

Abstract
Plasmodium falciparum infection can result in severe disease that is associated with elevated inflammation and vital organ dysfunction; however, malaria-endemic residents gain protection from lethal outcomes and manifest only mild symptoms during infection. To characterize host responses associated with this more effective antimalarial response, we characterized whole-blood transcriptional profiles in Rwandan adults during a mild malaria episode and compared them with findings from a convalescence sample. We observed transcriptional up-regulation in many pathways, including type I interferon, interferon γ, complement activation, and nitric oxide during malaria infection, which provide benchmarks of mild disease physiology. Transcripts encoding negative regulators of T-cell activation, such as programmed death ligand 1 (PD-L1), programmed death 1 ligand 2 (PD-L2), and the butyrophilin family member butyrophilin-like 2 (BTNL2) were also increased. To support an important functional role for BTNL2 during malaria infection, we studied chimeric mice reconstituted with BTNL2−/− or wild-type hematopoietic cells that were inoculated with Plasmodium berghei ANKA, a murine model of cerebral malaria. We found that BTNL2−/− chimeric mice had a significant decrease in survival compared with wild-type counterparts. Collectively these data characterize the immune responses associated with mild malaria and uncover a novel role for BTNL2 in the host response to malaria.
Keywords: Plasmodium falciparum, mild malaria, immune response, BTNL2, Plasmodium berghei, mouse model, experimental cerebral malaria, Rwanda HIV, malaria, antibody responses, atypical memory B cells
Fifty-seven percent of Africans reside in areas of Plasmodium falciparum transmission and infection, which can result in severe disease with high inflammation and significantly increased morbidity and mortality rates [1, 2]. Over time, malaria-endemic residents gain protection from severe disease and manifest mild clinical symptoms or are asymptomatic, despite having an ongoing bloodstream infection [3]. Robert Koch described this age-associated decrease in clinical symptoms, severe disease, and parasite burden >100 years ago, and studies have since demonstrated that repeated exposure to malaria parasites is associated with reduced clinical symptoms and inflammatory cytokines levels [4–7]. Further understanding of the mechanisms of antimalarial immunity and the reduction of malaria-related inflammation could inform a severe disease–modifying vaccine.
Malaria-infected individuals who develop protection from severe disease present with mild and nonspecific symptoms, such as fever, chills, sweats and headaches however, they do not develop encephalopathy, severe anemia or any of the other severe illness criteria [8]. Human and animal studies of malaria have demonstrated that antimalarial antibodies, interferon (IFN) γ, regulation of the inflammatory response, and the development of adaptive responses provide protection from severe disease [9]. To further study the host responses associated with mild malaria, we characterized host immune transcripts and pathways that were up-regulated in whole blood from a cohort of Rwandan adults during mild malaria compared with a convalescence sample obtained 30 days later.
We found an up-regulation of immune-related pathways, including response to type I IFN, cellular response to IFN-γ, complement activation, nitric oxide, tumor necrosis factor (TNF) α, interleukin 1, and others, which provide benchmarks of host physiology associated with mild malaria. We also identified up-regulation of the gene ontology (GO) pathway negative regulation of immune processes, which includes immune inhibitory genes, such as PD-L1, PD-L2, and butyrophilin-like 2 (BTNL2). BTNL2 was significantly up-regulated during mild malaria and is a member of the butyrophilin family, which includes important immune modulators that inhibit T-cell activation. To examine the functional significance of BTNL2 during malaria, we examined Plasmodium berghei ANKA infection outcomes in BTNL2−/− bone marrow chimeric C57BL/6 mice and identified a decrease in survival, suggesting that BTNL2 is a potential immunomodulator of host response and disease outcomes during malaria infection.
MATERIALS AND METHODS
Study Population
From January to April 2011, adult subjects with mild malaria were recruited from health clinics in Kigali City, Rwanda, which is hypoendemic for malaria transmission. Mild malaria was defined as any level of symptomatic P. falciparum parasitemia without evidence of vital organ dysfunction [8]. Subjects aged ≥18 years who presented with symptoms consistent with malaria and had a positive malaria smear were offered enrollment. The inclusion criteria required a positive thick blood smear and a positive malaria rapid diagnostic test (First Response Malaria Antigen Rapid Test; Premier Medical Corporation). All enrolled subjects provided informed consent and underwent a human immunodeficiency virus (HIV) test (Abbott Determine Rapid Test Strips for HIV-1/2 [Abbott Laboratories] and Uni-Gold HIV Rapid Test [Trinity Biotech]), symptom review, and physical examination and received artemether-lumefantrine for treatment of malaria. The percentage of parasitemia was calculated from 5 microscopic fields: [(No. of asexual parasites/No. of red blood cells) × 100]. Subjects were also examined and underwent venipuncture at day 30 of convalescence (after study enrollment). The study protocol was approved by the institutional review board of the Albert Einstein College of Medicine and the National Ethics Committee of Rwanda.
