

Significance
Vasodilation is impaired in the postischemic heart due to loss of endothelial nitric oxide synthase (eNOS) function; however, the central trigger of this endothelial dysfunction is unknown. We observe that near total depletion of the eNOS substrate NADPH occurs in postischemic endothelium, triggering eNOS dysfunction and limiting rescue of its essential cofactor BH4. This NADPH depletion was shown to be due to marked activation of the NAD(P)ase activity of CD38 and could be prevented by its inhibition or genetic knockdown. Thus, CD38 activation with pronounced NADPH depletion is first identified as a critical trigger of postischemic endothelial dysfunction, and this presents a novel therapeutic target for the treatment of unstable coronary syndromes and prevention of coronary disease.
Keywords: ischemia reperfusion injury, endothelial nitric oxide synthase, nitric oxide, endothelial dysfunction, tetrahydrobiopterin
Abstract
In the postischemic heart, coronary vasodilation is impaired due to loss of endothelial nitric oxide synthase (eNOS) function. Although the eNOS cofactor tetrahydrobiopterin (BH4) is depleted, its repletion only partially restores eNOS-mediated coronary vasodilation, indicating that other critical factors trigger endothelial dysfunction. Therefore, studies were performed to characterize the unidentified factor(s) that trigger endothelial dysfunction in the postischemic heart. We observed that depletion of the eNOS substrate NADPH occurs in the postischemic heart with near total depletion from the endothelium, triggering impaired eNOS function and limiting BH4 rescue through NADPH-dependent salvage pathways. In isolated rat hearts subjected to 30 min of ischemia and reperfusion (I/R), depletion of the NADP(H) pool occurred and was most marked in the endothelium, with >85% depletion. Repletion of NADPH after I/R increased NOS-dependent coronary flow well above that with BH4 alone. With combined NADPH and BH4 repletion, full restoration of NOS-dependent coronary flow occurred. Profound endothelial NADPH depletion was identified to be due to marked activation of the NAD(P)ase-activity of CD38 and could be prevented by inhibition or specific knockdown of this protein. Depletion of the NADPH precursor, NADP+, coincided with formation of 2’-phospho-ADP ribose, a CD38-derived signaling molecule. Inhibition of CD38 prevented NADP(H) depletion and preserved endothelium-dependent relaxation and NO generation with increased recovery of contractile function and decreased infarction in the postischemic heart. Thus, CD38 activation is an important cause of postischemic endothelial dysfunction and presents a novel therapeutic target for prevention of this dysfunction in unstable coronary syndromes.
Endothelial dysfunction is associated with a wide range of cardiovascular diseases including hypercholesterolemia, diabetes, atherosclerosis, hypertension, heart failure, and ischemic heart disease (1). In vivo coronary occlusion induces endothelial dysfunction with decreased endothelial nitric oxide synthase (eNOS)-dependent vasoreactivity, which persists upon reperfusion (2, 3). Persistent diminished flow through the coronary arteries upon reperfusion can lead to cardiac myocyte injury and heart failure (4). Endothelial dysfunction is induced by the marked oxidant stress that occurs following the onset of ischemia and reperfusion (I/R) (5).
Normally, vascular tone and coronary vasodilation are modulated by nitric oxide (NO). Synthesis of NO occurs within the endothelium via eNOS, which uses l-arginine and O2 to form NO and l-citrulline. This enzymatic process uses NADPH as the source of reducing equivalents and requires Ca2+/calmodulin, flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), heme, and tetrahydrobiopterin (BH4) as cofactors. eNOS regulates vasomotor tone and blood pressure by producing NO, which activates soluble guanylate cyclase in vascular smooth muscle, resulting in vasorelaxation (6, 7).
In postischemic myocardium, the critical redox-sensitive eNOS cofactor, BH4, is depleted, leading to eNOS uncoupling with production of superoxide (O2•–) instead of NO (8, 9). The essential role of BH4 in NO production has made it a primary target of therapeutic studies aimed at restoring endothelial function following I/R (10). Preventing loss of and/or replenishing BH4 has proven beneficial, although recovery of eNOS function post-BH4 replacement therapy remains incomplete (10, 11). Furthermore, it remains unclear why BH4 salvage pathways fail to adequately regenerate this critical cofactor. Thus, there is a great need to identify other factor(s) that lead to eNOS dysfunction in the postischemic heart and develop new therapeutic approaches to restore endothelium-dependent vasodilatory function.
