

Abstract
Ubiquitylation is an essential post-translational modification that regulates numerous cellular processes, most notably protein degradation. Ubiquitin can itself be phosphorylated at nearly every serine, threonine, and tyrosine residue. However, the effect of this modification on ubiquitin function is largely unknown. Here, we characterized the effects of phosphorylation of yeast ubiquitin at serine 65 in vivo and in vitro. We find this post-translational modification to be regulated under oxidative stress, occurring concomitantly with the restructuring of the ubiquitin landscape into a highly polymeric state. Phosphomimetic mutation of S65 recapitulates the oxidative stress phenotype, causing a dramatic accumulation of ubiquitylated proteins and a proteome-wide reduction of protein turnover rates. Importantly, this mutation impacts ubiquitin chain disassembly, chain linkage distribution, ubiquitin interactions, and substrate targeting. These results demonstrate that phosphorylation is an additional mode of ubiquitin regulation with broad implications in cellular physiology.
Keywords: oxidative stress, phosphorylation, protein turnover, proteomics, ubiquitin
Introduction
Ubiquitylation is a highly conserved protein post-translational modification (PTM) that regulates a wide array of cellular processes. Protein ubiquitylation is controlled by a multi-enzyme cascade of ubiquitin activation by an E1 enzyme, ubiquitin conjugation by an E2 enzyme, and transfer of ubiquitin to a lysine residue on the protein substrate by an E3 ligase associated with the E2. In addition to monomeric addition of ubiquitin, lysine residues on ubiquitin itself can serve as sites to form polymeric ubiquitin chains with distinct topologies and functions 1, 2. Different chain linkages have been associated with specific cellular processes, the most common of which is lysine 48 (K48)-linked branching resulting in proteasome-mediated degradation. Ubiquitylation can be reversed by deubiquitinating enzymes (DUBs) that both remove ubiquitin from substrates and disassemble ubiquitin polymer chains 3. Together, this suite of enzymes and chain structures results in a complex and highly dynamic ubiquitin signaling network 1.
Ubiquitin and other ubiquitin-like (UBL) proteins can themselves be modified by other PTMs, adding another layer of complexity that cannot occur on many other modifications. Previous work has documented that nearly every S/T/Y residue in ubiquitin can be phosphorylated 4, 5, 6, 7, 8, 9. Many of these phosphorylation events are conserved across the vast evolutionary distance from yeast to humans. Despite this knowledge, the functional consequences of ubiquitin’s phosphorylation sites remain largely unknown.
Only recently has a functional role for ubiquitin phosphorylation been described in human cells, in which S65 phosphorylation by PINK1 kinase contributes to the activation of the Parkin E3 ligase 10, 11, using an allosteric mechanism that is still being refined 12, 13. Interestingly, S65 phosphorylation also exists in yeast 5, despite the lack of orthologs to PINK1 and Parkin. Considering the lack of these two enzymes in lower eukaryotes and the central role of ubiquitin in protein processing, we hypothesized that ubiquitin S65 phosphorylation could have broader regulatory implications, extending well beyond activating an individual E3 ligase.
In fact, recent work has demonstrated that ubiquitin S65 phosphorylation affects its reactivity with a number of E2s and DUBs in vitro 13, findings that we independently made in this study. However, how S65 phosphorylation is regulated in vivo and what are the immediate molecular and phenotypic effects are open questions. Here, we have utilized the yeast model organism, phosphomimetic and phosphoinhibitory mutants, and quantitative proteomics approaches to facilitate the discovery of novel functional roles and substrates for ubiquitin S65 phosphorylation in vivo.
Results
Phosphorylation sites on ubiquitin
Since the first proteomic study of protein ubiquitylation in yeast reported phosphorylation on S57 4, large-scale phosphoproteomics experiments have documented additional sites of phosphorylation on human ubiquitin on T7 and T12 6, Y59 8, and S65 7 (Fig1A). We recently took a deep look at the crosstalk between phosphorylation and ubiquitylation, developing novel biochemical enrichment approaches that allowed us to confidently identify phosphorylation of yeast ubiquitin at T12, T14, S19, T22, T55, S57, S65, and T66 (S65 is shown in Fig1B) 5, whereas T7 and T9 showed ambiguous site localization (Fig1A). Combined, and assuming homology, these reports demonstrate that at least 77–82% (10 of 13 in yeast and 9 of 11 in human) of the S/T/Y residues on ubiquitin are phosphorylated. Based on the prevalence of phosphorylation on ubiquitin, we hypothesized that phosphorylation of ubiquitin represents a previously unappreciated mechanism of ubiquitin regulation.
Figure 1.
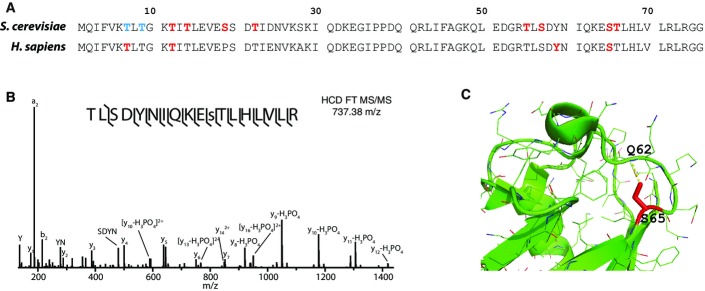
Phosphorylation sites on ubiquitin
- Sequence alignment of yeast and human ubiquitin showing identified phosphorylation sites (red: localized sites, blue: ambiguous localization).
- Tandem mass spectra displaying phosphorylation site localization to S65 (denoted by lower case “s”). This spectrum is the result of peptide fragmentation via HCD and represents five averaged scans detected with the Orbitrap mass analyzer.
- Zoomed in view of the α2-β5 loop of ubiquitin (PDB ID: 2ZCC) with S65 highlighted in red, and hydrogen bond and distance between S65 hydroxyl and Q62 amide are shown by yellow dashed line.
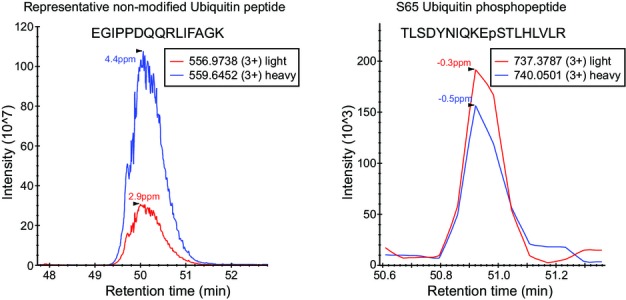
We selected S65 phosphorylation for further interrogation based on functional and structural criteria. Ubiquitin structure suggests this site can have effects on local structure and dynamics of the α2-β5 loop, particularly in light of the hydrogen bond between S65 hydroxyl and Q62 amide (Fig1C), with recent structural studies of S65 phosphoubiquitin confirming disruption of this hydrogen bond upon phosphorylation 13. We used AQUA mass spectrometry to measure the stoichiometry of this phosphorylation site in yeast cultures under exponential growth and determined it to be below 0.5% over total ubiquitin (Fig EV1).
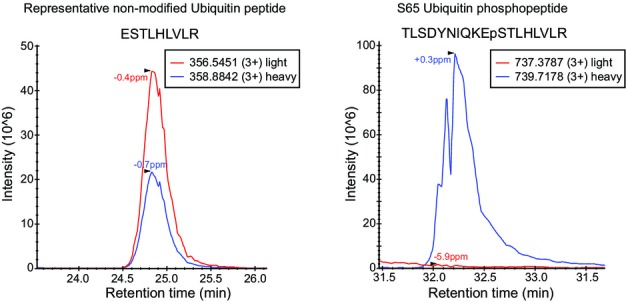
Phosphorylation stoichiometry of ubiquitin Ser65
Extracted ion chromatograms of a representative non-modified ubiquitin peptide (left) and S65 ubiquitin phosphopeptide (right). Red lines correspond to endogenous peptides, and blue lines are heavy-labeled AQUA peptides, which served as internal standards for quantification. S65 AQUA phosphopeptide was spiked in at 1% of the non-modified ubiquitin peptides prior to IMAC enrichment.
In vitro analysis of S65E ubiquitin phosphomimetic
The dynamic nature of protein phosphorylation presents challenges in the in vivo characterization of phosphorylation function. Thus, we sought to use a phosphomimetic mutant (S65E) to control the phosphorylation state of ubiquitin and permit robust characterization of its in vivo regulatory effects. Recent work has demonstrated that phosphomimetic mutation can recapitulate ubiquitin phosphorylation binding and activation of the Parkin E3 ligase 10, 11, 12. To further assess the degree of phosphomimicry for the S65E mutant, we carried out a series of in vitro reactions, examined the interaction with a few representative components of the ubiquitin machinery, and compared the results with the in vitro reactivity of S65 phosphoubiquitin 13.
We first evaluated the ability of ubiquitin mutants to load onto an E1 enzyme and found that S65E loaded onto the E1 enzyme with moderately reduced conjugation relative to that of WT ubiquitin; however, this reduced level was equivalent to that of the S65A mutant (Fig2A). Next, we introduced E2 ubiquitin-conjugating enzymes to determine whether S65E regulates ubiquitin chain formation. Here, we selected two E2 enzymes, which robustly generate un-anchored chains with WT ubiquitin. Specifically, we used Ubc1, which generates the canonical K48-linked chains, and the Ubc13/Mms2 complex, which primarily generates K63-linked chains. We found S65E mutants to be defective in ubiquitin chain formation with both E2 enzymes (Fig2B and C). Additionally, despite moderately reduced E1 loading with S65A, this mutant robustly generated ubiquitin polymers, indicating that the reduced E1 loading observed with S65E is not the cause of its inability to form ubiquitin chains in vitro.
