

Abstract
Special populations, including women (non-pregnant and pregnant), pediatrics, and the elderly, require additional consideration with regard to clinical research. There are very specific regulatory laws, which protect these special populations, that need to be understood and adhered to in order to perform clinical research. This review provides a broad overview of some of the physiological differences in special populations and discusses how these differences may affect study design and regulatory considerations. These various special populations, with respect to regulatory affairs, are clearly defined within the Code of Federal Regulations. The definition of “special population” exists to provide enhanced awareness of their vulnerabilities, thereby allowing the creation of regulatory guidance aimed to decrease injury or outright harm. Currently, progress is being made to be more inclusive of special populations in clinical trials. This reflects changing attitudes towards drug information, with it being more representative of those patients that will ultimately be prescribed or exposed to the therapy. However, all research undertaken in these populations should be performed in a manner that ensures all protections of each participant are upheld.
Keywords: Geriatrics, human subject research, pediatrics, pregnancy, regulations
Introduction
The motto “One size fits all” cannot be applied for anything intended to fit the depth and breadth of a population, and is clearly inadequate for the determination of dosing regimens across the human population. Accounting for the heterogeneity of the general population, numerous clinical trials are required to gather the appropriate information to make dosing recommendations for a single drug. Special populations include women, children (pediatrics), the elderly (geriatrics), pregnancy (obstetrics), and patients with concurrent disease states. Each of these populations has specific considerations that must be taken into account in relation to study design and regulations.
Often these critically important populations are left out of pre-marketing clinical trials in the US (1). The typical inclusion/exclusion criteria for phase I or II clinical trials primarily limit participation to Caucasian adults of 18–65 years with a body mass index (BMI) less than 25 mg/m2, while excluding pregnancy, hepatic dysfunction, and a variety of other typical conditions encountered within the general population. As trials progress to phase III, the criteria are somewhat broadened. For instance, later stage trials are more likely to include those older than 65 years of age, increased diversity in ethnic background, patients with minor hepatic dysfunction, and those who are overweight. Yet, until very recently, children, pregnant women, and those with severe comorbidities have been very rarely incorporated into even late stage trials and, therefore, information remains limited for these and other populations that present some complexity to trial design (2). This means that dosing guidance for these populations is not based on rigorous study and the prescriber must extrapolate from dosing guidance based on a population that may not share many characteristics that may impact drug pharmacokinetics, creating an environment with a higher risk for sub-optimal therapy or, conversely, toxicity from over-dosing.
FDA “Guidance for Industry” documents and decision trees provide recommendations for medication dosing in special populations (e.g. pregnancy, children, and elderly). These guides emphasize the importance of drug dosing information for special populations on drug labels (3). Increased participation of individuals from “special populations” in clinical trials is a critical step to appropriately study these individuals and interpret the data to serve as the basis for improving clinical pharmacologic therapies.
Moreover, a lack of pre-clinical translational models contributes to the gap in knowledge that exists for special populations. While most would not consider females as a special population, the traditional research approaches have excluded them and established a scientific culture in which the acceptable norm is biased heavily towards young, healthier male participants, culminating in a profound lack of knowledge regarding drug metabolism and kinetics for approximately half of the population. A high profile example of the disparity of drug disposition knowledge and testing between males and females was recently revealed, resulting in a FDA Med Watch Safety Alert in 2013, with the sleep aid zolpidem (Ambien®, Sanofi-Aventis, Bridgewater, NJ). Zolpidem was granted FDA approval in 1992. In this case, after millions of prescriptions, the dose given to women, being equivalent to men, was nearly twice what it should have been. In 1993, the FDA issued guidance recommending powering clinical studies to detect gender-specific differences and subsequently issued regulations in 1998 and 2000, with the force of law bearing on this subject (4). The zolpidem story has served to heighten awareness that gender differences are likely a much more significant factor in drug disposition than previously thought (5).
Pre-clinical animal models in drug studies come in many forms, but often have a male bias. Typically, they are comprised exclusively of male animals of two species and restrict the investigation to healthy young animals. Once the initial pre-clinical animal studies are complete, models that have been characterized as being representative of the disease state under study are employed, if available. In an effort to address the historic lack of incorporating gender difference within drug studies, the NIH has now begun implementing policies that will require applicants to report their plans for including a balance of male and female in pre-clinical animal studies (6). This will augment requirements that have been built into human clinical trials (7).
The objective of the following review is to give a broad overview of some of the physiological differences in several special populations and discuss how these differences may affect study design and regulatory considerations.
Special populations
Women
Variability in drug response and disease progression between men and women has been well documented. For example, women appear to be more sensitive to QT interval prolongation following anti-arrhythmic drug administration (8,9). Disease progression can also manifest differently between men and women. This phenomenon is evidenced in multiple sclerosis, a neurodegenerative autoimmune disorder that has a higher prevalence in women, but demonstrates a more progressive disease course in men. Murine models have demonstrated a genetic link that may explain this difference via the toll-like receptor 7 gene, which has increased expression in male mice (10).
Estrogen levels may play a role in altered drug pharmacokinetics in women by affecting gastric emptying. This effect can change drug transit times between healthy non-pregnant women, throughout pregnancy, and at menopause (11–13). Very few studies have been conducted in humans to demonstrate changes in pharmacokinetics and pharmacodynamics that occur during the menstrual cycle, but those that have been done suggest altered motility and absorption during different phases of the menstrual cycle (14). In addition to the influences of sex hormones, differences in body composition also influence the absorption and distribution of drugs. Women tend to have a lower body weight, higher percentage body fat, and lower plasma volumes than men.
Pregnant women
In the US, ∼4 million women give birth each year (15). An estimated 90% take at least one medication during each pregnancy (16,17), and more than half take at least one medication or more during the first trimester (16,18). Very few studies have examined drug dosing and consequences as it pertains to the physiologic changes and trans-placental factors during pregnancy (16).
The rate at which a drug is absorbed can be greatly impacted by many factors occurring in pregnancy, including patients who are experiencing nausea and vomiting, changes in gastric volume and pH, drug metabolizing enzyme or transporter expression changes (19,20), or altered gastrointestinal motility (21). Additional factors that may alter pharmacokinetics and pharmacodynamics during pregnancy include changes in weight and body composition, pregnancies with multiples (e.g. twins, triplets), increased blood volume, and abrupt changes in behaviors such as alcohol and tobacco use (19,21).
The kidneys and liver are also affected during pregnancy. Increased intravascular volume may lead to 50% increased glomerular filtration and increased renal clearance due to higher creatinine clearance. These changes can be altered during the course of pregnancy and persist into the postpartum period. There is conflicting evidence surrounding changes in hepatic metabolism which may increase, decrease, or stay the same (20,21).