Sample Collection
Venipuncture was performed with K2 ethylenediaminetetraacetic acid blood collection tubes (BD Vacutainer), and 3 mL of the blood collected was added to Tri-Reagent BD (Molecular Research Center), mixed thoroughly immediately after collection, and frozen at −80°C. Peripheral blood mononuclear cells (PBMCs) were collected using CPT tubes (BD Vacutainer), washed with Hank's buffered salt solution (Lonza), frozen at a cell concentration of 106/mL in 90% fetal bovine serum (Atlanta Biologicals), and 10% dimethyl sulfoxide and stored at −80°C (Sigma). A complete cell blood count was performer for each subject at the National Reference Laboratory in Kigali.
Assessing the Host Transcriptome During P. falciparum Infection by Microarray
RNA from the whole-blood sample was extracted following the manufacturer's protocol (Tri-Reagent BD, Molecular Research Center). Twenty paired RNA samples with an A260/A280 ratio >1.5 and an RNA integrity number >6 (Agilent 2100 BioAnalyzer; Agilent Technologies) were randomly selected for hybridization. Sense-strand complementary DNA was generated from total RNA using the Ambion WT Expression Kit (Life Technologies), and labeling was done with the GeneChip WT Terminal Labeling Kit (Affymetrix) for use with the GeneChip Human Gene 1.0 ST Arrays (Affymetrix). The array image was generated by a high-resolution GeneArray Scanner 3000 7G (Affymetrix).
Generation of BTNL2−/− Mice
To develop BTNL2−/− mice, a targeting vector was generated by the introduction of a neomycin-resistance gene as a positive selection marker and diphtheria toxin A as a negative selection marker (Supplementary Figure 1A). Targeted embryonic stem cell clones were selected by means of polymerase chain reaction analysis and injected into B6 blastocysts to generate chimeras. The homologous recombination was confirmed using the following primer pairs (Supplementary Figure 1B): 5ARM Knockout (KO) = 3K, 5ARM-F: 5′-TGAGGGAGAAATAGCCCTTG and 5ARM-R: 5′-ATGCTCCAGACTGCCTTGGGA; 3ARM KO = 5K, 3ARM-F: 5′-GCTGATCCGGAACCCTTAAT and 3ARM-R: 5′-TGCAGTCACCAACCTGAGAG. High-percentage chimeras were bred with B6 mice for germline transmission. BTNL2+/− mice were backcrossed with B6 for 10 generations and then interbred to generate BTNL2 KO mice. Mice were generated at The University of Texas MD Anderson Cancer Center. All efforts were made to minimize suffering and provide humane treatment to the animals included in the study. Animal experiments protocols were approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center and by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine.
Mice
To create the chimeras, 6–8-week-old C57BL/6 (B6) female mice (Harlan Laboratories) were given a lethal dose of 1200 rad with a Shepard Mark I cesium irradiator and then hematopoietically reconstituted by intravenous injection of either wild-type (WT) or BTNL2−/− bone marrow cells (1–2 × 106 donor bone marrow cells per recipient), and the hematopoietic compartment was allowed to reconstitute for >8 weeks [10].
Infection and Parasites
Plasmodium berghei ANKA cl15cy1 (MRA-871) parasites (Malaria Research and Reference Reagent Resource Center) were intraperitoneally injected into 1 WT B6 mouse and grown for 4 days. Once parasitemia reached 2%–5%, 106 P. berghei–infected red blood cells were infected intravenously into WT and BTNL2−/− bone marrow chimeras. We observed a reproducible experimental cerebral malaria (ECM) death rate of approximately 25% in our study groups of female B6 mice, consistent with prior findings showing lower susceptibility of female compared with male mice during malaria infections [11].
Parasitemia and ECM
Parasitemia was determined with flow cytometry, as described elsewhere [12]. Briefly 1 µL of blood obtained from the tail tip was fixed with glutaraldehyde, permeabilized, incubated in 1 mg/mL RNase A (Sigma), stained with 0.5 µM YOYO-1 (Invitrogen), and directly analyzed by a BD FACSCanto II cell sorter (Becton Dickinson). Red blood cells were gated on forward and side scatter and parasitemia was determined as the frequency of YOYO-1–positive cells among all cells and was confirmed with microscopy of Giemsa-stained blood smears. Clinical scoring of ECM induced by P. berghei infection was done using the rapid murine coma and behavior scale (RMCBS), developed by Carroll et al [13]. The RMCBS quantifies 10 parameters, including coordination, exploratory behavior, strength and tone, reflexes and self-preservation, and hygiene-related behavior. The 10 parameters are each scored 0, 1, or 2, with a score of 2 denoting a normal response.
Statistical Analysis
Demographic and clinical data were analyzed using Stata software, version 12 (StataCorp). Differences were considered statistically significant at P ≤ .05 (2 tailed).
RNA Microarray
To define relative transcript levels, we calculated model-based gene expression values and performed nonlinear normalization based on Li-Wong methods [14] using dChip software, resulting in a gene list of 28 869 probe sets. To identify gene sets that were differential between the 2 time points (malaria infection and convalescence) we performed gene set enrichment analysis (GSEA), which uses a weighted Kolmogorov–Smirnov-like statistic (Gene Pattern; Broad Institute) [15]. Microarray data have been deposited in the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/; accession No. GSE64338).
To determine which individual genes were differentially expressed between the 2 time points we performed a paired t test of log2-transformed data using R (P < .001; 2 tailed), and the cutoff for clinical significance was a mean difference of 0.263 at log2 scale, which corresponds to a fold change of 1.2. The estimated median false discovery rate based on permutation of the labels was 0.006. Ingenuity Pathway Analysis (IPA) was used to identify canonical pathways associated with mild malaria infection (Ingenuity Systems).
