

Abstract
Mutations in PINK1 and PARKIN cause recessive, early-onset Parkinson’s disease (PD). Together, these two proteins orchestrate a protective mitophagic response that ensures the safe disposal of damaged mitochondria. The kinase PINK1 phosphorylates ubiquitin (Ub) at the conserved residue S65, in addition to modifying the E3 ubiquitin ligase Parkin. The structural and functional consequences of Ub phosphorylation (pS65-Ub) have already been suggested from in vitro experiments, but its (patho-)physiological significance remains unknown. We have generated novel antibodies and assessed pS65-Ub signals in vitro and in cells, including primary neurons, under endogenous conditions. pS65-Ub is dependent on PINK1 kinase activity as confirmed in patient fibroblasts and postmortem brain samples harboring pathogenic mutations. We show that pS65-Ub is reversible and barely detectable under basal conditions, but rapidly induced upon mitochondrial stress in cells and amplified in the presence of functional Parkin. pS65-Ub accumulates in human brain during aging and disease in the form of cytoplasmic granules that partially overlap with mitochondrial, lysosomal, and total Ub markers. Additional studies are now warranted to further elucidate pS65-Ub functions and fully explore its potential for biomarker or therapeutic development.
Keywords: early-onset Parkinson’s disease, mitophagy, Parkin, phosphorylated ubiquitin, PINK1
Introduction
Mutations in PINK1 and PARKIN are the most common cause of recessive early-onset Parkinson’s disease (PD). Together, they coordinate a mitochondrial quality control pathway that ensures safe disposal of defective (mitophagy) and maintenance of healthy mitochondria 1. This stress-induced pathway is tightly controlled and underlies complex regulation at multiple steps of a sequential process 2. Upon mitochondrial damage, the protein kinase PINK1 is stabilized on the outer membrane and recruits the E3 ubiquitin (Ub) ligase Parkin from the cytosol 3. PINK1 has been shown to phosphorylate Parkin 4-6 in its N-terminal Ub-like (UBL) domain, which is required for Parkin’s structural 7 and functional activation 8. Parkin is “charged” with Ub by E2 co-enzymes that modulate its mitochondrial translocation and enzymatic functions, both of which are linked 9, 10. Parkin then labels mitochondrial substrate proteins with poly-Ub chains of distinct topologies to mediate their sequestration and/or degradation. Parkin and generated Ub conjugates are also subject to regulation by specific de-ubiquitinating enzymes (DUBs) 11. Removal of individual Ub moieties or chains from substrates modulates downstream functions that are decoded by Ub-binding adaptors.
PINK1 has just recently been identified to phosphorylate Ub, in addition to the Ub ligase Parkin, at a conserved serine 65 (S65) residue 12, 13, 14. Both phosphorylation events are required for full activation of Parkin by feed-forward mechanisms during mitophagy 15, 16, 17. While phosphorylation of the modifier protein further increases complexity, it also provides more selectivity and specificity for a seemingly universal ubiquitination process. In addition to activation of Parkin, consequences of pS65-Ub on structure, chain assembly, hydrolysis, and recognition have been reported in vitro 18. During preparation of this manuscript, another study suggested pS65-Ub as the Parkin receptor on damaged mitochondria 19. However, the (patho-)physiological significance of this posttranslational modification in particular in neurons and in brain remains unclear.
Here, we developed and carefully characterized two phospho-specific antibodies as tools to demonstrate the (patho-)physiological relevance of pS65-Ub. While one of the antibodies was specific to pS65-Ub, the other antibody recognized both pS65-Ub and pS65-Parkin. We confirmed that the obtained signals were: (i) specific to phosphorylated S65, (ii) induced by mitochondrial stress, (iii) dependent on PINK1 kinase, and (iv) reversible by and sensitive to phosphatase activity. For the first time, we corroborated the presence of pS65-Ub under endogenous conditions in stressed primary neurons and in vivo in human postmortem brains. Importantly, primary cells and brain tissue from PD patients carrying PINK1 mutations were largely devoid of pS65-Ub signal. Our findings suggest that pS65-Ub accumulates with stress, disease, or age, and highlight its significance and potential for future biomarker and/or therapeutic development.
Results
Validation of pS65-Ub antibodies in vitro
We sought to develop antibodies specific to Ub phosphorylated at Ser65 (pS65-Ub) to investigate its significance in primary neurons, in human brain, and in PD patient samples. Affinity purification yielded two selective and sensitive rabbit polyclonal antibodies (hereafter referred to as pS65-Ub#1 and pS65-Ub#2) as shown by dot blot with the immunogenic and an unmodified control peptide (Fig1A). Western blots (WBs) of synthetic or PINK1-phosphorylated pS65-Ub confirmed their selectivity for modified Ub only (Fig1B and C). Similar to monomeric pS65-Ub, antibodies also detected poly-Ub chains that had been phosphorylated by PINK1 wild-type (WT), but not kinase-dead (KD) mutant (Figs1D and EV1A). The slight preference for K48 over K63 linkage might be explained by the proximity of S65 to K63 (Fig1E) and distinct topologies of the respective chains (Fig EV1B). K63 linkage could equally affect PINK1 phosphorylation of S65 or binding of the pS65-Ub antibodies.
Figure 1.

Novel antibodies selectively detect phosphorylated Ub monomers and poly-Ub chains in vitro
- Dot blots were performed with the immunogen (pSer65, amino acids 59–71 of Ub: YNIQKE[pS]TLHLVL) and a corresponding, non-phosphorylated peptide (Ser65) and probed with sera and affinity-purified antibodies pS65-Ub#1 and #2.
- Western blots (WBs) with increasing amounts of Ub and synthetic pS65-Ub (both N-terminally biotinylated, N-biotin) were probed with anti-pS65-Ub antibodies or streptavidin coupled to horseradish peroxidase (SA-HRP) as indicated.
- Monomeric N-biotin-Ub was incubated with recombinant MBP-tagged PINK1 kinase wild-type (WT) or kinase-dead (KD) mutant for the indicated times in vitro prior to WB.
- N-biotin-tagged K48- and K63-linked poly-Ub chains (n = 2–7) were incubated with or without MBP-tagged PINK1 WT in vitro prior to WB. Membranes were also probed with SA-HRP and K48 or K63 linkage-specific anti-Ub antibodies as controls, which did not discriminate between phosphorylated or non-phosphorylated Ub conjugates.
- Ribbon diagram of the pS65-Ub monomer. pSer65 is shown in VdW representation, while all seven lysine (Lys) residues are shown in stick/licorice, both colored by atom type.

anti-pS65-Ub antibodies detect PINK1-phosphorylated poly-Ub chains
- Untagged K48- and K63-linked poly-Ub chains (n = 2–7) were incubated with recombinant MBP-PINK1 WT or KD for the indicated times in vitro prior to WB. Both, pS65-Ub#1 and #2 similarly recognized phosphorylated K48-linked poly-Ub chains, whereas pS65-Ub#2 appeared to react stronger with phosphorylated K63-linked Ub conjugates. Blots were probed with MBP and total Ub antibodies to show equal loading.
- Axial and lateral views of a K48-linked or K63-linked (8-mers) poly-Ub chains show the different topologies. In the more closed and compacted structure of K48-linked poly-Ub chains, S65 residues of the individual Ub moieties are faced outwards. However, in the rather extended conformation of K63-linked chains with larger spacing between Ub moieties, S65 residues are positioned more toward the inside of the chain. Each Ub moiety and corresponding S65 is shown in a different color. pS65 is shown in VdW representation. K48-Gly76 and K63-Gly76 linkages are shown in licorice rendering colored by atom type.
To determine a potential cross-reactivity of the pS65-Ub antibodies with Parkin, the other substrate of PINK1, we performed further experiments. Direct comparison of S65-phosphorylated Ub and Parkin peptides showed a minor detection of pS65-Parkin in addition to pS65-Ub, with one of two antibodies (pS65-Ub#2, FigEV2A). In vitro phosphorylation of Parkin with PINK1 confirmed some cross-reactivity of pS65-Ub#2 with phosphorylated full-length Parkin. However, compared to pS65-Ub (Fig1C), pS65-Parkin was detected only after longer kinase reactions (60 min) and longer exposures (Fig EV2B). As this could be consistent with the idea that Ub is the preferred substrate for PINK1 over Parkin, we generated equimolar amounts of both phosphorylated proteins (in a 2-day kinase reaction to ensure complete modification of all Ub and Parkin molecules). In this setting, pS65-Ub#2 showed a stronger signal for pS65-Parkin compared to pS65-Ub (FigEV2C).