Fetal vulnerability to drug pharmacodynamics and toxicity is of concern for many clinical researchers working with pregnant women, and special consideration must be taken with differences that occur during each trimester. For example, thalidomide exposure to the fetus during the first trimester can cause severe impairment of limb development (i.e. phocomelia) (21–23); use of NSAIDS during the first trimester increases risk of miscarriage and malformation (e.g. gastroschisis), while use after 30 weeks' (3rd trimester) gestation increases the risk for premature closure of the ductus arteriosus (24). The interaction between a drug and development during each trimester must be considered when determining appropriate dosing (19).
In addition to the physiologic changes that occur in the mother, it is important for clinical researchers to consider the distinct genetic differences of the mother and the fetus that may have a pharmacological impact on one or both. For example, codeine is metabolized by cytochrome P450 isoform 2D6 (CYP2D6). This enzyme is induced during pregnancy and can effectively shift the phenotype of the mother into an ultra-rapid metabolizer of codeine (25,26).
As research participants, pregnant women are generally willing to participate in non-invasive research (27); however, any research that imposes risk must take into account both the mother and the unborn (28). As with other populations, it is important to clearly inform potential participants about the nature of the study and address concerns early. Clinical research in pregnant participants generates complex ethical issues and requires special protection.
Regulatory affairs
The US Congress established the Office of Women's Health (OWH) within the FDA in 1994. One of the primary mandates of this office was for the investigation of gender differences related to medicinal product safety and efficacy. In addition to tracking the rate of women participating in clinical trials and providing education, this office funds a wide variety of studies to better understand the role of gender and health. For instance, OWH research funding has been used to develop pre-clinical animal models, as discussed above (29).
The OWH extends to or encompasses the mission of advancing the understanding of gender and health to the state of pregnancy (29). Pregnant women very often receive or take medication and, therefore, warrant protection from “off-label” experimentation by being included in the process of clinical trials where relevant.
The FDA hosts a list of Pregnancy Exposure Registries (30). These registries are a hub for information on drugs and vaccines for pregnant women. Although, the registries are a great resource to find what data exist and is being actively collected, drug information concerning pregnant women is frequently unavailable. Therefore, many registries are seeking pregnant women for participation in a clinical study.
Pediatrics
Pediatric patients were referred to as “therapeutic orphans” by Shirkey (31) in 1968, because of inherent differences in drug metabolism in children they are usually not included in drug development. The American College of Clinical Pharmacology (32), in 2008, published a white paper detailing the lack of drug studies conducted in the pediatric population. Historically drugs were initially evaluated in children who were poor, institutionalized, mentally ill, or physically disabled (32). Later, drugs were evaluated in children after being approved in adults. The metabolic profile between adults and children is striking, with maturational changes taking place as development advances from fetus to infant to adolescent. Ginsberg et al. (33) evaluated numerous pharmacokinetic studies in the literature to characterize the differences between adults and children in drug metabolism. For example, they reported that with some drugs there was a decreased clearance in the neonate and infant up to 2 months of age followed by a rise in metabolic capacity that could exceed that of an adult from ∼6 months to 2 years of age. Additionally, they also described a general phenomenon whereby many drugs, that are primarily excreted renally, are cleared at a faster rate than adults, in children between the ages of 2–12 years of age (33). The relative size and perfusion of organs (e.g. liver) is also different between children and adults, which impacts the pharmacokinetic profile of many drugs (34).
Hepatic drug metabolism and elimination is divided into two phases: oxidation, reduction, and hydrolysis (phase I); and hydroxylation and conjugation (phase II). Phase I metabolism at birth is ∼30% that of adults and then rapidly increases to exceed adult rates by age 3 years for some drugs (35). By early puberty Phase I metabolic capacity is trending toward adult levels and is attained by the end of puberty. Phase II metabolism has much greater variation that is independent of stage of growth and is due to different substrates involved in hydroxylation and conjugation (35–37).
From birth through infancy developmental processes are highly dynamic and can profoundly affect drug metabolism. As the body continues to mature to adulthood, there are drastic changes in body form and proportion, resulting in decreased total body water (TBW) and fat stores. These differences in TBW and body composition will directly affect the absorption and volume of distribution of many compounds. Moreover, children tend to have less acidic stomach contents, resulting in better absorption of acid drugs, and experience delayed gastric emptying, which impacts the kinetics of absorption (34). Infants can experience increased levels of active drug in the circulation due to lower levels of serum albumin and, consequently, lower levels of protein-bound drug. Moreover, well into infancy, the blood–brain barrier (BBB) is often not fully developed and, consequently, there can be increased and unintended central nervous system drug exposure.
A lack of research in pediatric pharmacology is in part due to the ethical issues related to conducting these studies and the lack of financial incentives for industry. There is a tremendous need for pediatric clinical trials. However, much time and expertise is required to adequately design a pediatric clinical trial. Historically, arguments from ethical, practical, and economic perspectives led to adult clinical trial data being extrapolated to children (38). Current guidelines do encourage the use of extrapolation for efficacy when it is appropriate to use data obtained from adults to children or from older to younger children (38). Additionally, the application of extrapolation, modelling, and simulation should be considered once the specific pediatric needs and study-related question(s) are answered. The limitations of extrapolation are generally related to dose-finding/PK studies and safety studies where these aspects are not transferable between adults and children (38).
The main limitation to undertaking PK studies in newborns and infants is related to blood collection (39). Obviously, the number of samples and blood volumes that can be taken in children, especially very sick children, will be far less than that of an adult. It is becoming accepted that, when developing assays for PK studies in children, there is a need to utilize sampling techniques that require as small a volume of blood as possible. There is a trend to develop micro-analytical methods suitable for doing PK studies in children and neonates. The most widely used micro-analytical methods use chromatographic methods for the analysis of drugs in plasma or serum (40). There have also been advances in the development of methods that utilize dried blood spots (DBS) (40). There has been a rapid increase in the use of DBS in pediatric PK studies over the last 2 years. Although promising, DBS is still developing in its clinical utility. Concerns related to questions of drug stability, the effect of hematocrit level, and best practice extraction techniques are still being addressed. Therefore, consensus has yet to be reached for the approach to the application of DBS analytical procedures for TDM and clinical studies (40).
Neonates
In neonates, drug pharmacokinetics are affected by the rapid dynamic physical and physiological changes (e.g. weight or increase in GFR), making it difficult to appropriately use many drugs during the neonatal period (41,42). Additionally, there are differences between neonates related to gestational age (GA) and birth weight (43). Extremely low birth weight (ELBW) neonates (<26 weeks GA) exhibit the composition of displaced fetuses, whose total-body water comprises nearly 92% body weight, extracellular fluid (ECF) volume is ∼25%, and body fat is less than 1% (44,45). In term neonates, total body water falls to ∼75% body weight and body fat increases to ∼15%. At 40 weeks gestation, ECF volume has been estimated to range from 350–440 mL/kg body weight. In term neonates, ECF has been found to correlate more closely to body weight than GA (46). Intracellular fluid volume increases from 25% body weight in premature neonates to 33% in term neonates and ∼37% at 4 months of age (44,45).