Studies were performed to characterize the previously unidentified factors that trigger and contribute to postischemic endothelial dysfunction. We observed that marked NADP(H) depletion occurs, resulting in impaired eNOS function and limited BH4 recycling through NADPH-dependent salvage pathways. Tandem NADPH and BH4 supplementation resulted in complete restoration of NOS-dependent coronary flow (CF). The profound endothelial NADP(H) depletion was shown to be due to activation of CD38. siRNA-mediated CD38 knockdown in endothelial cells prevented NADP(H) depletion. CD38 inhibition preserved endothelium-dependent CF, eNOS coupling, and NO generation, with enhanced recovery of cardiac contractile function and decreased infarction in the postischemic heart.
Results
Effects of I/R on Pyridine Nucleotides.
Depletion of NADPH could explain both the loss of eNOS function and failure to salvage BH4 in the postischemic heart. It is not known if alterations occur in the level of this critical reducing substrate required for eNOS activity and BH4 resynthesis through dihydrofolate reductase (10). The pyridine nucleotides NADP+ and NADPH as well as NAD+ and NADH were measured in control hearts and hearts subjected to I/R. In control hearts, NADP+ and NADPH levels were 23.5 ± 4.1 and 55.3 ± 4.3 nmol/g tissue, respectively, whereas NAD+ and NADH levels were 564.3 ± 31.6 and 5.7 ± 0.2 nmol/g tissue, respectively (Fig. 1 and SI Appendix, Fig. S1). By 10 min of ischemia, NADP+ declined with concomitant increase in NADPH, due to the reduced state of the myocardium that accompanies ischemia (12). By 30 min of ischemia, NADP+ and NADPH both declined below preischemic levels, with a further dramatic decrease following reperfusion. Importantly, although the total NADP(H) pool does not change during early ischemia, depletion occurs during later ischemia and following reperfusion, with NADP+ declining by ∼70% and NADPH levels declining by ∼50% by 30 min of reperfusion (Fig. 1A). In contrast, a much lower decrease was seen in the NAD(H) pool (∼25%; SI Appendix, Figs. S1 and S2).
Fig. 1.

NADP(H) whole-heart measurements. (A) NADP+ and NADPH preischemia (PI), then during I/R. (B) Representative chromatograms showing the NADP+ and NADPH peaks (ex. 330 nm; em. 460 nm) in nonischemic control hearts and hearts with 30 min of ischemia (I) and 30 min of ischemia/30 min reperfusion (I/R). For the entire chromatogram, see SI Appendix, Fig. S2. *P < 0.05 compared with preischemic values (mean ± SEM; n = 5–7).
Repletion of eNOS Cofactors/Substrates.
To determine if postischemic NADPH depletion contributed to the impaired NOS-dependent CF, experiments were performed to test if NADPH repletion could reverse this dysfunction. Liposomal NADPH administration performed in hearts subjected to 30 min of ischemia and 30 min of reperfusion (I/R) induced a prominent 30.9 ± 3.2% rise in CF compared with baseline. When combined with BH4, CF increased by 39.0 ± 5.7%, or >3 times the amount with BH4 alone (10.0 ± 1.0%; Fig. 2A). Coadministration of the eNOS inhibitor, nitro-l-arginine methyl ester (l-NAME), along with NADPH, completely blocked the increase seen with NADPH administration with a 18.6 ± 6.2% decrease in CF (Fig. 2A). BH4 treatment alone resulted in only ∼50% recovery of NOS-dependent CF. NADPH treatment resulted in ∼85% recovery, whereas combined BH4 and NADPH treatment resulted in full recovery (Fig. 2B).
Fig. 2.

Effect of eNOS cofactor/substrate repletion on CF and endothelial depletion of NADP(H) after I/R. (A, Top) Tracings showing change in CF after administration of control liposome (vehicle) or liposomal BH4 (50 µM), NADPH (175 µM), NADPH (175 µM) + BH4 (50 µM), and NADPH (175 µM) + l-NAME (1 mM), with the start of administration at time depicted by an arrow. (A, Bottom) Graph of the mean changes in CF. NADPH elicited a dramatic rise in CF, much greater than that of BH4. This increase in CF was NOS-dependent, as coinfusion of l-NAME abolished NADPH-mediated vasodilation with vasoconstriction seen. The effects of combined BH4 and NADPH infusion were additive. *P < 0.05 compared with vehicle; +P < 0.05 compared with BH4 (mean ± SEM; n = 5). (B) BH4 or NADPH treatment resulted in improved but incomplete restoration of NOS-dependent CF, whereas combined, BH4 + NADPH resulted in complete restoration. *P < 0.05 compared with vehicle; +P < 0.05 compared with BH4 (mean ± SEM; n = 5). (C) Endothelial NADP(H) content was measured from the effluent after endothelial permeabilization from hearts subjected to nonischemic perfusion (Control) or I/R. Endothelial NADP(H) levels were ∼90% lower in hearts subjected to I/R compared with hearts subjected to nonischemic perfusion. **P < 0.01 versus untreated I/R group (mean ± SEM; n = 5–7).