Figure 2.
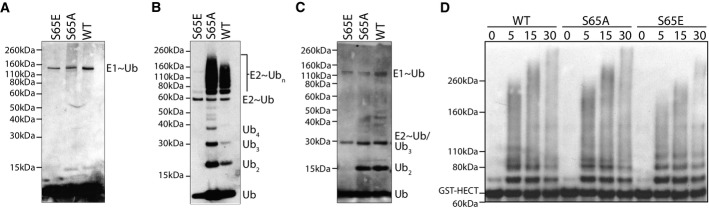
In vitro characterization of ubiquitin chain assembly by S65 ubiquitin mutants
- Loading of recombinant ubiquitin mutants onto an Uba1 E1 enzyme.
- Generation of unanchored ubiquitin chains using the Ubc1 E2 enzyme.
- Generation of unanchored ubiquitin chains using the Ubc13/Mms2 E2 enzyme complex.
- Rsp5 HECT domain auto-ubiquitylation using the Ubc4 E2 enzyme.
Next, we conducted ubiquitylation reactions in the presence of an E3 ubiquitin ligase. Specifically, we used the HECT domain of the E3 Rsp5 and the E2 Ubc4, and Rsp5 auto-ubiquitination was monitored over time (Fig2D). Auto-ubiquitylation readily occurs for WT and S65A ubiquitin. Interestingly, we observed comparable reaction kinetics and output for S65E mutant, indicating that Rsp5 seems to mitigate the deleterious effects of the S65E ubiquitin mutation on E2 ligase activity.
Lastly, we tested the effectiveness of DUBs against ubiquitin S65E in an in vitro deubiquitylation assay. In this assay, we generated WT and S65E ubiquitin dimers and compared them regarding their sensitivity to DUB disassembly. We used Usp5, the functional homolog of the yeast Ubp14 DUB which is primarily responsible for chain disassembly 14. Structural analysis of ubiquitin complexed with Usp5 indicated that ubiquitin S65 lies at the binding interface of this protein complex (PDB ID: 3IHP, Fig3A). Thus, we reasoned that phosphorylation of S65 could alter binding interactions. We used E1, Ubc1, and a mixture of WT and His-tagged S65E ubiquitin to generate di-ubiquitin species: WT-WT, S65-S65, and WT-S65, all of which can be distinguished in an SDS–PAGE gel due to the shift introduced by the histidine tag. Different concentrations of Usp5 were then used to disassemble the di-ubiquitin species (Fig3B and C). Results indicate that Usp5 has similar activity against WT-WT homodimer and WT-S65E heterodimer; nonetheless, S65E dimer seems to be resistant to the protease activity. Similar trends were observed in a time–course experiment in which we used a fixed concentration of Usp5 and monitored the extent of the reaction over time (Appendix Fig S1).
Figure 3.
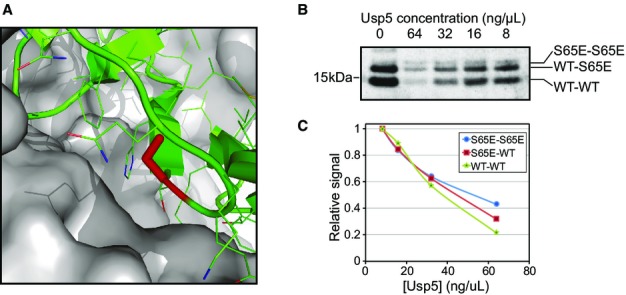
Usp5 sensitivity of S65 ubiquitin mutant chains
- Structure of ubiquitin in complex with Usp5 (PDB ID: 3IHP). S65 is highlighted in red on the cartoon representation of ubiquitin, while Usp5 surface is shown in gray.
- Anti-ubiquitin immunoblotting of in vitro generated mixed chains comprised of both WT and His-tagged S65E ubiquitin. Chains were subjected to disassembly with various concentrations of Usp5, and the abundance of the dimers was measured.
- Quantification of band signal of the Western blot shown in (B). Y-axis represents signal relative to the point of maximum signal for each series.
All in all, our observations of S65E inhibiting both free ubiquitin chain formation and DUB-mediated chain disassembly are analogous to recent work with phosphorylated ubiquitin 13 and suggest that S65E should be a biochemically faithful mimic of native S65 ubiquitin phosphorylation.
The effects of S65 mutation on ubiquitin interactions with ubiquitin-binding domains
Other than the biochemical consequences that ubiquitin S65 mutation or phosphorylation has on E2, E3 or DUB enzymes, this substitution may exert its role by modulating ubiquitin interactions with other proteins. In particular, there is a host of proteins containing ubiquitin-binding domains such as UBA, CUE and UIM that act as the decoders of ubiquitin-mediated cellular functions 15, 16 and can be affected by the mutation. To interrogate these effects in an unbiased manner, we carried out a quantitative pull-down proteomic experiment, using His-tagged (WT, S65A and S65E) ubiquitin as baits against a yeast whole-cell extract. We performed quantitative pairwise comparisons of proteins binding to WT versus S65A and WT versus S65E, as well as a WT versus no-bait control that served to define the set of interactors that are specific to ubiquitin.
In the WT versus no-bait experiment, we found many enriched proteins (high log2 ratio) involved in ubiquitin signaling, including E2s, E3s, DUBs, and proteasome components (Table1 and related source data). As expected, several of these proteins contain ubiquitin-binding domains (UBD) (Table1). Most of these proteins showed similar binding to wild-type ubiquitin and S65 ubiquitin mutants, as evidenced by log2 ratios of mutant versus WT around zero. Despite the lack of dramatic fold changes, a trend can readily be observed: in general, S65E mutant seems to be slightly disruptive to ubiquitin recognition. Some examples are as follows: Cue5, Vps27, and Tom1 (Table1 and related source data).
Table 1.
Interactions of ubiquitin and ubiquitin mutants with proteins containing UBD
| Protein | log2 (WT/S65A) | log2 (WT/65E) | log2 (WT/Ctrl) | Delta | No. of independent quantifications | UBD |
|---|---|---|---|---|---|---|
| Def1 | −0.30 | −0.11 | 0.58 | −0.19 | 34 | CUE |
| Vps27 | −0.15 | 0.27 | 0.77 | −0.42 | 10 | UIM |
| Ent2 | −0.02 | 0.03 | 0.92 | −0.05 | 9 | UIM |
| Rad23 | −0.24 | −2.02 | 1.13 | 1.78 | 10 | UBA |
| Hse1 | −0.24 | 0.14 | 1.69 | −0.38 | 8 | UIM |
| Cue5 | −0.12 | 0.28 | 1.99 | −0.40 | 29 | CUE |
| Dsk2 | −0.68 | −0.93 | n/a | 0.26 | 8 | UBA |
| Ede1 | −0.39 | −0.91 | n/a | 0.52 | 4 | UBA |
| Ubp14 | −0.28 | −0.52 | n/a | 0.23 | 24 | UBA |
| Rup1 | 1.57 | 0.63 | n/a | 0.94 | 6 | UBA |
Log2 quantifications for proteins containing ubiquitin-binding domains of the α-helix family as detected in the WT versus S65E, WT versus S65A, and WT versus no-bait control. Delta represents the preference of binding to S65E over S65A and is calculated as log2(WT/S65A)-log2(WT/S65E).
Source data are available online for this table.
A remarkable exception to this trend is Rad23, showing preferential binding to S65E ubiquitin over wild-type by four-fold. Interestingly, Dsk2—a protein with similar role to Rad23—and Png1—a known Rad23 interactor—are also significantly enriched in the S65E pulldown (Table1 and related source data). The primary function of both Rad23 and Dsk2 is to shuttle ubiquitylated proteins to the proteasome for degradation. They both share a domain architecture consisting of one (Dsk2) or two (Rad23) UBA domains that interact with ubiquitylated proteins, and an N-terminal ubiquitin-like domain (UBL) that interacts with a regulatory proteasome subunit (Rpn10, Rpn1 or Rpn2) and enables recruitment to the proteasome 17.
The substantial increase of Rad23 on the S65E ubiquitin pulldown could be the result of increased affinity toward phosphomimetic ubiquitin mutant or the consequence of accumulating S65E-ubiquitin conjugates that cannot be processed by the proteasome and bind more tightly to Rad23. We should note that the experiment was carried out in conditions where ubiquitin conjugation was not inhibited; thus, both non-covalent and covalent interactions may contribute to our observations.
The effect of stress on ubiquitin mutant cell viability
Ubiquitin is a highly conserved protein whose function is essential for cell survival, with previous work demonstrating that mutations to individual residues is sufficient to render cells inviable 18, 19. To screen for functional implications of S65 phosphorylation, we tested how mutation of S65 regulated the viability of yeast upon a selection of stresses, including osmotic stress (NaCl), oxidative stress (H2O2), proteasome inhibition (bortezomib), DNA damage (MMS), and protein misfolding (canavanine). In these experiments, we used yeast strains that express a single form of ubiquitin, either WT, a phosphorylation inhibitory S65A, or a phosphomimetic S65E mutant (Appendix Table S1). Of the conditions tested, S65E ubiquitin cells showed reduced viability under two stresses which both cause mitochondrial damage and require well functioning degradation machinery for cell survival: protein misfolding with canavanine and oxidative stress with hydrogen peroxide (H2O2) (Fig4A).