Animal Model Experiments
The statistical significance of the difference in survival between groups was determined using a log-rank (Mantel–Cox) test. For RMCBS scores, Student t test was used. Both tests were performed using GraphPad Prism software, version 6.0a for Macintosh (GraphPad Software).
RESULTS
Demographics and Clinical Characteristics of the Study Cohort
To examine the host response during mild malaria in adults, we enrolled 57 HIV-negative adults with mild P. falciparum malaria who underwent clinical evaluation and venipuncture at enrollment and day 30 of convalescence. Subjects presented with myalgias, headache, joint pain, and gastrointestinal symptoms. The majority were afebrile at presentation and displayed minimal laboratory abnormalities with normal hematocrit (41%; interquartile range, 38%–46%) and a modest increase in neutrophils and monocytes and reduction in lymphocytes and platelets compared with convalescence (Supplementary Table 1). All patients were well at the convalescent visit and smear negative for parasites.
Up-regulation of Immune Responses During Mild Malaria Infection
To gain a global perspective of the immune response to P. falciparum mild malaria we performed whole-blood transcriptional profiling on 20 paired infected and matched convalescence samples. There were no significant differences in patient's laboratory features between the mild malaria episode and the convalescence visit except for a decreased platelet count at the time of malaria infection (Table 1). Using GSEA and an immune-specific GO library, we found up-regulation of 86 gene sets (P < .05; false discovery rate, ≤0.006) at the time of malaria infection, compared with convalescence (Supplementary Table 2). Table 2 displays selected up-regulated gene sets that reflect a broad range of immune responses, including IFN-γ production, response to type 1 IFN, nitric oxide biosynthetic process, humoral immune response, phagocytosis, complement activation, and negative regulation of immune system processes.
Table 1.
Clinical and Laboratory Characteristics of 20 Rwandan Adults During Mild Malaria Infection and at Day 30 of Convalescencea
| Characteristic | Median (Interquartile Range)b |
|
|---|---|---|
| Mild Malaria Infection | Convalescence | |
| Age, y | 25 (23–35) | … |
| Sex, % female | 47 | … |
| Duration of illness, d | 3 (3–6) | … |
| Temperature, °C | 37.0 (36.5–37.5) | … |
| Laboratory values | ||
| Parasitemia, % | 0.4 (0.3–0.5) | 0 |
| Blood cell count, ×109/L | ||
| Leckocytes | 4.9 (3.9–6.3) | 5.4 (4.8–6.2) |
| Neutrophils | 3.0 (1.5–3.7) | 2 (1.7–2.9) |
| Lymphocytes | 1.5 (0.8–1.8) | 2.3 (2.2–2.7) |
| Monocytes | 0.5 (0.4–0.6) | 0.4 (0.3–0.5) |
| Plateletsc | 149 (63–205) | 215 (165–240) |
| Hematocrit, % | 41.1 (37.4–43.8) | 41.8 (39–46.8) |
a Data are provided for the 20 sample pairs who underwent whole-blood transcriptional profiling.
b Values represent medians, except for the percentage of female patients.
c Statistically significant difference from time of infection to convalescence (P = .01; Wilcoxon matched-pairs signed rank test).
Table 2.
Selected Gene Sets Up-regulated During Mild Malaria Infection Compared With Day 30 of Convalescence in Rwandan Adultsa
| Gene Set | Description | Size | P Value | FDR |
|---|---|---|---|---|
| Inflammation | ||||
| GO.0006954 | Inflammatory response | 319 | <.001 | 0.006 |
| GO.0006809 | Nitric oxide biosynthetic process | 38 | <.001 | 0 |
| Cytokines | ||||
| GO.0034340 | Response to type I IFN | 59 | <.001 | 0 |
| GO.0032609 | IFN-γ production | 44 | .004 | 0.047 |
| GO.0032680 | Regulation of TNF production | 40 | .003 | 0.014 |
| GO.0032611 | IL-1β production | 20 | .003 | 0.013 |
| GO.0032612 | IL-1 production | 24 | <.001 | 0.012 |
| GO.0032635 | IL-6 production | 45 | <.001 | 0.002 |
| GO.0032637 | IL-8 production | 23 | .02 | 0.034 |
| GO.0032615 | IL-12 production | 27 | .006 | 0.048 |
| GO.0032620 | IL-17 production | 16 | .003 | 0.017 |
| Innate immune response pathways | ||||
| GO.0045087 | Innate immune response | 354 | <.001 | 0.001 |
| GO.0042116 | Macrophage activation | 21 | <.001 | 0.001 |
| GO.0002275 | Myeloid cell activation involved in immune response | 26 | <.001 | 0.011 |
| GO.0002444 | Myeloid leukocyte–mediated immunity | 25 | .01 | 0.046 |
| GO.0006909 | Phagocytosis | 66 | <.001 | 0.005 |
| Humoral pathways | ||||
| GO.0019724 | B-cell–mediated immunity | 72 | <.001 | 0.019 |
| GO.0006959 | Humoral immune response | 86 | <.001 | 0.024 |
| Adaptive immune response pathways | ||||
| GO.0002250 | Adaptive immune response | 120 | <.001 | 0.027 |
| Regulation | ||||
| GO.0002683 | Negative regulation of immune system process | 93 | <.001 | 0.022 |
| GO.0032872 | Regulation of stress-activated MAPK cascade | 15 | .02 | 0.047 |
| Coagulation pathways | ||||
| GO.0006956 | Complement activation | 42 | <.001 | 0.001 |
| GO.0002576 | Platelet degranulation | 76 | <.001 | 0.004 |
Abbreviations: FDR, false discovery rate; GO, gene ontology; IFN, interferon; IL-1, interleukin 1; IL-1β, interleukin 1β; IL-6, interleukin 6; IL-8, interleukin 8; IL-12, interleukin 12; IL-17, interleukin 17; MAPK, mitogen-activated protein kinase; TNF, tumor necrosis factor.