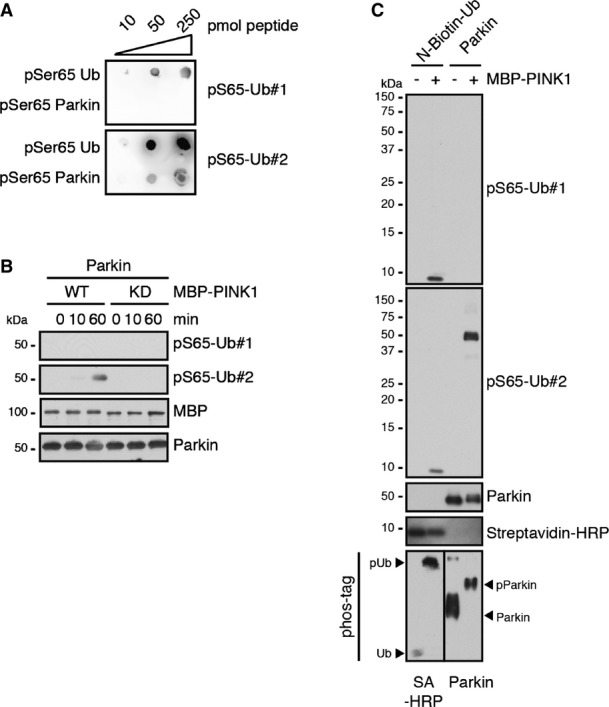
pS65-Ub#1 is highly specific, whereas pS65-Ub#2 recognizes both PINK1-modified substrates, pS65-Ub and pS65-Parkin
- Dot blots were performed with the immunogen (pSer65-Ub, amino acids 59–71 of Ub: YNIQKE[pS]TLHLVL) and a phosphorylated Parkin peptide (pSer65-Parkin, amino acids 60–71 of Parkin: DLDQQ[pS]IVHIVQ) and probed with affinity-purified antibodies pS65-Ub#1 and #2. While pS65-Ub#1 was specific to the phosphorylated Ub peptide, pS65-Ub#2 showed reactivity toward Ub and Parkin phospho-peptides.
- Recombinant untagged Parkin was incubated with MBP-tagged PINK1 WT or KD for the indicated times in vitro. PINK1-phosphorylated Parkin is detected by pS65-Ub#2, but not #1, although at much lower level than pS65-Ub (compared to Fig1C).
- Equimolar amounts of recombinant N-biotin-Ub or untagged Parkin were incubated with MBP-PINK1 for 2 days to achieve complete phosphorylation as shown by phos-tag gel electrophoresis. Under these conditions, pS65-Ub#1 specifically detected pS65-Ub, while pS65-Ub#2 cross-reacted somewhat stronger with pS65-Parkin. Both antibodies did not recognize unphosphorylated Ub or Parkin, respectively.
Cellular pS65-Ub signal is induced by stress and amplified by functional Parkin
Next, we tested pS65-Ub antibodies on samples from human HeLa cells. Parental cells, which lack detectable amounts of endogenous Parkin, or cells stably overexpressing native, untagged Parkin, were treated with the mitochondrial uncoupler carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (Fig2A). WB of lysates revealed almost no signal in untreated cells, but a robust increase in pS65-Ub signal with mitochondrial damage over time. Presence of functional Parkin WT amplified the pS65-Ub levels, likely through enhanced formation of poly-Ub chains that in turn serve as substrates for PINK1. While pS65-Ub signal steadily increased over longer times CCCP treatment in cells without Parkin, it never reached levels observed in the presence of functional Parkin. Here, a peak was reached already around 4 h upon CCCP treatment, after which pS65-Ub levels decreased possibly due to substrate degradation. In addition to the prominent high molecular weight (HMW) smear, we also detected monomers and dimers of pS65-modified Ub. Interestingly, monomeric pS65-Ub accumulated more in HeLa cells lacking Parkin and appeared to be utilized in Parkin overexpressing cells for the formation of HMW conjugates. Immunofluorescence (IF) staining of HeLa cells expressing GFP-Parkin corroborated lack of pS65-Ub signal at basal conditions and robust induction upon CCCP treatment. Moreover, IF revealed an exclusive co-localization of pS65-Ub with Parkin on mitochondria as expected (Fig2B and C).
Figure 2.

Cellular pS65-Ub signal is stress-induced and amplified by functional Parkin on mitochondria
- A Parental HeLa cells and cells stably overexpressing untagged, native human Parkin WT were treated with CCCP for the indicated times, and lysates were analyzed by WB as indicated. HeLa cells without or with functional Parkin both showed an increase in pS65-Ub signal over time of CCCP treatment; however, the signal was strongly amplified in the presence of Parkin. Inlay WB (16%) for the stronger pS65-Ub#2 antibody shows phosphorylated mono- and di-Ub (as indicated by one or two black circles). Note the increase of both species over time in parental cells. In the presence of Parkin, these appear to be utilized for conjugation of higher molecular weight (HMW) phosphorylated poly-Ub chains as indicated by enhanced overall total Ub levels.
- B, C HeLa cells stably overexpressing GFP-tagged Parkin (green) were treated with CCCP as indicated, fixed and stained with anti-TOM20 (mitochondria, cyan) and anti-pS65-Ub#1 (B) or anti-pS65-Ub#2 (C) (red) as well as Hoechst (nucleus, blue). Upon CCCP treatment, pS65-Ub signal forms on mitochondria in the presence of functional Parkin. Scale bars, 10 μm.
pS65-Ub#2 recognizes both PINK1 kinase products, pS65-Ub and pS65-Parkin
To further investigate the PINK1- and Parkin-dependent amplification of pS65-Ub signal, we stably expressed a catalytically dead Parkin C431S mutant in HeLa cells. Similar to parental cells that lack Parkin, expression of this inactive variant did not result in amplification of pS65-Ub signal and appearance of HMW species as compared to cells expressing Parkin WT (FigEV3A). The Parkin C431S mutant, which traps the Ub moiety on the catalytic center in a stable oxyester, but not WT, was recognized by the antibody pS65-Ub#2 as a discrete doublet band upon CCCP treatment. The upper band of these was indeed sensitive to NaOH cleavage, suggesting the accumulation of Ub-charged pS65-Parkin C431S over time (FigEV3B). Of note, pS65-Ub#2 did not recognize the Parkin S65A mutant, suggesting its specificity for the S65-phosphorylated form of Parkin (FigEV3C). To exclude a major contribution of pS65-Parkin detection to the overall cellular signal observed with pS65-Ub#2, we studied GFP-Parkin WT and C431S HeLa cells. We confirmed that the pS65-Ub signal is amplified only in the presence of functional Parkin (Appendix Fig S1). Yet, after longer times of CCCP incubation (8 h), we noticed a very weak signal in the presence of non-functional C431S Parkin with the antibody pS65-Ub#1 and, to a stronger extent, with pS65-Ub#2. However, Parkin C431S did not translocate to damaged mitochondria and remained evenly distributed throughout the cell (Appendix Fig S1). The signal obtained with pS65-Ub#2 rather resembled the intracellular localization of mitochondria.

Cross-reactivity of the pS65-Ub#2 antibody with cellular pS65-Parkin
- Parental HeLa cells and cells stably expressing 3× FLAG-tagged Parkin WT or C431S mutants were treated with CCCP for the indicated times, lysed and analyzed by WB. In comparison with parental HeLa cells and with cells overexpressing the ligase-dead Parkin mutant C431S, cells overexpressing functional Parkin show strongly enhanced pS65-Ub signal. Similarly, enhanced overall ubiquitination is present in WT overexpressing cells as reflected by total Ub signal. In addition to pS65-Ub, pS65-Ub#2 also recognizes Parkin as observed with the inactive Parkin mutant C431S, where it labels two discrete bands, which likely correspond to pS65-Parkin (open arrowhead) and pS65-Parkin charged with Ub or pS65-Ub (filled arrowhead).
- HeLa cells stably expressing 3× FLAG-tagged Parkin C431S were incubated with 4 or 10 μM CCCP for 2 h, lysed and left untreated or treated with NaOH to strip off Ub from “Ub-charged” Parkin. Chemical cleavage of the Ub moiety from C431S by NaOH results in the collapse of the upper band (Ub-charged pS65-Parkin, filled arrowhead) into the lower band (pS65-Parkin, open arrowhead) as shown by FLAG and pS65-Ub#2 antibodies.
- HeLa cells were transiently transfected with FLAG-Parkin C431S or a C431S+S65A double mutant as an additional specificity control. Cells were challenged with CCCP, lysed, and left untreated or treated with NaOH. Of note, pS65-Ub#2 does only recognize a discrete band where Parkin WT but not the phospho-dead variant Parkin S65A was expressed.
To finally confirm the nature of the cellular signal obtained with the pS65-Ub antibodies, we performed immunoprecipitation (IP) of denatured lysates. Both pS65-Ub antibodies pulled down increasing amounts of Ub conjugates over time following CCCP treatment from HeLa cells overexpressing Parkin WT (Appendix Fig S2A). Yet, Parkin was not detectable from either pS65-Ub IP (not shown). Reciprocal anti-FLAG IP, this time under non-denaturing, but stringent RIPA buffer conditions, pulled down substantial amounts of phosphorylated poly-Ub conjugates with 3× FLAG-tagged Parkin WT, but not C431S mutant (Appendix Fig S2B). Using an anti-FLAG antibody, Parkin protein did not show a major shift into HMW species (Appendix Fig S2B). This suggests that Parkin strongly binds to pS65-Ub chains rather than it is overtly modified by them.
pS65-Ub is specific to mitochondrial damage in all cells
Given that our novel tools allowed detection of a mitochondrial stress-induced signal, we aimed to corroborate the mitochondrial co-localization of pS65-Ub with Parkin. Subcellular fractionations from HeLa cells stably expressing 3× FLAG-Parkin WT or C431S identified stabilized PINK1 protein and functional Parkin in mitochondrial samples upon CCCP treatment (Fig3A). In contrast to WT, Parkin C431S remained in the cytosol and was detected only with pS65-Ub#2 as the characteristic doublet band. In both cells, polymeric HMW pS65-Ub species accumulated in the mitochondrial fraction, at much higher levels in the presence of functional Parkin as seen with a total Ub antibody. However, monomeric pS65-Ub and dimeric pS65-Ub were exclusively detected in the cytoplasmic fraction and were much more abundant in the absence of functional Parkin.