Figure 4.
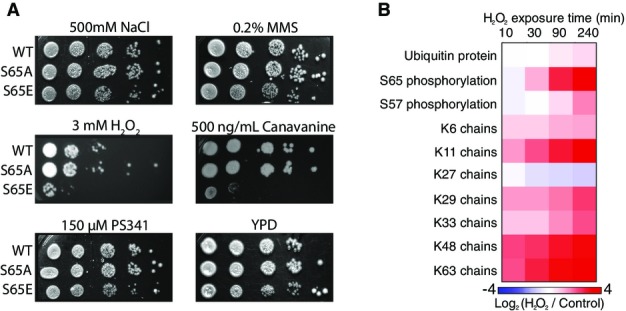
The effect of stress on ubiquitin signaling and cell viability
- Viability assay by serial dilution (1:10) of yeast strains exclusively expressing either WT or S65 mutant ubiquitin. Strains were spotted on YPD plates with or without different stressors (as indicated).
- Log2 (H2O2/control) heat maps displaying the response to 5 mM H2O2 exposure for global ubiquitin protein, S65 phosphorylation, and ubiquitin chain abundances.
Canavanine is an arginine analog, which is readily incorporated into proteins causing protein misfolding. Subsequently, protein quality control pathways degrade misfolded proteins 20. It has been shown that both deficiencies in ubiquitin signaling (via E2 deletion), or more commonly, deficiencies in protein degradation (via DUB deletion) render yeast sensitive to canavanine 21, 22. The reduced viability of S65E cells when required to utilize canavanine indicates that this mutation may impact ubiquitin homeostasis in vivo.
Cells with compromised degradation systems are more sensitive to hydrogen peroxide exposure as oxidative damage causes an increased demand for the degradative turnover of damaged proteins. Additionally, oxidation inhibits proteasomal processing by direct inhibition of DUB catalytic activity 23. Here, we observed that S65E ubiquitin mutants showed a concentration-dependent reduction in viability under H2O2 stress (FigEV2A). At lower concentrations, all strains were similarly viable, but as the concentration increased to 3 mM, S65E shows a loss in viability relative to WT or S65A, and by 5 mM H2O2, the viability of the strain with WT ubiquitin was also affected (Fig4A and Appendix Fig S1A). These results suggest that ubiquitin phosphorylation at S65, either native (WT) or via mimetic mutation (S65E), reduces the capacity of cells to tolerate H2O2-induced oxidative stress. Note, the sensitivity of S65E mutant strains to both canavanine and H2O2 was found to be specific for this phosphomimetic, as mutation of a neighboring serine (S57E) did not display such sensitivities (FigEV2B).
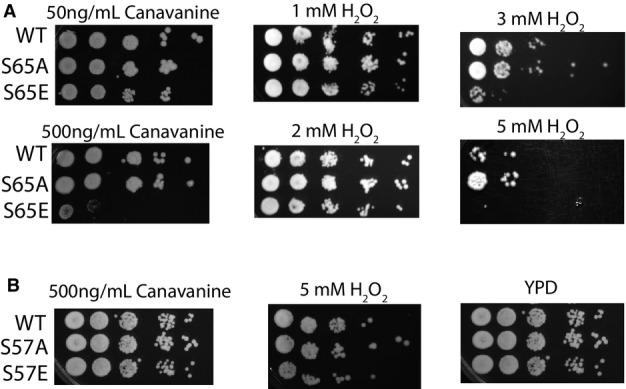
The effect of H2O2 and canavanine on the cell viability of ubiquitin S65 mutant strains
- Viability assay by serial dilution (1:10) of yeast strains exclusively expressing either WT or S65 mutant, ubiquitin. Strains were spotted on plates at different concentrations of the stressors hydrogen peroxide and canavanine (as indicated).
- For two conditions under which S65E strains were sensitive, the spotting assays were also performed with mutants to an alternate phosphorylation site, S57.
Ubiquitin is regulated under hydrogen peroxide stress
Yeast spotting assays indicated that S65E phosphomimetic mutation has phenotypic consequences under hydrogen peroxide exposure. To further characterize the molecular consequences of this stress, we exposed wild-type yeast to H2O2 stress and used a quantitative proteomics approach to measure how this stress regulated global levels of ubiquitin protein, ubiquitin chain structures, and ubiquitin phosphorylation at S65 (Fig4B and Appendix Table S2). This experiment revealed a nearly universal increase in abundance of the ubiquitin chains that exceeded the modest increase in global ubiquitin protein abundance. One exception to this was K27-linked chains, which decreased in abundance. The most highly regulated components were S65 phosphorylation, and K11- and K63-linked chains, which all increased in abundance by 14-fold (Fig4B). In contrast, S57 phosphorylation showed only a modest change (Fig4B and Appendix Table S2). These findings indicate that ubiquitin S65 phosphorylation can be dynamically regulated upon oxidative stress and this phosphorylation correlates with the restructuring of the ubiquitin landscape into a highly polymeric state.
A similar experiment exposing yeast to canavanine for 8 h revealed a significant decrease in the stoichiometry of S65 phosphorylation, resulting from a three-fold up-regulation of total ubiquitin, while the levels of S65 phosphorylation remained more or less constant (FigEV3). The stoichiometry of S57 did not change with canavanine.
Quantification of ubiquitin and S65 ubiquitin upon canavanine treatment
Extracted ion chromatograms of a representative non-modified ubiquitin peptide (left) and S65 ubiquitin phosphopeptide (right). Blue lines correspond to heavy-labeled peptides from the canavanine treated culture, and red lines correspond to light peptides from the control culture.
Phosphomimetic S65E ubiquitin increases global ubiquitylation
We next sought to characterize the regulatory effects of S65 phosphorylation on the ubiquitylation landscape. We used strains that express a single form of ubiquitin, either WT (with normally occurring S65 ubiquitin phosphorylation stoichiometry), phosphorylation inhibitory S65A (representing no phosphorylation), or a phosphomimetic S65E mutant (representing 100% phosphorylation), and compared these strains via anti-ubiquitin Western blotting and quantitative proteomics.
Western blot analysis of in vivo ubiquitylation revealed a dramatic proteome-wide increase in ubiquitylation in the S65E phosphomimetic strain relative to the WT or S65A strain (Fig5A), indicating an imbalance in ubiquitin flux in S65E cells. Additionally, we observed enrichment in unanchored ubiquitin chains and a corresponding decrease in monomeric ubiquitin. Further analysis of individual chain types by quantitative proteomics illustrated that all chain types increased significantly, with the exception of K27 ubiquitin chains (Fig5B).
Figure 5.
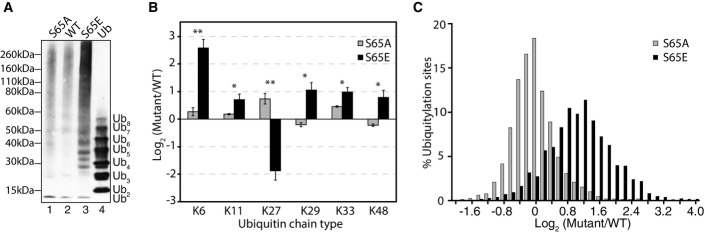
In vivo characterization of S65 ubiquitin mutants
- Anti-ubiquitin immunoblotting of yeast cell lysates from S65A, WT, and S65E mutant ubiquitin yeast strains (lanes 1–3). Lane 4 shows human K48-linked polyubiquitin chains as a control for co-migration of unanchored ubiquitin chains.
- Log2 quantification of ubiquitin chain abundance in S65A and S65E mutant strains relative to WT. Here, quantification was normalized for total ubiquitin protein expression. Error bars indicate the standard deviation of biological triplicate measurements. Statistical significance was assessed between S65A/WT and S65E/WT using two-sided pairwise t-test, where *P < 0.05 and **P < 0.005. Note, K63 chains could not be quantified due to the presence of the mutated S65 amino acid.
- Log2 distribution of ubiquitylation sites in S65A and S65E mutant strains relative to WT (n = 2,636). Distributions represent the average of biological triplicate measurements.
Source data are available online for this figure.
Proteomics analysis of ubiquitylation substrates, via enrichment with a di-glycyl lysine antibody 24, 25, 26, 27, resulted in the identification of 2,636 ubiquitylation sites on 1,088 proteins (Fig5C and related source data). A pronounced accumulation of ubiquitylation sites on substrate proteins was observed in the S65E strain relative to WT or S65A ubiquitin (Fig5C). This increase in ubiquitylation was greater than has been observed upon proteasome inhibition 5, indicating that the S65E mutation has a broad and potent impact on ubiquitin processing. This large increase was not the result of protein accumulation, as individual protein levels among all three strains were similar (Fig EV4A).