a Selected GO pathways were enriched in samples obtained during malaria infection compared with an uninfected 30-day convalescent sample. Gene sets were identified from whole-blood transcriptomes using gene set enrichment analysis with a library of GO functions for 891 immune-related gene sets (P < .05; FDR, ≤0.05).
Up-regulation of Inhibitory Molecules During Malaria Infection, Including BTNL2
We then examined genes that were up-regulated during malaria infection. Between the time of infection and convalescence, 263 genes demonstrated changes that were both statistically significant (P < .001; 2 tailed) and clinically significant (mean increase [log2 scale], 0.263; false discovery rate, ≤0.006) (Supplementary Table 3). Many of the individual transcripts up-regulated during mild malaria were consistent with the GSEA findings, including genes involved in the complement system (C1QA, C1QB, and SERPING1) and in the inflammatory response (IFITM3, CXCL10, matrix metalloproteinase 9, and IFI30). The inflammatory response biofunction (Figure 1) was the top up-regulated network (P = 1.3 × 10−8–3.5 × 10−2) using IPA.
Figure 1.

Up-regulation of the inflammatory response biofunction in Rwandan adults during mild malaria compared with day 30 of convalescence. The Ingenuity Pathway Analysis inflammatory response canonical pathway was significantly up-regulated (P = 1.25 × 10−8 –3.48 × 10−2) during infection and was generated from the differentially abundant transcripts at the time of malaria infection compared with day 30 of convalescence (P < .001; false discovery rate, ≤0.006). Pink denotes up-regulation; blue, down-regulation. Abbreviations: IFN, interferon; IgG, immunoglobulin G; IL1RL1, interleukin 1 receptor–like 1; IL5RA, interleukin 5 receptor, alpha subunit; IL-6; interleukin 6; IL-12, interleukin 12; IL-12B, interleukin 12B; IL-13, interleukin 13; MAPK, mitogen-activated protein kinase; MHC, major histocompatibility complex; MMP9, matrix metalloproteinase 9; PD-L1, programmed death ligand 1; TGF, transforming growth factor; TLR1, Toll-like receptor 1.
The cohort also demonstrated significant up-regulation of genes in the GO pathway termed Negative Regulation of Immune System Processes, which included PD-L1, PD-L2 and BTNL2. Other inhibitory molecules, including sialic acid–binding immunoglobulinlike lectin 5 (SIGLEC5) and members of the leukocyte immunoglobulin-like ligand family, were also up-regulated during infection (Figure 2, Supplementary Table 3) [16]. We focused our analysis on BTNL2 because its allelic diversity has been associated with inflammatory conditions, such as sarcoidosis, the gene has important immunomodulatory functions, and this is the first association between BTNL2 and the host response during malaria infection [17, 18].
Figure 2.

Induction of immune inhibitory genes during Plasmodium falciparum infection compared with day 30 of convalescence. Scatterplots show selected immune inhibitory transcripts from the microarray data for 20 study subjects assessed at the time of mild malaria infection and 30 days later. A, Butyrophilin-like 2 (BTNL2). B, Programmed death ligand 1 (PD-L1). C, Programmed death ligand 2 (PD-L2). D, Sialic acid–binding immunoglobulinlike lectin 5 (SIGLEC5). E, leukocyte immunoglobulin-like receptor, subfamily B (LILRB1). F, LILRB4. G, LILRB5. *P < .05; †P < .005; ‡P < .001. All P values were calculated using Wilcoxon matched-pairs signed rank test; bars represent median values for each scatterplot.
To confirm the BTNL2 transcript data and determine its cellular expression, we examined BTNL2 expression on T and B cells from PBMCs from malaria-infected subjects compared with uninfected US controls (Supplementary Methods). BTNL2 staining was detected on CD3+ T cells only in malaria-infected patients and not in US controls (P = .02; Mann–Whitney test) (Figure 3A). In contrast, BTNL2 was expressed similarly on CD19+ B cells in malaria-infected and uninfected US controls (Figure 3B) suggesting that BTNL2 may be functionally important on T cells.
Figure 3.