To confirm that the observed NADPH depletion limits eNOS activity, l-arginine to l-citrulline conversion assays were performed in heart tissue with and without addition of NADPH. In both control and postischemic tissues, NADPH addition was required to observe maximum activity. With NADPH addition, postischemic NOS activity was increased ∼fivefold to levels similar to those in control tissue (SI Appendix, Fig. S3).
To verify that NADPH repletion-mediated restoration of postischemic CF is eNOS-dependent, similar repletion studies were performed in wild-type (WT) and eNOS−/− mouse hearts. Although liposomal NADPH repletion increased CF in WT hearts by ∼30%, similar to that in the rat model, this effect was totally lost in eNOS−/− hearts. Thus, the NADPH-mediated increase in postischemic CF is eNOS-dependent (SI Appendix, Fig. S4).
Endothelial Levels of NADP(H).
Due to the marked impairment in NOS-dependent CF, we hypothesized that the depletion of NADP(H) in the endothelium would be more severe relative to the depletion in the myocardium. To measure I/R effects on pyridine nucleotides within the endothelium, we permeabilized the endothelium of the heart as previously reported (13). NADP(H) efflux from the endothelium in control hearts was measured with 0.45 ± 0.10 nmol detected, whereas with hearts subjected to I/R only 0.069 ± 0.050 nmol of NADP(H) was detected (Fig. 2C). Thus, I/R depletes the endothelial NADP(H) pool by almost 90%, accounting for impairment in endothelial vasodilatory function, which is restored by NADPH administration. Endothelial levels of NAD(H) also trended downward after I/R, but the depletion was not as severe as with the NADP(H) pool (SI Appendix, Fig. S5).
Expression, Activity, and Localization of CD38.
NADP+ was preferentially depleted during reperfusion, with the NADP(H) pool almost totally depleted from the endothelium. Therefore, this depletion was not simply due to oxidation of NADPH to NADP+ but to depletion of NADP+ as well. This implies that there must be a process consuming NADP+ causing depletion of the NADP(H) pool. CD38 preferentially uses NADP+ over NAD+ with >sixfold lower Km and higher Vmax (14). We hypothesized that CD38, with NAD(P)ase activity, which metabolizes NADP+ to 2’-phospho-ADP ribose (2’-P-ADPR) and nicotinamide (NA), may be of importance in this process of NADP(H) depletion (Fig. 3).
Fig. 3.

Multiple reactions of CD38. CD38 catalyzes several reactions including (1) formation of the Ca2+-liberating molecule 2’-P-cADPR; (2) hydrolysis of 2’-P-cADPR, forming 2’-P-ADPR; and (3) NAD(P)ase activity directly generating 2’-P-ADPR from NAD(P)+.
To determine the presence and location of CD38 in the heart, immunoblotting and immunohistology were performed in control hearts and hearts subjected to 30 min of ischemia or 30 min of ischemia followed by 30 min of reperfusion (I/R). Control hearts showed clear bands corresponding to CD38, and no change in CD38 expression was seen with ischemia or I/R (Fig. 4A). Immunohistochemistry demonstrated that high levels of CD38 are present in the endothelium of the coronary vasculature and colocalize with the endothelial markers von Willebrand factor (vWf) and eNOS (Fig. 4B). Lower levels of staining were seen in cardiac myocytes with an ∼eightfold lower intensity.
Fig. 4.

CD38 in the isolated rat heart. (A) Immunoblotting demonstrated that CD38 expression was present in control heart tissue (control), and this was unchanged by 30 min of ischemia (I) or 30 min of ischemia and 30 min of reperfusion (I/R). (B) CD38 was found predominately within the vasculature of isolated hearts. Confocal microscopy (60×) revealed CD38 colocalized with other endothelium-specific proteins, vWf, and eNOS (images in the Merge column also include DAPI nuclear stain). The presence and localization of CD38 was unchanged after ischemia or I/R. (C) A basal level of CD38 activity is measured in control hearts; however, activity with ischemia and I/R is significantly increased. **P < 0.01 versus Control (mean ± SEM; n = 5).