Figure 3.

Levels of cellular pS65-Ub species are dependent on PINK1 kinase activity
- HeLa cells stably expressing 3× FLAG-tagged Parkin WT or C431S were treated for 2.5 h with CCCP and subjected to subcellular fractionation. Post-nuclear supernatant (PNS, as the total lysate) as well as mitochondrial and cytosolic fractions are shown. Functional Parkin showed enhanced levels of phosphorylated poly-Ub chains in the mitochondrial fraction and reduced levels of phosphorylated mono- and di-Ub levels in the cytoplasmic fraction (indicated by one or two black circles), compared to the inactive C431S mutant. Upon CCCP, PINK1 and Parkin WT accumulate in the mitochondrial fraction. Note: PINK1 has generally higher levels in the absence of functional Parkin and vice versa, consistent with the literature. pS65-Parkin (open arrowhead) and “Ub-charged” Parkin (filled arrowhead) are only detectable for C431S mutant, but not for WT. Both species are prominently detected by pS65-Ub#2, but not the Ub-specific pS65-Ub#1. Anti-VDAC1 and p38 MAPK antibodies served as controls for fractionation into mitochondria and cytosol, respectively.
- HeLa Parkin cells were transfected with control or PINK1 siRNA and treated with CCCP as indicated. Phosphorylated mono-, di-, and poly-Ub species were almost undetectable upon knockdown of PINK1. Loss of kinase activity also suppressed the amplification of total poly-Ub signal otherwise observed in the presence of functional Parkin. In agreement, ubiquitination/degradation of the control mitophagy substrate mitofusin 2 was not observed upon silencing of PINK1.
To further show that pS65-Ub is specific to mitochondrial damage, we challenged cells by ER (tunicamycin) or DNA (etoposide) stress (Appendix Fig S3A). Only CCCP treatment robustly induced the PINK1-/Parkin-dependent formation of phosphorylated poly-Ub chains, whereas unfolded protein response or DNA damage response did not. This increase of pS65-Ub upon mitochondrial damage was not cell type specific since it occurred in several non-neuronal and neuronal-like cells (Appendix Fig S3B).
pS65-Ub depends on PINK1 kinase activity and is reversible
Lysates from CCCP-treated HeLa cells that were incubated with alkaline phosphatase had greatly reduced anti-pS65-Ub signal, while total Ub levels remained unchanged (FigEV4A), showing phosphorylation dependence. The swift reversion of the pS65-Ub signal became apparent in CCCP washout experiments (FigEV4A). HeLa cells stably expressing 3× FLAG-Parkin WT were stressed for 2 h and then washed with fresh medium lacking the mitochondrial depolarizer to allow their recovery over time. pS65-Ub signals started to fade already after 30 min of washout and were followed by de-conjugation of Ub polymers as judged by the total Ub signal. While levels of pS65-Ub obviously declined shortly after CCCP washout, quantification of the de-phosphorylation events from WB experiments reached significance only after 3 h recovery (FigEV4C). Comparable kinetics upon CCCP washout were observed for de-phosphorylation of Parkin using the C431S mutation (Appendix Fig S4). In addition, we also measured (de-)phosphorylation of pS65-Ub over time using high content imaging (HCI) (FigEV4D). HeLa cells stably expressing native, untagged Parkin showed significant increases in pS65-Ub levels over 2 or 4 h CCCP treatment. Given higher sensitivity of imaging vs. WB, de-phosphorylation of pS65-Ub was significant already after 15 min of recovery from CCCP.
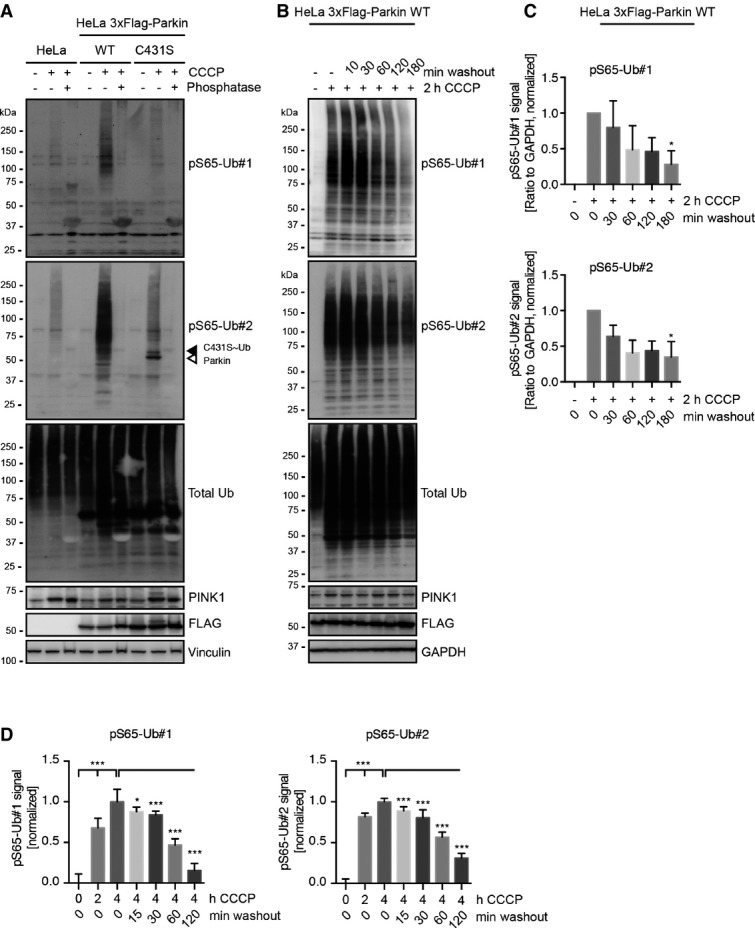
pS65-Ub is sensitive to phosphatase and is quickly de-phosphorylated during recovery from mitochondrial stress
- Parental HeLa cells or HeLa cells stably expressing 3× FLAG-tagged Parkin WT or C431S were treated for 2.5 h with CCCP, lysed and incubated with or without alkaline phosphatase. Treatment with phosphatase results in removal of phospho-moieties from poly-Ub chains and Parkin, which are no longer recognized by the anti-pS65-Ub antibodies.
- HeLa cells stably expressing 3× FLAG-tagged Parkin WT were treated with CCCP for 2 h. CCCP was washed out for different times in medium lacking the uncoupler. Over time of washout, pS65-Ub levels dropped considerably, as did total poly-Ub levels that were initially induced by CCCP.
- Quantification of pS65-Ub de-phosphorylation from WB experiments shown in (B). n = 5 independent experiments; shown are mean values ± SD (one-way ANOVA with Tukey post hoc; *P < 0.05) for each pS65-Ub antibody.
- (De-)phosphorylation of pS65-Ub signal upon CCCP treatment and subsequent recovery was quantified by high content imaging (HCI) 7, 9. HeLa Parkin WT cells were incubated with CCCP for 0, 2, or 4 h. Then, CCCP was washed out for the indicated times prior to fixation and staining with pS65-Ub antibodies and a nuclear counterstain. Mean intensity of pS65-Ub in a cytoplasmic ring around the nucleus was measured and normalized to untreated cells and cells that had been treated with CCCP for 4 h. n = 4 experiments; shown are mean values ± SEM (one-way ANOVA with Tukey post hoc; *P < 0.05; ***P < 0.0005).
Consistent with the PINK1-dependent phosphorylation of Ub, siRNA-mediated loss of the kinase activity almost entirely suppressed the induction of pS65-Ub upon CCCP treatment (Fig3B). This was observed for phosphorylated HMW as well as mono- and di-Ub species. Knockdown of PINK1 also suppressed formation of poly-Ub chains (see total Ub antibody) and degradation of the Parkin substrate mitofusin 2 (Mfn2). Next, we analyzed human dermal fibroblasts from healthy control subjects or PD patients harboring a homozygous PINK1 Q456X mutation 20. pS65-Ub was strongly induced by CCCP over time in control human fibroblasts and was absent in PINK1 mutant cells or in cells prior to mitochondrial damage (Fig4A; for valinomycin treatment, see Fig EV5A). In line with these findings, control cells showed a strong increase in pS65-Ub and almost exclusive co-localization with clustered mitochondria when analyzed by IF (Fig4B). However, in PINK1 mutant fibroblasts, pS65-Ub antibodies showed only faint background signal under both non-stress conditions and CCCP treatment. Similar to HeLa cells (Fig EV4B), primary human fibroblasts showed swift reversion of pS65-Ub signal in response upon CCCP washout (FigEV5B). PINK1 was again de-stabilized as early as 10 min after CCCP washout. pS65-Ub levels strongly declined within 30 min of recovery from mitochondrial stress reaching statistical significance after 1 h as measured from WB experiments (FigEV5C). De-phosphorylation of pS65-Ub was accompanied by re-establishment of mitofusin 1 protein levels shortly after.
Figure 4.

pS65-Ub signal is absent in PD patients’ fibroblasts with a PINK1 mutation
Primary skin fibroblasts derived from healthy controls or PD patients with a homozygous PINK1 Q456X mutation were incubated with CCCP as indicated.
- Cells were lysed and analyzed by WB with antibodies against pS65-Ub and total Ub. Mitofusin 1 serves as a control substrate that is ubiquitinated and degraded in response to CCCP in the presence of functional PINK1. For treatment of fibroblasts with valinomycin, see FigEV5A.