In neonates, anatomical and functional immaturity of the kidney limits glomerular and tubular functional capacity. Renal maturation has been shown to be related to GA (47). The result is that GFR and absolute CL is decreased with decreasing GA (48). Hence, neonates <30 weeks GA have a lower CL and this results in a longer half-life of renally cleared drugs than neonates >30 weeks GA (49,50). The GFR in term neonates is ∼25% (20mL/min/1.73 m2) of that of an adult, and is even lower in extremely premature neonates (51). Comparatively, GFR can be reported as follows: at birth GFR for full-term neonates is 2–4 mL/min, while GFR for pre-term neonates is 0.6–0.8 mL/min (52). Normal adult GFR is ∼125 mL/min (53). GFR in neonates is increased rapidly during the first 6 h of life compared to the second or third day of life in full-term neonates, but the degree of change in GFR is more difficult to estimate in extremely premature neonates (54,55). Increasing GFR is associated with increasing postnatal age (PNA); however, in pre-term neonates the trajectory of GFR increase is slower than in full-term neonates (49,50,55).
The relationship between PNA and a neonates composition (i.e. percent body water) is one of the most important considerations when determining drug dosage in these patients (48). Water-soluble drugs have a higher volume of distribution (VD) so that a higher dose per kilogram is needed to achieve the same serum concentration comparable to older infants and children (56,57). Consequently, the dose of some drugs needs to be assessed and changed repeatedly to account for rapid changes in body composition during development (42,54). Then, as the neonate ages, his/her dosing requirement may be reduced to reflect the changing body composition.
It has been recognized that there are many issues that need to be addressed when undertaking drug studies in neonates and children. Some of the proposed solutions include expansion of the use of modeling and simulation by in silico investigations. These methods can provide a systematic manner of balancing factors to design a drug regimen that achieves target concentrations to optimize efficacy and safety (58). Marsot et al. (39) reviewed population PK studies done in children during the first 2 years of life. The span of children per study was 25–100, with ∼3–5 samples collected per patient. The majority of studies were prospective (60%), with the rest (40%) using retrospective data from therapeutic drug monitoring. Only 10% of the models that were described had been evaluated by an external evaluation. What is concerning is that only 2% of the prospective studies used optimal design/sampling. In the studies reviewed it was seen that the volume of blood collected was the limiting factor, despite the emergence of new assay techniques such as mass spectrometry which uses smaller sample volumes (39). This review clearly revealed that there is room for improvement on the methodological side of clinical research, but also that harnessing the power of newer modeling techniques may provide a deep insight into ways to improve dosing for neonates (39).
Regulatory affairs
Regulatory oversight of protecting human subjects in research was established in 1970 when the Department of Health and Human Services developed regulations for research with additional protections for children (59). The Code of Federal Regulations (CFR) states that research may include children if “(a) No greater than minimal risk to children is presented; and (b) Adequate provisions are made for soliciting the assent of the children and the permission of their parents or guardians as set forth in CFR 50.55” (60). It is important to note that the CFR does not specifically define minimal risk for a child. What may be considered minimal risk in an adult (e.g. venipuncture for obtaining a blood sample) may be considered “greater than minimal risk” for a child, depending on, for instance, the child's age, health status, or volume of blood to be collected.
Often pharmaceutical studies are determined to be “greater than minimal risk”, but are allowed because the research offers the prospect of direct benefit or may contribute to the well-being of the individual child (61,62). There are two more categories of research as it applies to children: (1) Research involving interventions or procedures that present a minor increase over minimal risk without the prospect of direct benefit to the individual participant, but is likely to produce generalizable knowledge about the child's disorder or condition (63,64). (2) Research which presents an opportunity to understand, prevent, or alleviate a serious problem affecting the health or welfare of children. Research in this category must be reviewed and approved by the Secretary of DHHS or the Commissioner of the FDA (65,66).
The history of regulation specific to pediatric use goes back to 1979, when product labels first started to include pediatric use. Since that time there has been a number of changes (Figure 1), with the most significant changes occurring in 2002 and 2003, with the Best Pharmaceuticals for Children Act (BPCA) and Pediatric Research Equity Act (PREA). The BPCA (2002) facilitated clinical trials for on-and off-patent drugs in children by providing an additional 6 months of patent exclusivity for on-patent drugs tested for pediatric use, and established a program for pediatric drug development. In 2012, the Food and Drug Administration Safety and Innovation Act (FDASIA) replaced the BPCA and PREA Acts and became a permeant law (67).
Figure 1.
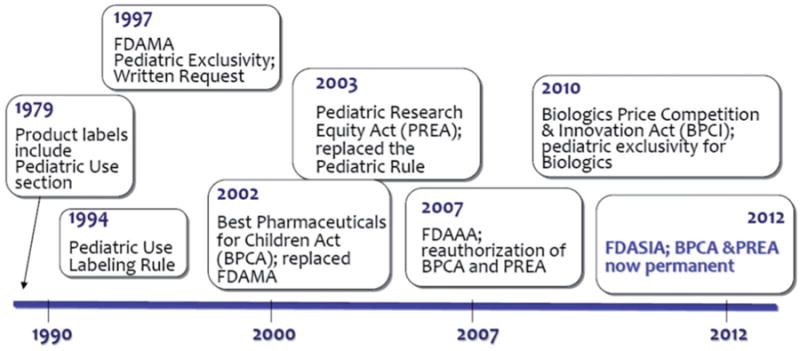
Timeline of the changes to the regulations related to pediatric product development (67).
Geriatric
Advances in medicine in the last 100 years have resulted in a substantially increased life expectancy and a burgeoning population of geriatric patients. It is estimated that, by 2030, there will be over 70 million people over the age of 65 years, which will account for 20% of the population in the US (68). In the US, between the years 2007–2008 more than 75% of people 60 years and older used two or more prescription drugs and 37% used five or more prescription drugs (69). The older patient, while receiving more drugs on average than younger patients, often poses a greater challenge to dose appropriately as the geriatric patient potentially has altered kinetics due to declining organ function and concurrent disease, and drug–drug interactions are more likely and hard to predict. Adverse drug events occur in ∼15% of elderly patients. Of which, about half of the events are estimated as being preventable (70).
Older people with multiple disease processes requiring treatment with multiple medications increase the risk for drug–drug interactions that may cause an increased or decreased bioavailability of the drug. Drugs such as phenytoin and estrogens that are metabolized by the cytochrome P450 pathway, when used concomitantly, require higher doses to produce the desired effect because their metabolism is increased. In some cases, only one drug may undergo rapid metabolism while the other does not. Geriatric patients will often have decreased stomach acid, reduced intestinal blood flow and slower gastric-emptying time. These alterations tend to perturb the rate of drug absorption, but not the amount of drug that is absorbed. Therefore, there may be a delay in the onset of action and peak effect of medications. Some examples of medications affected by changes in the gastrointestinal tract include anti-epileptic drugs (phenytoin, carbamazepine) (71), indomethacin, prazosin, and digoxin (72).