When CD38 activity was measured (as production of 2’-P-ADPR) from these hearts, activity was low in control (nonischemic) hearts. CD38 activity increased 2.2-fold after 30 min of ischemia and 5.5-fold after 30 min of reperfusion (Fig. 4C and SI Appendix, Fig. S6A). Furthermore, levels of 2’-P-ADPR in control hearts were barely detectable (1.68 ± 0.42 nmol/g tissue) but greatly increased after I/R to 9.11 ± 0.43 nmol/g tissue (SI Appendix, Fig. S6B).
CD38 Inhibition Salvages NADPH and Restores NOS-Dependent CF.
As CD38 activity and 2’-P-ADPR levels rise in the postischemic heart, we would expect NADP+ to be consumed. To confirm the role of CD38 in the process of NADP(H) depletion, experiments were performed with the CD38 inhibitor, α-NAD (15). In hearts subjected to I/R, administration of α-NAD immediately before the onset of ischemia resulted in near total salvage of endothelial NADP(H) (Fig. 5A). In parallel with this, near total recovery of NOS-dependent CF was seen (Fig. 5B). This confirms the role of CD38 in NADP(H) depletion as well as the importance of NADP(H) levels in modulating NOS-dependent CF in the postischemic heart (Fig. 5B).
Fig. 5.

Endothelial NADP(H) levels, correlation with NOS-dependent CF recovery, BH4 salvage, and NO production. Levels of endothelial NADP(H) were determined by endothelial membrane permeabilization with measurement of the effluent via HPLC. (A) ∼90% depletion of endothelial NADP(H) occurred with I/R compared with control nonischemic hearts. CD38 inhibition with α-NAD salvaged NADP(H). **P < 0.01 versus untreated I/R group (mean ± SEM; n = 5–7). (B) Prevention of NADP(H) loss through CD38 inhibition by α-NAD correlated with preservation of NOS-dependent CF. **P < 0.01 versus untreated I/R group (mean ± SEM; n = 5–7). (C) Levels of BH4 from whole-heart tissue were depleted after 30 min of ischemia followed by 30 min of reperfusion. When 5 mM α-NAD was administered just before ischemia, the loss of BH4 was blunted. **P < 0.01 versus untreated I/R group (mean ± SEM; n = 4). (D) NO production from hearts was measured by EPR spin trapping. NO levels dramatically decreased with I/R but were preserved with 5 mM α-NAD treatment. **P < 0.01 versus untreated I/R group (mean ± SEM; n = 4).
CD38 Inhibition Improves BH4 Recovery.
Due to the NADPH dependence of the BH4 salvage pathway (9), we hypothesized that CD38 inhibition with resultant NADP(H) preservation would lead to enhanced BH4 salvage. In hearts pretreated with α-NAD, ∼twofold higher BH4 levels were observed after I/R compared with those in untreated hearts (Fig. 5C). Thus, CD38 inhibition enhanced both postischemic NADP(H) and BH4 salvage.
CD38 Inhibition Preserves NO and Decreases O2•– Production.
EPR spin trapping was performed to directly measure eNOS coupling and function in the heart before and after I/R. NO production was measured using Fe2+-N-methyl-d-glucamine dithiocarbamate (Fe-MGD) and O2•– using 5,5′-dimethyl-1-pyrroline-N-oxide (DMPO). Following I/R, a >sixfold decrease in NO generation was seen, and this was fully restored with α-NAD treatment (Fig. 5D and SI Appendix, Fig. S7). Following I/R, a large >10-fold increase in O2•– generation was seen compared with preischemic control, and this was decreased ∼fourfold by the NOS inhibitor l-NAME. α-NAD pretreatment blocked this NOS-dependent O2•– by ∼85% (SI Appendix, Fig. S8). Together these measurements demonstrate that CD38 inhibition restored eNOS function and coupling.
CD38 Knockdown Enhances NADP(H) Recovery in Rat Aortic Endothelial Cells.
To further demonstrate the role of CD38 in NADP(H) depletion, cultured rat aortic endothelial cells (RAECs) with or without CD38 knockdown by shRNA were subjected to normoxic treatment or hypoxia/reoxygenation (H/R). The RAECs with CD38 knockdown showed preserved NADP(H) levels following H/R compared with RAECs receiving the control plasmid lacking CD38 shRNA (empty plasmid) (SI Appendix, Fig. S9), providing additional evidence that H/R-associated CD38 activation causes endothelial NADP(H) depletion.
Role of Oxidative Stress in CD38 Activation with Conversion of NADP(H) to 2’-P-ADPR.