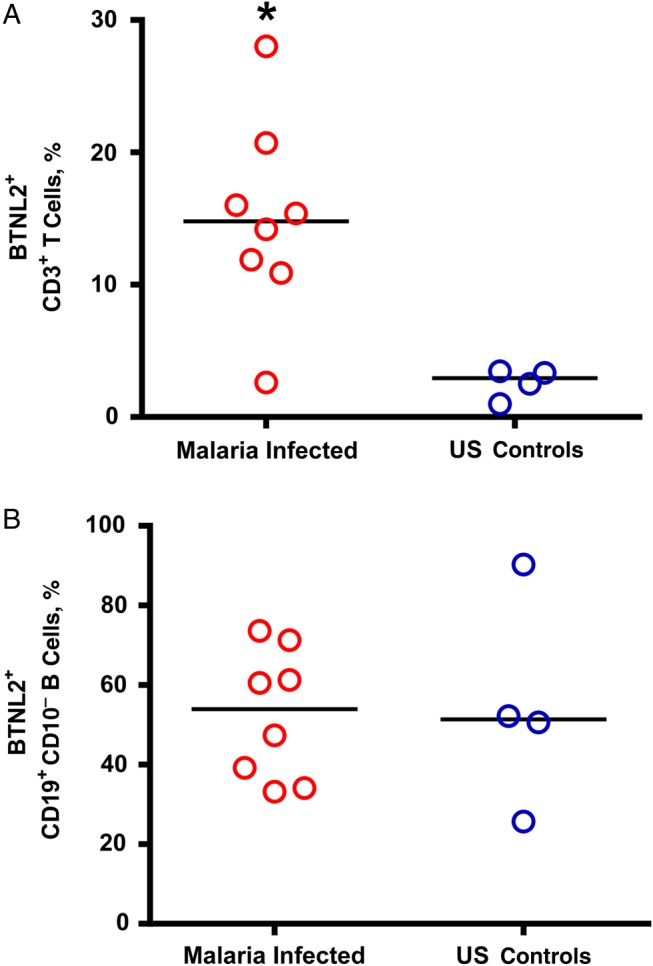
Percentage expression of butyrophilin-like 2 (BTNL2) on CD3+ T cells and CD19+ CD10- B cells in Rwandan adults with mild malaria compared with malaria-naive US controls. Surface expression of BTNL2 on peripheral blood mononuclear cells isolated from study subjects during infection were analyzed with flow cytometry. Surface expression of BTNL2 on CD3+ T cells and CD19+ B cells were measured in malaria-infected patients (n = 8) and malaria-naive US controls (n = 4). *P = .02 (Mann–Whitney test); bars represent median values.
Susceptibility of BTNL2-Deficient (BTNL2−/−) Mice to Blood-Stage Infection With P. berghei
To investigate the functional significance of BTNL2 in the P. berghei ANKA model of ECM, we lethally irradiated WT C57BL/6 (B6) female mice and reconstituted their hematopoietic compartment with either BTNL2−/− or control WT B6 bone marrow cells (Supplementary Figure 2, Figure 4). We observed 90% of donor-derived hematopoietic cells in both groups, with similar proportions of lymphoid and myeloid cell subsets, suggesting no obvious inherent defects in the hematopoietic compartment of BTNL2−/− chimeric or WT mice.
Figure 4.

Butyrophilin-like 2–deficient (BTNL2−/−) mice are more susceptible to Plasmodium berghei ANKA infection than wild-type (WT) mice. WT or BTNL2−/− bone marrow chimera mice were infected with 106 P. berghei ANKA–infected red blood cells. A, Kinetics of mice survival, B, Rapid murine coma and behavior scale (RMCBS) scores measured starting 4 days after infection, C, Kinetics of blood parasitemia. Data represent the pool of 2 independent replicate experiments (n = 15–16 mice; error bars show standard errors of the mean). *P < .05; †P < .01.
After 6–8 weeks, reconstituted mice were infected with P. berghei, and their survival, and blood parasitemia were monitored (Figure 4). BTNL2−/− chimera mice were significantly more susceptible to P. berghei infection than their WT counterparts, with 80% developing ECM and dying of infection, versus only 25% of WT female mice (Figure 4A). WT chimeras had only a modest drop in the RMCBS at day 6 after infection, from 12.5 to 12.0 (Figure 4B) [12]. In contrast, BTNL2−/− chimera RMCBS scores dropped significantly during the same period, from 10.1 to 5 (Figure 4B). Thus, the increased susceptibility of BTNL2−/− compared with WT chimeras was reflected in a significantly lower RMCBS. Despite differences in survival and ECM scores, levels of parasitemia did not differ between WT and BTNL2−/− chimeras (Figure 4C). Clinical parameters, such as body weight decrease and temperature loss, did not differ significantly between the groups (data not shown). Collectively, these data suggest that expression of the inhibitory ligand BTNL2 on hematopoietic cells significantly protected P. berghei–infected mice from developing ECM.
Increased Proliferation and Frequencies of T Cells in BTNL2−/− Versus WT Mice During P. berghei Infection
To gain further insights into the possible causes of increased ECM and death in BTNL2−/− mice infected with P. berghei, we analyzed the innate and adaptive immune response 6.5 days after infection, just as mice start experiencing severe symptoms of ECM (Figure 5 and Supplementary Figures 3 and 4). We found higher frequencies (and numbers, not shown) of conventional CD4+ T cells, Foxp3+ regulatory T cells, and CD8+ T cells in BTNL2−/− compared with WT chimeras (>15% overall increase), in both the spleens and the brains of infected mice (Figure 5A). A similar trend was also detected among Ly6C+ monocytes and neutrophils but not in B cells or natural killer cells (Supplementary Figure 3A). We observed very modest differences of activation for CD8+ T cells (CD25, CXCR3, Tbet), regulatory T cells (ICOS), and B cells (GL7) (Supplementary Figure 3B). We noted a 30% to 50% increase in the proportion of actively proliferating T cells (Ki67+) in BTNL2−/− compared with WT chimeras (Figure 5B and Supplementary Figure 4). Overall, these results support the idea that BTNL2 helps dampen T-cell proliferation and possibly T-cell activation in the P. berghei model of ECM.
