, Necas E, Andera L, Stopka T. TRAIL (Apo2L) suppresses growth of primary human leukemia and myelodysplasia progenitors. Leukemia. 2002;16:67–73. doi: 10.1038/sj.leu.2402338. [DOI] [PubMed] [Google Scholar]
- Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–821. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- Unterkircher T, Cristofanon S, Vellanki SH, Nonnenmacher L, Karpel-Massler G, Wirtz CR, Debatin KM, Fulda S. Bortezomib primes glioblastoma, including glioblastoma stem cells, for TRAIL by increasing tBid stability and mitochondrial apoptosis. Clin Cancer Res. 2011;17:4019–4030. doi: 10.1158/1078-0432.CCR-11-0075. [DOI] [PubMed] [Google Scholar]
- Yin S, Xu L, Bandyopadhyay S, Sethi S, Reddy KB. Cisplatin and TRAIL enhance breast cancer stem cell death. Int J Oncol. 2011;39:891–898. doi: 10.3892/ijo.2011.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Wu X, Mao Y, Bao W, Gao L, Zhou P, Xie R, Zhou L, Zhu J. Dual-targeted antitumor effects against brainstem glioma by intravenous delivery of tumor necrosis factor-related, apoptosis-inducing, ligand-engineered human mesenchymal stem cells. Neurosurgery. 2009;65:610–624. doi: 10.1227/01.NEU.0000350227.61132.A7. discussion 624. [DOI] [PubMed] [Google Scholar]
- Menon LG, Kelly K, Yang HW, Kim SK, Black PM, Carroll RS. Human bone marrow-derived mesenchymal stromal cells expressing S-TRAIL as a cellular delivery vehicle for human glioma therapy. Stem Cells. 2009;27:2320–2330. doi: 10.1002/stem.136. [DOI] [PubMed] [Google Scholar]
- Ciavarella S, Grisendi G, Dominici M, Tucci M, Brunetti O, Dammacco F, Silvestris F. In vitro anti-myeloma activity of TRAIL-expressing adipose-derived mesenchymal stem cells. Br J Haematol. 2012;157:586–598. doi: 10.1111/j.1365-2141.2012.09082.x. [DOI] [PubMed] [Google Scholar]
- Reagan MR, Seib FP, McMillin DW, Sage EK, Mitsiades CS, Janes SM, Ghobrial IM, Kaplan DL. Stem cell implants for cancer therapy: TRAIL-expressing mesenchymal stem cells target cancer cells in situ. J Breast Cancer. 2012;15:273–282. doi: 10.4048/jbc.2012.15.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniri MR, Sun XY, Rayat J, Dai D, Ao Z, He Z, Verchere CB, Dai LJ, Warnock GL. TRAIL-engineered pancreas-derived mesenchymal stem cells: characterization and cytotoxic effects on pancreatic cancer cells. Cancer Gene Ther. 2012;19:652–658. doi: 10.1038/cgt.2012.46. [DOI] [PubMed] [Google Scholar]
- Loebinger MR, Sage EK, Davies D, Janes SM. TRAIL-expressing mesenchymal stem cells kill the putative cancer stem cell population. Br J Cancer. 2010;103:1692–1697. doi: 10.1038/sj.bjc.6605952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, Zhang H, Mountz JD, Koopman WJ, Kimberly RP, et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med. 2001;7:954–960. doi: 10.1038/91000. [DOI] [PubMed] [Google Scholar]
- Kaliberov S, Stackhouse MA, Kaliberova L, Zhou T, Buchsbaum DJ. Enhanced apoptosis following treatment with TRA-8 anti-human DR5 monoclonal antibody and overexpression of exogenous Bax in human glioma cells. Gene Ther. 2004;11:658–667. doi: 10.1038/sj.gt.3302215. [DOI] [PubMed] [Google Scholar]
- Oliver PG, LoBuglio AF, Zinn KR, Kim H, Nan L, Zhou T, Wang W, Buchsbaum DJ. Treatment of human colon cancer xenografts with TRA-8 anti-death receptor 5 antibody alone or in combination with CPT-11. Clin Cancer Res. 2008;14:2180–2189. doi: 10.1158/1078-0432.CCR-07-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner JA, Willey CD, Yang ES, Dobelbower MC, Sanford LL, Bright SJ, Buchsbaum DJ, Raisch KP. Treatment of small cell lung cancer with TRA-8 in combination with cisplatin and radiation. Radiother Oncol. 2011;101:183–189. doi: 10.1016/j.radonc.2011.05.083. [DOI] [PubMed] [Google Scholar]
- Yada A, Yazawa M, Ishida S, Yoshida H, Ichikawa K, Kurakata S, Fujiwara K. A novel humanized anti-human death receptor 5 antibody CS-1008 induces apoptosis in tumor cells without toxicity in hepatocytes. Ann Oncol. 2008;19:1060–1067. doi: 10.1093/annonc/mdn015. [DOI] [PubMed] [Google Scholar]