I/R is associated with marked oxidative stress with production of O2•–. Because CD38 activation has been reported to be associated with oxidative stress (16–18), we questioned if I/R-associated oxidant stress triggers CD38 activation. We observed that CD38 is activated following I/R (Fig. 4C), and this leads to a marked increase in 2’-P-ADPR levels in the myocardium. We observed a >fivefold increase in 2’-P-ADPR from 1.68 ± 0.42 to 9.12 ± 0.44 nmol/g tissue following I/R (Fig. 6). When hearts were treated with the superoxide dismutase (SOD) mimetic Mn(III) tetrakis(4-benzoic acid)porphyrin chloride (MnTBAP), this increase was largely blocked. The I/R-associated increase in myocardial 2’-P-ADPR level was also totally abolished by the CD38 inhibitor α-NAD (Fig. 6). Thus, O2•–-derived oxidant stress is a key trigger of CD38 activation with NADP(H) degradation.
Fig. 6.
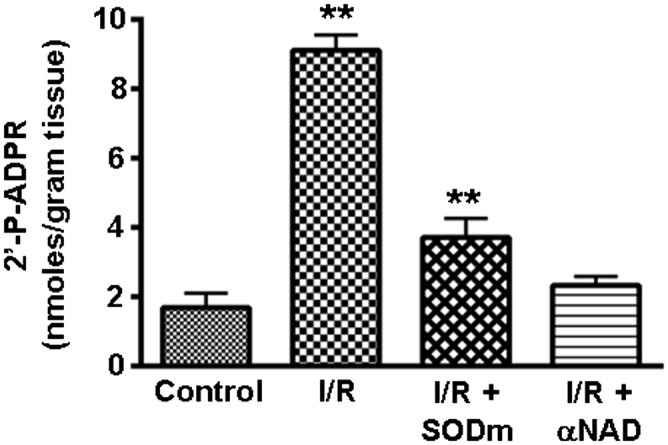
Regulation of CD38 activity measured by 2’-P-ADPR levels. With activation of CD38 activity by I/R, there is an increase in 2’-P-ADPR. The rise in 2’-P-ADPR level was largely abolished by pretreatment with a SOD mimetic (MnTBAP), suggesting activation was triggered by I/R-associated oxidant stress. The CD38 inhibitor α-NAD decreased 2’-P-ADPR formation. **P < 0.01 versus control (mean ± SEM; n = 4).
NADPH Supplementation and CD38 Inhibition Enhance Recovery of Contractile Function and Decrease Infarction.
To determine if the normalization of eNOS-mediated endothelial function seen with NADPH supplementation or CD38 inhibition is associated with myocardial protection as evidenced by improved recovery of contractile function and decreased infarction, additional studies were performed in isolated hearts from WT and eNOS−/− mice subjected to I/R. In WT hearts, the recovery of both left ventricular (LV) developed pressure (LVDP) and its peak derivative (dP/dt) were ∼75% higher with liposomal NADPH treatment (N) and ∼125% higher with α-NAD treatment than in untreated hearts (Fig. 7). Significantly lower LV end diastolic pressure (LVEDP) values were seen, indicating improved diastolic relaxation. Infarct size was also reduced by ∼30% in the treated WT hearts. In contrast, in eNOS−/− hearts there were no significant functional changes with either treatment. In eNOS−/−, NADPH treatment did not alter infarction and α-NAD treatment exerted only a small decrease in infarct size that did not reach significance, whereas in WT a significant decrease was seen (Fig. 7). Thus, the observed normalization of endothelial function through NADPH supplementation or CD38 inhibition is accompanied by myocardial protection.
Fig. 7.

Effect of NADPH supplementation or CD38 inhibition on recovery of contractile function and infarction in WT and eNOS−/− hearts. Studies were performed on isolated hearts that were pretreated with liposomal NADPH (N) or the CD38 inhibitor (αNAD) and then subjected to I/R. Results are shown for LVDP, maximum derivative of LVDP versus time (dP/dt), LVEDP, and infarct size as percentage of the LV. In WT hearts, either NADPH or α-NAD treatment enhanced the recovery of contractile function, with higher LVDP and dP/dt seen with lower LVEDP; whereas in eNOS−/− no significant changes were seen with either treatment. Liposomal NADPH decreased infarct size only in WT but not in eNOS−/− hearts. α-NAD decreased infarct size in WT with only a small effect in eNOS−/− hearts that did not reach significance. **P < 0.01 and *P < 0.05 versus respective untreated group (mean ± SEM; n = 7–9).
