

Abstract
Utilizing an induced-fit model and taking advantage of rotatable acetylenic C(sp)–C(sp2) bonds, we disclose the synthesis and solid-state structures of a series of conformationally diverse bis-sulfonamide arylethynyl receptors using either pyridine, 2,2′-bipyridine, or thiophene as the core aryl group. Whereas the bipyridine and thiophene structures do not appear to bind guests in the solid state, the pyridine receptors form 2 + 2 dimers with water molecules, two halides, or one of each, depending on the protonation state of the pyridine nitrogen atom. Isolation of a related bis-sulfonimide derivative demonstrates the importance of the sulfonamide N–H hydrogen bonds in dimer formation. The pyridine receptors form monomeric structures with larger guests such as BF4− or HSO4−, where the sulfonamide arms rotate to the side opposite the pyridine N atom.
INTRODUCTION
Induced-fit guest recognition agents and conformationally flexible hosts provide a complement to traditional lock-and-key approaches to molecule and ion recognition.1–3 In traditional lock-and-key approaches, a molecule or ion binding site on a host molecule is exquisitely tailored to be selective for the size, shape, charge, polarity, binding preferences, etc. of the target guest.4–8 Conformationally flexible and adaptable hosts, on the other hand, allow a guest substrate to induce a specific organization of the host to create a recognition site,9–12 a motif frequently encountered in biological molecular recognition.13 In essence, the guest is able to choose the ideal binding site in these flexible hosts either by altering the host’s conformation or even reorganizing a dynamic host into a new structure.
The adaptation of arylethynyl scaffolds to supramolecular chemistry has yielded a surprising array of host–guest and coordination complexes.14–17 This rigid and linear motif provides suitable geometric dimensions for both macrocyclic and acyclic receptors. Furthermore, alteration of the aryl moiety (e.g., inclusion of N-based heterocycles such as pyridine) has enabled applications in coordination chemistry, such as selective transition metal and small molecule complexation as well as fluorescence sensing.18–21 Addition of simple synthetic handles to the arenes in these systems has allowed for substitution of chiral functional groups, yielding macrocycles capable of saccharide complexation as well as asymmetric catalysis and chiral recognition.22,23 Of all of the benefits of using an arylethynyl foundation on which to build a receptor, the rigidity, and hence preorganization, they lend to the structure is particularly important. Furthermore, in the case of acyclic receptors, the axial rotation around the linear substituted alkynyl bonds provides receptors that are also conformationally flexible. Although a wide array of guests have been targeted by arylethynyl-based hosts, until our entry into this field, receptors of this type targeting anions were lacking. Given the elegant metal cation sensors that have been developed using cores such as this,24–26 our modular receptor class featuring fluorescent cores has opened up a new area of anion sensing and molecular probe development and shown that these receptors exhibit a variety of binding conformations depending on the identity of the guests. Herein, we describe a complete series of bis(sulfonamide) receptors built off of a variety of arylethynyl cores. Each receptor shows a propensity to form strong hydrogen bonds with various small molecule and ion guests, and the axial flexibility imparted by the alkynes provides a rich diversity in host–guest geometries.
In our initial report, we disclosed that sulfonamides 1 and 2 (Figure 1) exhibit an unusual 2 + 2 dimeric self-assembly motif consisting of two receptors stitched together by either two water molecules, two halides, or one of each, depending on the protonation state of the pyridine nitrogen in the receptor.27 The isostructural 2 + 2 dimers exist not only in the crystalline state but also in solution (vide infra), thus complicating accurate determination of anion binding. We switched to urea derivatives such as 3–5, as these molecules exhibited simpler 1:1 speciation with anions in solution because of the four hydrogen bonds that the two urea binding groups afford.28–32 The crystal structure of 3·HCl corroborates 1:1 complex formation as well in the solid state.28 Interestingly, the urea derivatives display switchable fluorescent and colorimetric responses upon protonation: 4 showed an on–off fluorescence behavior in the presence of chloride in organic solvents, whereas 5 displayed the reverse off–on fluorescence behavior.29 The magnitude of the fluorescence response was dictated by the anion, resulting in a rare, fully organic turn-on fluorescent sensor for chloride, which typically quenches fluorescence. Sulfonamides 2 and 6 also exhibit an analogous, if slightly muted, switching response.29 Subsequent studies of a small library of urea derivatives showed that a similar turn-on behavior is observable even in relatively polar solvents such as MeCN.31
Figure 1.

Previously reported bis(2-anilinoethynyl)pyridine sulfonamides/ureas 1–8 and related new structures 9–12.
Through these studies, it became apparent that the bis(2-anilinoethynyl)pyridine ureas possess a rich and varied array of receptor conformations with and without guests. Three different conformations are observed about the cores of 3–5, 7, and 8: S, U, and W, depending upon the guests or solvents of crystallization.28,30 The receptors each have rotatable C(sp)–C(sp2) bond(s) that can potentially adopt a variety of topological structures, and the interaction of the urea groups with themselves and with potential guests dictate the conformations that are observed in the crystal structures. In fact, some ligands exhibit all three conformations (S, U, and W), depending on the choice of single, binary, or ternary solvent systems for crystallization.30,33–37
Because of this rich conformational diversity, we decided to revisit the sulfonamide class of receptors and thus re-evaluate their potential use as hosts for neutral molecules and anions. Herein, we report the synthesis and crystal structures of pyridine-based sulfonamide 9 and sulfonimide 10. We highlight the modularity of this ligand class by replacing the pyridine moiety with other heteroaromatic rings to tailor the size of the binding cavity, generating expanded core structures based on either thiophene (11) or 2,2-bipyridine (12). Finally, we examine the solid-state structures of the sulfonamides with neutral and anionic guests.