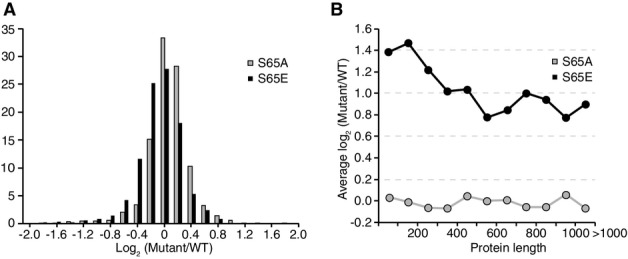
Quantification of S65 ubiquitin mutant proteins
- Log2 distribution of protein abundance in S65A and S65E mutant strains relative to WT.
- Histogram displaying the average of the log2 relative ubiquitylation site abundance (mutant/WT) in each protein length bin.
Evaluation of the substrate targets of ubiquitylation revealed enrichment in ubiquitylation sites on ribosomal proteins within the top 10% of increased sites (P = 5.0 × 10−30, Appendix Table S3). This observation is likely due to a conflict of the fast protein turnover rate of ribosomal proteins 28 with the inhibition of DUB processing by S65E. Smaller proteins were also found to be more highly ubiquitylated in the S65E strain (FigEV4B), although this correlation could simply be due to the small size of most ribosomal proteins (< 300 amino acids, FigEV2B) 29. Further analysis of additional protein properties, including protein abundance, subcellular localization, Pfam domain presence, ubiquitylation site position, and frequency, revealed no additional enriched characteristics.
Targets of ubiquitin phosphorylation
The global increase in ubiquitylation induced by exclusive expression of S65E ubiquitin revealed its potential regulatory roles, but made it difficult to distinguish between substrates that increased in ubiquitylation because they are preferred targets and those that increased due to the degradation inhibitory effects of S65E. Thus, to identify the preferred targets of ubiquitin phosphorylation, we devised an alternative approach. As no antibodies exist for the specific purification of phosphorylated ubiquitin species, we instead applied a phosphomimetic affinity purification strategy. Here, we used yeast strains that simultaneously expressed two forms of ubiquitin: the endogenous WT ubiquitin as well as a plasmid-based His-tagged ubiquitin (Fig6A). In our quantitative proteomics approach, we varied the sequence of the His-tagged ubiquitin in each strain to be WT sequence, phosphorylation inhibitory (S65A), or phosphomimetic (S65E). As displayed in Fig6A, we purified His-tagged ubiquitin (HisUb) conjugates and used proteomics to detect and quantify the preference of ubiquitin conjugate proteins for different ubiquitin mutants. From this purification, we identified 958 ubiquitin conjugate proteins and 960 ubiquitylation sites (Fig 6 and related source data). Additionally, we quantified protein and ubiquitylation site abundances in the His-tag depleted portion of the sample. These measurements were used as a control to ensure that the introduction of mutant ubiquitin into the yeast strains did not alter global protein abundances or ubiquitylation signaling interactions with endogenous ubiquitin.
Figure 6.

Targets and effects of S65E ubiquitin
- A A schematic of the experimental design used here.
- B, C Distribution of protein quantification ratios (B) and ubiquitylation site ratios (C) of the different samples analyzed.
- D Log2 quantification of ubiquitin chain abundance in S65E mutant strain relative to WT. Both the endogenous ubiquitin and plasmid-based His-tagged ubiquitin were compared between the S65E and WT strains. Quantification was normalized for total ubiquitin protein expression.
- E Protein–protein interactions between SNARE proteins and vacuolar sorting proteins enriched in S65E ubiquitin-associated proteins. The log2 (S65E/WT) ratio is represented in the box interior, while the log2 (S65A/WT) ratio is represented in the border color of each protein box.
Data information: Box limits in (B, C) indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, outliers are represented by dots. Error bars in (D) indicate the standard deviation of biological duplicate measurements. The statistical significance of the difference between His-tagged and endogenous ubiquitin expression in the S65E or WT strains was assessed using two-sided pairwise t-test, where *P < 0.05. Note, K63 chains could not be quantified due to the presence of the mutated S65 amino acid. Source data are available online for this figure.
At a global level, we found the majority of ubiquitin conjugate proteins identified from His-tag purifications were of equal abundance between ubiquitin mutants, suggesting that S65E has a limited set of specific binding partners (Fig6B). Despite this, the abundance of ubiquitylation on S65E ubiquitin conjugates was significantly increased (P = 3.0 × 10−216), indicating that phosphomimetic S65E increases in the frequency of ubiquitylation on its target protein substrates (Fig6C). This increased ubiquitylation state was highly specific to proteins that were conjugated to S65E ubiquitin, as the levels of endogenous ubiquitylation in this yeast strain were comparable to strains that contained S65A or WT His-tagged ubiquitin (Fig6C). A similar trend was observed when evaluating ubiquitin chain abundance. Integration of S65E into ubiquitin chains was significantly preferred for K6 and K11 chain types, as indicated by their increased abundance in S65E conjugates, while K27 chains were disfavored (Fig6D). Note that in this experiment, due to the mutations, we were not able to measure the relative abundance of K63 chains. Interestingly, it has recently been reported that in human cells, mitochondrial damage promotes Parkin to preferentially assemble K6, K11, and K63 ubiquitin chain types 30. This suggests that despite the lack of a Parkin ortholog in yeast, there may be similar mechanisms with an E3 sensing mitochondrial damage and regulating the ligation of S65E ubiquitin chains.
Two classes of proteins were found to be preferentially targeted by S65E ubiquitin. Specifically, we observed a two-fold preference for S65E to associate with histone H2B protein (HTB2), and correlating with increased ubiquitylation of K50 (9.4-fold enrichment), a residue that is adjacent to the HTB2 N-terminal nuclear localization signal. Increased ubiquitylation was also observed on two other HTB2 sites, K112 and K124 (2.4- and 1.9-fold enrichment, respectively). Histone-associated proteins were also enriched in association with the S65E mutant, including the histone H2B nuclear import protein Kap95 31, and the histone deacetylase Rpd3. It was also recently reported that mammalian histone H2B can be modified by K6- or K48-acetylated ubiquitin 32. The association of both phosphorylated and acetylated ubiquitin with histone H2B suggests ubiquitin post-translational modifications may serve as an additional mode of histone regulation, expanding significantly the canonical histone code 33.
Another class of proteins preferentially conjugated to S65E ubiquitin was the soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) proteins (P = 3.3 × 10−5). These are membrane proteins that facilitate vesicle fusions responsible for the passage of cargo from one cellular component to another. STRING analysis of proteins enriched in S65E conjugates revealed high-confidence protein interactions between many of these SNARE proteins, as well as two vacuolar sorting proteins Vps21 and Pep1 34 (Fig6E). Interestingly, while S65 phosphorylation has been shown to activate Parkin E3 ligase activity, reduced Parkin activity and dysfunction in vesicle transport have been reported to contribute to neurodegenerative disease, such as Parkinson’s 35, 36, 37. Thus, the preference of SNARE proteins points toward a conserved role for S65 phosphorylation in yeast.
Phosphomimetic ubiquitin decreases protein degradation and turnover
Ubiquitylated proteins can be de-ubiquitylated and subsequently degraded by the proteasome. The significant accumulation of ubiquitylated substrates and ubiquitin chains in cells expressing S65E ubiquitin and the results of in vitro DUB disassembly assays led us to ask whether this modification interferes with protein degradation and turnover in vivo. To investigate this, we performed a global quantitative proteomics experiment in which we simultaneously measured protein synthesis, degradation, and turnover for WT and the two mutant ubiquitin (S65A and S65E) strains (Fig7A) using a pulse SILAC triple labeling approach 38. This analysis revealed differences in protein flux between WT and S65E strains that could not be attributed to differences in cell growth (Appendix Fig S2). Relative to S65A or WT cells, S65E cells had significantly slower protein degradation and turnover rates (Fig7B and related source data). While synthesis rates in S65E cells were also slower, this process was found to be the most similar to S65A and WT strains. These results indicate that S65E inhibits protein degradation, and may be used as a mechanism to protect substrates from protein turnover.
Figure 7.
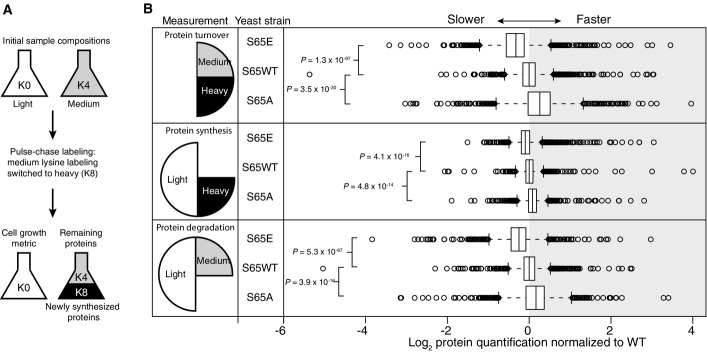
Analysis of protein synthesis, degradation, and turnover
- The experiment begins with equal populations of light (K0)-labeled and medium (K4)-labeled cells. The light population serves as a control to account for cell growth. Medium-labeled cells are then transferred to media containing heavy lysine (K8). Incorporation of heavy label permits quantification of synthesis, loss of medium label permits quantification of degradation, and the ratio between heavy and medium measures protein turnover.
- Log2 of protein quantification measured for proteome-wide protein turnover (heavy/medium), protein synthesis (heavy/light), and protein degradation (medium/light). Center lines show the median of each distribution (S65E n = 1,765, WT n = 1,666, S65A n = 1,718), and all distributions have been normalized such that the median of the WT sample is centered at zero. Strains with rates slower than WT have median values < 0, while those with faster rates have median values > 0. Box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, outliers are represented by empty circles. P-values displayed are the result of a two-sided t-test assuming unequal variance.