Discussion
Loss of eNOS cofactors and substrates can trigger endothelial dysfunction (13, 19). Our laboratory (10) and others (20) have demonstrated that the NOS cofactor, BH4, is lost during ischemic injury, contributing to endothelial dysfunction. Although BH4 repletion can ameliorate endothelial dysfunction, the recovery is incomplete (10). A recent clinical study highlighted the limitations in recovery of endothelial function with BH4 repletion where patients with coronary artery disease were unresponsive to BH4 treatment (11). The unsuccessful recovery of endothelial function was reported to be due to the oxidative environment that accompanies coronary artery disease with oxidation of BH4 to BH2. This inability to maintain BH4 in a reduced state suggests possible impairment of NADPH-dependent BH4 salvage.
We observed that NADPH, the essential reducing substrate for eNOS, was severely depleted during I/R and that this limited eNOS activity in the heart. NADPH is required for reduction of the FAD cofactor of eNOS, enabling catalytic conversion of l-arginine and O2 to l-citrulline and NO (21). In addition, NADPH is required for the function of the critical BH4 salvaging enzyme, dihydrofolate reductase (22).
Measurements from heart homogenates revealed that NADPH is depleted during I/R (Fig. 1). This loss of NADPH could not be explained by oxidation, as levels of NADP+ decreased more than NADPH. In contrast, the NAD(H) pool was much less affected. This preferential depletion of NADP(H) as well as the more marked NADP+ depletion relative to NADPH demonstrates that the loss of NADP(H) cannot be explained by washout from damaged cells; rather, this occurs due to a specific process that depletes NADP+ with corresponding diminution of the NADP(H) pool.
The significance of this NADPH depletion on endothelial function was demonstrated in studies in which repletion with liposomal NADPH resulted in marked restoration of postischemic CF (Fig. 2A). Repletion of NADPH was ∼threefold more potent than similar BH4 treatment, and combined NADPH and BH4 treatment achieved full restoration of NOS-dependent CF (Fig. 2B). The marked potency of NADPH in restoration of endothelial vasodilatory function suggested that endothelial NADPH depletion may be more severe than that in the myocardium. Measurements of the endothelial NADP(H) pool demonstrated near total depletion with I/R (Fig. 2C). Thus, I/R triggered severe depletion of endothelial NADP(H), leading to severe impairment of endothelium-dependent vasodilation.
Enzymes that catabolize pyridine nucleotides include poly-ADP ribose polymerase (PARP), sirtuin deacetylases, and CD38 (22). Of these, CD38 is unique in its substrate preference for NADP+ over NAD+ with >sixfold lower Km and twofold higher Vmax (14). CD38 is a complex and multifunctional enzyme (Fig. 3) with wide distribution in many tissues (14, 23–25). Originally identified as an antigen marker on B cells (26), CD38 was later found to contain ADP ribosyl cyclase, cADPR hydrolase, and NAD(P)ase activities. The conversion of NADP+ to 2’-P-ADPR and NA can be achieved through the cyclization of NADP+ to 2’-P-cADPR, followed by the hydrolysis of 2’-P-cADPR to 2’-P-ADPR. CD38 can also act as an NAD(P)ase, directly hydrolyzing NADP+ to 2’-P-ADPR and NA.
We observed that CD38 is present in the heart, with high levels of expression in the endothelium (Fig. 4B). Although CD38 expression did not change following the onset of I/R, its activity greatly increased, accounting for the decrease in NADP+ levels with a corresponding increase in 2’-P-ADPR (Fig. 4C). The increased activity of CD38 with ischemia and I/R was shown to cause the depletion of the endothelial NADP(H), as inhibition of CD38 led to complete salvage of endothelial NADP(H) and a near-complete recovery of NOS-dependent CF and NO generation following I/R (Fig. 5 A, B, and D). This was confirmed in studies of cultured endothelial cells subjected to H/R where genetic knockdown of CD38 preserved the levels of NADP(H) (SI Appendix, Fig. S9). Thus, CD38 activation was shown to have a profound effect on endothelial levels of NADP(H) and eNOS function after I/R.
CD38 activation in the postischemic heart was inhibited by a SOD mimetic, suggesting that O2•– can trigger activation. Thus, oxidative modification of CD38 may contribute to its activation. CD38 has essential cysteines that must be oxidized to form disulfide bonds for maximum activity. Mutations of these cysteines decrease or abrogate the enzymatic function of CD38 (27). It has also been reported that endogenous or exogenous sources of O2•– can activate CD38 by internalization of membrane-associated enzyme, leading to exposure to its intracellular substrates, NADP+ and NAD+ (16, 18).