RESULTS AND DISCUSSION
Receptor Synthesis
Treatment of previously utilized bis-aniline 1327–33 in pyridine with an excess of 4-bromophe-nylsulfonyl chloride gave receptor 9 in excellent yield (Scheme 1). The syntheses of bis-sulfonamides 1,27 2,27 and 629 (performed analogously) have been described previously. As a whole, this afforded a series of molecules where the electronic nature of the terminal phenyl rings could be tuned from donating (6, R = OMe) to withdrawing (4, R = NO2).
Scheme 1.

Synthesis of Sulfonamide Receptor 9 and Unanticipated Sulfonimide 10
We next considered a 3,5-bis(trifluoromethyl)-phenylsulfonamide receptor to examine solid-state and solution effects of a different substitution pattern and increased electron deficiency of the peripheral phenyl rings. Surprisingly, product isolation afforded tetra-substituted bis-sulfonimide 10 (Scheme 1) along with a small amount (<10%) of a trisubstituted mixed sulfonamide/-imide (not shown), the result of enhanced reactivity of the electron-deficient bis(trifluoromethyl)-phenylsulfonyl unit. Subsequent efforts to control substitution via stoichiometric treatment with the sulfonyl chloride afforded only a mixture of mono-, di-, and trisubstituted adducts, out of which the desired product was impossible to separate from monosubstituted byproduct.
We also sought to examine the effect of changing the size and shape of the receptor cleft by replacing the central pyridine unit with other heteroarenes. Simple modeling of both pyrrole and bipyridine cores indicated the potential to target larger guest molecules, a direct result of the change in the bite angle of the functionalized phenylacetylene arms. Dihalo analogues of both heterocycles are known; however, 2,5-dibromopyrrole has been reported to decompose rapidly upon concentration.38 Because of the well-established chemistry of 2,5-difunctionalized thiophenes,39 a thiophene moiety was utilized as a simple substitute to model the pyrrole core and determine if further examination of pyrrole analogues was warranted, specifically via examination of the solid-state structure.
Synthesis of thiophene analogue 11 began with desilylation of 1440,41 followed by 2-fold Sonogashira cross-coupling with commercially available 2,5-dibromothiophene to furnish bis-aniline 15 (Scheme 2). Known bis-aniline 16 was similarly prepared from 6,6′-dibromo-2,2′-bipyridine.34 Independent treatment of 15 and 16 with p-toluenesulfonyl chloride in pyridine provided expanded receptors 11 and 12 in moderate to very good yield.
Scheme 2.
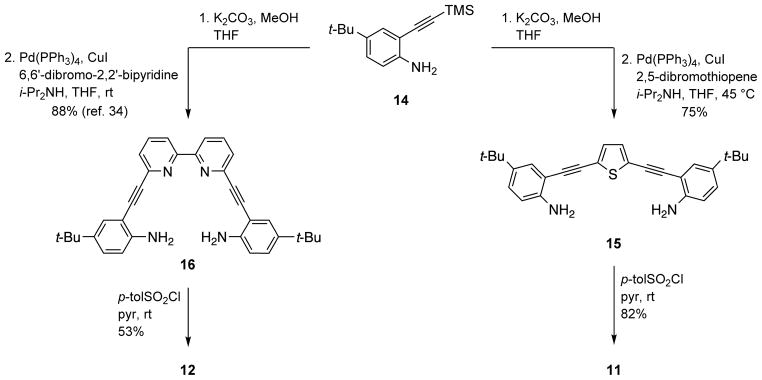
Synthesis of Alternative Core Bis-Sulfonamide Analogues 11 and 12
Neutral Pyridine-Core Crystal Structures
While the 1H NMR signals of pyridine-based receptors 1, 2, 6, and 9 exhibited considerable concentration dependence in organic solvents, we were especially intrigued by the behavior of the residual water signal in CDCl3. Normally this resonance appears as a sharp singlet at ca. 1.55 ppm; however, the peak broadened significantly and would appear as far downfield as ca. 4 ppm, depending on concentration. This observation strongly implicated the hydrogen-bonding capacity of the pyridine-sulfonamide receptors in solution, a result that was also corroborated in their solid-state structures. Fortunately, the sulfonamide class of receptors crystallized easily, and we were able to obtain eight new structures for this study; all of the key crystal details and structure parameters are compiled in Table 1.
Table 1.