The distribution of a drug often depends on the nutritional status of the patient. Geriatric patients typically have reduced lean body mass and may have an increase in body weight resulting in some drugs distributing more easily, such as opiates (i.e. hydrocodone, oxycodone) (73) and long-acting benzodiazepines (i.e. diazepam, flurazepam). Additional factors that can affect a drug's distribution are a reduction in drug binding, such as to serum albumin, and reduced cardiac output.
Reduced hepatic blood flow and mass can lead to a decrease in drug metabolism, contributing to increased concentrations of circulating drug and, possibly, drug–drug interactions in geriatric patients. Pathophysiologic factors such as thyroid disease, cancer, congestive heart failure, or smoking may influence the pharmacokinetic profile of drugs. With age there are also typically decreases in the expression level of the phase I and II enzymes, particularly in patients older than 60 years of age (74–76).
Renal function also tends to decline with age with a decrease in renal blood flow and consequent decrease in filtration rate. As with decreased hepatic blood flow a reduction in blood flow to the kidney can result in the accumulation of some drugs such as cardiac agents like digoxin and antibiotics (72). With altered gastrointestinal, hepatic, and renal metabolism and elimination capacity, there are several drug categories that should be used with caution in the elderly.
Elderly patients with reduced organ function are often excluded from clinical trials, making it difficult for clinicians to determine safe doses. However, including elderly patients with comorbidities in trials is a difficult problem as this population is, by definition, highly heterogeneous and would require the enrollment of many more subjects to determine safety and efficacy of a drug. Moreover, experience has shown that, among elderly patients with comorbid conditions, they are less likely to complete a trial due to decreased follow-up, impaired cognition, and higher mortality rates. Therefore, “recruitment bias” results in healthier individuals with higher cognitive function representing the “elderly population” in the drug development process (77,78).
Regulatory affairs
FDA recommends that drug sponsors include elderly people (65 years and older) in clinical trials, as well as requiring that drug sponsors report data by age. The US Government Accountability Office found that ∼25% of clinical trials reviewed by medical officers documented the sufficiency (or lack thereof) of representation of elderly people (79).
Individuals with Decisional Impairment are those who have diminished capacity for understanding information or making a reasoned decision due to a cognitive or emotional disorder. A legally authorized representative (LAR) may consent for these individuals to participate in research studies. The LAR may be the spouse of a married person or any adult child (80,81).
Animal models
The need for developing better translational animal models is an ongoing challenge. Many species differences, a lack of knowledge on age specific maturation in different organ systems, and a scarcity of appropriate disease models hinder the ability to effectively predict human drug doses from animal studies.
In situations where human studies are not feasible (e.g. represent too much risk), animal studies are pivotal in providing the information necessary to determine the appropriate dosing and efficacy of a drug before it is used in the human population of interest. The Animal Rule of 2014 is a draft guidance for industry for product development published by the FDA (82). This guidance provides recommendations for drug development when human efficacy studies are not ethical or feasible. The Animal Rule clearly states that FDA will rely on evidence from animal studies to provide substantial evidence of effectiveness only when all of the following four criteria, quoted below, are met (82, p. 3):
There is a reasonable well-understood pathophysiological mechanism of toxicity of the substance and its prevention or substantial reduction by the product;
The effect is demonstrated in more than one animal species expected to react with a response predictive for humans, unless the effect is demonstrated in a single animal species that represents a sufficiently well-characterized animal model for predicting the response in humans;
The animal study end-point is clearly related to the desired benefit in humans, generally the enhancement of survival or prevention of major morbidity; and
The data or information on the kinetics and pharmacodynamics of the product or other relevant data or information, in animals and humans, allows selection of an effective dose in humans.
When focusing on women and the influence of hormones, one needs to consider the differences in reproductive cycles. Animal with estrus cycles include rodents, dogs, cats, swine, and sheep, all spontaneous ovulators, while cats and rabbits are induced ovulators. In contrast, non-human primates, such as rhesus macaques, have a menstrual cycle similar to humans. Placentation varies considerably amongst species, with the mouse and non-human primate being similar to the human. Sheep have a cotyledonary placenta, which allows for poor transfer of substrate, rendering it an undesirable model for human placental transfer, whereas guinea pigs are a well-established model for placental transfer and fetal growth restriction (83).
Age-specific maturation of drug metabolizing enzymes and transporters as well as physiological alterations need to be comparable to that of the human population being investigated to assess the probability that drug absorption, distribution, metabolism, and excretion will follow similar pathways. A number of studies have described the hepatic ontogeny of phase I and II enzymes mainly in rodents (84,85) and also in sheep (86). However, limitations with such models exist. For example, the sheep is a ruminant, and once the young animal begins eating solid food it develops a multi-compartment stomach and relies on fermentation for digestion, thus greatly altering the disposition of orally administered compounds. Roth et al. (87) demonstrated that juvenile pigs can serve as a human pediatric surrogate for pre-clinical pharmacokinetic testing of oral compounds as demonstrated with rifampicin.
No animal model is perfect and an abundance of differences exist between humans and the animals we use to model them; however, the information we gain from these studies will guide us to evidence-based dosing strategies, ultimately resulting in improved patient care.
Discussion
In special populations there are often significant deficits in the quality and efficacy of studies used to determine PK, PD, appropriate dosing, application in different age periods, and many questions surrounding the derivation of doses for neonatal to children from adult studies. The majority of data related to the safety of interventions and medicinal products given to neonates or children are obtained from adult studies (88). A common assumption is that safety and efficacy will be the same for children as it is in adults, and this is typically accepted (58). However, this assumption is based on little, if any, evidence, and this means that children, particularly those in the newborn group, are constantly participants in uncontrolled N-of-1 trials (38). In circumstances where data is limited the use of a particular drug for a given indication/condition can put children at increased risk (38). There are two contradictory ethical requirements that need to be balanced with respect to children involved in research; (1) there is a need to obtain evidence of efficacy and safety for the medication(s) and (2) there is the need for respect and protection of the child in the research environment (88).
It is well known that PK in pregnant women, children, and neonates is very different than in adults with respect to drug absorption, distribution, metabolism, and elimination (39). These specific characteristics impact the general fundamentals of clinical pharmacology, clinical research, and clinical trials and warrant a population-focused approach for drug administration and patient-specific PK and PD (89). Those involved in drug development should consider different study designs that account for the PK characteristics of pregnant women, children, and neonates as well as comply with the regulatory and ethical concerns of clinical research in these populations (89).
One set of tools that can be applied by clinical pharmacologists are the application of modeling and simulation techniques (89). Population modeling has been driven by the need for accurate PK models of numerous drugs. Non-linear mixed effects modeling is commonly used for a population-based approach, with inclusion of estimates of intra- and inter-individual variability which allows for simulation of drug dosing regimens. The technique also allows for the inclusion of covariates for disease state, bodyweight, and age, which are of particular relevance to pediatric and neonatal populations, to be taken into account in the PK analysis (55).