- Fibroblasts were fixed and stained with anti-TOM20 (mitochondria, red), anti-pS65-Ub (green), and Hoechst (nuclei, blue). The right panels represent enlarged views of the boxed areas. Upper set: anti-pS65-Ub#1 and lower set: anti-pS65-Ub#2. pS65-Ub signal was induced in CCCP-treated control fibroblasts with clear co-localization to clustered mitochondria. As expected, inducible pS65-Ub signal was absent in PD patients’ cells lacking PINK1 kinase. Scale bars, 10 μm.
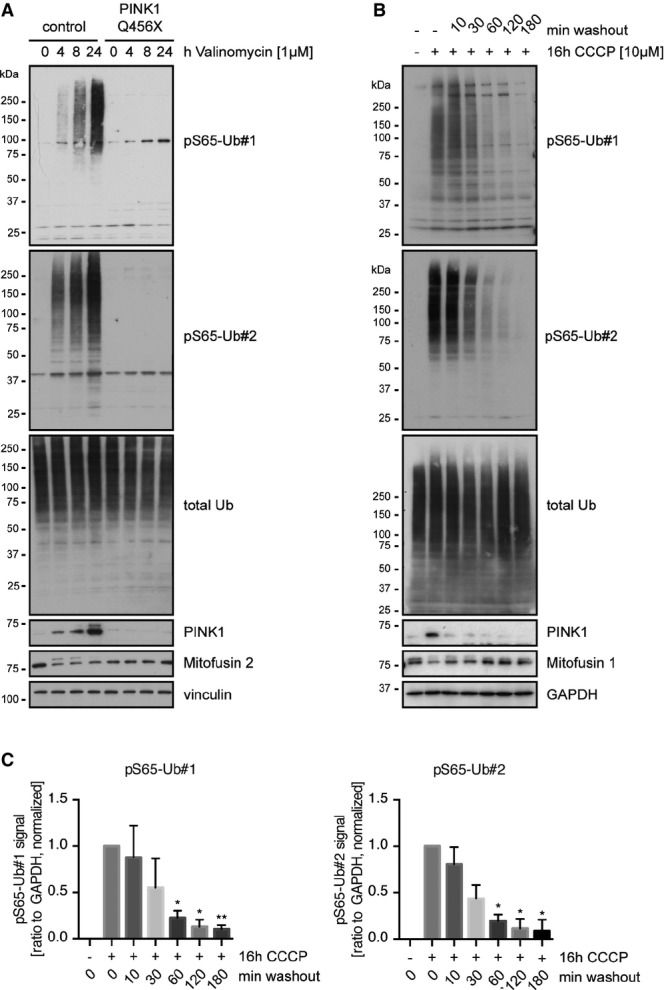
pS65-Ub is PINK1 dependent and reversible in human primary fibroblasts
- Primary human fibroblasts derived from healthy controls or PD patients harboring a PINK1 Q456X mutation were incubated with the alternative mitochondrial damaging agent valinomycin as indicated. Cells were lysed and analyzed by WB with antibodies against pS65-Ub and total Ub.
- Human control fibroblasts were left untreated or were treated with CCCP for 16 h and then subjected to washout using medium lacking CCCP for the times indicated. Within 30 min after medium change, pS65-Ub signal was drastically diminished.
- Quantification of pS65-Ub de-phosphorylation from WB experiments shown in (B). n = 3 independent experiments; shown are mean values ± SD (one-way ANOVA with Tukey post hoc; *P < 0.05; **P < 0.005) for each pS65-Ub antibody.
pS65-Ub in human patients’ fibroblast-derived and primary mouse neurons
To demonstrate the relevance of pS65-Ub in neuronal cells, primary human fibroblasts were directly converted into induced neurons (iNeurons) using short hairpin RNA targeting polypyrimidine-tract-binding protein (shPTBP) 21, 22. iNeurons differentiated from control or PINK1 Q456X fibroblasts were treated with valinomycin, and cell lysates were analyzed by WB. Similar to other cell types, mitochondrial stress robustly induced pS65-Ub in iNeurons from controls, concomitant with stabilization of PINK1 (Fig5A). Induction of pS65-Ub signals was not evident from PINK1 mutant iNeurons. IF analysis of iNeurons confirmed significantly elevated pS65-Ub levels upon valinomycin treatment specifically in control cells (Fig5B and C). Again, PINK1 mutant iNeurons showed only background pS65-Ub antibody signal that was not enhanced upon mitochondrial damage.
Figure 5.
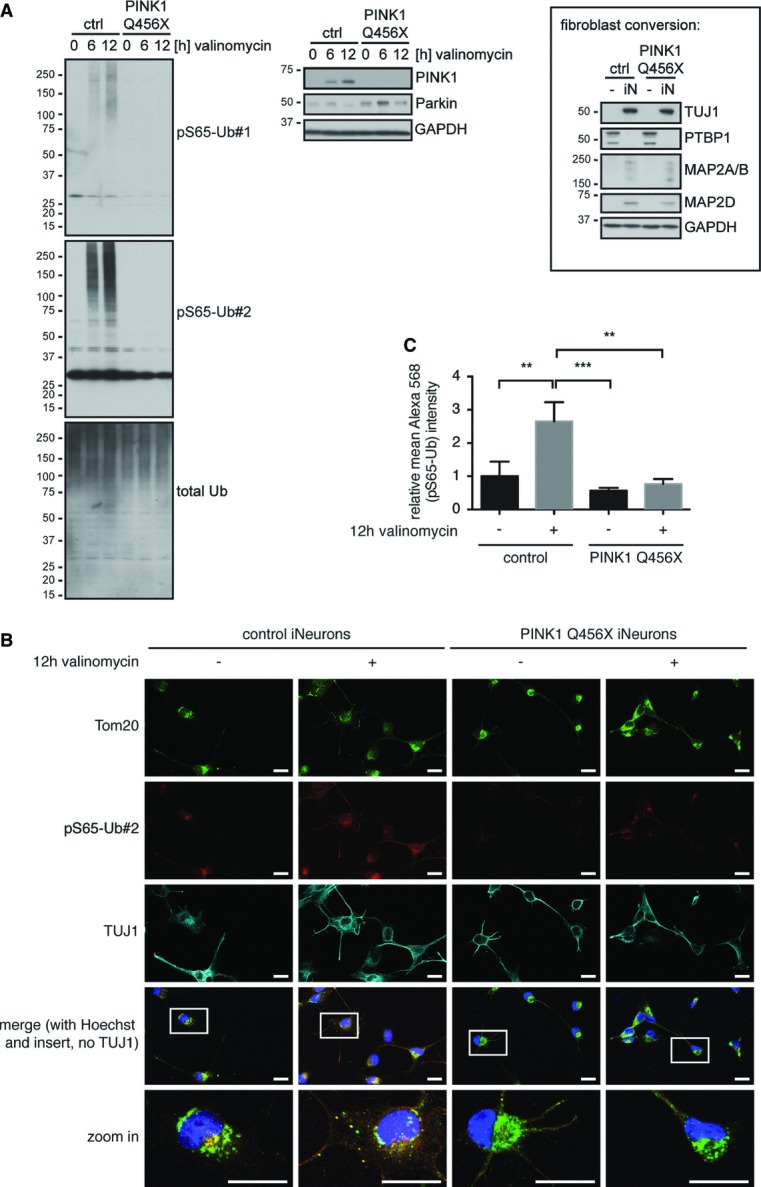
pS65-Ub is induced in human iNeurons from controls but not from PINK1 patients
Primary human fibroblasts were directly converted into iNeurons by viral transduction with shPTBP 21, 22.
- iNeurons were treated with valinomycin as indicated, lysed, and analyzed by WB. Inducible pS65-Ub was detectable in controls, but not in iNeurons from PINK1 patients. Loss of PTBP and appearance of several neuronal markers (TUJ1, MAP2A/B, and MAP2D) show the successful conversion of fibroblasts (-) to iNeurons (iN).
- iNeurons were treated with valinomycin, fixed and stained with anti-TOM20 (mitochondria, green), anti-pS65-Ub#2 (red), anti-TUJ1 (neuronal marker, cyan), and Hoechst (nuclei, blue). Scale bars, 20 μm. Bottom images represent enlarged view of the boxed areas.
- Quantification of pS65-Ub signal in iNeurons. TUJ1 staining was used to generate a mask outlining the total area of the iNeurons. Mean pS65-Ub signal intensity was measured in four images per condition from three independent experiments (one-way ANOVA with Tukey post hoc; **P < 0.005, ***P < 0.0005). Shown are mean values ± SD.
Since Ub and in particular S65 are highly conserved across all species, we tested the ability of the antibodies to recognize pS65-Ub in primary mouse neurons. IF of untreated vs. valinomycin-treated mouse embryonic cortical cultures demonstrated strongly induced levels of pS65-Ub upon mitochondrial damage (Fig6A). Indeed, valinomycin treatment significantly enhanced pS65-Ub levels in primary mouse neurons (see mask in Fig6A and quantification of intensity in Fig6B). Of note, most of the pS65-Ub signal was derived from cytosolic granules (see Fig6A and quantification of spots in Fig6C) that were largely co-localized with fragmented mitochondria. In summary, we show the induction of pS65-Ub upon mitochondrial damage in primary and neuronal cells and absence in cells from PINK1 mutation carriers.
Figure 6.

pS65-Ub is induced in mouse primary neurons upon mitochondrial damage
Mouse primary neuronal cultures were left untreated or challenged with valinomycin as indicated.