X-ray Crystallographic Parameters for the Eight New Structures in this Article
| crystal parameters | (6·H2O)2 | (9·H2O)2 | 2 | 10 | 1·HBF4 | 1·H2SO4 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|
| structure code | ob135 | mh11 | ob100 | mh16 | ob138 | ob156 | ob116 | ob102 |
| empirical formula | C43H45N3O7S2 | C41H39Br2N3O5S2 | C83H75Cl3N10O16S4 | C85H95F24N3O8S4 | C43H44BF4N3O4S2 | C98H114N6O19S6 | C42H42N2O4S3 | C48H46N4O4S2 |
| formula weight | 779.94 | 877.69 | 1703.12 | 1870.88 | 817.74 | 1872.31 | 734.96 | 807.01 |
| T (K) | 173(2) | 173(2) | 153(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| crystal size (mm) | 0.80 × 0.12 × 0.08 | 0.32 × 0.26 × 0.12 | 0.30 × 0.20 × 0.15 | 0.45 × 0.06 × 0.03 | 0.35 × 0.20 × 0.10 | 0.52 × 0.11 × 0.01 | 0.10 × 0.08 × 0.01 | 0.10 × 0.08 × 0.02 |
| crystal system | Triclinic | Triclinic | Orthorhombic | Monoclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| space group | P1̄ | P1̄ | Pbca | C2/c | P1̄ | P1̄ | P1̄ | P1̄ |
| a (Å) | 9.8389(19) | 10.0725(7) | 27.226(4) | 9.986(3) | 11.6480(18) | 14.727(4) | 11.748(5) | 7.960(3) |
| b (Å) | 13.094(3) | 12.7166(8) | 20.765(3) | 54.639(16) | 13.432(2) | 15.082(4) | 12.750(5) | 10.860(4) |
| c (Å) | 17.479(3) | 17.3538(11) | 31.191(4) | 31.711(10) | 15.472(2) | 21.992(6) | 13.028(6) | 12.852(4) |
| α (deg) | 68.463(3) | 111.3340(10) | 90 | 90 | 105.643(2) | 83.367(5) | 97.514(9) | 101.699(7) |
| β (deg) | 75.262(3) | 96.3670(10) | 90 | 99.004(6) | 109.855(2) | 89.980(5) | 95.518(10) | 97.680(7) |
| γ (deg) | 80.671(3) | 101.7700(10) | 90 | 90 | 98.858(3) | 84.521(5) | 94.730(8) | 95.720(7) |
| V (Å3) | 2019.7(7) | 1984.8(2) | 17634(4) | 17090(9) | 2110.4(6) | 4829(2) | 1916.8(15) | 1068.9(6) |
| Z | 2 | 2 | 8 | 16 | 2 | 2 | 2 | 1 |
| Dc (M g m−3) | 1.282 | 1.469 | 1.283 | 1.454 | 1.287 | 1.288 | 1.273 | 1.254 |
| μ (mm−1) | 0.185 | 2.195 | 0.267 | 0.221 | 0.188 | 0.212 | 0.237 | 0.173 |
| F(000) | 824 | 896 | 7088 | 7760 | 856 | 1984 | 776 | 426 |
| 2θmax (deg) | 24.998 | 54 | 56 | 56.96 | 55.98 | 56 | 50 | 50 |
| no. reflns. | 14 695 | 22 389 | 99 288 | 98 547 | 18 362 | 41 922 | 13 920 | 7795 |
| no. ind. reflns. | 7058 | 8599 | 20 858 | 20 434 | 9488 | 21758 | 6690 | 3745 |
| Rint | 0.0218 | 0.0183 | 0.0685 | 0.1520 | 0.0172 | 0.0902 | 0.1307 | 0.0452 |
| R1 (I > 2σ(I)) | 0.0437 | 0.0356 | 0.0782 | 0.0839 | 0.0514 | 0.0902 | 0.1223 | 0.0611 |
| wR2 (I > 2σ(I)) | 0.1078 | 0.0930 | 0.2096 | 0.1849 | 0.1367 | 0.1682 | 0.2586 | 0.1297 |
| GOF | 1.026 | 1.053 | 1.025 | 0.867 | 1.025 | 1.001 | 1.056 | 1.041 |
| max/min residual e−density (e Å3) | 0.439/−0.328 | 0.686/−0.890 | 1.171/−1.088 | 0.669/−0.407 | 0.856/−0.436 | 0.715/−0.504 | 0.404/−0.435 | 0.339/−0.296 |
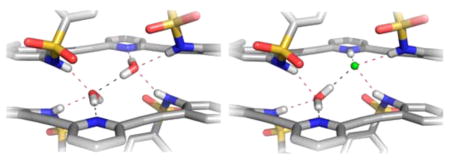
Interestingly, the neutral pyridine-core sulfonamides all exhibit the same propensity to crystallize as hydrogen-bonded 2 + 2 dimers with water in the solid state. The structures are very similar throughout the series of receptors, differing only in minor adjustments to the hydrogen-bonding and π-stacking distances regardless of the electronic nature of the para substituent on the terminal phenyl rings. In fact, the structures are so alike that unit cell dimensions could not be used to screen each sample reliably. Figure 2 illustrates the similarity among receptors 1, 2, 6, and 9 in the solid state, with four different representations of the single-crystal X-ray structures.
Figure 2.

Four different representations of the 2 + 2 water dimer formed when sulfonamides 1, 2, 6, and 9 are crystallized in their neutral form.
Close-in examination shows how the dimers are held together (Figure 3); hydrogen-bond lengths and bond angles for all four structures are compiled in Table 2. The respective pyridine nitrogen atoms accept hydrogen bonds from different water molecules (2.789(2)–2.804(2) Å, O–H⋯N angles 172(3)–175(3)°), with one water–water hydrogen bond present in the binding pocket of the U conformation (2.866(4)–3.006(7) Å, O–H⋯O angles 147(5)–178(6)°). All of the N-substituted sulfonamides adopt the energetically most favored staggered conformation.42 Each sulfonamide proton donates a hydrogen bond from the receptors to a different water molecule (2.852(3)–2.879(2) Å, 157(2)–164(3)° and 2.963(3)–3.039(3) Å, 158(2)–164(3)°) such that the 2 + 2 dimer structure is held together by four sulfonamide–water hydrogen bonds, two pyridine–water hydrogen bonds, one water–water hydrogen bond, and two π-stacking interactions between the receptors ranging from 3.2774(5) to 3.617(2) Å.
Figure 3.

Close-up view of hydrogen-bonding interactions within the sulfonamide–water dimers.
Table 2.