Figure 5.

Butyrophilin-like 2–deficient (BTNL2−/−) mice exhibit increased proportion and proliferation of T cells compared with wild-type (WT) mice during experimental cerebral malaria. WT or BTNL2−/− chimeras mice were infected with 106 Plasmodium berghei ANKA–infected red blood cells. After 6.5 days, spleens and brains were harvested, stained for indicated markers and analyzed by flow cytometry. A, Frequency of indicated T-cell subsets among CD45+ hematopoietic cells in spleen and brain. B, Representative dot plots of indicated T-cell subsets (CD4, regulatory T cells [Tregs], CD8) expressing the marker of proliferation Ki67. Numbers represent the percentages of Ki67+ cells in the gate shown. In all bar graphs, dots represent individual mice, and bar shows the mean. Data represent the pool of 2 replicate experiments (n = 5–6 mice). *P < .05; †P < .01; ‡P < .0001 (unpaired t test); where values are not specified, differences are not significant.
DISCUSSION
We characterized the transcriptional responses in adult patients with mild malaria and identified a role for the inhibitory ligand BTNL2 during malaria infection. The up-regulated gene sets during mild malaria represent diverse arms of the immune system and reflect processes that are associated with mild clinical disease. We also found an up-regulation of immune inhibitory molecules such as PD-L1, PD-L2, SIGLEC5 and BTNL2, which may play a role in dampening inflammation during malaria. We demonstrated that BTNL2−/− mice in the P. berghei ANKA ECM model were more likely to develop lethal disease than WT mice despite similar parasite burdens, suggesting that BTNL2 has an important immunomodulatory role during malaria infection.
This cohort of Rwandan adults manifest the clinical symptoms of mild malaria which reflect exposure-related clinical immunity [5]. Patients were typically afebrile at the time of enrollment with nonspecific symptoms and normal hematocrits, which is in contrast to severe malarial disease, which involves vital organ dysfunction and significant levels of mortality [6]. To examine the global host responses associated with mild malaria, we carried out whole-blood transcriptional profiling and identified up-regulation of nitric oxide, phagocytosis, humoral, and adaptive immune response pathways and the IFN-γ, TNF-α, interleukin 1, and type I IFN cytokine pathways [19–21]. There is abundant evidence documenting the importance of IFN-γ, TNF-α, and nitric oxide and the role of antimalarial antibodies in parasite clearance in human and/or animal models [22–25]. The collective up-regulation of cytokine pathways in turn is controlled by regulatory pathways, which we found to be up-regulated (regulation of cytokine secretion and regulation of chemokine production).
Studies of children with mild malaria from Kenya, Benin, and Malawi have also shown up-regulation of inflammation, TNF-α, and complement pathways, despite differences in age and geographic location compared with the current study [19, 26, 27]. Direct comparisons of severe to mild transcriptomes are needed to determine the specificity of these transcriptional pathways with mild malaria. However, a prior whole-blood transcriptional study associated the up-regulation of response to type I IFN to mild compared with severe malaria, suggesting a role of type I IFN in mild disease [19]. The up-regulation of the type 1 IFN response during mild infection adds to a growing literature on the importance of this pathway in malaria [28, 29].
We then examined individual transcripts to identify specific mechanisms of host response (Supplementary Table 3). Up-regulation of the inflammatory response biofunction was the most significantly associated IPA network generated from the significantly differential gene list, reflecting the induction of inflammatory pathways even though patients displayed only minimal signs of clinical inflammation. To explore potential mechanisms that dampen inflammation during infection, we further examined genes that were significantly up-regulated in the negative regulation of immune system processes gene set. Up-regulation of members of leukocyte immunoglobulin-like receptor, subfamily B member 4 (LILRB4), BTNL2, SIGLEC5, and PD-L1, and PD-L2 were identified (Figure 2 and Supplementary Table 3). The majority of these inhibitory genes function to reduce T-cell activation; likewise, we found that genes involved in T-cell activation (CD1c and CD40LG) were down-regulated (Figure 1) [17, 30–32]. Only PD-L1 has been previously associated with malaria infection [30, 33].
We further examined a potential role of BTNL2, a negative regulator of T-cell proliferation, which can induce regulatory T-cell development and suppress T-cell proliferation and cytokine production [17, 34]. BTNL2 genetic polymorphisms are associated with sarcoidosis, ulcerative colitis, and systemic lupus erythematosus, also suggesting an important role in inflammation [18, 35, 36]. We found BTNL2 expression on CD19+ B cells in both the infected and uninfected control samples, reflective of prior findings of BTNL2 expression on many cell types including B lymphocytes [10, 17]. However, BTNL2+ CD3+ T cells were found only in the malaria-infected samples, suggesting that malaria infection results in T-cell up-regulation of BTNL2, consistent with the observation that BTNL2 is detected on concanavalin A–activated T cells and not on unstimulated T cells [10].