- Rajeshkumar NV, Rasheed ZA, Garcia-Garcia E, Lopez-Rios F, Fujiwara K, Matsui WH, Hidalgo M. A combination of DR5 agonistic monoclonal antibody with gemcitabine targets pancreatic cancer stem cells and results in long-term disease control in human pancreatic cancer model. Mol Cancer Ther. 2010;9:2582–2592. doi: 10.1158/1535-7163.MCT-10-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londono-Joshi AI, Oliver PG, Li Y, Lee CH, Forero-Torres A, LoBuglio AF, Buchsbaum DJ. Basal-like breast cancer stem cells are sensitive to anti-DR5 mediated cytotoxicity. Breast Cancer Res Treat. 2011;133:437–445. doi: 10.1007/s10549-011-1763-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman RT, Ricci MS, Hart LS, Sun SY, El-Deiry WS. Chemotherapy-resistant side-population of colon cancer cells has a higher sensitivity to TRAIL than the non-SP, a higher expression of c-Myc and TRAIL-receptor DR4. Cancer Biol Ther. 2007;6:1490–1495. doi: 10.4161/cbt.6.9.4905. [DOI] [PubMed] [Google Scholar]
- Li M, Knight DA, Smyth MJ, Stewart TJ. Sensitivity of a novel model of mammary cancer stem cell-like cells to TNF-related death pathways. Cancer Immunol Immunother. 2012;61:1255–1268. doi: 10.1007/s00262-012-1200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton A, Young LS, Murray PG. The role of cellular FLICE inhibitory protein (c-FLIP) in the pathogenesis and treatment of cancer. Expert Opin Ther Targets. 2006;10:27–35. doi: 10.1517/14728222.10.1.27. [DOI] [PubMed] [Google Scholar]
- Shirley S, Micheau O. Targeting c-FLIP in cancer. Cancer Lett. 2013;332:141–150. doi: 10.1016/j.canlet.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Zobalova R, Stantic M, Prokopova K, Dong LF, Neuzil J. Cancer cells with high expression of CD133 exert FLIP upregulation and resistance to TRAIL-induced apoptosis. BioFactors. 2009;34:231–235. doi: 10.3233/BIO-2009-1076. [DOI] [PubMed] [Google Scholar]
- Zobalova R, McDermott L, Stantic M, Prokopova K, Dong LF, Neuzil J. CD133-positive cells are resistant to TRAIL due to up-regulation of FLIP. Biochem Biophys Res Commun. 2008;373:567–571. doi: 10.1016/j.bbrc.2008.06.073. [DOI] [PubMed] [Google Scholar]
- Ding L, Yuan C, Wei F, Wang G, Zhang J, Bellail AC, Zhang Z, Olson JJ, Hao C. Cisplatin restores TRAIL apoptotic pathway in glioblastoma-derived stem cells through up-regulation of DR5 and down-regulation of c-FLIP. Cancer Invest. 2011;29:511–520. doi: 10.3109/07357907.2011.605412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggott L, Omidvar N, Marti Perez S, Eberl M, Clarkson RW. Suppression of apoptosis inhibitor c-FLIP selectively eliminates breast cancer stem cell activity in response to the anti-cancer agent, TRAIL. Breast Cancer Res. 2011;13:R88. doi: 10.1186/bcr2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llobet D, Eritja N, Yeramian A, Pallares J, Sorolla A, Domingo M, Santacana M, Gonzalez-Tallada FJ, Matias-Guiu X, Dolcet X. The multikinase inhibitor Sorafenib induces apoptosis and sensitises endometrial cancer cells to TRAIL by different mechanisms. Eur J Cancer. 2010;46:836–850. doi: 10.1016/j.ejca.2009.12.025. [DOI] [PubMed] [Google Scholar]
- Pennarun B, Kleibeuker JH, Boersma-van Ek W, Kruyt FA, Hollema H, de Vries EG, de Jong S. Targeting FLIP and Mcl-1 using a combination of aspirin and sorafenib sensitizes colon cancer cells to TRAIL. J Pathol. 2013;229:410–421. doi: 10.1002/path.4138. [DOI] [PubMed] [Google Scholar]
- Yoon MJ, Kang YJ, Kim IY, Kim EH, Lee JA, Lim JH, Kwon TK, Choi KS. Monensin, a polyether ionophore antibiotic, overcomes TRAIL resistance in glioma cells via endoplasmic reticulum stress, DR5 upregulation and c-FLIP downregulation. Carcinogenesis. 2013;34:1918–1928. doi: 10.1093/carcin/bgt137. [DOI] [PubMed] [Google Scholar]