Selected Hydrogen-Bond Distances (Å) and Bond Angles (deg) within the 2 + 2 Water Dimer Structures of Sulfonamides 1, 2, 6, and 9
| 1 (R = Me) | 2 (R = NO2) | 6 (R = OMe) | 9 (R = Br) | |
|---|---|---|---|---|
| O–H⋯N | 2.804(2) | 2.797(4) | 2.789(2) | 2.792(2) |
| O–H⋯N | 175(3) | 172(4) | 172(3) | 174(3) |
| O–H⋯O | 2.917(5) | 3.006(7) | 2.866(4) | 2.897(4) |
| O–H⋯O | 164(4) | 178(6) | 151(5) | 147(5) |
| N–H⋯O | 2.860(3) | 2.855(4) | 2.879(2) | 2.852(3) |
| N–H⋯O | 157(2) | 164(3) | 157(2) | 157(3) |
| N–H⋯O | 3.039(3) | 3.028(4) | 2.963(3) | 3.036(3) |
| N–H⋯O | 158(2) | 164(3) | 161(3) | 158(3) |
In one isolated instance, a polymorph of receptor 2 was obtained while attempting to generate co-crystals of 2 and Bu4NI under CHCl3/pentane diffusion conditions. In this structure, 2 crystallizes in the Pcba space group with eight molecules per unit cell. The receptor forms a dimer, with each molecule exhibiting the same helical arrangement. A single sulfonamide functionality from each receptor is interlocked with the neighboring molecule (Figure 4), whereas the second sulfonamide functionality hydrogen bonds to a separate dimeric pair in the solid state (2.904(4)–2.989(4) Å, N–H⋯O angles 155(4)–159(5)°). The key driving forces for the solid-state structure are the hydrogen bonds between the pyridine nitrogen and sulfonamide hydrogens from the adjacent receptor (2.902(4)–2.919(4) Å, N–H⋯N angles 166(4)–167(5)°) as well as intermolecular π-stacking (3.479(15) Å). The CHCl3 solvent molecule occupies a void between the aromatic rings of three different molecules of 2 and the sulfonamide functionality of another molecule of 2. This structure remains the only example of dimer formation in these receptors without assisting water molecules. In every other crystal structure studied to date, the sulfonamide receptors crystallize as 2 + 2 dimers with H2O in solvents dried over 3 Å molecular sieves and even in the presence of other potential guest molecules (e.g., MeOH, EtOH, i-PrOH, MeCN, DMSO, THF, acetone, tetra-n-butylammonium halides), guests that do co-crystallize with the corresponding urea class of receptors.28,30
Figure 4.

Polymorph of 2 dimer notably lacking assisting water molecules, with each molecule colored differently. ORTEPs are drawn at 50% probability, and non hydrogen-bonding hydrogens and CHCl3 solvent molecule are removed for clarity.
The fortuitous isolation of bis-sulfonimide 10, which does not contain sulfonamide hydrogens, provides crucial solution evidence for the importance of water–sulfonamide hydrogen bonds in the self-assembly of 1, 2, 6, and 9. Whereas the residual water resonance in the 1H NMR spectra for the sulfonamide receptors shifts markedly downfield to δ3–4 ppm (depending upon receptor concentration), no such shift is observed in the proton spectrum of sulfonimide 10, as the residual water resonance in CDCl3 is unchanged (δ1.55 ppm). These contrasting results clearly support the presence of a solution-based hydrogen-bonding phenomena in the corresponding sulfonamides.
The importance of the sulfonamide hydrogen was also corroborated by the solid-state structure of 10 (Figure 5), which showed no water in the crystal lattice. Sulfonimide 10 crystallizes in the C2/c space group with 16 molecules per unit cell. Additionally, with no hydrogen bond donor present, this molecule does not form strong intermolecular hydrogen bonds in the solid state. The steric bulk of the two sulfonimide functionalities enforces a W-shaped conformation in the solid state, with the pyridine nitrogen directed away from the sulfonimides. Compound 10 crystallizes in a close pack conformation that places one fluorine from a –CF3 group directly above the electron-deficient aromatic ring of another molecule of 10 (F⋯centroid 3.079(5) Å). Attractive interactions between electron-rich species and electron-deficient aromatic rings are an area of current interest.43–49
Figure 5.
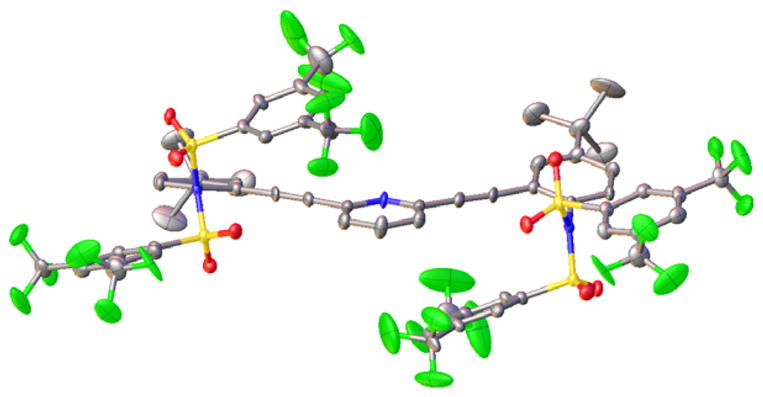
ORTEP (30%) representation of bis-sulfonimide 10, which exhibits no interactions with water molecules in the solid state.
Expanded-Core Crystal Structures
Substitution of thiophene and bipyridine into the ligand backbone clearly increases the binding cavity size and will allow for exploration of binding larger polyatomic ions and small molecules compared to that for the pyridine-based system. Single crystals of 11 were isolated from evaporation of hexane/EtOAc mixtures. Receptor 11 crystallizes with two molecules per unit cell in the P1̄ space group and exhibits a solid-state W conformation, with both receptor arms rotated away from the binding cavity due to their participation in intermolecular hydrogen bonds (2.962(12) and 2.999(15) Å, N–H⋯O angles 130.1(6)–133.4(6)°) with the arms of two adjacent receptors (Figure 6). This zigzag ribbon packing arrangement propagates through the crystal structure, forming hydrogen-bonding sheets.
Figure 6.

ORTEP (50%) representation of the crystal structure of sulfonamide 11 with expanded thiophene core. Three molecules are shown to highlight the intermolecular hydrogen-bonding in the solid state; non-hydrogen-bonding hydrogens atoms are removed for clarity.