Source data are available online for this figure.
Considering our findings of S65E-induced increases in ubiquitylation on ribosomal proteins, and the established role of S65 phosphorylation in mitochondria function, we evaluated if the mutational status of ubiquitin S65 impacted the turnover rate of proteins associated with specific cellular compartments. We compared the relative turnover between S65E, WT, and S65A for a selection of cellular compartments. Here, we find that the relative differences between these mutants are somewhat compartment specific. For example, when all proteins are considered, protein turnover rates in the WT ubiquitin strains are between that of S65E and S65A (Fig7B). However, for individual cellular compartments, including the mitochondria, vacuoles, and the plasma membrane, protein turnover rates in S65A and WT cells are statistically indistinguishable (Fig EV5).
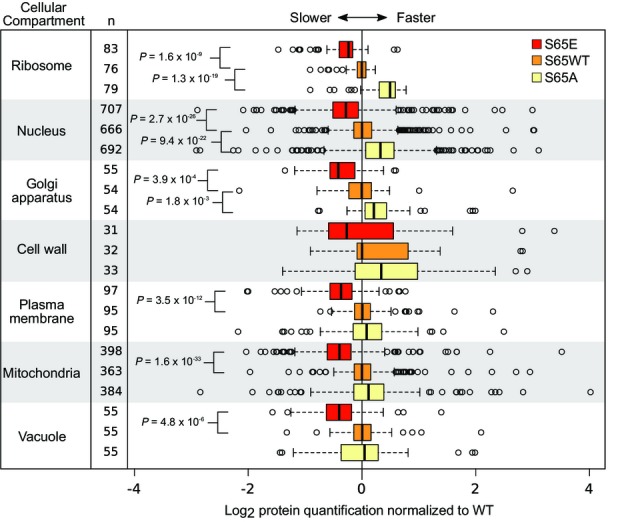
Protein turnover rates in different cellular compartments
Comparison of global turnover rates for different compartments, measured as log2 (heavy/medium). In the box plot, center lines show the median of each distribution, and all distributions have been normalized such that the median of the WT sample is centered at zero. Strains with rates slower than WT have median values < 0, while those with faster rates have median values > 0. Box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, and outliers are represented by empty circles. P-values displayed are the result of a two-sided t-test assuming unequal variance. The number of proteins on each compartment is also provided (n).
Lastly, we evaluated those proteins with the greatest difference in turnover between S65E and S65A/WT, being much slower on the S65E. For these proteins, STRING analysis of protein–protein interactions revealed a network of proteins related to mRNA processing and ribosome biogenesis (Appendix Fig S3). This network included 2 of the 3 components of the nonsense mRNA decay complex (Upf3 and Nmd2). This complex has been shown to associate with both aberrant mRNA and the resulting truncated poly-peptide, and facilitates the ubiquitin-mediated degradation of the aberrant peptide 39.
Discussion
Our finding of phosphorylation on most Ser/Thr/Tyr residues of ubiquitin prompted us to interrogate the broad regulatory implications of ubiquitin phosphorylation on the ubiquitin signaling network. Here, we have provided evidence that S65 phosphorylation of ubiquitin is a biologically important modification that influences nearly every step of the in vivo ubiquitin life cycle.
Defects were observed in the ability of the S65E phosphomimetic mutant to generate polyubiquitin chains in vitro, pointing toward a potential role for S65 phosphorylation in regulating ubiquitin chain assembly by modulating the ubiquitylation output of particular E2 and E3 ligases. However, under in vivo conditions, when provided the full repertoire of ubiquitin machinery, the S65E mutant produced higher levels of substrate ubiquitylation and ubiquitin chains. While we cannot rule out that enhanced ubiquitin conjugation machinery activity (i.e. individual E2 or E3s) contributes to this increase, as seen with Parkin in human cells 10, 11, 12, 13, we attribute this increase primarily to an imbalance in ubiquitin flux caused by impairment of DUB processing and protein turnover. This is supported by in vivo protein turnover and yeast phenotypic spotting experiments, which both indicate that S65E mutant ubiquitin has defects in protein degradation, as well as by recent in vitro observations demonstrating a defect in phosphorylated ubiquitin chains to be disassembled by a variety of DUBs 13.
Indeed, previous experiments have shown that mutations to both the hydrophobic core and the C-terminal region of ubiquitin can render chains resistant to disassembly by DUBs and proteasomal degradation 40, 41, 42, 43. Our results indicate that such inhibition can be achieved endogenously by phosphorylation of the S65 residue and suggest that ubiquitin phosphorylation represents a novel mechanism to dynamically regulate ubiquitin recycling and protein degradation. Serine 65 lies at the Usp5 binding interface and its phosphorylation or phosphomimetic mutation interferes with Usp5 activity. Interestingly, structural analysis indicates that additional ubiquitin phosphorylation sites su ch as T12, T14, and T66 also lie at the Usp5 binding interface. Considering that many DUBs share this USP fold and ubiquitin recognition mode, phosphorylation of ubiquitin may serve as a more general mechanism to regulate protein degradation 41. Indeed, our pull-down experiment shows that Ubp3 and Ubp15 have less affinity toward S65E ubiquitin than S65A or WT ubiquitin.
Another possibility for the impaired degradation of S65 phosphoubiquitin lays in the allosteric regulation of Rad23. Our quantitative ubiquitin pull-down experiments show that S65E mutation may enhance the interaction or conjugation of ubiquitin to Rad23, and could potentially inactivate Rad23 shuttling function. This is a possibility given that under certain conditions, Rad23 has been shown to inhibit deubiquitylation 44, 45. S65E ubiquitin could also promote the formation of Rad23 dimers with impaired shuttling activity. In either case, the ultimate result would be that Rad23 is effectively depleted from the cell, thereby reducing substrate processing by the proteasome and leading to longer protein half-lives and accumulation of ubiquitin conjugates. A similar phenotype has been observed with Rad23 deletion strains 46 and is consistent with our observations.
The low level abundance of ubiquitin phosphorylation sites has resulted in their sporadic detection. Only the most abundant, S57 phosphorylation, has been routinely observe in proteomics studies; however, the function of this site remains unknown. For S65, it has been reported that ∼1% of global ubiquitin is phosphorylated in human cells; however, this concentration is highly dependent on cellular localization, with ∼10% of mitochondrial ubiquitin being phosphorylated at S65 12. In yeast, we find that < 0.5% of the total ubiquitin pool is phosphorylated on S65. Therefore, the effects observed using phosphomimetic mutants (100% is S65E ubiquitin), while informative, probably represent extreme phenotypes, and the comparison with the S65A may be more relevant.
Considering the breadth of the ubiquitin signaling network, it is not unexpected that global levels of ubiquitin phosphorylation are low, as this allows for ubiquitin phosphorylation to exert specificity and function as a regulatory signal in localized or highly dynamic environments. A low phosphorylation stoichiometry is also expected given the polymeric nature of ubiquitin, where the modification of a single moiety in a polymer could be sufficient to elicit a response. Finally, low stoichiometry would also be relevant in phosphoubiquitin’s role of imparting allosteric regulation to other proteins, such as Parkin, or possibly Rad23.
In the context of allosterically regulating other proteins, phosphorylation of ubiquitin seems to have functional implications beyond ubiquitin regulation. Interestingly, many of the phosphorylated residues of ubiquitin are conserved, Ser and Thr, across other UBL domains and proteins, and several of the UBLs have an Asp or Glu on those positions (e.g. Smt3—yeast SUMO—and Rad23 for S65), mimicking a phosphorylated state. It is plausible then that phosphorylation of ubiquitin competes for binding with some of these UBLs. As it has recently been reported, not only does PINK1 phosphorylate ubiquitin S65 10, 11, 12, 13, but it also phosphorylates the UBL domain of Parkin at S65 47. This phosphorylation occurs upon mitochondrial damage and activates the E3 ligase Parkin, which in turn preferentially ubiquitylates substrates via K6-, K11-, K48-, and K63-linked chains and promotes mitophagy 12. This presents an interesting connection to our study, in which H2O2, which is known to induce mitochondrial damage, caused a > 10-fold increase in K11-, K48-, and K63-linked chains and ubiquitin S65 phosphorylation.
Defects in both mitochondrial turnover and protein turnover are associated with pathological phenotypes. Thus, the association of S65 phosphorylation with both of these processes presents an additional interesting therapeutic strategy route, as attested by the number of DUB, and E2 and E3 inhibitors in current use or clinical trials 48. Considering the multitude of additional phosphorylation sites on ubiquitin, we anticipate that future research expanding beyond S65 phosphorylation will undoubtedly reveal novel dimensions to ubiquitin signaling with direct implications for our understanding of basic biology and disease treatment.