- Chen X, Wang T, Yang D, Wang J, Li X, He Z, Chen F, Che X, Song X. Expression of the IAP protein family acts cooperatively to predict prognosis in human bladder cancer patients. Oncol Lett. 2013;5:1278–1284. doi: 10.3892/ol.2013.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzybowska-Izydorczyk O, Cebula B, Robak T, Smolewski P. Expression and prognostic significance of the inhibitor of apoptosis protein (IAP) family and its antagonists in chronic lymphocytic leukaemia. Eur J Cancer. 2010;46:800–810. doi: 10.1016/j.ejca.2009.11.023. [DOI] [PubMed] [Google Scholar]
- Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- Wu TY, Wagner KW, Bursulaya B, Schultz PG, Deveraux QL. Development and characterization of nonpeptidic small molecule inhibitors of the XIAP/caspase-3 interaction. Chem Biol. 2003;10:759–767. doi: 10.1016/s1074-5521(03)00157-1. [DOI] [PubMed] [Google Scholar]
- Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ, Pedersen IM, Kitada S, Scott FL, Bailly-Maitre B, et al. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell. 2004;5:25–35. doi: 10.1016/s1535-6108(03)00332-5. [DOI] [PubMed] [Google Scholar]
- Loeder S, Drensek A, Jeremias I, Debatin KM, Fulda S. Small molecule XIAP inhibitors sensitize childhood acute leukemia cells for CD95-induced apoptosis. Int J Cancer. 2010;126:2216–2228. doi: 10.1002/ijc.24816. [DOI] [PubMed] [Google Scholar]
- Checinska A, Hoogeland BS, Rodriguez JA, Giaccone G, Kruyt FA. Role of XIAP in inhibiting cisplatin-induced caspase activation in non-small cell lung cancer cells: a small molecule Smac mimic sensitizes for chemotherapy-induced apoptosis by enhancing caspase-3 activation. Exp Cell Res. 2007;313:1215–1224. doi: 10.1016/j.yexcr.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Engesaeter BO, Sathermugathevan M, Hellenes T, Engebraten O, Holm R, Florenes VA, Maelandsmo GM. Targeting inhibitor of apoptosis proteins in combination with dacarbazine or TRAIL in melanoma cells. Cancer Biol Ther. 2011;12:47–58. doi: 10.4161/cbt.12.1.15714. [DOI] [PubMed] [Google Scholar]
- Vogler M, Walczak H, Stadel D, Haas TL, Genze F, Jovanovic M, Bhanot U, Hasel C, Moller P, Gschwend JE, et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res. 2009;69:2425–2434. doi: 10.1158/0008-5472.CAN-08-2436. [DOI] [PubMed] [Google Scholar]
- Mohr A, Albarenque SM, Deedigan L, Yu R, Reidy M, Fulda S, Zwacka RM. Targeting of XIAP combined with systemic mesenchymal stem cell-mediated delivery of sTRAIL ligand inhibits metastatic growth of pancreatic carcinoma cells. Stem Cells. 2010;28:2109–2120. doi: 10.1002/stem.533. [DOI] [PubMed] [Google Scholar]
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellanki SH, Grabrucker A, Liebau S, Proepper C, Eramo A, Braun V, Boeckers T, Debatin KM, Fulda S. Small-molecule XIAP inhibitors enhance gamma-irradiation-induced apoptosis in glioblastoma. Neoplasia. 2009;11:743–752. doi: 10.1593/neo.09436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahana S, Finniss S, Cazacu S, Xiang C, Lee HK, Brodie S, Goldstein RS, Roitman V, Slavin S, Mikkelsen T, et al. Proteasome inhibitors sensitize glioma cells and glioma stem cells to TRAIL-induced apoptosis by PKCepsilon-dependent downregulation of AKT and XIAP expressions. Cell Signal. 2011;23:1348–1357. doi: 10.1016/j.cellsig.2011.03.017. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985;229:1390–1393. doi: 10.1126/science.3929382. [DOI] [PubMed] [Google Scholar]
- Mandik L, Katsumata M, Erikson J. Effects of altered Bcl-2 expression on B lymphocyte selection. Ann N Y Acad Sci. 1997;815:40–54. doi: 10.1111/j.1749-6632.1997.tb52043.x. [DOI] [PubMed] [Google Scholar]