To examine the function of BTNL2 during malaria, we used the P. berghei ECM model. We chose a severe disease animal model as inflammation plays an important role in ECM outcomes. We identified decreased survival in the BTNL2−/− mice and found that these mice succumbed to ECM at a higher rate than the WT mice. Moreover, the BTNL2−/− mice had higher coma scores, suggesting that the function of BTNL2 in this infection model is to act as a key immunomodulator. Interestingly, we detected no difference in parasite load between BTNL2−/− and WT mice.
Our analysis of the immune response in the BTNL2−/− chimeras showed a significant increase in T-cell proliferation and possibly frequency, for conventional CD4+, Foxp3+ regulatory, and CD8+ T cells. This supports the idea that BTNL2 has an essential inhibitory function during malaria infection. Whether it prevents expansion of regulatory T cells during infection or directly inhibits T-cell function will require further study of P. berghei in BTNL2−/− mice. The lack of BTNL2 also leads to a modest increase in CD8+ T-cell activation and T-helper 1 differentiation (Tbet), consistent with a role for CD8+ T cells in the pathologic mechanism of ECM [37]. Furthermore, because BTNL2 is also expressed on B cells, its role in humoral responses to malaria needs to be investigated.
In summary, this study provides a transcriptomic analysis of the immune responses during mild P. falciparum malaria infection in adults. We identified the up-regulation of innate and adaptive immune pathways known to be important in the host defense to malaria. Concomitantly, there is an up-regulation of inhibitory immune molecules that may act to modulate the inflammation associated with malaria infection. BTNL2 may be involved in a newly described mechanism that mediates the host response to malaria. Future studies of BTNL2 and other immune inhibitory molecules in human severe disease and in animal models of non-lethal malaria are needed to fully define their functions in clinical immunity.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We acknowledge the study subjects for their time and commitment in participation in this study. We particularly acknowledge 3 research assistants: Marcus Bushaku, Gahongayire Fatuma, and Uwihanganye Jean Chrisostome for their contribution to this study. Albert Einstein College of Medicine Center for AIDS Research.
Author contributions. K. S. S. was involved in human study design and execution, human sample handling, RNA isolation, peripheral blood mononuclear cell fluorescence-activated cell sorting analysis, and manuscript preparation; E. S. and G. L. designed and carried out the rodent model experiments and were involved in manuscript preparation; E. I., E. M., C. M. F., and K. A. were involved in human study design and execution and manuscript preparation; X. L. and C. D. developed the BTNL2−/− mouse and were involved in manuscript preparation; X. Z. contributed BTNL2 expertise and manuscript development and J. P. D. was involved in human and mouse model study design, data analysis, and manuscript preparation.
Financial support. This work was supported by the National Institutes of Health (1RC1AI086224 to J. P. D.) and an Albert Einstein Global Health Center pilot grant. K. S. S. was supported by a Geographic Medicine and Emerging Infections training grant to the Albert Einstein College of Medicine from the National Institute of Allergy and Infectious Disease (NIH-NIAID T32 AI046985). E. S. was supported by NIH-NIAID T32 AI070117.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Grau GE, Taylor TE, Molyneux ME et al. Tumor necrosis factor and disease severity in children with falciparum malaria. N Engl J Med 1989; 320:1586–91. [DOI] [PubMed] [Google Scholar]
- 2.Lyke KE, Burges R, Cissoko Y et al. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect Immun 2004; 72:5630–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen E, Hogh B, Marbiah NT, David K, Hanson AP. Development of immunity against Plasmodium falciparum malaria: clinical and parasitologic immunity cannot be separated. J Infect Dis 1991; 164:949–53. [DOI] [PubMed] [Google Scholar]
- 4.Harrison G. Mosquitoes, malaria and man: a history of hostilities since 1880. London: John Murray, 1978:172–4. [Google Scholar]
- 5.Baird JK. Host age as a determinant of naturally acquired immunity to Plasmodium falciparum. Parasitol Today 1995; 11:105–11. [DOI] [PubMed] [Google Scholar]
- 6.Walther M, Jeffries D, Finney OC et al. Distinct roles for FOXP3 and FOXP3 CD4T cells in regulating cellular immunity to uncomplicated and severe Plasmodium falciparum malaria. PLoS Pathog 2009; 5:e1000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Portugal S, Moebius J, Skinner J et al. Exposure-dependent control of malaria-induced inflammation in children. PLoS Pathog 2014; 10:e1004079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.World Health Organization. Guidelines for the treatment of malaria. 2nd ed Geneva, Switzerland: World Health Organization, 2010. [Google Scholar]