Materials and Methods
Expression and purification of recombinant ubiquitin and ubiquitin mutants
The ubiquitin sequence was subcloned into pET-24a vector, expressed in E. coli and purified as described elsewhere 49. Site-directed mutagenesis was performed to generate mutant S65A and S65E ubiquitin sequences. Expression was carried out using transformed BL21 DE3, grown to a density of 0.8 OD600, and induced overnight at 30°C with 1 mM IPTG. Harvested cells were resuspended in a 50 mM Tris, 100 mM NaCl, and pH 7.5 buffer containing protease inhibitors (Roche, complete) and benzonase and lysed by sonication. Lysate was subjected to acidic precipitation by the addition of perchloric acid to 0.7% (v/v). Supernatant was dialyzed against 50 mM sodium acetate, pH 4.5, and purified using a Resource S cation exchange column a gradient from 0 to 300 mM NaCl. Ubiquitin-containing fractions were pooled and concentrated with a 2,000 MWCO centricon and further purified over a Superdex S75 size exclusion column in 50 mM phosphate, 50 mM NaCl, and pH 7.5 buffer.
Generation of mutant ubiquitin S. cerevisiae strains
The Leu marker was removed from the p425GPD plasmid and replaced with a Hyg cassette to generate the p42HGPD 50. Next, a truncated version of the Ubi3 gene (YLR167W) corresponding to the cleaved ubiquitin product (amino acids 1–76) was cloned into the plasmid. Site-directed mutagenesis was performed on this plasmid to generate mutant S65A and S65E ubiquitin sequences. The WT and mutant plasmids were individually transformed into a yeast strain exclusively expressing ubiquitin from a synthetic gene. This strain was strain LSY207 (MATa lys2-801, ura3-52, leu2-3,112, his3Δ200, trp1-1, ubi1ΔTRP1, ubi2Δura3, ubi3Δ, ubi4ΔLEU2, pdr5::KanMX [pUB221] [pUB100]). pUB221 is a URA3-marked plasmid that expresses 6His-myc-ubiquitin under a CUP1 promoter, whereas pUB100 expresses the essential ribosomal protein encoded by the UBI1 gene 5, 51.
The 6His-myc-ubiquitin plasmid (pUB221) was shuffled out by plating on 5FOA, such that the only remaining expression of ubiquitin was from the p42HGPD plasmid, with expression of WT ubiquitin in the DS147 strain, expression of S65A ubiquitin in the DS181 strain, and expression of S65E ubiquitin mutant in the DS182 strain. To verify removal of the pUB221 plasmid, cells were grown overnight in YPD. Cells were harvested by centrifugation and resuspended in 500 μl of native buffer comprised of 50 mM Tris, and 100 mM NaCl, pH 8.2. After lysis by bead beating, lysates were clarified by centrifugation at 10,000 × g. Perchloric acid was added to a final concentration of 0.7% v/v, and samples were kept at 4°C for 3 h to precipitate larger proteins. Lysates were then spun at 10,000 × g for 15 min, and the ubiquitin-containing supernatant was transferred to a new tube. The pH was adjusted by the addition of 50 μl of 1 M ammonium bicarbonate and 100 μl of 1 M Tris, pH 8.2. After the addition of 5 μl of acetonitrile, the proteins were digested by the addition of 5 μg of trypsin (Promega), and overnight incubation at 37°C. Each sample was desalted on a 50 mg tC18 SepPak cartridge (Waters), and the eluant was lyophilized. Samples were reconstituted in 4% formic acid and 3% acetonitrile solution, and ∼1 μg was analyzed on a Velos Orbitrap. In all cases, the proper ubiquitin sequence (WT, S65A, or S65E) was observed and no evidence of the 6His-myc-ubiquitin was detected.
Quantitative proteomic analysis of His-tagged ubiquitin pulldowns
Native whole-cell lysate was produced by bead beating of BY4742 yeast cells in a buffer containing 25 mM Tris, 50 mM NaCl, 1 mM β-mercaptoethanol, and 5 mM MgCl2, pH 7.5. About 250 μg of purified recombinant His-tagged ubiquitin (WT and S65A, S65E mutants) was incubated with 4 mg of whole-cell lysate and immobilized onto 25 μl of Cobalt TALON metal affinity resin (Clontech). Resin was washed with 10 column volumes of lysis buffer, 10 column volumes of 25 mM Tris, 50 mM NaCl, 1 mM β-mercaptoethanol, 5 mM MgCl2, and 10 mM imidazole, pH 7.5, and eluted with the same buffer at 300 mM imidazole.
About 50 mM ammonium bicarbonate was added to eluate and digested overnight at 37°C with trypsin at 5 μg/μl. Resulting peptides were acidified, desalted on a 50 mg tC18 SepPak cartridge (Waters), and the eluant was lyophilized. Reductive dimethylation was carried out on peptides using a 100 mM citrate, 500 mM phosphate buffer at pH 5.8, 0.4% formaldehyde, and 60 mM sodium cyanoborohydride. WT His-ubiquitin pull-down samples were labeled using deuterated formaldehyde and NaCNBD3 (Isotec, Sigma), and the other samples (ubiquitin mutants and no-bait control) were labeled with the light (non-deuterated) counterparts. Reaction was stopped by the addition of 10% TFA, and heavy and light peptides were then mixed and further desalted on 50 mg tC18 SepPak cartridge prior to mass spectrometry (MS) analysis.
His-tagged ubiquitin mutant proteomics
Yeast was grown in lysine-free synthetic complete media supplemented with 436 μM lysine. Culture for strain DSD209 was supplemented with K0 lysine, DS208 was supplemented with K4 lysine (4,4,5,5-D4), and DS210 was supplemented with K8 lysine (13C6, 15N2) 52. Cells were grown in a 320 ml volume in biological duplicate, and harvested at a density of ∼1.0 (OD600). Samples were lysed and ubiquitin protein conjugates were purified by His-tag affinity chromatography exactly as previously described, with the exception that the enrichment of peptides containing the di-glycyl lysine remnant (indicative of mostly ubiquitylation) was also performed on the His-tag flow-through sample which contained endogenous ubiquitin 5. Additionally, prior to His-tag purification, a 100-μg aliquot was removed from each sample and diluted five-fold in 50 mM Tris, pH 8.9. This aliquot, which was used for the analysis of cell mixing normalization and ubiquitin protein quantification, was reduced and alkylated prior to digestion with lysyl endopeptidase (LysC) (Wako) at RT overnight. The resulting peptides were desalted on 50 mg tC18 SepPak cartridges (Waters) prior to MS analysis.
H2O2 proteomics
LSY207 yeast was grown in lysine-free synthetic complete media supplemented with 436 μM K0 lysine, or K8 lysine 52. Cells were grown to a density of ∼0.8 (OD600) and then 5 mM H2O2 was added to the K8 culture. During H2O2 exposure, additional media was added to cultures as needed to maintain log-phase growth. Cells were harvested by centrifugation at 10, 30, 90, and 240 min after H2O2 addition. At each time point, a volume of 90 ml was collected from both the K0 and K8 labeled cultures. Samples were lysed and ubiquitin protein conjugates were purified by His-tag affinity chromatography exactly as previously described, with the exception that no reduction and alkylation was performed on purified ubiquitin-conjugated proteins 5. Additionally, prior to His-tag purification, a 100-μg aliquot was removed from each sample and diluted five-fold in 50 mM Tris, pH 8.9. This aliquot, which was used for the analysis of cell mixing normalization and ubiquitin protein quantification, was reduced and alkylated prior to digestion with lysyl endopeptidase (LysC) at RT overnight. The resulting peptides were desalted on 50 mg tC18 SepPak cartridges prior to MS analysis.
In vitro ubiquitylation reactions
In vitro reactions were performed in a 25-μl volume and contained 0.5 μM Uba1 E1 enzyme from wheat, 0.5 μM E2 enzyme, 20 μM recombinant ubiquitin (S65A, WT, or S65E), 2 mM ATP, 2 mM DTT, 50 mM Tris, pH 7.6, 10 mM MgCl2, and 100 mM NaCl. The wheat Uba1 E1 enzyme was a gift from the Rachel Klevit laboratory, and Ubc13/Mms2 was purchased from LifeSensors. Ubc1 was subcloned into PGEX-4T1, expressed in E. coli, and purified with glutathione agarose affinity purification and size exclusion chromatography. For the E1 loading assay, E2 enzyme was omitted from the reaction. Immunoblotting of in vitro reactions was performed using an anti-ubiquitin antibody (P4D1, Enzo Life Sciences).
GST-Rsp5 HECT domain 425–809 was subcloned into PGEX-6P1 plasmid, expressed in E. coli, and purified with glutathione agarose affinity purification. Auto-ubiquitination reactions were carried using 0.1 μM Uba1, 0.2 μM Ubc4, 0.3 μM GST-Rsp5 HECT, 20 μM ubiquitin (S65A, WT, or S65E), 2 mM ATP, 2 mM DTT, 50 mM Tris, pH 7.6, 10 mM MgCl2, and 100 mM NaCl. Samples were taken at several time points and immunoblotting was performed using a GST-tag antibody (mouse monoclonal, Cat# A00865-40, GenScript).
Usp5 disassembly assays
Ubiquitin dimers were generated in a 40-μl volume containing 0.5 μM Uba1 E1 enzyme, 1 μM Ubc1 enzyme, 10 μM recombinant WT ubiquitin, 20 μM His-ubiquitin mutant S65E, 2 mM ATP, 2 mM DTT, 50 mM Tris, pH 7.6, 10 mM MgCl2, and 100 mM NaCl. Reaction proceeded for 1 h at 37°C, and 40 units of CIP were added to the reaction for 10 min to deplete ATP. Usp5 (Enzo Life Sciences) was pre-activated at 32 ng/μl for 10 min at room temperature in 50 mM HEPES, pH 7.8, 100 mM NaCl, and 5 mM DTT. About 7 μl of Usp5 solution was reacted with 7 μl of the dimers from above for 15 min at 37°C. Disassembly reaction was stopped with reducing LDS buffer, boiled, and run on an SDS–PAGE gel. Immunoblotting of in vitro reactions was performed using an anti-ubiquitin antibody (P4D1, Enzo Life Sciences).