- Hockenbery DM, Zutter M, Hickey W, Nahm M, Korsmeyer SJ. BCL2 protein is topographically restricted in tissues characterized by apoptotic cell death. Proc Natl Acad Sci USA. 1991;88:6961–6965. doi: 10.1073/pnas.88.16.6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domen J, Cheshier SH, Weissman IL. The role of apoptosis in the regulation of hematopoietic stem cells: overexpression of Bcl-2 increases both their number and repopulation potential. J Exp Med. 2000;191:253–264. doi: 10.1084/jem.191.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domen J, Weissman IL. Hematopoietic stem cells need two signals to prevent apoptosis; BCL-2 can provide one of these, Kitl/c-Kit signaling the other. J Exp Med. 2000;192:1707–1718. doi: 10.1084/jem.192.12.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- Matsuzaki Y, Nakayama K, Nakayama K, Tomita T, Isoda M, Loh DY, Nakauchi H. Role of bcl-2 in the development of lymphoid cells from the hematopoietic stem cell. Blood. 1997;89:853–862. [PubMed] [Google Scholar]
- Ma A, Pena JC, Chang B, Margosian E, Davidson L, Alt FW, Thompson CB. Bclx regulates the survival of double-positive thymocytes. Proc Natl Acad Sci USA. 1995;92:4763–4767. doi: 10.1073/pnas.92.11.4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- Motoyama N, Kimura T, Takahashi T, Watanabe T, Nakano T. bcl-x prevents apoptotic cell death of both primitive and definitive erythrocytes at the end of maturation. J Exp Med. 1999;189:1691–1698. doi: 10.1084/jem.189.11.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- Huang HM, Huang CJ, Yen JJ. Mcl-1 is a common target of stem cell factor and interleukin-5 for apoptosis prevention activity via MEK/MAPK and PI-3K/Akt pathways. Blood. 2000;96:1764–1771. [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–249. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Ziegler A, Luedke GH, Fabbro D, Altmann KH, Stahel RA, Zangemeister-Wittke U. Induction of apoptosis in small-cell lung cancer cells by an antisense oligodeoxynucleotide targeting the Bcl-2 coding sequence. J Natl Cancer Inst. 1997;89:1027–1036. doi: 10.1093/jnci/89.14.1027. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Rocchi P, Miyake H, Fazli L, Vessella B, Zangemeister-Wittke U, Gleave ME. A novel antisense oligonucleotide inhibiting several antiapoptotic Bcl-2 family members induces apoptosis and enhances chemosensitivity in androgen-independent human prostate cancer PC3 cells. Mol Cancer Ther. 2005;4:1689–1698. doi: 10.1158/1535-7163.MCT-05-0064. [DOI] [PubMed] [Google Scholar]
- Jansen B, Schlagbauer-Wadl H, Brown BD, Bryan RN, van Elsas A, Muller M, Wolff K, Eichler HG, Pehamberger H. bcl-2 antisense therapy chemosensitizes human melanoma in SCID mice. Nat Med. 1998;4:232–234. doi: 10.1038/nm0298-232. [DOI] [PubMed] [Google Scholar]
- Julien T, Frankel B, Longo S, Kyle M, Gibson S, Shillitoe E, Ryken T. Antisense-mediated inhibition of the bcl-2 gene induces apoptosis in human malignant glioma. Surg Neurol. 2000;53:360–368. doi: 10.1016/s0090-3019(00)00178-6. discussion 368-369. [DOI] [PubMed] [Google Scholar]
- Kantarjian HM, Talpaz M, Santini V, Murgo A, Cheson B, O’Brien SM. Homoharringtonine: history, current research, and future direction. Cancer. 2001;92:1591–1605. doi: 10.1002/1097-0142(20010915)92:6<1591::aid-cncr1485>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Chen Y, Hu Y, Michaels S, Segal D, Brown D, Li S. Inhibitory effects of omacetaxine on leukemic stem cells and BCR-ABL-induced chronic myeloid leukemia and acute lymphoblastic leukemia in mice. Leukemia. 2009;23:1446–1454. doi: 10.1038/leu.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan EK, Holyoake TL, Craig AR, Jorgensen HG. Omacetaxine may have a role in chronic myeloid leukaemia eradication through downregulation of Mcl-1 and induction of apoptosis in stem/progenitor cells. Leukemia. 2011;25:985–994. doi: 10.1038/leu.2011.55. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Lauria F, Raspadori D, Rondelli D, Ventura MA, Fiacchini M, Visani G, Forconi F, Tura S. High bcl-2 expression in acute myeloid leukemia cells correlates with CD34 positivity and complete remission rate. Leukemia. 1997;11:2075–2078. doi: 10.1038/sj.leu.2400854. [DOI] [PubMed] [Google Scholar]