- Two representative images are shown for each condition. Cells were fixed and stained with anti-cyclophilin F (mitochondria, red), anti-pS65-Ub#2 (green), anti-TUJ1 (cyan), and Hoechst (blue). Scale bars, 10 μm.
- Quantification of the mean intensity of the pS65-Ub signal in (A).
- The pS65-Ub mask shown in black/white in (A) is a binary image that was generated to quantify the area of the pS65-Ub spots.
Data information: Analyses were performed on five images for each condition for three independent experiments (two-sided unpaired t-test; *P < 0.05). Shown are mean values ± SD.
pS65-Ub in human postmortem brain
To further demonstrate the pathophysiological relevance of pS65-Ub, we stained human postmortem brain sections of the substantia nigra (SN) from neurologically normal, young (< 40 years, n = 4), and old (> 60 years, n = 6) individuals as well as from PD patients (n = 6) (Fig7; for details on brain samples, see Appendix Table S1). Very few cytoplasmic granules were recognized in human SN from younger neurologically normal individuals. In contrast, SN from older healthy individuals showed much more cytoplasmic granules, here often within optically clear vacuoles (Fig7A). These granules were exclusively detectable with bleeds upon immunization and were absent from sections stained with pre-immune sera (Fig7B). Importantly, brain sections from a compound heterozygous PINK1 mutation carrier 23 showed only SN characteristic neuromelanin pigment, but were in large parts devoid of specific pS65-Ub signal (indicated by arrows, Fig7C).
Figure 7.

pS65-Ub is detected in human brain and increases with age and disease
Human postmortem brains were stained with pS65-Ub antibodies. Representative images of pS65-Ub#2 are shown. Similar results were obtained for both antibodies.
- A Representations of human substantia nigra (SN) sections from young and old brain stained with pS65-Ub antibodies. Only few cytoplasmic granules are present in a 37-year-old man, while many granules, often within optically clear vacuoles, are present in a 93-year-old man.
- B Test of (pre-)immune sera and affinity-purified pS65-Ub antibodies in human brains. Pre-immune serum and affinity-purified pS65-Ub antibody were tested on brain sections from a 67-year-old woman with ALS. Basal nucleus neurons are shown with only non-specific lipofuscin staining of the pre-immune serum, but many cytoplasmic granules detected with anti pS65-Ub. Scale bar, 6 μm.
- C SN sections from a PD patient with compound heterozygous mutations in PINK1 (c.1252_1488del and c.1488+1G>A) and age-matched control brain. Both PINK1 mutations should lead to in-frame loss of exon 7 (which encodes parts of the kinase domain) either by direct deletion or splicing-mediated exon skipping, respectively 23. Control brain: 37-year-old man (neurologically normal, type 1 diabetes, end-stage renal disease); note sparse cytoplasmic granules and granules in cell processes (arrows). PINK1 mutant brain: 39-year-old man; note only non-specific neuromelanin staining, characteristic for SN, is observed. The asterisk indicates an unstained Lewy body.
- D, E Double staining of pS65-Ub with different antibody markers by classical IHC and by IF labeling. (D) Representative image of many cytoplasmic granules adjacent to Lewy body (asterisk) in a 84-year-old man with PD. Double IF staining with anti-α synuclein corroborates non-overlap, but localization of pS65-Ub adjacent to Lewy neurites and bodies. (E) Upper panel: Double stain (cathepsin D for lysosomes, blue; pS65-Ub, brown) in 78-year-old man with PD (pS65-Ub granules next to Lewy body (asterisk)). IF corroborates partial co-localization of pS65-Ub with a lysosomal marker (cathepsin D). Middle panel: Double stain (M117 for mitochondria, blue; pS65-Ub, brown) in 78-year-old man with PD. IF corroborates partial co-localization of pS65-Ub with a mitochondrial marker (cyclophilin F). Lower panel: Double stain (total Ub, blue; pS65-Ub, brown) in a granular axonal spheroid in 78-year-old man with PD. IF corroborates partial co-localization of pS65-Ub with total Ub. Scale bar, 6 μm for IHC and 10 μm for IF images.
Interestingly, in PD patient brain, pS65-Ub signals were detected in granules adjacent to, but not within PD characteristic Lewy bodies (LBs) and Lewy neurites (Fig7D). Double-label IHC and IF of PD brains with pS65-Ub and different markers (Fig7E) showed a strong overlap with lysosomes (Fig7E, upper panel) and a partial co-localization with mitochondria (Fig7E, middle panel), as would be expected for a mitophagy label. While pS65-Ub staining also overlapped with total Ub signal (Fig7E, lower panel), both patterns were largely distinct from each other. Taken together, pS65-Ub-positive mitophagy granules accumulate with age and disease in human brain.
Discussion
Here, for the first time, we show that pS65-Ub is present in primary neurons under endogenous conditions and is inducible by and specific to mitochondrial damage. Using two novel rabbit antibodies, we confirmed that pS65-Ub is dependent on PINK1 kinase activity as it was absent from primary cells and brain tissue of PD patients with PINK1 mutations. We demonstrated the (patho-)physiological relevance for pS65-Ub as a specific label for mitochondrial stress, as it accumulates with organelle damage, age, and disease. Mitochondrial stress-induced phosphorylation of Ub was observed in all analyzed cell types including mouse primary neurons and human iNeurons. Monomeric pS65-Ub and dimeric pS65-Ub were identified in the cytoplasm, whereas most of the pS65-Ub signal was derived from higher molecular weight species conjugates to mitochondrial proteins. In human brain, we found pS65-Ub within cytoplasmic structures that partially co-localized with ubiquitin, mitochondria, and particularly with lysosomes. Though adjacently localized, LBs characteristic for PD were not stained with pS65-Ub antibodies. Rather pS65-Ub-positive granules were reminiscent of pathology in granulovacuolar degeneration. Enhanced, persistent levels of the pS65-Ub mitophagy label are indicative of increased mitochondrial damage and/or decreased flux through the degradation systems. However, the nature and composition of the pS65-Ub-positive structures must now be exactly determined from larger case series.
Together with PINK1, functional Parkin amplified the pS65-Ub signal in cells in response to mitochondrial damage as would be expected from the involved feed-forward and feedback loop mechanisms 24. Parkin itself is likely an (pS65-)Ub substrate to some extent, but strongly binds to phosphorylated poly-Ub chains, consistent with their role as the Parkin receptor 19. Further functional effects of pS65 on Ub structure, chain assembly, and hydrolysis have already been shown in vitro 18. Of note, the reversibility of both posttranslational modifications allows complex control over mitophagy and modulation over time. Upon washout of CCCP, pS65-Ub quickly disappeared and was followed by disassembly of earlier formed poly-Ub conjugates, consistent with the idea that phosphorylation of poly-Ub blocks their cleavage by DUBs 18. Several studies indicate a role of the mitochondrial phosphatase PGAM5 for PD. In mice, loss of PGAM5 was found to result in PD-like phenotypes, potentially by dysregulating PINK1 during mitophagy 25. In flies, however, PGAM5 deficiency rescued loss of PINK1, but not Parkin, suggesting a functional role downstream of PINK1 either between PINK1 and Parkin or independently of Parkin 26. Going forward it will be important to determine the exact function(s) of PGAM5 and if these are mediated through de-phosphorylation of the PINK1 substrates Ub and/or Parkin.
Both of our pS65-Ub antibodies recognized phosphorylated forms of Ub, but not unmodified, across several applications. One of the antibodies (pS65-Ub#2) additionally cross-reacted with pS65-Parkin, which was only detectable by using recombinant proteins or in cells expressing the inactive Parkin C431S mutant, but not WT. In vitro, PINK1 phosphorylated much more Ub moieties over shorter time, compared to Parkin as a substrate. The preference of PINK1 for Ub as a substrate, vast cellular levels of Ub and relatively low expression of Parkin, together with an amplification of the pS65-Ub signal by PINK1 and Parkin cooperation, highly suggests a much greater cellular abundance of pS65-Ub over pS65-Parkin. Consistently, the staining pattern of both pS65-Ub antibodies was identical in all applications and from all samples tested. Although not noticeable, we cannot formally exclude some minor contribution of pS65-Parkin cross-signal to the staining obtained with pS65-Ub#2. Regardless, Ub and Parkin are both PINK1 substrates that accumulate on damaged mitochondria and jointly act as important regulators of the mitochondrial quality control pathway.
Taken together, we have investigated the pathophysiological significance of pS65-Ub signaling in neurons and in disease pathogenesis. Our findings show that pS65-Ub antibodies can be used as a tool to monitor mitochondrial damage in various applications from cell models and brain tissue. Given that the epitope is conserved across all species, further analyses of pS65-Ub in different genetic or environmental animal models of PD are now warranted. Moreover, PINK1-/Parkin-dependent mitochondrial quality control has recently been linked to other human diseases besides PD 27. Since mitochondrial and autophagic dysfunctions emerge as a common leitmotif during aging and disease, pS65-Ub might have far-reaching implications for biomarker and therapeutic development.