Single crystals of 12 were grown by slowly diffusing hexane into EtOAc solutions of receptor. Compound 12 crystallizes in the P1̄ space group with one molecule contained in the unit cell. In the solid state, the bipyridine core of 12 is planar with anti N atoms, a factor that results in rotation of a single sulfonamide arm away from the proposed binding cavity (Figure 7, top).34 Intermolecular π-stacking interactions are observed between the electron-deficient pyridine ring of one receptor and the electron-rich p-toluene ring of another (3.658(5) Å). Like thiophene 11, each sulfonamide functionality of 12 hydrogen bonds in a head-to-tail fashion with different adjacent molecules (2.939(4) Å, N–H⋯O angle 170(3)°), forming an infinite hydrogen-bonding chain in the solid state (Figure 7, bottom). Also present are nontraditional intermolecular C–H hydrogen bonds34,35,50–52 between a p-toluene ring and an adjacent sulfonamide oxygen (3.187(4) Å, C–H⋯O angle 134.35(19)°). This weak hydrogen bond is likely enforced from the packing arrangement of this molecule.
Figure 7.

ORTEP (50%) representations of the single-crystal X-ray structure of sulfonamide 12 with expanded bipyridine core (top). The extended hydrogen-bonding chain is displayed below; some hydrogen atoms have been removed for clarity.
Protonated Pyridine-Core Crystal Structures
As we demonstrated in the initial sulfonamide communication27 and subsequently with amide33 and urea derivatives,28,29,32 protonation of the pyridine nitrogen activates the anion binding capacity of these receptors. In the specific case of the sulfonamides, the resultant complexes with anions all resemble the 2 + 2 water dimer structures. For example, (H2+·Cl−)2 (Figure 8, right) is held together by four sulfonamide hydrogen bonds (3.156(2)–3.229(2) Å, N–H⋯Cl angles 151(2)–171(3)°), two pyridinium N–H hydrogen bonds to the chlorides (3.022(2) Å, 175(3)°), two Caryl–H⋯Cl hydrogen bonds (3.688(3) Å), and two π-stacking interactions between receptors (3.495(3) Å). The numerous hydrogen bonds and unique dimerization bring the negatively charged chlorides into close proximity with halide–halide distance of 3.9204(13) Å.
Figure 8.
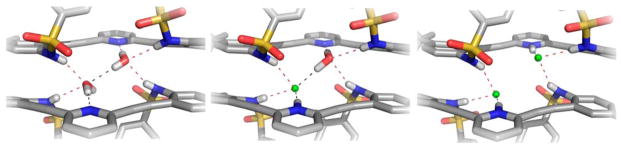
Stick representations of the crystal structures of (1·H2O)2 (left), (1·H2O)·(H1+·Cl−) (middle), and (H2+·Cl−)2 (right), highlighting the interchangeable role that halides and water play in the dimerization of the sulfonamide class of receptors. Hydrogen bonds are illustrated as dashes.
The surprise isolation of heterodimer (1·H2O)·(H1+·Cl−), which crystallized in the presence of concentrated aqueous HCl (Figure 8, center), shows that water and hydrogen chloride are freely exchangeable in this binding pocket and provide intermediate structural features to the H2O and hydrogen halide dimers. As before, the heterodimer is stabilized by π-stacking interactions between the two receptors (3.454(5) Å) and a series of seven guest-assisted hydrogen bonds. Each guest molecule, water and chloride, accepts two sulfonamide N–H hydrogen bonds (3.157(3)–3.181(3) Å, N–H⋯X angles 158(3)–167(3)°) and additionally forms a hydrogen-bonding pattern running among the pyridinium, chloride, water, and pyridine heteroatoms (2.926(3)–3.099(2) Å, 164(3)–176(4)°). Two Caryl–H⋯Cl hydrogen bonds (3.935(4) Å) also stabilize the dimer. This is one of the first examples of both hydrogen halides and water molecules serving the same structural hydrogen-bonding roles in a synthetic self-assembled system. This work also sheds light on the complex self-assembly capabilities of water molecules and anions acting in tandem, a synergy only recently appreciated as being an important structural feature in some proteins.53
While the sulfonamide receptors form 2 + 2 dimers in the solid state with water, halides, or both water and halides, we found that use of larger anionic guest molecules generates only monomeric species in the solid state. For example, single crystals of H1+·BF4− are isolated by dissolving receptor 1 in EtOAc and then adding aqueous HBF4 with vigorous stirring followed by layering with hexane. H1+·BF4− crystallizes in the P1̄ space group with two molecules per unit cell. As shown in Figure 9 (top), the larger size of the BF4− anion forces the receptor arms to the opposite side of the pyridine N and thus into a W conformation to maintain a strong pyridinium hydrogen bond with the BF4− anion (2.658(2) Å, N–H⋯F angle 167(2)°). This interaction is also complemented by a weak C–H⋯F hydrogen bond (3.431(3) Å, C–H⋯F angle 166.85(12)°) from an adjacent phenyl ring. One receptor sulfonamide forms a head-to-tail intermolecular hydrogen bond (2.916(2) Å, N–H⋯O angle 161.0(19)°) with an adjacent receptor molecule, whereas the other sulfonamide hydrogen bonds to the BF4− anion of a second receptor (2.907(2) Å, N–H⋯F angle 163(2)°). This sequence of hydrogen bonds forms a polymeric hydrogen-bonding chain in the solid state (Figure 9, bottom). This structure is somewhat unusual considering that fluorine is typically a poor hydrogen-bond acceptor and BF4 is typically used as a noncoordinating anion.54 The short distances, nearly linear X–H⋯F angles, and the change in molecular conformation to accommodate the BF4− anion are consistent with the weak hydrogen bonds suggested here,55 although the existence and frequency of occurrence of hydrogen bonds to organic fluorine is not without some controversy in the literature.56–59
Figure 9.

(Top) Molecular structure of H1+·BF4− showing the N–H⋯ F hydrogen bond; (Bottom) ORTEP representation of the polymeric hydrogen-bonding chain present in H1+·BF4− in the solid state.