Marsot et al. (39), in reviewing population PK studies in newborns and infants, found that 51% of studies that used these software tool and advanced clinical trial designs and techniques reported an estimate for PK parameters in that population, with 49% concluding with dosing recommendations, despite the fact these studies had not made a clinical evaluation of the new proposed dosing regimen (11,90).
Over the last 10 years there has been an increase in the use of modeling techniques to improve not only drug development efficiency, but also for the design and conduct of clinical studies in neonates, pediatric patients, and pregnant women which has always been challenging (91). Population PK modeling offers many advantages to doing drug studies in these special populations (54). The increasing number of studies correlates with the use of spares data, which reduces the need for large blood volumes and invasiveness, making it increasingly possible to do robust PK studies in special populations (92). This approach is of immense benefit to not only clinicians treating special populations, but to those undertaking clinical trials. Consideration of the specific requirements of special populations will continue to allow more studies to be done where one study can use different doses, times of sampling, numbers of samples, and occasions, and be conducted under “daily life” conditions (93).
Future considerations for clinical research
The various groups that make up special populations are clearly defined within the Code of Federal Regulations. The definition of special population exists to provide enhanced awareness of their vulnerabilities, thereby allowing the creation of regulatory guidance aimed to decrease injury or outright harm. This definition attempted to define and support the ethical position needed to afford this protection, yet has been used to justify the avoidance of much needed research in the very same special populations that the regulations were intended to protect.
It is clear that the intent of these regulations was not to cripple or hinder the attainment of knowledge, but rather to allow data to be gathered in an ethical and respectful manner. The dualism of obtaining assent from a cognizant, yet legally immature participant, while obtaining parental or guardian permission to satisfy the legal need for consent, places the necessary choices in the hands of those who have the most at stake—the individual and their guardian(s). While this does indeed take additional time and effort, it provides the assurance that the highest quality data can be obtained without placing undue burdens upon the participant. This is no different than clinical care or behavior within the school system, yet it has a disproportionate effect on research that is not seen in either of the two other situations.
A balance that allows the ethical gathering of information from pregnant women, children, prisoners, and those with impaired cognition is necessary and long overdue. Special populations should not be forced to remain “special” out of ignorance of their conditions, treatments, adverse events, and outcomes. Research must be conducted to enhance our knowledge and provide better care for each individual within those populations, for the same ethical reason that they are considered special. For those individuals who do this type of research, adhering to the regulations is second nature as there is an intrinsic understanding of those populations and the importance of the protections. Often, those who do not understand the need for the regulations or the populations cite those regulations as reasons to not conduct research in those groups that it is most needed. This is clearly seen when it comes to decisions of funding of research, recruitment of potential participants, and publication of results.
To make the progress in research that these special populations deserve, a change in attitude needs to occur. The uniqueness of these populations requires research to be done, and when done, performed in a manner that ensures all protections of each participant are upheld. These populations deserve the ability to understand their own health, care, and well-being based upon actual data, just as other groups that are receiving optimal care.
Conclusions
The frequently dynamic physiology (e.g. state of pregnancy or processes of maturation) presents a formidable challenge for testing drugs in special populations. Nonetheless, these patients, in whom the drugs will be used, deserve dosing recommendations that are based on adequate and accurate information derived from controlled trials. All of the populations outlined have increased inter-patient variability and confounders that necessitate the need for larger sample size when studies are undertaken. All populations deserve to be included in research studies, to increase understanding of the benefit and risks of a medication. The principle of “respect for persons” embodies the concept that individuals be invited and not deprived of the opportunity to participate in research.
Acknowledgments
N. L. Mihalopoulos was supported by a career development award from the National Heart, Lung and Blood Institute (HL092069).
Footnotes
Declaration of interest: The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Atuah KN, Hughes D, Pirmohamed M. Clinical pharmacology: Special safety considerations in drug development and pharmacovigilance. Drug Safety Int J Med Toxicol Drug Experience. 2004;27:535–54. doi: 10.2165/00002018-200427080-00006. [DOI] [PubMed] [Google Scholar]
- 2.Taylor HA. Monitoring adherence to the NIH policy on the inclusion of women and minorities as subject in clinical research. In: Committee NTI, editor. Comprehensive report. Silver Springs, MD, USA: United States Federal Agency; 2008. 2011–2012. [Google Scholar]
- 3.FDA. Guidance for industry. United States Federal Agency; 2001. [accessed 9 Nov 2014]. Available online at: www.fda.gov. [Google Scholar]
- 4.GAO. Report to congressional requestors, women sufficiently represented in new drug testing, but fda oversight needs improvement. [accessed 9 Nov 2014];2001 Available online at: www.gao.gov/new.items/d01754.pdf.
- 5.FDA. Safety Announcement 5-14-2013, Drug Safety Communication. Silver Springs, MD, USA: United States Federal Agency; 2013. FDA approves new label changes and dosing for zolpidem products and a recommendation to avoid driving the day after using ambien cr. [Google Scholar]
- 6.Clayton JA, Collins FS. Policy: NIH to balance sex in cell and animal studies. Nature. 2014;509:282–3. doi: 10.1038/509282a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.NIH. Human clinical trials. [accessed 3 Nov 2014];2001 Available online at: http://grants.nih.gov/grants/funding/women_min/guidelines_amended_10_2001.htm.
- 8.Rodriguez I, Kilborn MJ, Liu XK, et al. Drug-induced qt prolongation in women during the menstrual cycle. JAMA. 2001;285:1322–6. doi: 10.1001/jama.285.10.1322. [DOI] [PubMed] [Google Scholar]
- 9.El-Eraky H, Thomas SH. Effects of sex on the pharmacokinetic and pharmacodynamic properties of quinidine. Br J Clin Pharmacol. 2003;56:198–204. doi: 10.1046/j.1365-2125.2003.01865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du S, Itoh N, Askarinam S, et al. Xy sex chromosome complement, compared with xx, in the cns confers greater neurodegeneration during experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2014;111:2806–11. doi: 10.1073/pnas.1307091111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadik R, Abrahamsson H, Stotzer PO. Gender differences in gut transit shown with a newly developed radiological procedure. Scand J Gastroenterol. 2003;38:36–42. doi: 10.1080/00365520310000410. [DOI] [PubMed] [Google Scholar]
- 12.Hutson WR, Roehrkasse RL, Wald A. Influence of gender and menopause on gastric emptying and motility. Gastroenterology. 1989;96:11–17. doi: 10.1016/0016-5085(89)90758-0. [DOI] [PubMed] [Google Scholar]
- 13.Singer AJ, Brandt LJ. Pathophysiology of the gastrointestinal tract during pregnancy. Am J Gastroenterol. 1991;86:1695–712. [PubMed] [Google Scholar]
- 14.Heitkemper MM, Chang L. Do fluctuations in ovarian hormones affect gastrointestinal symptoms in women with irritable bowel syndrome? Gender Med. 2009;6:152–67. doi: 10.1016/j.genm.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.CDC. Births and natality. [accessed 1 November 2014];2013 Available online at: http://www.cdc.gov/nchs/fastats/births.htm.