Materials and Methods
pS65-Ub peptide and antibody generation
Polyclonal anti-phospho serine 65 ubiquitin (pS65-Ub) antibodies were generated by immunization of rabbits with two phosphorylated Ub peptides C-Ahx-YNIQKE[pS]TLHLVL-amide and Ac-YNIQKE[pS]TLHLVL-Ahx-C-amide (pSer65-Ub) corresponding to aa 59–71 of Ub (21st Century Biochemicals, Inc., Marlborough, MA, USA). Affinity purification (AP) and immunodepletion (ID) were performed using phosphorylated and non-phosphorylated peptides (C-Ahx-YNIQKESTLVLHL, Ser65-Ub), respectively. A phosphorylated Parkin-specific peptide (C-Ahx-DLDQQ[pS]IVHIVQ-amide pSer65-Parkin) corresponding to aa 60–71 or 61–73 was used to analyze cross-reactivity of the antibodies.
N-terminally biotinylated, synthetic pS65-Ub (Biotin-AHXMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKE[pS]TLHLVLRLRGG) was generated by Thinkpeptides (Oxford, UK).
Western blots
Protein was subjected to SDS–PAGE using 14, 16, or 8–16% Tris–glycine gels (Invitrogen, Carlsbad, CA, USA) and transferred onto polyvinylidene fluoride (PVDF) or nitrocellulose (NC) membranes (both Millipore, Billerica, MA, USA). Membranes were incubated with primary antibodies overnight at 4°C followed by HRP-conjugated secondary antibodies (1:10,000; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Bands were visualized with Immobilon Western Chemiluminescent HRP Substrate (Millipore) on Blue Devil Lite X-ray films (Genesee Scientific, San Diego, CA, USA) or on a Fuji LAS-3000 (Fujifilm Life Science USA, Stamford, CT, USA) system. For densitometric analysis, ImageJ software version 1.46R was used. Total ubiquitin blots were either prepared on NC that was microwaved 10 min in ddH2O or on PVDF washed 5 min in 4% paraformaldehyde (PFA) before incubation with primary antibody for antigen retrieval.
In vitro phosphorylation efficiency of Parkin and Ub was determined by sample separation on 14% (Ub) or 10% (Parkin) Tris–glycine gels containing 50 μM phos-tag acrylamide AAL-107 (Wako Chemicals, Richmond, VA, USA) and 100 μM ZnCl2. Prior transfer onto PVDF membranes, gels were incubated for 20 min in transfer buffer containing 0.5 mM EDTA and 0.01% of SDS and twice in the absence of EDTA.
Dot blots
pUb immunization, Ub immunodepletion, and pParkin peptides were dissolved in ddH2O and spotted onto 0.02-μm NC membranes that were dried overnight and incubated with primary antibodies for 2 h at room temperature followed by secondary antibody incubation. Dot blots were imaged on a FUJI LAS-3000 system.
Antibodies
Following antibodies were used for Western blot analysis (WB), dot blots (DBs), immunofluorescence (IF), or immunohistochemistry (IHC): mouse anti-cathepsin D (IHC/IF, 1:5,000, IM03, Millipore), rabbit anti-mouse anti-cyclophilin F (IF, 1:1,000, ab110324, Abcam, Cambridge, UK), mouse anti-FLAG (WB, 1:250,000, F3165, M2, Sigma), mouse anti-FLAG-HRP (WB, 1:200,000, A8592, M2, Sigma), mouse anti-GAPDH (WB, 1:150,000, H86504M, Meridian Life science, Memphis, TN, USA), mouse anti-human mitochondria (IHC, 1:15, M117, Leinco Technologies, Saint-Louis, MO, USA), rabbit anti-maltose-binding protein (MBP, WB, 1:800,000, E8030S, New England Biolabs Inc., Ipswich, MA, USA), rabbit anti-MAP2 (WB, 1:1,000, M3696, Sigma), mouse anti-mitofusin 1 (WB, 1:5,000, ab57602, Abcam), mouse anti-mitofusin 2 (WB, 1:5,000, ab56889, Abcam), mouse anti-Parkin (WB, 1:1,000–1:800,000, #4211, Prk8, Cell Signaling, Beverly, MA, USA), rabbit anti-PINK1 (WB, 1:500–2,000, BC100-494, Novus Biologicals, Littleton, CO, USA), goat anti-PTBP1 (WB, 1:500, ab5642, Abcam), mouse anti-α synuclein (IF, 1:50, 18-0215, LB509, Invitrogen), rabbit anti-TUJ1 (WB, 1:1,000, β3-tubulin, D71G9, CST), chicken anti-TUJ1 (IF, 1:500, β3-tubulin, AB9354, Millipore), rabbit anti-Ub K48-linked (WB, 1:50,000, #05-1307, Apu2, Millipore), rabbit anti-Ub K63-linked (WB, 1:50,000, #05-1308, Apu3, Millipore), mouse anti-Ub (WB, 1:2,000–1:500,000, IHC/IF, 1:1,000–30,000, MAB1510, ubi-1, Millipore), mouse anti-TOM20 (IF, 1:1,000, sc-17764, Santa Cruz Biotechnology, Dallas, TX, USA), mouse anti-vinculin (WB, 1:100,000–1:500,000, V9131, Sigma). pS65-Ub antibodies were used 1:1,000 for WB and DB, 1:200 for IF, and 1:100–1:1,000 for IHC. Streptavidin-HRP (Jackson ImmunoResearch 016-030-084) was diluted 1:5,000,000.
In vitro kinase assays
MBP-tagged insect (Tribolium castaneum) wild-type (WT, 66-0043-050) and kinase-dead (KD, D359A, 66-0044-050) PINK1 as well as untagged human Parkin (63-0048-025) were purchased from Ubiquigent (Dundee, UK). Untagged Ub chains (UC-240, UC-340), N-terminally biotinylated Ub (UB-560), and Ub chains (UCB-230, UCB-330) were from Boston Biochem (Cambridge, MA, USA). Reactions were performed in kinase buffer (20 mM Hepes pH 7.4, 10 mM DTT, 0.1 mM EGTA, 100 μM ATP and 10 mM MgCl2) and incubated at 30°C for the indicated times. For standard reactions, 1 μg PINK1 protein was used per μg of substrate. To achieve complete phosphorylation of equimolar amounts of ubiquitin and Parkin, reactions containing 1 μg of MBP-PINK1 per 1 μg of Parkin or 0.17 μg of N-terminally biotinylated Ub (both 19 pmol) were incubated for 2 days at 30°C. All reactions were stopped by addition of SDS sample buffer and denaturing at 95°C. 15–300 ng was loaded per lane.
Cell culture
All cells were grown at 37°C under humidified conditions in 5% CO2/air. Human epithelial cancer (HeLa), human neuroglioma H4, and human neuroblastoma BE(2)-M17 (M17) cells were obtained from the ATCC (American Type Culture Collection, Manassas, VA, USA) and human embryonic kidney (Hek293E) cells from Invitrogen. Parental HeLa cells or clonal cells stably expressing untagged Parkin (HeLa Parkin), EGFP-Myc Parkin (HeLa GFP-Parkin), or 3× FLAG-Parkin WT or C431S 9 were grown in Dulbecco’s modified Eagle medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Gemini Bio-Products, West Sacramento, CA, USA). Human neuroglioma and M17 cells were grown in Opti-MEM (Invitrogen) supplemented with 10% FBS. Control fibroblasts were obtained from Cell Applications (San Diego, CA, USA). PINK1 Q456X fibroblasts have been described previously 20. Fibroblasts were grown in DMEM supplemented with 10% FBS, 1% PenStrep, and 1% non-essential amino acids (both Invitrogen). Fibroblasts were collected under approved Mayo Clinic IRB protocols.
Chemical treatments, DNA, and siRNA transfection of cells
Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) and tunicamycin were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA) and valinomycin and etoposide from Axxora (Axxora, Farmingdale, NY, USA). If not stated otherwise, CCCP and valinomycin were used in a final concentration of 10 or 1 μM, respectively. Washout of CCCP was performed by three washes in 1× PBS. Cells were incubated further in medium lacking CCCP.
siRNA transfections were performed on two consecutive days using HiPerfect with 20 nM control (all stars negative control) or PINK1-specific siRNA (5′-GACGCTGTTCCTCGTTATGAA-3′, both from Qiagen, Valencia, CA, USA).
HeLa cells were transfected with either FLAG-Parkin C431S or the double mutant C431S/S65A using Lipofectamine 2000 according to manufacturer’s protocol, and medium was replaced 4 h later.
Cell lysates and subcellular fractionation
For regular Western blot analysis, cells were lysed in RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS) plus Complete protease and PhosSTOP phosphatase inhibitor (Roche Applied Science, Penzberg, DE). Protein concentration was determined by the use of bicinchoninic acid (Pierce Biotechnology, Rockford, IL, USA). For phosphatase treatment, lysates prepared in RIPA buffer without PhosSTOP and were incubated in the presence of FastAP thermosensitive alkaline phosphatase (1U/μg protein, Thermo Scientific, Waltham, MA, USA) in 1× enzyme buffer for 24 h at 37°C.
For Oxyester analysis, cells were harvested in preheated (95°C) SDS lysis buffer (50 mM Tris pH 7.6, 150 mM NaCl, 1% SDS). Lysates were homogenized by 10 strokes through a 20-G needle. To verify the band shift by oxyester formation, aliquots of lysates were treated with or without NaOH (final concentration: 100 mM) for 1 h at 37°C before loading onto SDS gels.
For subcellular fractionation, cells were harvested in fractionation buffer containing 20 mM HEPES pH 7.4, 1 mM EDTA, 250 mM sucrose, and cocktail of protease and phosphatase inhibitors (Roche). After twenty strokes through a 22-G needle, nuclei and cell debris were removed by centrifugation at 1,500 g for 5 min. The post-nuclear supernatant was transferred into a new tube and centrifuged at 12,000 g for 10 min. The mitochondria-enriched pellet was resuspended in cell fractionation buffer. Cytoplasmic fractions were isolated as supernatant after ultracentrifugation at 100,000 g for 30 min.