The structure of H1+·HSO4− also deviates from the 2 + 2 arrangement observed for smaller guests. Single crystals of H1+·HSO4− were grown by diffusing pentane into THF solutions of receptor mixed with H2SO4. The H1+·HSO4− salt crystallizes in the P1̄ space group with two receptor molecules and three THF solvent molecules per unit cell. Similar to the H1+·BF4− structure, the counteranion HSO4− forms a strong hydrogen bond with the pyridinium nitrogen (2.668(5) Å, N–H⋯O angle 173(6)°), resulting once again in a W conformation that puts the receptor arms on the opposite side of the molecule (Figure 10). The HSO4− anion is also stabilized by three additional hydrogen bonds. One hydrogen bond is donated from the sulfonamide N–H of an adjacent molecule of H1+ (2.981(6) Å, N–H⋯O angle 162(4)°). A second molecule of H1+ forms a tight head-to-tail dimer with this same HSO4− anion (N–H⋯O of 2.856(6) Å, angle 168(5)° and O–H⋯O of 2.717(6) Å, angle 178.3(3)°). Additionally, the H1+·HSO4− structure is complicated by additional solvents of crystallization. The second HSO4− anion is stabilized by four different hydrogen bonds. The strongest interaction is with another pyridinium N–H (2.637(5) Å, N–H⋯O angle 170(5)°). Two adjacent molecules of H1+ also donate sulfonamide N–H hydrogen bonds (2.864(5) and 2.945(6) Å, N–H⋯O angle 171(5) and 177(5)°). The HSO4− anion also donates a hydrogen bond to an adjacent THF molecule (2.568(6) Å, OH⋯O angle 176.3(3)°).
Figure 10.
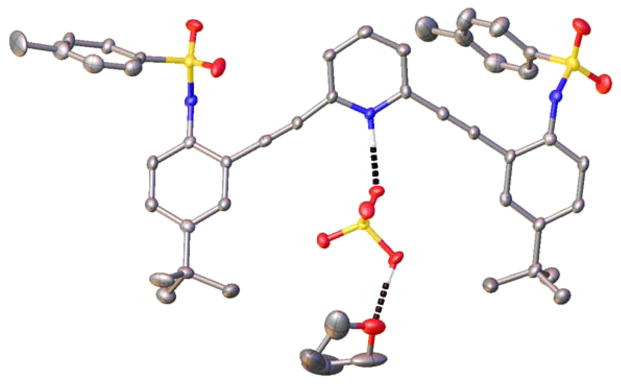
ORTEP representation (30%) of a portion of the H1+·HSO4− crystal structure highlighting the pyridinium and HSO4− hydrogen bonds present in the solid state.
CONCLUSIONS
In summary, we have presented a series of heteroarylacetylene ligands designed for small molecule and ion recognition. The receptors are prepared in a modular fashion capable of simple exchange of binding substituents and core motifs. The facile synthesis has produced a series of sulfonamide-bearing receptors that show a predilection for interacting with small molecules (H2O) or anions (Cl−, Br−, BF4−, and HSO4−) in solution and the solid state. Facile axial rotation about the acetylene C(sp)–C(sp2) bonds allows these molecules to function as induced-fit receptors. A variety of conformationally different binding motifs are observed that depend on the size and shape of the guest present as well as the heterocyclic core. Solid-state conformations are largely dominated by hydrogen-bonding interactions and π-stacking. The 2,6-bis(2-anilinoethynyl)pyridine sulfonamides form 2 + 2 dimers in solution and the solid state with H2O, halides, or both H2O and halides. Sulfonamide receptors based on larger heterocyclic core molecules exhibit larger cavity sizes in the solid state and show no propensity to form 2 + 2 dimers. The efficiency of our new system, directly attributed to a straightforward synthesis and facile derivatization, bodes well for future receptor development to target remediation and sensing applications for environmental contaminants and biologically relevant anions.
EXPERIMENTAL SECTION
Receptor Synthesis
General Crystallographic Data
Diffraction intensities for all structures were collected at low temperature (153 K for 2 and 173 K for others) on a Bruker Apex CCD diffractometer using Mo Kα radiation (λ = 0.71073 Å). Space group was determined based on systematic absences (2 and 10) and intensity statistics. Absorption corrections were applied by SADABS.60 Structures were solved by direct methods and Fourier techniques and refined on F2 using full matrix least-squares procedures. All non-H atoms were refined with anisotropic thermal parameters. All H atoms in (6·H2O)2 and (9·H2O)2 were found from the residual density map and refined with isotropic thermal parameters. All H atoms in 10 and 11 were refined in calculated positions in a rigid group model. In structures 2, 12, 1·HBF4, and 1·H2SO4, H atoms involved in hydrogen bonds were found on the residual density maps and refined with isotropic thermal parameters; all other H atoms were refined in calculated positions in a rigid group model. Highly disordered solvent hexane molecules, C6H14, in 10 were treated by SQUEEZE.61 The correction of the X-ray data by SQUEEZE (215 electrons) is close to the required value of 200 electrons for four solvent molecules in a symmetrically independent part of the unit cell. The main crystallographic data and details of data collection and refinement are given in Table 1. All calculations have been performed by the SHELXTL-6.10 package.62
Supplementary Material
Acknowledgments
This work was funded initially by the National Science Foundation (CHE-0718242) and subsequently supported by the National Institutes of Health (R01-GM087398). This publication was made possible, in part, by the Mass Spectrometry Facilities and Services Core of the Environmental Health Sciences Center, Oregon State University, grant no. P30 ES00210, National Institute of Environmental Health Sciences, National Institutes of Health.
Footnotes
X-ray crystallographic data for H1+·BF4−, H1+·HSO4−, 2, (6·H2O)2, (9·H2O)2, and 10–12 in CIF format; experimental procedures for and copies of 1H and 13C NMR spectra of 9–12 and 15. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare the following competing financial interest(s): NIH grant R01-GM087398 funded early stage intellectual property that was licensed by SupraSensor Enterprises, a company co-founded by D.W.J. and M.M.H. O.B.B. and C.A.J. are also shareholders in this company.