- 16.Mazer-Amirshahi M, Samiee-Zafarghandy S, Gray G, van den Anker JN. Trends in pregnancy labeling and data quality for us-approved pharmaceuticals. Am J Obstet Gynecol. 2014;211:690.e1–e11. doi: 10.1016/j.ajog.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 17.Fine JS. Reproductive and perinatal principles. In: Nelson LS, editor. Goldfrank's toxicologic emergencies. New York, NY: McGraw Hill; 2010. pp. 423–46. [Google Scholar]
- 18.Mitchell AA, Gilboa SM, Werler MM, et al. National Birth Defects Prevention Study. Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am J Obstet Gynecol. 2011;205:e51–8. doi: 10.1016/j.ajog.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cleary KL, Roney K, Costantine M. Challenges of studying drugs in pregnancy for off-label indications: Pravastatin for preeclampsia prevention. Sem Perinatol. 2014;38:523–7. doi: 10.1053/j.semperi.2014.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abduljalil K, Furness P, Johnson TN, et al. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: A database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2012;51:365–96. doi: 10.2165/11597440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, Hebert MF, Venkataramanan R. Basic obstetric pharmacology. Sem Perinatol. 2014;38:475–86. doi: 10.1053/j.semperi.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito T, Handa H. Deciphering the mystery of thalidomide teratogenicity. Congen Anom. 2012;52:1–7. doi: 10.1111/j.1741-4520.2011.00351.x. [DOI] [PubMed] [Google Scholar]
- 23.Taussig HB. Phocomelia and thalidomide. Am J Obstet Gynecol. 1962;84:979. doi: 10.1016/0002-9378(62)90079-0. [DOI] [PubMed] [Google Scholar]
- 24.Antonucci R, Zaffanello M, Puxeddu E, et al. Use of non-steroidal anti-inflammatory drugs in pregnancy: Impact on the fetus and newborn. Curr Drug Metab. 2012;13:474–90. doi: 10.2174/138920012800166607. [DOI] [PubMed] [Google Scholar]
- 25.Campbell SC, Spigarelli MG. Pharmacology and pharmacogenomics of neurological medications used in pregnancy. Clin Obstet Gynecol. 2013;56:305–16. doi: 10.1097/GRF.0b013e31828f241d. [DOI] [PubMed] [Google Scholar]
- 26.Lam J, Woodall KL, Solbeck P, et al. Codeine-related deaths: The role of pharmacogenetics and drug interactions. Forensic Sci Int. 2014;239:50–6. doi: 10.1016/j.forsciint.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 27.Nechuta S, Mudd LM, Biery L, et al. Michigan Alliance for the National Children's Study. Attitudes of pregnant women towards participation in perinatal epidemiological research. Paediat Perinat Epidemiol. 2009;23:424–30. doi: 10.1111/j.1365-3016.2009.01058.x. [DOI] [PubMed] [Google Scholar]
- 28.Rodger MA, Makropoulos D, Walker M, et al. Participation of pregnant women in clinical trials: Will they participate and why? Am J Perinatol. 2003;20:69–76. doi: 10.1055/s-2003-38318. [DOI] [PubMed] [Google Scholar]
- 29.FDA. Womens health research. Office of Women's Health (OWH); 1994. [accessed 1 Nov 2014]. Available online at: http://www.fda.gov/ScienceResearch/SpecialTopics/WomensHealthResearch. [Google Scholar]
- 30.FDA. Pregnancy exposure registries. [accessed 1 Nov 2014];2011 Available online at: http://www.fda.gov/scienceresearch/specialtopics/womenshealthresearch/ucm251314.htm.
- 31.Shirkey H. Therapeutic orphans. J Pediat. 1968;72:119–20. doi: 10.1016/s0022-3476(68)80414-7. [DOI] [PubMed] [Google Scholar]
- 32.American College of Clinical Pharmacy. Cheang KI, Ott C, et al. Research in women and special populations. Pharmacotherapy. 2008;28:93E–113E. doi: 10.1592/phco.28.9.1203. [DOI] [PubMed] [Google Scholar]
- 33.Ginsberg G, Hattis D, Sonawane B, et al. Evaluation of child/adult pharmacokinetic differences from a database derived from the therapeutic drug literature. Toxicol Sci Offic J Soc Toxicol. 2002;66:185–200. doi: 10.1093/toxsci/66.2.185. [DOI] [PubMed] [Google Scholar]
- 34.Jacqz-Aigrain E, Choonara I, editors. Paediatric clinical pharmacology. New York: CRC Press; Taylor & Francis; 2006. [Google Scholar]
- 35.Pineiro-Carrero VM, Pineiro EO. Liver. Pediatrics. 2004;113:1097–106. [PubMed] [Google Scholar]
- 36.Kennedy MJ, Davis DA, Smith N, et al. Reduced activities of cytochrome p450 1a2 and xanthine oxidase in children with growth hormone deficiency. Clin Pharmacol Ther. 2008;84:674–8. doi: 10.1038/clpt.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tayman C, Rayyan M, Allegaert K. Neonatal pharmacology: Extensive interindividual variability despite limited size. J Pediat Pharmacol Ther: JPPT: Offic L PPAG. 2011;16:170–84. doi: 10.5863/1551-6776-16.3.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rocchi F, Tomasi P. The development of medicines for children. Part of a series on pediatric pharmacology, guest edited by gianvincenzo zuccotti, emilio clementi, and massimo molteni. Pharmacol Res Offic J It Pharmacol Soc. 2011;64:169–75. doi: 10.1016/j.phrs.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 39.Marsot A, Boulamery A, Bruguerolle B, Simon N. Population pharmacokinetic analysis during the first 2 years of life: An overview. Clin Pharmacokin. 2012;51:787–98. doi: 10.1007/s40262-012-0015-8. [DOI] [PubMed] [Google Scholar]
- 40.Millership JS. Microassay of drugs and modern measurement techniques. Paediatr Anaesth. 2011;21:197–205. doi: 10.1111/j.1460-9592.2011.03535.x. [DOI] [PubMed] [Google Scholar]
- 41.Stolk LML, Degraeuwe PLJ, Nieman FHM, et al. Population pharmacokinetics and relationship between demographic and clinical variables and pharmacokinetics of gentamicin in neonates. Ther Drug Monit. 2002;24:527–31. doi: 10.1097/00007691-200208000-00011. [DOI] [PubMed] [Google Scholar]
- 42.Shevchuk YM, Taylor DM. Aminoglycoside volume of distribution in pediatric patients. DICP, Ann Pharmacother. 1990;24:273–6. doi: 10.1177/106002809002400313. [DOI] [PubMed] [Google Scholar]
- 43.Paap CM, Nahata MC. Clinical pharmacokinetics of antibacterial drugs in neonates. Clin Pharmacokin. 1990;19:280–318. doi: 10.2165/00003088-199019040-00003. [DOI] [PubMed] [Google Scholar]
- 44.Friis-Hansen B. Water distribution in the foetus and newborn infant. Acta Paediatr Scand Suppl. 1983;305:7–11. doi: 10.1111/j.1651-2227.1983.tb09852.x. [DOI] [PubMed] [Google Scholar]
- 45.Butte NF, Hopkinson JM, Wong WW, et al. Body composition during the first 2 years of life: An updated reference. Pediat Res. 2000;47:578–85. doi: 10.1203/00006450-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 46.Bauer LA. Clinical pharmacokinetics handbook. New York: McGraw-Hill; 2006. [Google Scholar]