Immunoprecipitation
For pS65-Ub IP, cells were lysed in SDS lysis buffer. DNA was sheared by 22-G needle, and samples were boiled at 95°C. Lysates were 10× diluted with NP-40-containing buffer (50 mM Tris pH 7.6, 150 mM NaCl, and 0.5% NP-40). Five hundred microgram of total cell lysate was incubated overnight at 4°C with 5 μg of control rabbit IgG (Jackson ImmunoReseach) or pS65-Ub antibodies. Formed immunocomplex was captured by addition of 40 μl Protein G Agarose Fast Flow (Millipore 16-266) for 2 h at 4°C and eluted with 2× SDS sample buffer.
For FLAG IP, HeLa cells stably expressing 3× FLAG-Parkin WT or C431S were left untreated or treated with 10 μM CCCP for 2 or 4 h and lysed in RIPA buffer plus protease and phosphatase inhibitors. One thousand microgram protein lysate was immunoprecipitated using 20 μl of anti-FLAG EZview beads (Sigma F2426) at 4°C for 4 h. Beads were washed three times in cold RIPA buffer and eluted with 150 μl 2× SDS sample buffer and boiled at 95°C. Ten microgram of input cell lysates and 15 μl of immunoprecipitates were analyzed by Western blot.
Differentiation of iNeurons
iNeurons were generated as described previously with modifications 21, 22. Briefly, human PTBP shRNA (shPTBP) cloned into lentiviral vector pLKO.1 (Sigma), a kind gift from Dr. Fu (University of California, San Diego), was packaged in HEK293FT cells using Virapower (Invitrogen). Viral particles were collected 48 and 72 h post-transfection. Fibroblasts were seeded on a poly-D-lysine (PDL)-coated surface and transduced with lentiviral particles containing shPTBP for 16–18 h in the presence of 5 μg/ml Polybrene (Sigma). Transduced cells were selected with 1.5 μg/ml puromycin (Invitrogen) starting 2 days after transduction. Two days later, 10 ng/ml basic fibroblast growth factor (bFGF, GenScript Z02734, Piscataway, NJ, USA) was added to the fibroblast media and cultivation continued for additional two days. From day 6 on, cells were maintained in differentiation medium containing DMEM/F12 (Invitrogen), 5% FBS (further reduced to 2% after 2 days), 25 μg/ml insulin, 100 nM putrescine, 50 μg/ml transferrin, 30 nM sodium selenite (all from Sigma), and 15 ng/ml bFGF. After 4 days, B27 supplement without antioxidants (Invitrogen) and 10 ng/ml each of brain-derived neurotrophic factor (BDNF, R&D Systems 248-BD-025, Minneapolis, MN, USA), glial cell-derived neurotrophic factor (GDNF, R&D Systems 212-GD-050), neurotensin-3 (NT3, Peprotech AF-450-03, Rocky Hill, NJ, USA), and ciliary neurotrophic factor (CNTF, Peprotech 450-13) were added to the differentiation medium. The cells were used for experiments 2–4 days later. Neuronal differentiation was confirmed by expression of the neuronal marker TUJ1 and MAP2.
Preparation of mouse cortical cultures
Primary cortical cultures were prepared from embryonic day 18 (E18) C57BL6/J mouse pups as previously described 28 with slight modifications. Briefly, fetal cortices were dissected in Hibernate A media without calcium (BrainBits, Springfield, IL, USA) and subsequently transferred into 10 ml of growth media consisting of Neurobasal A media supplemented with antioxidant-free B27, GMAX, gentamicin, and bFGF (all from Invitrogen). Cells were dissociated in growth media and strained through a 40-μm Falcon cell strainer (Becton Dickinson Biosciences, San Jose, CA, USA) to remove cellular debris and to obtain a uniform, single-cell suspension. For IF studies, cells were plated onto PDL-coated coverslips in 24-well tissue culture plates at a density of 3 × 104 cells/well. Media was partially replaced every 2–3 days until experiments were performed at 7 days in vitro. All procedures using animals were approved by the Institutional Animal Care and Use Committees at Mayo Clinic. Cells were mock-treated or treated with 1 μM valinomycin in the presence of 50 μM z-VAD-FMK (Apex Bio, Houston, TX, USA) as previously described 29.
Immunofluorescence staining of cells
Cells were grown on glass coverslips coated with PDL (Sigma P0899). Cells were fixed with 4% (w/v) PFA and permeabilized with 1% Triton X-100. Cells were incubated with primary antibodies followed by incubation with secondary antibodies (anti-mouse IgG Alexa Fluor 488, 568, or 647 and anti-rabbit Alexa Fluor 488 or 568 and anti-chicken Alexa Fluor 647—all Invitrogen) diluted 1:1,000. Nuclei were stained with Hoechst 33342 (Invitrogen) diluted 1:5,000. Coverslips were mounted onto microscope slides using fluorescent mounting medium (Dako, Carpinteria, CA, USA). Confocal fluorescence images were taken with an AxioObserver microscope equipped with an ApoTome Imaging System or a laser scanning microscope 510 META equipped with 405-, 488-, 543-, and 633-nm lasers (Zeiss, Jena, DE).
Immunohistochemistry (IHC) and IF of brain tissue
IHC has been described before 30. In brief, sections from paraffin-embedded tissue were cut at a thickness of 5 μm, mounted on onto positively charged glass slides, and allowed to dry overnight in a 60°C oven. Following deparaffinization and rehydration, target retrieval was performed by steaming the sections for 30 min in deionized water. Endogenous peroxidase was blocked for 5 min with 0.03% hydrogen peroxide. Sections were then treated with 5% normal goat serum for 20 min, followed by incubation in the primary antibodies for 45 min at room temperature. Immunostaining was performed on the Dako Autostainer using Envision Plus or Envision G/2 Doublestain kit (Dako, Carpinteria, CA). Horseradish peroxidase (HRP) labeling of immune-depleted pS65-Ub was visualized using 3,3′-diaminobenzidine (DAB+) as the chromogen. Alkaline phosphatase labeling of mouse monoclonal antibodies M117, Ubi-1, and cathepsin D was visualized using Vector Blue Alkaline Phosphatase Substrate kit III (Vector Laboratories, Burlingame, CA). After dehydration, sections were coverslipped with or without hematoxylin counterstaining. For IF, sections were blocked with serum-free protein block and incubated overnight with primary antibodies in background-reducing antibody diluent (both Dako) after deparaffinization and steaming. Sections were washed and incubated with secondary antibodies plus Hoechst for 2 h. Signal was quenched in 3% Sudan black in 70% MeOH solution for 2 min before sections were washed and coverslipped.
High content imaging (HCI)
HeLa Parkin cells were seeded with 7,000 cells/100 μl per well in 96-well imaging plates (BD Biosciences). Twenty-four hours after plating, cells were treated with 10 μM CCCP for 0, 2, or 4 h. For CCCP washout, cells were washed three times with media lacking the uncoupler and further incubated for the indicated times. Cells were then fixed for 20 min in 4% PFA and stained with pS65-Ub antibodies and Hoechst 33342 (1:5,000, Invitrogen). Plates were imaged on a BD Pathway 855 system (BD Biosciences) with a 20× objective using a 3 × 3 montage (no gaps) with laser autofocus every second frame. Raw images were processed using the built-in AttoVision V1.6 software. Regions of interest (ROIs) was defined as cytoplasm using the built-in “RING 2 outputs” segmentation for the Hoechst channel after applying a shading algorithm. Resulting mean intensity values were normalized.
Image quantification and statistical analysis
Images were analyzed using ImageJ (1.48v). The mean intensity of pS65-Ub signal was analyzed over the whole image (primary neurons) or using a binary mask created from the corresponding TUJ signal (iNeurons). For area analysis of pS65-Ub spots, ImageJ built-in “analyze particles” was used after applying a uniform threshold over all images. Spots with < 4 pixels were excluded from the analysis. For quantification of pS65-Ub signal on WB, the mean value of the entire lane (25–250 kDa) was used and normalized to the corresponding loading control (GAPDH). Statistical analysis of three to five independent experiments was performed with one-way ANOVA or two-sided t-test, as indicated.
Modeling ubiquitin structures with interpolation and minimization
The sequence of human ubiquitin proteins (PDB codes: 2KDF and 3H7S) 31, 32, a cellular protein encoded by the polyubiquitin-C gene, was taken from the NCBI Reference Sequence NP_066289.3. The modeling was built using the above PDB structures and cleaned using protein preparation wizard within Maestro console of Schrodinger 33, 34. The connectors between K48- and K63-linked chains were built using Maestro, and then, superposition was applied on duplicate structures to generate transformations in X, Y, Z Cartesian coordinate space for rotation and translation to generate octa-mers lysine-linked chains. Structures were inspected using Yasara program WHAT-IF 35 and have valid conformation consistent with good phi-psi space 36, 37, 38, which includes packing (1D and 3D), dihedrals, phi-psi, Backbone, and Packing3 (WHAT-IF implementation), plus force field considerations. The superposition and subsequent minimization of each structure was completed to generate didactic models for various linked forms of pS65-Ub protein. The final model was subjected to energy optimization with PR conjugate gradient with an R-dependent dielectric. Model manipulation was done with Maestro (Macromodel, version 9.8, Schrödinger, LLC, New York, NY, 2010). Previous models have been successfully generated using these techniques 7, 28, 39.