- 9.Crompton PD, Moebius J, Portugal S et al. Malaria immunity in man and mosquito: insights into unsolved mysteries of a deadly infectious disease. Annu Rev Immunol 2014; 32:157–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen T, Liu XK, Zhang Y, Dong C. BTNL2, a butyrophilin-like molecule that functions to inhibit T cell activation. J Immunol 2006; 176:7354–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benten WP, Bettenhaeuser U, Wunderlich F, Van Vliet E, Mossmann H. Testosterone-induced abrogation of self-healing of Plasmodium chabaudi malaria in B10 mice: mediation by spleen cells. Infect Immun 1991; 59:4486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jimenez-Diaz MB, Rullas J, Mulet T et al. Improvement of detection specificity of Plasmodium-infected murine erythrocytes by flow cytometry using autofluorescence and YOYO-1. Cytometry A 2005; 67:27–36. [DOI] [PubMed] [Google Scholar]
- 13.Carroll RW, Wainwright MS, Kim KY et al. A rapid murine coma and behavior scale for quantitative assessment of murine cerebral malaria. PLoS One 2010; 5:e13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol 2001; doi:10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanian A, Tamayo P, Mootha VK et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avril T, Freeman SD, Attrill H, Clarke RG, Crocker PR. Siglec-5 (CD170) can mediate inhibitory signaling in the absence of immunoreceptor tyrosine-based inhibitory motif phosphorylation. J Biol Chem 2005; 280:19843–51. [DOI] [PubMed] [Google Scholar]
- 17.Arnett HA, Escobar SS, Gonzalez-Suarez E et al. BTNL2, a butyrophilin/B7-like molecule, is a negative costimulatory molecule modulated in intestinal inflammation. J Immunol 2007; 178:1523–33. [DOI] [PubMed] [Google Scholar]
- 18.Wennerstrom A, Pietinalho A, Lasota J et al. Major histocompatibility complex class II and BTNL2 associations in sarcoidosis. Eur Respir J 2013; 42:550–3. [DOI] [PubMed] [Google Scholar]
- 19.Krupka M, Seydel K, Feintuch CM et al. Mild Plasmodium falciparum malaria following an episode of severe malaria is associated with induction of the interferon pathway in Malawian children. Infect Immun 2012; 80:1150–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berry MP, Graham CM, McNab FW et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010; 466:973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lill M, Koks S, Soomets U et al. Peripheral blood RNA gene expression profiling in patients with bacterial meningitis. Front Neurosci 2013; 7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Favre N, Ryffel B, Bordmann G, Rudin W. The course of Plasmodium chabaudi chabaudi infections in interferon-gamma receptor deficient mice. Parasite Immunol 1997; 19:375–83. [DOI] [PubMed] [Google Scholar]
- 23.Cohen S, Mc GI, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature 1961; 192:733–7. [DOI] [PubMed] [Google Scholar]
- 24.Clark IA, Hunt NH, Butcher GA, Cowden WB. Inhibition of murine malaria (Plasmodium chabaudi) in vivo by recombinant interferon-gamma or tumor necrosis factor, and its enhancement by butylated hydroxyanisole. J Immunol 1987; 139:3493–6. [PubMed] [Google Scholar]
- 25.Rockett KA, Awburn MM, Cowden WB, Clark IA. Killing of Plasmodium falciparum in vitro by nitric oxide derivatives. Infect Immun 1991; 59:3280–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Idaghdour Y, Quinlan J, Goulet JP et al. Evidence for additive and interaction effects of host genotype and infection in malaria. Proc Natl Acad Sci U S A 2012; 109:16786–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffiths MJ, Shafi MJ, Popper SJ et al. Genomewide analysis of the host response to malaria in Kenyan children. J Infect Dis 2005; 191:1599–611. [DOI] [PubMed] [Google Scholar]
- 28.Sharma S, DeOliveira RB, Kalantari P et al. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity 2011; 35:194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aucan C, Walley AJ, Hennig BJ et al. Interferon-alpha receptor-1 (IFNAR1) variants are associated with protection against cerebral malaria in the Gambia. Genes Immun 2003; 4:275–82. [DOI] [PubMed] [Google Scholar]
- 30.Butler NS, Moebius J, Pewe LL et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol 2012; 13:188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci U S A 1992; 89:6550–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.del C Salamone M, Mendiguren AK, Salamone GV, Fainboim L. Membrane trafficking of CD1c on activated T cells. J Leukoc Biol 2001; 70:567–77. [PubMed] [Google Scholar]
- 33.Hafalla JC, Claser C, Couper KN et al. The CTLA-4 and PD-1/PD-L1 inhibitory pathways independently regulate host resistance to Plasmodium-induced acute immune pathology. PLoS Pathog 2012; 8:e1002504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swanson RM, Gavin MA, Escobar SS et al. Butyrophilin-like 2 modulates B7 costimulation to induce Foxp3 expression and regulatory T cell development in mature T cells. J Immunol 2013; 190:2027–35. [DOI] [PubMed] [Google Scholar]
- 35.Pathan S, Gowdy RE, Cooney R et al. Confirmation of the novel association at the BTNL2 locus with ulcerative colitis. Tissue Antigens 2009; 74:322–9. [DOI] [PubMed] [Google Scholar]
- 36.Orozco G, Eerligh P, Sanchez E et al. Analysis of a functional BTNL2 polymorphism in type 1 diabetes, rheumatoid arthritis, and systemic lupus erythematosus. Hum Immunol 2005; 66:1235–41. [DOI] [PubMed] [Google Scholar]
- 37.Belnoue E, Kayibanda M, Vigario AM et al. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J Immunol 2002; 169:6369–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.