- 47.Besunder JB, Reed MD, Blumer JL. Principles of drug biodisposition in the neonate. A critical evaluation of the pharmacokinetic-pharmacodynamic interface (part 2) Clin Pharmacokin. 1988;14:261–86. doi: 10.2165/00003088-198814040-00001. [DOI] [PubMed] [Google Scholar]
- 48.van den Anker JN. Pharmacokinetics and renal function in preterm infants. Acta Paediatr. 1996;85:1393–9. doi: 10.1111/j.1651-2227.1996.tb13942.x. [DOI] [PubMed] [Google Scholar]
- 49.Miall LS, Henderson MJ, Turner AJ, et al. Plasma creatinine rises dramatically in the first 48 hours of life in preterm infants. Pediatrics. 1999;104:e76. doi: 10.1542/peds.104.6.e76. [DOI] [PubMed] [Google Scholar]
- 50.Wahl EF, Lahdes-Vasama TT, Churchill BM. Estimation of glomerular filtration rate and bladder capacity: The effect of muturation, ageing, gender and size. BJU Int. 2003;91:255–62. doi: 10.1046/j.1464-410x.2003.04053.x. [DOI] [PubMed] [Google Scholar]
- 51.Schwartz GJ, Feld LG, Langford DJ. A simple estimate of glomerular filtration rate in full-term infants during the first year of life. J Pediatr. 1984;104:849–54. doi: 10.1016/s0022-3476(84)80479-5. [DOI] [PubMed] [Google Scholar]
- 52.Coulthard MG. Maturation of glomerular filtration in preterm and mature babies. Early Human Dev. 1985;11:281–92. doi: 10.1016/0378-3782(85)90082-9. [DOI] [PubMed] [Google Scholar]
- 53.Assael B, Rusconi F. Aminoglycoside antibotics. In: Yaffe SJ, Aranda JV, editors. Pediatric pharmacology: Therapeutic principles in practice. Philadelphia, PA: Saunders Company; 1992. pp. 244–9. [Google Scholar]
- 54.Prober CG, Stevenson DK, Benitz WE. The use of antibiotics in neonates weighing less than 1200 grams. Pediatr Infect Dis J. 1990;9:111–21. doi: 10.1097/00006454-199002000-00009. [DOI] [PubMed] [Google Scholar]
- 55.Arant BS., Jr Developmental patterns of renal functional maturation compared in the human neonate. J Pediatr. 1978;92:705–12. doi: 10.1016/s0022-3476(78)80133-4. [DOI] [PubMed] [Google Scholar]
- 56.Alcorn J, McNamara PJ. Pharmacokinetics in the newborn. Adv Drug Deliv Rev. 2003;55:667–86. doi: 10.1016/s0169-409x(03)00030-9. [DOI] [PubMed] [Google Scholar]
- 57.Weber W, Kewitz G, Rost KL, et al. Population kinetics of gentamicin in neonates. Eur J Clin Pharmacol. 1993;44:S23–5. doi: 10.1007/BF01428387. [DOI] [PubMed] [Google Scholar]
- 58.Jacqz-Aigrain E. Drug policy in Europe research and funding in neonates: Current challenges, future perspectives, new opportunities. Early Hum Dev. 2011;87:S27–30. doi: 10.1016/j.earlhumdev.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 59. [accessed 5 Nov 2014];US Department of Health & Human Services Regulations. 2014 Available online at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=7.55.
- 60.United States Federal Agency. 21CFR § 50.55: Protection of human subjects subpart d - additional safeguards for children in clinical investigations, requirements for permission by parents or guardians and for assent by children. Washington, DC: U.S. Department of Health and Human Services; [accessed 13 Nov 2014]. Revised 2014. Available online at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=50&showFR=1&subpartNode=21:1.0.1.1.20.4. [Google Scholar]
- 61.United States Federal Agency. 45CFR § 46.405: Protection of human subjects subpart d – additional protections for children involved as subjects in research, research involving greater than minimal risk but presenting the prospect of direct benefit to the individual subjects. Washington, DC: U.S. Department of Health and Human Services; 2009. [accessed 13 Nov 2014]. Available online at: http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.html. [Google Scholar]
- 62.United States Federal Agency. 21CFR § 50.52: Protection of human subjects subpart d – additional safeguards for children in clinical investigations, clinical investigations involving greater than minimal risk but presenting the prospect of direct benefit to individual subjects. Washington, DC: U.S. Department of Health and Human Services; [accessed 13 Nov 2014]. Revised 2014. Available online at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=50&showFR=1&subpartNode=21:1.0.1.1.20.4. [Google Scholar]
- 63.United States Federal Agency. 45CFR § 46.406: Protection of human subjects subpart d – additional protections for children involved as subjects in research, research involving greater than minimal risk and no prospect of direct benefit to individual subjects, but likely to yield generalizable knowledge about the subject's disorder or condition. Washington, DC: U.S. Department of Health and Human Services; 2009. [accessed 13 Nov 2014]. Available online at: http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.html. [Google Scholar]
- 64.United States Federal Agency. 21CFR § 50.53: Protection of human subjects, part d – additional safeguards for children in clinical investigations, clinical investigations involving greater than minimal risk and no prospect of direct benefit to individual subjects, but likely to yield generalizable knowledge about the subjects' disorder or condition. Washington, DC: U.S. Department of Health and Human Services; [accessed 13 Nov 2014]. Revised 2014. Available online at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPar=50&showFR=1&subpartNode=21:1.0.1.1.20.4. [Google Scholar]
- 65.United States Federal Agency. 45CFR § 46.407: Protection of human subjects subpart d – additional protections for children involved as subjects in research, research not otherwise approvable which presents an opportunity to understand, prevent, or alleviate a serious problem affecting the health or welfare of children. Washington, DC: U.S. Department of Health and Human Services; 2009. [accessed 13 Nov 2014]. Available online at: http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.html. [Google Scholar]
- 66.United States Federal Agency. 21CFR § 50.54: Protection of human subjects, part d – additional safeguards for children in clinical investigations, clinical investigations not otherwise approvable that present an opportunity to understand, prevent, or alleviate a serious problem affecting the health or welfare of children. Bethesda, MD, USA: Department of Health and Human Services; [accessed 13 Nov 2014]. Revised 2014. Available online at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=50&showFR=1&subpartNode=21:1.0.1.1.20.4. [Google Scholar]
- 67.NICHD/NIH. Best pharmaceuticals for children act. [accessed 6 Nov 2014];2002 Available online at: http://bpca.nichd.nih.gov/about/Pages/Index.aspx.