References
- 1.Rebek J, Jr, Killoran AM, Nemeth D, Lin FT. J Am Chem Soc. 1987;109:2426–2431. [Google Scholar]
- 2.Hamilton AD, Engen DV. J Am Chem Soc. 1987;109:5035–5036. [Google Scholar]
- 3.Fujita M, Nagao S, Ogura K. J Am Chem Soc. 1995;117:1649–1650. doi: 10.1021/ja00110a026. [DOI] [PubMed] [Google Scholar]
- 4.Fischer E. Ber Dtsch Chem Ges. 1894;27:2984–2993. [Google Scholar]
- 5.Behr J-P, editor. The Lock-and-Key Principle: The State of the Art–100 Years On. Wiley; Chichester: 1994. [Google Scholar]
- 6.Steed JW, Atwood JL. Supramolecular Chemistry. 2. Wiley; Chichester: 2009. [Google Scholar]
- 7.Holyoak T. Molecular Recognition: Lock-and-Key, Induced Fit, and Conformational Selection. In: Roberts GCK, editor. Encyclopedia of Biophysics. Springer; New York: 2013. pp. 1584–1588. [Google Scholar]
- 8.Yatsimirsky AK. The Lock and Key Principle. In: Atwood JL, Steed JW, editors. Encyclopedia of Supramolecular Chemistry. Dekker; New York: 2004. pp. 809–815. [Google Scholar]
- 9.Haino T, Rudkevich DM, Shivanyuk A, Rissanen K, Rebek J., Jr Chem–Eur J. 2000;6:3797–3805. doi: 10.1002/1521-3765(20001016)6:20<3797::aid-chem3797>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 10.Turner DR, Paterson MJ, Steed JW. J Org Chem. 2006;71:1598–1608. doi: 10.1021/jo052339f. [DOI] [PubMed] [Google Scholar]
- 11.Yoo J, Jeoung E, Lee CH. Supramol Chem. 2009;21:164–172. [Google Scholar]
- 12.Azov VA, Schlegel A, Diederich F. Angew Chem, Int Ed. 2005;44:4635–4638. doi: 10.1002/anie.200500970. [DOI] [PubMed] [Google Scholar]
- 13.Bosshard HR. News Physiol Sci. 2001;16:171–173. doi: 10.1152/physiologyonline.2001.16.4.171. [DOI] [PubMed] [Google Scholar]
- 14.Vonnegut CL, Tresca BW, Johnson DW, Haley MM. Chem–Asian J. 2015 doi: 10.1002/asia.201403212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carroll CN, Naleway JJ, Haley MM, Johnson DW. Chem Soc Rev. 2010;39:3875–3888. doi: 10.1039/b926231h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leininger S, Olenyuk B, Stang PJ. Chem Rev. 2000;100:853–907. doi: 10.1021/cr9601324. [DOI] [PubMed] [Google Scholar]
- 17.Romero F, Ziessel R, Dupont-Gervais A, van Dorsselaer A. Chem Commun. 1996:551–553. [Google Scholar]
- 18.Reger D, Gardinier J, Bakbak S, Semeniuc R, Bunz U, Smith M. New J Chem. 2005;29:1035–1043. [Google Scholar]
- 19.Orita A, Nakano T, An D, Tanikawa K, Wakamatsu K, Otera J. J Am Chem Soc. 2004;126:10389–10396. doi: 10.1021/ja048327d. [DOI] [PubMed] [Google Scholar]
- 20.Stang P, Olenyuk B, Muddiman D, Smith R. Organometallics. 1997;16:3094–3096. [Google Scholar]
- 21.Massena CJ, Riel AMS, Neuhaus GF, Decato DA, Berryman OB. Chem Commun. 2015;51:1417–1420. doi: 10.1039/c4cc09242b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Droz AS, Diederich F. J Chem Soc, Perkin Trans. 2000;1:4224–4226. [Google Scholar]
- 23.Fang J, Selvi S, Liao J, Slanina Z, Chen C, Chou P. J Am Chem Soc. 2004;126:3559–3566. doi: 10.1021/ja039237w. [DOI] [PubMed] [Google Scholar]
- 24.Zucchero A, Wilson J, Bunz U. J Am Chem Soc. 2006;128:11872–11881. doi: 10.1021/ja061112e. [DOI] [PubMed] [Google Scholar]
- 25.Lewis J, Moore J. Phys Chem Chem Phys. 2004;6:4595–4606. [Google Scholar]
- 26.Tolosa J, Zucchero A, Bunz U. J Am Chem Soc. 2008;130:6498–6506. doi: 10.1021/ja800232f. [DOI] [PubMed] [Google Scholar]
- 27.Berryman OB, Johnson CA, II, Zakharov LN, Haley MM, Johnson DW. Angew Chem, Int Ed. 2008;47:117–120. doi: 10.1002/anie.200703971. [DOI] [PubMed] [Google Scholar]
- 28.Carroll CN, Berryman OB, Johnson CA, II, Zakharov LN, Haley MM, Johnson DW. Chem Commun. 2009:2520–2522. doi: 10.1039/b901643k. [DOI] [PubMed] [Google Scholar]
- 29.Carroll CN, Coombs BA, McClintock SP, Johnson CA, II, Berryman OB, Johnson DW, Haley MM. Chem Commun. 2011;47:5539–5541. doi: 10.1039/c1cc10947b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engle JM, Lakshminarayanan PS, Carroll CN, Zakharov LN, Haley MM, Johnson DW. Cryst Growth Des. 2011;11:5144–5152. doi: 10.1021/cg201074v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engle JM, Carroll CN, Johnson DW, Haley MM. Chem Sci. 2012;3:1105–1110. doi: 10.1039/C2SC00975G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engle JM, Singh PS, Vonnegut CL, Zakharov LN, Johnson DW, Haley MM. Cryst Eng Comm. 2014;16:3703–3706. doi: 10.1039/c3ce42307g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson CA, Berryman OB, Sather AC, Zakharov LN, Haley MM, Johnson DW. Cryst Growth Des. 2009;9:4247–4249. [Google Scholar]