- Tothova E, Fricova M, Stecova N, Kafkova A, Elbertova A. High expression of Bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Neoplasma. 2002;49:141–144. [PubMed] [Google Scholar]
- Campos L, Rouault JP, Sabido O, Oriol P, Roubi N, Vasselon C, Archimbaud E, Magaud JP, Guyotat D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993;81:3091–3096. [PubMed] [Google Scholar]
- Madjd Z, Mehrjerdi AZ, Sharifi AM, Molanaei S, Shahzadi SZ, Asadi-Lari M. CD44+ cancer cells express higher levels of the anti-apoptotic protein Bcl-2 in breast tumours. Cancer Immun. 2009;9:4. [PMC free article] [PubMed] [Google Scholar]
- Tagscherer KE, Fassl A, Campos B, Farhadi M, Kraemer A, Bock BC, Macher-Goeppinger S, Radlwimmer B, Wiestler OD, Herold-Mende C, et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene. 2008;27:6646–6656. doi: 10.1038/onc.2008.259. [DOI] [PubMed] [Google Scholar]
- Voss V, Senft C, Lang V, Ronellenfitsch MW, Steinbach JP, Seifert V, Kogel D. The pan-Bcl-2 inhibitor (−)-gossypol triggers autophagic cell death in malignant glioma. Mol Cancer Res. 2010;8:1002–1016. doi: 10.1158/1541-7786.MCR-09-0562. [DOI] [PubMed] [Google Scholar]
- Beverly LJ, Varmus HE. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene. 2009;28:1274–1279. doi: 10.1038/onc.2008.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aichberger KJ, Mayerhofer M, Krauth MT, Skvara H, Florian S, Sonneck K, Akgul C, Derdak S, Pickl WF, Wacheck V, et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood. 2005;105:3303–3311. doi: 10.1182/blood-2004-02-0749. [DOI] [PubMed] [Google Scholar]
- Mak DH, Wang RY, Schober WD, Konopleva M, Cortes J, Kantarjian H, Andreeff M, Carter BZ. Activation of apoptosis signaling eliminates CD34+ progenitor cells in blast crisis CML independent of response to tyrosine kinase inhibitors. Leukemia. 2012;26:788–794. doi: 10.1038/leu.2011.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DJ, Recart AC, Sadarangani A, Chun HJ, Barrett CL, Krajewska M, Leu H, Low-Marchelli J, Ma W, Shih AY, et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell. 2013;12:316–328. doi: 10.1016/j.stem.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–341. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurlet S, Omidvar N, Gorombei P, Krief P, Le Pogam C, Setterblad N, de la Grange P, Leboeuf C, Janin A, Noguera ME, et al. BCL-2 inhibition with ABT-737 prolongs survival in an NRAS/BCL-2 mouse model of AML by targeting primitive LSK and progenitor cells. Blood. 2013;122:2864–2876. doi: 10.1182/blood-2012-07-445635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, Deangelo DJ, Frattini MG, Letai A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151:344–355. doi: 10.1016/j.cell.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Palorini R, Votta G, Balestrieri C, Monestiroli A, Olivieri S, Vento R, Chiaradonna F. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J Cell Biochem. 2013;115:368–379. doi: 10.1002/jcb.24671. [DOI] [PubMed] [Google Scholar]
- Morfouace M, Lalier L, Bahut M, Bonnamain V, Naveilhan P, Guette C, Oliver L, Gueguen N, Reynier P, Vallette FM. Comparison of spheroids formed by rat glioma stem cells and neural stem cells reveals differences in glucose metabolism and promising therapeutic applications. J Biol Chem. 2012;287:33664–33674. doi: 10.1074/jbc.M111.320028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian C, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci USA. 2011;108:16062–16067. doi: 10.1073/pnas.1106704108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, Radovanovic I, Rheinbay E, Provero P, Stamenkovic I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26:1926–1944. doi: 10.1101/gad.188292.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvero AB, Montagna MK, Holmberg JC, Craveiro V, Brown D, Mor G. Targeting the mitochondria activates two independent cell death pathways in ovarian cancer stem cells. Mol Cancer Ther. 2011;10:1385–1393. doi: 10.1158/1535-7163.MCT-11-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–156. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT, Evans RM, Suda T, Lee CH, et al. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18:1350–1358. doi: 10.1038/nm.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, Rosenblatt J, Avigan DE, Teruya-Feldstein J, Pandolfi PP. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–1078. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sax JK, El-Deiry WS. p53 downstream targets and chemosensitivity. Cell Death Differ. 2003;10:413–417. doi: 10.1038/sj.cdd.4401227. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Cleary K, Bigner SH, Davidson N, Baylin S, Devilee P, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342:705–708. doi: 10.1038/342705a0. [DOI] [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol. 2010;80:724–730. doi: 10.1016/j.bcp.2010.04.031. [DOI] [PubMed] [Google Scholar]
- Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, Xu Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- Meletis K, Wirta V, Hede SM, Nister M, Lundeberg J, Frisen J. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363–369. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Zuber J, Diaz-Flores E, Lintault L, Kogan SC, Shannon K, Lowe SW. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010;24:1389–1402. doi: 10.1101/gad.1940710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spike BT, Wahl GM. p53, stem cells, and reprogramming: tumor suppression beyond guarding the genome. Genes Cancer. 2011;2:404–419. doi: 10.1177/1947601911410224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, Brisken C, Minucci S, Di Fiore PP, Pelicci PG. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- Li L, Wang L, Li L, Wang Z, Ho Y, McDonald T, Holyoake TL, Chen W, Bhatia R. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell. 2012;21:266–281. doi: 10.1016/j.ccr.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Zhang XM, Tavaluc RT, Hart LS, Dicker DT, Wang W, El-Deiry WS. The combination of 5-fluorouracil plus p53 pathway restoration is associated with depletion of p53-deficient or mutant p53-expressing putative colon cancer stem cells. Cancer Biol Ther. 2009;8:2186–2193. doi: 10.4161/cbt.8.22.10446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2011;148:244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Monville F, Wicinski J, Cabaud O, Cervera N, Josselin E, Finetti P, Guille A, Larderet G, Viens P, et al. Mevalonate metabolism regulates Basal breast cancer stem cells and is a potential therapeutic target. Stem Cells. 2012;30:1327–1337. doi: 10.1002/stem.1122. [DOI] [PubMed] [Google Scholar]
- Hartwell KA, Miller PG, Mukherjee S, Kahn AR, Stewart AL, Logan DJ, Negri JM, Duvet M, Jaras M, Puram R, et al. Niche-based screening identifies small-molecule inhibitors of leukemia stem cells. Nat Chem Biol. 2013;9:840–848. doi: 10.1038/nchembio.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22:436–448. doi: 10.1101/gad.1627008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carra E, Barbieri F, Marubbi D, Pattarozzi A, Favoni RE, Florio T, Daga A. Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures. Cell Cycle. 2013;12:491–500. doi: 10.4161/cc.23372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firat E, Weyerbrock A, Gaedicke S, Grosu AL, Niedermann G. Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote gamma-irradiation-induced cell death in primary stem-like glioma cells. PLoS ONE. 2012;7:e47357. doi: 10.1371/journal.pone.0047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai R, Wakimoto H, Martuza RL, Rabkin SD. A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells. Clin Cancer Res. 2011;17:3686–3696. doi: 10.1158/1078-0432.CCR-10-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003;102:972–980. doi: 10.1182/blood-2002-11-3429. [DOI] [PubMed] [Google Scholar]