The activity of CD38 is typically measured by production of the signaling molecules 2’-P-cADPR/cADPR, 2’-P-ADPR/ADPR, or nicotinic acid adenine dinucleotide phosphate (NAADP) (14). Both 2’-P-cADPR and cADPR signal Ca2+ release via intracellular stores (14). NAADP modulates lysosomal Ca2+ release, and ADPR increases Ca2+ levels through activation of the transient receptor potential cation channel, subtype M (28, 29). Because Ca2+ overload is a major mechanism of postischemic injury, inhibiting the formation of these signaling molecules could confer added benefit through moderation of Ca2+ release into the cytosol in endothelial cells and cardiac myocytes.
Oxidative stress and Ca2+ overload are central mechanisms of cardiac reperfusion injury. These two mechanisms interact and potentiate each other with oxygen radicals inducing Ca2+ overload, which in turn activates pathways of oxygen radical generation (30–32). In prior reports, CD38 activity was shown to be redox-sensitive, and exposure to oxidative stress and reactive oxygen species is reported to increase CD38 activity (18, 24). Consistent with this, we observed that treatment with a SOD mimetic decreased postischemic CD38 activation with a 2.5-fold decrease in levels of 2’-P-ADPR (Fig. 6).
Beyond its critical role in NOS function, NADPH is a requisite substrate for a number of enzymes of central importance in redox regulation and metabolism. NADPH provides the reducing equivalents to protect against oxidative stress and is the substrate regulating the glutathione redox state through glutathione reductase (33). It functions as a reducing substrate for dihydrofolate reductase that is required for the de novo synthesis of purines, thymidylic acid, and certain amino acids, in addition to serving as the major BH4 salvage pathway (22). We observed that inhibition of CD38 with preservation of NADPH resulted in increased levels of BH4, highlighting the importance of NADPH for BH4 salvage in I/R injury (Fig. 5C).
It was observed that supplementation of NADPH or inhibition of CD38 not only restored postischemic endothelial function but also conferred myocardial protection with enhanced recovery of contractile function and decreased infarction. This protection was shown to be eNOS-dependent, as it was lost in eNOS−/− hearts, suggesting that normalization of endothelial function can result in generalized myocardial protection (Fig. 7).
In summary, we observe that the critical eNOS reducing substrate NADPH is depleted in the postischemic heart. This loss is most severe in the endothelium and occurs due to activation of the NAD(P)ase function of CD38, thereby depleting the NADP(H) pool and salvage of BH4, triggering eNOS dysfunction and uncoupling. Thus, NADPH depletion is identified as a major cause of postischemic endothelial dysfunction, with NADPH repletion or CD38 inhibition restoring normal endothelial vasodilation. This restoration of endothelial function is particularly important, as it was also shown to be accompanied by enhanced recovery of contractile function and decreased infarction. These observations suggest that therapeutics that restore NADPH and/or prevent its degradation may be beneficial in the treatment of unstable coronary syndromes and myocardial infarction. In view of the central importance of oxidant-induced eNOS-mediated endothelial dysfunction in the pathogenesis of a broad spectrum of disease, identification of this novel pathway provides critical therapeutic insights for disease prevention and treatment.
Methods
Isolated Heart Studies.
Hearts from male, Sprague–Dawley rats (275–300 g) or C57BL/6J mice (WT and eNOS−/−) were prepared as described (10). eNOS−/− mice, strain B6.129P2-Nos3tm1Unc/J, were purchased from Jackson Laboratory. Hearts were excised, aorta cannulated, and perfused retrograde with Krebs buffer [119 mM NaCl, 17 mM Glucose, 25 mM NaHCO3, 5.9 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 0.5 mM EDTA, and 2 mM pyruvate (mice only)]. A balloon connected to a pressure transducer was placed in the LV to measure contractile function. An in-line flow probe and flowmeter (Transonic) were used to measure CF. Measurements of infarct size were performed after 120 min of reperfusion as described (34) (SI Appendix, SI Methods). Animal protocols were approved by the Institutional Animal Care and Use Committee of The Ohio State University.
Materials.
Unless otherwise noted, all chemicals as well as ADP ribosyl cyclase from Aplysia californica were purchased from Sigma.
Experimental Protocols.
For pyridine nucleotide measurements, hearts were subjected to either control perfusion, periods of ischemia (10, 20, or 30 min), or ischemia (30 min) followed by reperfusion (10, 20, or 30 min). Control hearts were perfused for 60 min, whereas those undergoing global ischemia or I/R were first perfused for a 20-min equilibration period. At the designated time, hearts were freeze-clamped and stored in liquid nitrogen.