Proteomic analysis of mutant ubiquitin strains
Yeast was grown in lysine-free synthetic complete media supplemented with 436 μM lysine. Culture for strain DS181 was supplemented with K0 lysine, DS147 was supplemented with K4 lysine (4,4,5,5-D4), and DS182 was supplemented with K8 lysine (13C6, 15N2) 52. Cells were grown in a 100 ml volume in biological triplicate and harvested at a density of ∼1.0 (OD600). Cells were resuspended at 4°C in proteomics lysis buffer consisting of 8 M urea, 75 mM NaCl, 50 mM Tris, pH 8.2, 50 mM NaF, 50 mM Na-β-glycerophosphate, 10 mM Na-pyrophosphate, 1 mM Na-orthovanadate, and 1× mini protease inhibitor (Roche). Cell suspensions were lysed by four repetitions of bead beating (1 min beating, 1.5 min rest). Tubes were spun first at 100 × g to remove beads and subsequently at 10,000 × g to pellet cellular debris. Supernatants were mixed 1:1:1 for each biological replicate. Each sample was reduced in 3 mM DTT at RT for 45 min and alkylated at RT in 8 mM iodoacetamide for 45 min in the dark, and finally, the reaction was quenched by the addition of 8 mM DTT for 15 min at RT. A 100-μg aliquot was removed from each replicate, diluted five-fold in 50 mM Tris, pH 8.9, and digested with 5 μg of LysC at RT overnight. This aliquot was used for the analysis of protein differences between the yeast strains. The remaining ∼20 mg of protein from each aliquot was diluted five-fold with 50 mM Tris, pH 8.2, and digested with 100 μg of trypsin at 37°C overnight (Promega). This larger aliquot was used for the characterization of differences in ubiquitylation between the yeast strains. After digestion, all samples were desalted on tC18 SepPak cartridges (Waters). LysC-digested peptides were directly analyzed by mass spectrometry, while tryptic peptides were enriched for di-glycyl peptides prior to MS analysis, as described elsewhere 25, 26. Eluates enriched in diGly peptides were loaded onto styrene divinylbenzene stage tips (3M) and washed with 50 μl of 0.15% TFA. Peptides were fractionated by stepwise elution from the stage tip under basic pH reversed-phase conditions. Four fractions were collected after the addition of 30 μl volumes of 20 mM ammonia with increasing acetonitrile concentration (6, 12, 18 and 80% acetonitrile, respectively). Fractions were lyophilized, resuspended in 4% formic acid and 3% acetonitrile, and directly analyzed by mass spectrometry.
Proteomic analysis of yeast turnover
The approach used here was based on a previously described experimental design 38. Each strain (DS147, DS181, and DS182) was individually cultured in lysine-free synthetic complete media supplemented with K0 or K4 lysine (4,4,5,5-D4) at 436 μM concentration 52. After a minimum of six doublings, cells were pelleted and the supernatant removed. Cultures grown in K0 media were resuspended in 20 ml of K0 media again, and cultures previously grown in K4 media were resuspended in media containing K8 lysine (13C6, 15N2) at an OD of ∼0.3. After 4 h of culture, cells had reached a density of ∼1.0, and 1.5 ml of culture was harvested by centrifugation. Cells from the same strain were mixed and resuspended at 4°C in proteomics lysis buffer (see above). Cell suspensions were lysed, reduced, alkylated, and digested with LysC as described above for the “Proteomic analysis of mutant ubiquitin strains”. After digestion, all samples were acidified to pH 2 with the addition of TFA and desalted on 100 mg styrene divinylbenzene solid phase extraction cartridges (Phenomenex). Peptides were fractionated by stepwise elution from the cartridge using basic pH reversed-phase conditions. Four fractions were collected after the addition of 500 μl volumes of 20 mM ammonia with increasing acetonitrile concentrations (6, 12, 18 and 80% acetonitrile, respectively). Fractions were lyophilized, resuspended in 4% formic acid and 3% acetonitrile, and directly analyzed by mass spectrometry.
Yeast spotting assay
Yeast cultures were grown to saturation in YPD overnight, pelleted, and diluted in water to a density of 1.0 OD600. Five 10-fold serial dilutions were prepared, and 4 μl of each dilution was plated onto a pre-warmed plate of either YPD or YPD supplemented with H2O2 (oxidative stress inducing), canavanine (arginine analog), 150 μM PS341 (proteasome inhibitor), 500 mM NaCl (osmotic stress), or 0.2% MMS (methyl methanesufonate, DNA damaging agent). Plates were imaged after 72–96 h of incubation at 30°C, depending upon cell growth of each particular stress condition.
Mass spectrometry analysis
Peptides were resuspended in 4% formic acid and 3% acetonitrile and loaded onto a 100 μm ID × 3 cm pre-column packed with Reprosil C18 1.9 μm, 120 Å particles (Dr. Maisch). Peptides were eluted over a 100 μm ID × 30 cm analytical column packed with the same material. Peptides were eluted into either a Velos Orbitrap or a Q-Exactive (Thermo Fisher) mass spectrometer by gradient elution delivered by an EasyII nLC or Easy1000 nLC system (Thermo Fisher). The exact gradient conditions were tailored to the complexity and chemical properties of each sample. Generally, the gradient was 8–30% acetonitrile in 0.15% formic acid over the course of 90–120 min. All MS spectra were collected with orbitrap detection, while MS/MS spectra of the 20 most abundant ions were collected either with ion trap detection and resonant excitation collision-activated dissociation (CAD), or with Orbitrap detection and beam-type CAD. The mass spectrometry proteomics data (raw and search result files) have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifiers ranging from PXD002197 to PXD002205 53.
AQUA mass spectrometry experiments used a mixture of heavy isotope peptides as internal standards for quantification. Non-modified ubiquitin peptides (ESTLHLVLR, TLSDYNIQK, and LIFAGK) were spiked in and measured after tryptic digestion, and S65 ubiquitin phosphopeptides (EpSTLHLVLR, TLSDYNIQKEpSTLHLVLR) were spiked in after digestion at a ratio of 1% over the non-modified peptides and measured after IMAC phosphopeptide enrichment. Samples were run on the Q-Exactive using a 60-min data-dependent acquisition method with similar settings as above. Quantifications were done by peak area integration of the precursor ion using the Skyline software package.
Identification and quantification of peptides, proteins, and modifications
All data were searched against the Saccharomyces cerevisiae database supplemented as appropriate with either the His-tagged ubiquitin sequence and/or the S65A- and S65E-mutated ubiquitin sequences. Searches and peptide quantifications were performed with the MaxQuant data analysis software 54. All peptide, protein, and PTM site identifications were filtered to a 1% false-discovery rate 55. To account for any errors in mixing of cells, all quantitative values were normalized such that the median of the protein quantification distribution was centered at log2 = 0. One exception was for the analysis of protein turnover data; here, normalization was performed such that the intensity of the light signal (K0) was equal to sum of the medium (K4) and the heavy (K8) signal. Another exception was the ubiquitin pull-down experiments; for these experiments, WT to mutant ubiquitin ratios were normalized to ubiquitin to account for differences in the amount of bait used.
For experiments involving biological replicates, only ubiquitylation sites identified in at least two replicates were included, and the statistical significance of quantitative measurements was calculated by performing a two-sided pairwise t-test between replicate results from S65A/WT and replicate results of S65E/WT.
Statistical analysis of differences between different yeast strain rates of protein degradation, synthesis, and turnover was performed using a two-sided t-test assuming unequal variance.
Bioinformatics
Box plots were generated using the BoxPlotR web-tool (http://boxplot.tyerslab.com/) 56. Gene ontology enrichment analysis was performed using the STRING database (http://string-db.org/) 34. For the turnover experiment, proteins in S65E ubiquitin yeast strains with the largest delay in turnover relative to S65A or S65WT ubiquitin yeast strains were analyzed for protein–protein interactions via STRING. Proteins within the primary interaction group were manually classified into mRNA decay, DEAD box helices, ribosomal, ribosome biogenesis and synthesis, or other proteins.
Acknowledgments
We thank S. Fields, L. Starita, R. Klevit, P. Brzovic, M. MacCoss, R. Gardner, D. Miller (University of Washington), and S. Beausoleil (Cell Signaling Techno-logy) for yeast strains and reagents, and S. Fields, R. Gardner, R. Klevit, and members of the Villén laboratory for critical discussions and reading of the manuscript. This work was supported in part by NIH/NCI grant R00CA140789 and the Ellison Medical Foundation New Scholar Award AG-NS- 0953-12 (to J.V.).