Modifications modeling
Modifications for amino acids, such as phosphorylation of serine 65 (pSer65), was achieved using the 2D sketcher and importing as an new amino acid type which can be parameterized using the Schrödinger force fields. Additionally, ubiquitin linkages were also obtainable in this manner. Using OPLS2005 or YASARA2, one can parameterize these modification and import into existing molecular dynamics integration engines, as the parameters for the modification are well documented 34, 40, 41, 42, 43.
Acknowledgments
We thank Dr. Fu for the lentiviral shPTBP plasmid. We are grateful to the patients and their families for providing tissues. This study was supported by NIH/NINDS R01NS085070, the Michael J. Fox Foundation for Parkinson’s Research and the Foundation for Mitochondrial Medicine, Mayo Clinic Foundation and the Center for Individualized Medicine, the Marriott Family Foundation, and a Gerstner Family Career Development Award to WS. Mayo Clinic Florida is a Morris K. Udall Parkinson’s Disease Research Center of Excellence (NINDS P50 #NS072187; DWD, ZKW, and OAR). LC, VLD, and TMD are supported by grants from the NIH/NINDS Udall Center P50 NS38377, MDSCRF 2007-MSCRFI-0420-00, 2009-MSCRFII-0125-00, MDSCRF 2012-MSCRFII-0268-00, MDSCRF 2013-MSCRFII-0105-00, and the JPB Foundation. TMD and VLD acknowledge the joint participation by the Adrienne Helis Malvin and the Diana Helis Henry Medical Research Foundations through their direct engagement in the continuous active conduct of medical research in conjunction with the Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation’s Parkinson’s Disease Programs, M-1 and H-2014. TMD is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. Further support: The Uehara Memorial Foundation (MA), Mayo Clinic Center for Regenerative Medicine (POB), ALS Association (POB), NIH/NINDS R01NS078086 (OAR).
Author contributions
FCF and WS conceived the study. FCF, DWD, and WS designed the experiments. FCF, MA, RH, ARH, ELML, TRC, and MCC performed experiments. FCF, DWD, and WS analyzed the data. FCF and WS wrote the manuscript. POB, JNS, ZKW, OAR, IF, JMA, OLB, JS, LC, VLD, and TMD provided reagents and discussion.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Appendix
Expanded View Figures PDF
Review Process File
References
- Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014;37:315–324. doi: 10.1016/j.tins.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The Roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, Suzuki N, Uchiyama S, Tanaka K, Matsuda N. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem. 2013;288:22019–22032. doi: 10.1074/jbc.M113.467530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2:1002. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield TR, Fiesel FC, Moussaud-Lamodiere EL, Dourado DF, Flores SC, Springer W. Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLoS Comput Biol. 2014;10:e1003935. doi: 10.1371/journal.pcbi.1003935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K, Inoshita T, Hattori N, Imai Y. PINK1-mediated phosphorylation of Parkin boosts Parkin activity in Drosophila. PLoS Genet. 2014b;10:e1004391. doi: 10.1371/journal.pgen.1004391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiesel FC, Moussaud-Lamodiere EL, Ando M, Springer W. A specific subset of E2 ubiquitin-conjugating enzymes regulate Parkin activation and mitophagy differently. J Cell Sci. 2014;127:3488–3504. doi: 10.1242/jcs.147520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kelly V, Johnson C, Baillie C, Hastie CJ, Peggie M, Macartney T, Woodroof HI, Alessi DR, Pedrioli PG, et al. Phosphorylation of Parkin at Serine65 is essential for activation: elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open Biol. 2014a;4:130213. doi: 10.1098/rsob.130213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I, Bremm A. DUBs counteract parkin for efficient mitophagy. EMBO J. 2014;33:2442–2443. doi: 10.15252/embj.201490101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014b;460:127–139. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56:360–375. doi: 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K, Arano T, Matsumoto G, Inoshita T, Yoshida S, Ishihama Y, Ryu KY, Nukina N, Hattori N, Imai Y. Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS Genet. 2014a;10:e1004861. doi: 10.1371/journal.pgen.1004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Lee S, Peng Y, Bunker E, Giaime E, Shen J, Zhou Z, Liu X. PINK1 triggers autocatalytic activation of Parkin to specify cell fate decisions. Curr Biol. 2014;24:1854–1865. doi: 10.1016/j.cub.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Swatek KN, Wagstaff JL, Gladkova C, Pruneda JN, Michel MA, Gersch M, Johnson CM, Freund SM, Komander D. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 2014;34:307–325. doi: 10.15252/embj.201489847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, Matsuda N. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol. 2015;209:111–128. doi: 10.1083/jcb.201410050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuda J, Jasinska-Myga B, Boczarska-Jedynak M, Opala G, Fiesel FC, Moussaud-Lamodiere EL, Scarffe LA, Dawson VL, Ross OA, Springer W, et al. Early-onset Parkinson’s disease due to PINK1 p. Q456X mutation–clinical and functional study. Parkinsonism Relat Disord. 2014;20:1274–1278. doi: 10.1016/j.parkreldis.2014.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, Fostvedt E, Jansen-West K, Belzil VV, Desaro P, et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 2014;83:1043–1050. doi: 10.1016/j.neuron.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, Wang G, Wu Q, Wei C, Bi Y, et al. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell. 2013;152:82–96. doi: 10.1016/j.cell.2012.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, Pastor MA, Marrero C, Isla C, Herrera-Henriquez J, et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain. 2010;133:1128–1142. doi: 10.1093/brain/awq051. [DOI] [PubMed] [Google Scholar]
- Riley BE, Olzmann JA. A polyubiquitin chain reaction: parkin recruitment to damaged mitochondria. PLoS Genet. 2015;11:e1004952. doi: 10.1371/journal.pgen.1004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Karuppagounder SS, Springer DA, Allen MD, Zheng L, Chao B, Zhang Y, Dawson VL, Dawson TM, Lenardo M. Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson’s-like movement disorder. Nat Commun. 2014;5:4930. doi: 10.1038/ncomms5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kanao T, Sawada T, Kobayashi Y, Moriwaki Y, Ishida Y, Takeda K, Ichijo H, Lu B, Takahashi R. The loss of PGAM5 suppresses the mitochondrial degeneration caused by inactivation of PINK1 in Drosophila. PLoS Genet. 2010;6:e1001229. doi: 10.1371/journal.pgen.1001229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Tavernarakis N. Mitophagy in neurodegeneration and aging. Front Genet. 2012;3:297. doi: 10.3389/fgene.2012.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Caulfield T, Xu YF, Gendron TF, Hubbard J, Stetler C, Sasaguri H, Whitelaw EC, Cai S, Lee WC, et al. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum Mol Genet. 2013;22:3112–3122. doi: 10.1093/hmg/ddt166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Zakaria HM, Simone A, Sheng ZH. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol. 2012;22:545–552. doi: 10.1016/j.cub.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WL, Castanedes-Casey M, Dickson DW. Transactivation response DNA-binding protein 43 microvasculopathy in frontotemporal degeneration and familial Lewy body disease. J Neuropathol Exp Neurol. 2009;68:1167–1176. doi: 10.1097/NEN.0b013e3181baacec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks SD, Grasty KC, Hernandez-Cuebas L, Loll PJ. Crystal structures of Lys-63-linked tri- and di-ubiquitin reveal a highly extended chain architecture. Proteins. 2009;77:753–759. doi: 10.1002/prot.22568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wang Q, Ehlinger A, Randles L, Lary JW, Kang Y, Haririnia A, Storaska AJ, Cole JL, Fushman D, et al. Structure of the s5a:k48-linked diubiquitin complex and its interactions with rpn13. Mol Cell. 2009;35:280–290. doi: 10.1016/j.molcel.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- Krieger E, Joo K, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins. 2009;77(Suppl 9):114–122. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Joosten RP, Salzemann J, Bloch V, Stockinger H, Berglund AC, Blanchet C, Bongcam-Rudloff E, Combet C, Da Costa AL, Deleage G, et al. PDB_REDO: automated re-refinement of X-ray structure models in the PDB. J Appl Crystallogr. 2009;42:376–384. doi: 10.1107/S0021889809008784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E, Nabuurs SB, Vriend G. Homology modeling. Methods Biochem Anal. 2003;44:509–523. doi: 10.1002/0471721204.ch25. [DOI] [PubMed] [Google Scholar]
- Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. 29. [DOI] [PubMed] [Google Scholar]
- Caulfield TR, Devkota B, Rollins GC. Examinations of tRNA Range of Motion Using Simulations of Cryo-EM Microscopy and X-Ray Data. J Biophys. 2011;2011:219515. doi: 10.1155/2011/219515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E, Dunbrack RL, Jr, Hooft RW, Krieger B. Assignment of protonation states in proteins and ligands: combining pKa prediction with hydrogen bonding network optimization. Methods Mol Biol. 2012;819:405–421. doi: 10.1007/978-1-61779-465-0_25. [DOI] [PubMed] [Google Scholar]
- Schrödinger. Maestro. New York, NY: Schrödinger, LLC; 2010. version 9.1. [Google Scholar]
- Schrödinger. Suite 2012. New York, NY: Schrödinger, LLC; 2012. [Google Scholar]
- Schrödinger. Biologics Suite: BioLuminate. New York, NY: Schrödinger, LLC; 2013. version 1.1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File