- 68.US Census Bureau. The next four decades the older population in the United States: 2010 to 2050. [accessed 11 Nov 2014];May 2010; Available online at: www.census.gov/…/2010pubs/p25-1138.pdf.
- 69.Gu Q, Dillon CF, Burt VL. Prescription drug use continues to increase: U.S. Prescription drug data for 2007–2008. NCHS Data Brief. 2010;42:1–8. [PubMed] [Google Scholar]
- 70.Pretorius RW, Gataric G, Swedlund SK, Miller JR. Reducing the risk of adverse drug events in older adults. Am Fam Physician. 2013;87:331–6. [PubMed] [Google Scholar]
- 71.Gidal BE. Drug absorption in the elderly: Biopharmaceutical considerations for the antiepileptic drugs. Epilepsy Res. 2006;68:S65–9. doi: 10.1016/j.eplepsyres.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 72.Turnheim K. When drug therapy gets old: Pharmacokinetics and pharmacodynamics in the elderly. Exp Gerontol. 2003;38:843–53. doi: 10.1016/s0531-5565(03)00133-5. [DOI] [PubMed] [Google Scholar]
- 73.Chau DL, Walker V, Pai L, Cho LM. Opiates and elderly: Use and side effects. Clin Intervent Aging. 2008;3:273–8. doi: 10.2147/cia.s1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schmucker DL. Liver function and phase i drug metabolism in the elderly: A paradox. Drugs Aging. 2001;18:837–51. doi: 10.2165/00002512-200118110-00005. [DOI] [PubMed] [Google Scholar]
- 75.Robertson DR, Waller DG, Renwick AG, George CF. Age-related changes in the pharmacokinetics and pharmacodynamics of nifedipine. Br J Clin Pharmacol. 1988;25:297–305. doi: 10.1111/j.1365-2125.1988.tb03307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klotz U. Pharmacokinetics and drug metabolism in the elderly. Drug Metab Rev. 2009;41:67–76. doi: 10.1080/03602530902722679. [DOI] [PubMed] [Google Scholar]
- 77.Denson AC, Mahipal A. Participation of the elderly population in clinical trials: Barriers and solutions. Cancer Cont J Moffitt Cancer Center. 2014;21:209–14. doi: 10.1177/107327481402100305. [DOI] [PubMed] [Google Scholar]
- 78.Inouye SK, Studenski S, Tinetti ME, Kuchel GA. Geriatric syndromes: Clinical, research, and policy implications of a core geriatric concept. J Am Geriat Soc. 2007;55:780–91. doi: 10.1111/j.1532-5415.2007.01156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. [accessed 9 Nov 2014];GAO-07-47R: Elderly persons in clinical drug trials. 2007 Available online at: http://www.gao.gov/products/GAO-07-47R.
- 80.United States Federal Agency. 45CFR § 46.102 Protection of human subjects, subpart a – basic hhs policy for protection of human research subjects, definitions. Washington, DC: U.S. Department of Health and Human Services; 2009. [accessed 13 Nov 2014]. Available online at: http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.html. [Google Scholar]
- 81.United States Federal Agency. 21CFR § 50.3: Protection of human subjects, part a – general provisions, definitions. Washington, DC: U.S. Department of Health and Human Services; [accessed 13 Nov 2014]. Revised 2014. Available online at: http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.html. [Google Scholar]
- 82.FDA. [accessed 15 Nov 2014];Animal rule of 2014 is a draft guidance for industry for product development. 2014 Available online at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm399217.pdf.
- 83.Carter AM. Animal models of human placentation—a review. Placenta. 2007;28:S41–7. doi: 10.1016/j.placenta.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 84.Peng L, Cui JY, Yoo B, et al. Rna-sequencing quantification of hepatic ontogeny of phase-i enzymes in mice. Drug Metab Disposit Biol Fate Chem. 2013;41:2175–86. doi: 10.1124/dmd.113.054635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barroso M, Dargouge O, Lechner MC. Expression of a constitutive form of cytochrome p450 during rat-liver development and sexual maturation. Eur J Biochem FEBS. 1988;172:363–9. doi: 10.1111/j.1432-1033.1988.tb13895.x. [DOI] [PubMed] [Google Scholar]
- 86.Pretheeban M, Hammond G, Bandiera S, et al. Ontogenesis of phase i hepatic drug metabolic enzymes in sheep. Reprod Fertil Dev. 2012;24:425–37. doi: 10.1071/RD11159. [DOI] [PubMed] [Google Scholar]
- 87.Roth WJ, Kissinger CB, McCain RR, et al. Assessment of juvenile pigs to serve as human pediatric surrogates for preclinical formulation pharmacokinetic testing. AAPS J. 2013;15:763–74. doi: 10.1208/s12248-013-9482-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gill D. Ethical principles and operational guidelines for good clinical practice in paediatric research. Recommendations of the ethics working group of the confederation of European specialists in paediatrics (cesp) Eur J Pediatr. 2004;163:53–7. doi: 10.1007/s00431-003-1378-5. [DOI] [PubMed] [Google Scholar]
- 89.Allegaert K, van de Velde M, van den Anker J. Neonatal clinical pharmacology. Paediat Anaesth. 2014;24:30–8. doi: 10.1111/pan.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kawamura A, Graham J, Mushtaq A, et al. Eukaryotic arylamine n-acetyltransferase. Investigation of substrate specificity by high-throughput screening. Biochem Pharmacol. 2005;69:347–59. doi: 10.1016/j.bcp.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 91.Rocchi F, Tomasi P. The development of medicines for children. Part of a series on pediatric pharmacology, guest edited by Gianvincenzo Zuccotti, Emilio Clementi, and Massimo Molteni. Pharmacol Res Offic J It Pharmacol Soc. 2011;64:169–75. doi: 10.1016/j.phrs.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 92.Rybak MJ, Abate BJ, Kang SL, et al. Prospective evaluation of the effect of an aminoglycoside dosing regimen on rates of observed nephrotoxicity and ototoxicity. Antimicrob Agents Chemother. 1999;43:1549–55. doi: 10.1128/aac.43.7.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Winter ME. Basic clinical pharmacokietics. Baltimore, MD: Lippincott Williams & Wilkins; 2004. [Google Scholar]