- 34.Gavette JV, Mills NS, Zakharov LN, Johnson CA, II, Johnson DW, Haley MM. Angew Chem, Int Ed. 2013;52:10270–10274. doi: 10.1002/anie.201302929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tresca BW, Zakharov LN, Carroll CN, Johnson DW, Haley MM. Chem Commun. 2013;49:7240–7242. doi: 10.1039/c3cc44574g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gavette JV, Evoniuk CJ, Zakharov LN, Carnes ME, Haley MM, Johnson DW. Chem Sci. 2014;5:2899–2905. doi: 10.1039/c4sc00950a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gavette JV, Klug CM, Zakharov LN, Shores MP, Haley MM, Johnson DW. Chem Commun. 2014;50:7173–7175. doi: 10.1039/c4cc02297a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gilow HM, Burton DE. J Org Chem. 1981;46:2221–2225. [Google Scholar]
- 39.Gronowitz S, editor. The Chemistry of Heterocyclic Compounds: Thiophene and Its Derivatives. Vol. 44 Wiley and Sons; New York: 1992. [Google Scholar]
- 40.Wan WB, Haley MM. J Org Chem. 2001;66:3893–3901. doi: 10.1021/jo010183n. [DOI] [PubMed] [Google Scholar]
- 41.Kimball DB, Weakley TJR, Haley MM. J Org Chem. 2002;67:6395–6405. doi: 10.1021/jo020229s. [DOI] [PubMed] [Google Scholar]
- 42.Hirsch AKH, Lauw S, Gersbach P, Schweizer WB, Rohdich F, Eisenreich W, Bacher A, Diederich F. Chem Med Chem. 2007;2:806–810. doi: 10.1002/cmdc.200700014. [DOI] [PubMed] [Google Scholar]
- 43.Berryman OB, Bryantsev VS, Stay DP, Johnson DW, Hay BP. J Am Chem Soc. 2006;129:48–58. doi: 10.1021/ja063460m. [DOI] [PubMed] [Google Scholar]
- 44.Berryman OB, Hof F, Hynes MJ, Johnson DW. Chem Commun. 2006:506–508. doi: 10.1039/b511570a. [DOI] [PubMed] [Google Scholar]
- 45.Berryman OB, Sather AC, Hay BP, Meisner JS, Johnson DW. J Am Chem Soc. 2008;130:10895–10897. doi: 10.1021/ja8035652. [DOI] [PubMed] [Google Scholar]
- 46.Berryman OB, Johnson DW. Chem Commun. 2009:3143–3153. doi: 10.1039/b823236a. [DOI] [PubMed] [Google Scholar]
- 47.Gamez P, Mooibroek TJ, Teat SJ, Reedijk J. Acc Chem Res. 2007;40:435–444. doi: 10.1021/ar7000099. [DOI] [PubMed] [Google Scholar]
- 48.Hay BP, Bryantsev VS. Chem Commun. 2008:2417–2428. doi: 10.1039/b800055g. [DOI] [PubMed] [Google Scholar]
- 49.Schottel BL, Chifotides HT, Dunbar KR. Chem Soc Rev. 2008;37:68–83. doi: 10.1039/b614208g. [DOI] [PubMed] [Google Scholar]
- 50.Vargas R, Garza J, Dixon DA, Hay BP. J Am Chem Soc. 2000;122:4750–4755. [Google Scholar]
- 51.Juwarker H, Lenhardt JM, Pham DM, Craig SL. Angew Chem, Int Ed. 2008;47:3740–3743. doi: 10.1002/anie.200800548. [DOI] [PubMed] [Google Scholar]
- 52.Hua Y, Flood AH. Chem Soc Rev. 2010;39:1262–1271. doi: 10.1039/b818033b. [DOI] [PubMed] [Google Scholar]
- 53.For an example of water and halides occupying the same binding sites in a protein, see: Fiedler TJ, Davey CA, Fenna RE. J Biol Chem. 2000;275:11964–11971. doi: 10.1074/jbc.275.16.11964.
- 54.Restorp P, Berryman OB, Sather AC, Ajami D, Rebek J., Jr Chem Commun. 2009:5692–5694. doi: 10.1039/b914171e. [DOI] [PubMed] [Google Scholar]
- 55.For the current IUPAC definition and criteria for establishing the existence of a hydrogen bond, see: Arunan E, Desiraju GR, Klein RA, Sadlej J, Scheiner S, Alkorta I, Clary DC, Crabtree RH, Dannenberg JJ, Hobza P, Kjaergaard HG, Legon AC, Mennucci B, Nesbitt DJ. Pure Appl Chem. 2011;83:1637–1641.
- 56.Dunitz JD, Taylor R. Chem–Eur J. 1997;3:89–98. [Google Scholar]
- 57.Dunitz JD. ChemBio Chem. 2004;5:614–621. doi: 10.1002/cbic.200300801. [DOI] [PubMed] [Google Scholar]
- 58.Althoff G, Ruiz J, Rodriguez V, Lopez G, Perez J, Janiak C. Cryst Eng Comm. 2006;8:662–665. [Google Scholar]
- 59.Schneider HJ. Chem Sci. 2012;3:1381–1394. [Google Scholar]
- 60.Sheldrick GM. SADABS. Bruker AXS; Madison, WI: 1998. [Google Scholar]
- 61.Van der Sluis P, Spek AL. Acta Crystallogr. 1990;A46:194–201. [Google Scholar]
- 62.SHELXTL, 6.10. BRUKER AXS; Madison, WI: [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.