- Xu Q, Thompson JE, Carroll M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood. 2005;106:4261–4268. doi: 10.1182/blood-2004-11-4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaitzidis D, Sykes SM, Wang Z, Punt N, Tang Y, Ragu C, Sinha AU, Lane SW, Souza AL, Clish CB, et al. mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell. 2012;11:429–439. doi: 10.1016/j.stem.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JA, Ikenoue T, Nakada D, Lee JY, Guan KL, Morrison SJ. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell. 2012;11:415–428. doi: 10.1016/j.stem.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, Nakao S, Motoyama N, Hirao A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463:676–680. doi: 10.1038/nature08734. [DOI] [PubMed] [Google Scholar]
- Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, Ferraro F, Mercier F, Singh H, Brumme KM, et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146:697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez E, Agliano A, Prior C, Nguewa P, Redrado M, Gonzalez-Zubeldia I, Plano D, Palop JA, Sanmartin C, Calvo A. The quinoline imidoselenocarbamate EI201 blocks the AKT/mTOR pathway and targets cancer stem cells leading to a strong antitumor activity. Curr Med Chem. 2012;19:3031–3043. doi: 10.2174/092986712800672076. [DOI] [PubMed] [Google Scholar]
- Kumar D, Shankar S, Srivastava RK. Rottlerin induces autophagy and apoptosis in prostate cancer stem cells via PI3K/Akt/mTOR signaling pathway. Cancer Lett. 2013;343:179–189. doi: 10.1016/j.canlet.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Liu H, Han D, Liu Y, Hou X, Wu J, Li H, Yang J, Shen C, Yang G, Fu C, et al. Harmine hydrochloride inhibits Akt phosphorylation and depletes the pool of cancer stem-like cells of glioblastoma. J Neurooncol. 2013;112:39–48. doi: 10.1007/s11060-012-1034-x. [DOI] [PubMed] [Google Scholar]
- Wang JY, Chang CC, Chiang CC, Chen WM, Hung SC. Silibinin suppresses the maintenance of colorectal cancer stem-like cells by inhibiting PP2A/AKT/mTOR pathways. J Cell Biochem. 2012;113:1733–1743. doi: 10.1002/jcb.24043. [DOI] [PubMed] [Google Scholar]
- Shimura T, Noma N, Oikawa T, Ochiai Y, Kakuda S, Kuwahara Y, Takai Y, Takahashi A, Fukumoto M. Activation of the AKT/cyclin D1/Cdk4 survival signaling pathway in radioresistant cancer stem cells. Oncogenesis. 2012;1:e12. doi: 10.1038/oncsis.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore N, Lyle S. Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J Oncol. 2011 doi: 10.1155/2011/396076. doi: 10.1155/2011/396076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal JA, Lleonart ME. MicroRNAs and cancer stem cells: therapeutic approaches and future perspectives. Cancer Lett. 2013;338:174–183. doi: 10.1016/j.canlet.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, Jacks T. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA. 2008;105:3903–3908. doi: 10.1073/pnas.0712321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, Weidhaas JB, Brown D, Bader AG, Slack FJ. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7:759–764. doi: 10.4161/cc.7.6.5834. [DOI] [PubMed] [Google Scholar]
- Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21-mediated tumor growth. Oncogene. 2007;26:2799–2803. doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- Huynh C, Segura MF, Gaziel-Sovran A, Menendez S, Darvishian F, Chiriboga L, Levin B, Meruelo D, Osman I, Zavadil J, et al. Efficient in vivo microRNA targeting of liver metastasis. Oncogene. 2011;30:1481–1488. doi: 10.1038/onc.2010.523. [DOI] [PubMed] [Google Scholar]
- Ji J, Yamashita T, Budhu A, Forgues M, Jia HL, Li C, Deng C, Wauthier E, Reid LM, Ye QH, et al. Identification of microRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells. Hepatology. 2009;50:472–480. doi: 10.1002/hep.22989. [DOI] [PMC free article] [PubMed] [Google Scholar]