Author contributions
DS and RR-M performed experiments and analyzed data. JV supervised the research. All authors designed the research, discussed the results, and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 5C
Source Data for Figure 6
Source Data for Figure 7
Source Data for Table 1
References
- Finley D, Ulrich HD, Sommer T, Kaiser P. The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics. 2012;192:319–360. doi: 10.1534/genetics.112.140467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol. 2009;10:755–764. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, Clague MJ, Urbé S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- Swaney DL, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan NJ, Villen J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat Methods. 2013;10:676–682. doi: 10.1038/nmeth.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-J, Na K, Kwon M-S, Kim H, Kim KS, Paik Y-K. Quantitative analysis of phosphopeptides in search of the disease biomarker from the hepatocellular carcinoma specimen. Proteomics. 2009;9:3395–3408. doi: 10.1002/pmic.200800943. [DOI] [PubMed] [Google Scholar]
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, Kornhauser J, Sprott K, Zhou J, Possemato A, et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal. 2010;3:ra64. doi: 10.1126/scisignal.2000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MMK. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460:127–139. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo J-M, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56:360–375. doi: 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Swatek KN, Wagstaff JL, Gladkova C, Pruneda JN, Michel MA, Gersch M, Johnson CM, Freund SMV, Komander D. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 2015;34:307–325. doi: 10.15252/embj.201489847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amerik AYu, Swaminathan S, Krantz BA, Wilkinson KD, Hochstrasser M. In vivo disassembly of free polyubiquitin chains by yeast Ubp14 modulates rates of protein degradation by the proteasome. EMBO J. 1997;16:4826–4838. doi: 10.1093/emboj/16.16.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol. 2009;10:659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem. 2012;81:291–322. doi: 10.1146/annurev-biochem-051810-094654. [DOI] [PubMed] [Google Scholar]
- Su V, Lau AF. Ubiquitin-like and ubiquitin-associated domain proteins: significance in proteasomal degradation. Cell Mol Life Sci. 2009;66:2819–2833. doi: 10.1007/s00018-009-0048-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloper-Mould KE, Jemc JC, Pickart CM, Hicke L. Distinct functional surface regions on ubiquitin. J Biol Chem. 2001;276:30483–30489. doi: 10.1074/jbc.M103248200. [DOI] [PubMed] [Google Scholar]
- Lee SY, Pullen L, Virgil DJ, Castañeda CA, Abeykoon D, Bolon DNA, Fushman D. Alanine scan of core positions in ubiquitin reveals links between dynamics, stability, and function. J Mol Biol. 2014;426:1377–1389. doi: 10.1016/j.jmb.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossé L, Harley ME, Pires IMS, Hughes DA. Fission yeast Dss1 associates with the proteasome and is required for efficient ubiquitin-dependent proteolysis. Biochem J. 2006;393:303. doi: 10.1042/BJ20051238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seufert W, McGrath JP, Jentsch S. UBC1 encodes a novel member of an essential subfamily of yeast ubiquitin-conjugating enzymes involved in protein degradation. EMBO J. 1990;9:4535. doi: 10.1002/j.1460-2075.1990.tb07905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amerik AY, Li S-J, Hochstrasser M. Analysis of the deubiquitinating enzymes of the yeast Saccharomyces cerevisiae. Biol Chem. 2000;381:981–992. doi: 10.1515/BC.2000.121. [DOI] [PubMed] [Google Scholar]
- Lee J-G, Li W, Baek K, Boykins RA, Soetandyo N, Backlund PS, Ye Y, Wang G, Chen H-C. Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nat Commun. 2013;4:1568. doi: 10.1038/ncomms2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Paige JS, Jaffrey SR. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat Biotechnol. 2010;28:868–873. doi: 10.1038/nbt.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10:M111.013284. doi: 10.1074/mcp.M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele MJ, Elia AEH, Xu Q, Thoma CR, Izhar L, Leng Y, Guo A, Chen Y-N, Rush J, Hsu PW-C, et al. Global identification of modular cullin-RING ligase substrates. Cell. 2011;147:459–474. doi: 10.1016/j.cell.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig AO, Daran-Lapujade P, van Maris AJA, de Hulster EAF, de Ridder D, Pronk JT, Heck AJR, Slijper M. The diversity of protein turnover and abundance under nitrogen-limited steady-state conditions in Saccharomyces cerevisiae. Mol BioSyst. 2011;7:3316–3326. doi: 10.1039/c1mb05250k. [DOI] [PubMed] [Google Scholar]
- Belle A, Tanay A, Bitincka L, Shamir R, O’Shea EK. Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci USA. 2006;103:13004–13009. doi: 10.1073/pnas.0605420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, Kirkpatrick DS, Bingol B, Corn JE. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol. 2015;17:160–169. doi: 10.1038/ncb3097. [DOI] [PubMed] [Google Scholar]
- Mosammaparast N, Jackson KR, Guo Y, Brame CJ, Shabanowitz J, Hunt DF, Pemberton LF. Nuclear import of histone H2A and H2B is mediated by a network of karyopherins. J Cell Biol. 2001;153:251–262. doi: 10.1083/jcb.153.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake F, Saeki Y, Sakamoto K, Ohtake K, Nishikawa H, Tsuchiya H, Ohta T, Tanaka K, Kanno J. Ubiquitin acetylation inhibits polyubiquitin chain elongation. EMBO Rep. 2015;16:192–201. doi: 10.15252/embr.201439152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Franceschini A, Franceschini A, Szklarczyk D, Szklarczyk D, Frankild S, Frankild S, Kuhn M, Kuhn M, Simonovic M, Simonovic M, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2012;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gao J, Chung KKK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland G-L, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33:282–295. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Ana Clara F, Verstreken P. Synaptic vesicle trafficking and Parkinson’s disease. Dev Neurobiol. 2011;72:134–144. doi: 10.1002/dneu.20916. [DOI] [PubMed] [Google Scholar]
- Boisvert F-M, Ahmad Y, Gierlinski M, Charrière F, Lamont D, Scott M, Barton G, Lamond AI. A quantitative spatial proteomics analysis of proteome turnover in human cells. Mol Cell Proteomics. 2012;11:M111.011429. doi: 10.1074/mcp.M111.011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroha K, Ando K, Nakagawa R, Inada T. The Upf factor complex interacts with aberrant products derived from mRNAs containing a premature termination codon and facilitates their proteasomal degradation. J Biol Chem. 2013;288:28630–28640. doi: 10.1074/jbc.M113.460691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Békés M, Okamoto K, Crist SB, Jones MJ, Chapman JR, Brasher BB, Melandri FD, Ueberheide BM, Lazzerini Denchi E, Huang TT. DUB-Resistant Ubiquitin to Survey Ubiquitination Switches in Mammalian Cells. Cell Rep. 2013;5:826–838. doi: 10.1016/j.celrep.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst A, Avvakumov G, Tong J, Fan Y, Zhao Y, Alberts P, Persaud A, Walker JR, Neculai A-M, Neculai D, et al. A strategy for modulation of enzymes in the ubiquitin system. Science. 2013;339:590–595. doi: 10.1126/science.1230161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang Y, Zhou L, Zhou L, Rouge L, Rouge L, Phillips AH, Phillips AH, Lam C, Lam C, et al. Conformational stabilization of ubiquitin yields potent and selective inhibitors of USP7. Nat Chem Biol. 2013;9:51–58. doi: 10.1038/nchembio.1134. [DOI] [PubMed] [Google Scholar]
- Haririnia A, Verma R, Purohit N, Twarog MZ, Deshaies RJ, Bolon D, Fushman D. Mutations in the hydrophobic core of ubiquitin differentially affect its recognition by receptor proteins. J Mol Biol. 2008;375:979–996. doi: 10.1016/j.jmb.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raasi S, Pickart CM. Rad23 ubiquitin-associated domains (UBA) inhibit 26 S proteasome-catalyzed proteolysis by sequestering lysine 48-linked polyubiquitin chains. J Biol Chem. 2003;278:8951–8959. doi: 10.1074/jbc.m212841200. [DOI] [PubMed] [Google Scholar]
- Hartmann-Petersen R, Hendil KB, Gordon C. Ubiquitin binding proteins protect ubiquitin conjugates from disassembly. FEBS Lett. 2003;535:77–81. doi: 10.1016/s0014-5793(02)03874-7. [DOI] [PubMed] [Google Scholar]
- Lambertson D, Chen L, Madura K. Pleiotropic defects caused by loss of the proteasome-interacting factors Rad23 and Rpn10 of Saccharomyces cerevisiae. Genetics. 1999;153:69–79. doi: 10.1093/genetics/153.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattern MR, Wu J, Nicholson B. Ubiquitin-based anticancer therapy: Carpet bombing with proteasome inhibitors vs surgical strikes with E1, E2, E3, or DUB inhibitors. Biochim Biophys Acta. 2012;1823:2014–2021. doi: 10.1016/j.bbamcr.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM, Raasi S. Controlled synthesis of polyubiquitin chains. Meth Enzymol. 2005;399:21–36. doi: 10.1016/S0076-6879(05)99002-2. [DOI] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- Spence J, Gali RR, Dittmar G, Sherman F, Karin M, Finley D. Cell cycle-regulated modification of the ribosome by a variant multiubiquitin chain. Cell. 2000;102:67–76. doi: 10.1016/s0092-8674(00)00011-8. [DOI] [PubMed] [Google Scholar]
- Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, et al. The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Spitzer M, Wildenhain J, Rappsilber J, Tyers M. BoxPlotR: a web tool for generation of box plots. Nat Methods. 2014;11:121–122. doi: 10.1038/nmeth.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 5C
Source Data for Figure 6
Source Data for Figure 7
Source Data for Table 1