For measurements of CF restitution, hearts were subjected to 30 min of ischemia followed by 30 min of reperfusion and infused with either control liposomes (25 mg/mL), liposomal NADPH (175 µM), liposomal BH4 (50 µM), liposomal NADPH (175 µM) + BH4 (50 µM), or liposomal NADPH (175 µM) + l-NAME (1 mM) for 10 min in Krebs buffer.
An additional set of hearts was used to measure endothelial NADP(H) after 30 min of ischemia followed by 30 min of reperfusion. Endothelial permeabilization was performed using the procedure of Giraldez et al., which selectively permeabilizes the endothelium, abrogating endothelial function, with minimal effects on cardiac myocytes (13). At the desired time, hearts received a brief (∼1 s) bolus of 0.25% Triton X-100, permeabilizing the endothelium allowing washout of endothelial substrates including the NADP(H) pool. The effluent was collected for three 1-min intervals. Additional hearts received either α-NAD (5 mM) or vehicle (Krebs buffer) infused for 3 min before the onset of ischemia. After 30 min of reperfusion, endothelial permeabilization was performed and effluent collected.
A final set of hearts was used to measure the effect of oxidant stress on 2’-P-ADPR production. Before ischemia, hearts were administered either α-NAD or the SOD mimetic MnTBAP (30 µM) for 3 min. Following 30 min of ischemia and 30 min of reperfusion, hearts were prepared for HPLC.
Detection of Pyridine Nucleotides by HPLC.
NAD(H) and NADP(H) detection was performed in hearts by modification of the method of Klaidman et al. (35) (SI Appendix, SI Methods). Samples were injected onto a TSKgel ODS-80TM column (25 cm × 4.6 mm) (Supelco). Separation was achieved with a flow rate of 1.0 mL/min and methanol gradient (0.2%/min for 25 min). Analytes were detected via fluorescence (ex. 330 nm; em. 460 nm).
Detection of Pyridine Nucleotides Following Endothelial Permeablization.
Before HPLC analysis, the heart effluent absorption spectrum was measured (Cary 50 Bio UV/VIS Spectrophotometer, Varian) and demonstrated strong absorption at 260 nm and 340 nm [for NAD(P)+ and NAD(P)H, respectively], with a lack of absorption from the α/β bands of heme, confirming a lack of myocyte leakage of myoglobin. Triton X-100 was removed from samples with addition of Surfactaway (BioTech Support Group) in a 1:4 dilution. Samples were concentrated using a SpeedVac (Thermo Scientific) and resuspended in 250 µL of potassium cyanide (KCN) buffer (KCN 0.2 M, KOH 0.06 M, and DTPA 1 mM). Surfactaway was again added at 1:4 dilution to ensure complete removal of Triton X-100, concentrated, and then HPLC measurements were performed as described above.
Immunoblotting for CD38.
Hearts were homogenized in 150 mM NaCl, 10 mM Tris, 1 mM EDTA with protease inhibitors, and 1% Triton X-100. Homogenates were separated on 10% (wt/vol) Tris-glycine polyacrylamide gels. Protein from gels was transferred to nitrocellulose membranes and blocked for 1 h at room temperature (RT) in Tris-buffered saline containing 0.075% Tween-20 (TBST), with 5% (wt/vol) milk. Membranes were incubated overnight with anti-CD38 antibody (Santa Cruz, M-19) diluted at 1:1,000 at 4 °C. Membranes were washed in TBST and incubated for 1 h with HRP-conjugated anti-rabbit IgG in TBST at RT. Imaging was performed with ECL immunoblotting detection reagents (Amersham Biosciences) and bands quantified with NIH ImageJ. Membranes were stripped and similarly immunoblotted with an anti-GAPDH antibody (Cell Signaling) diluted to 1:10,000.
Immunohistochemistry of CD38, vWF, and eNOS.
Isolated hearts were embedded in optimal cutting temperature compound and sectioned using a cryotome at –20 °C. Sections (5 µm) were attached to coverslips, fixed with 3.7% (wt/vol) paraformaldehyde (10 min), permeabilized with 0.25% Triton X-100 in TBST (0.01% Tween; 5 min), and blocked for 30 min with 1% BSA in TBST and incubated with anti-CD38 (Santa Cruz Biotechnology), anti-eNOS (BD Biosciences), and anti-vWF (Abcam) primary antibodies at dilutions of 1:300, 1:500, and 1:200, respectively, in TBST containing 1% BSA (1 h; RT). Sections were then incubated with secondary antibodies: Alexa Fluor 488 (anti-goat), 568 (anti-mouse), and 647 (anti-rabbit) (Life Technologies) at 1:1,000 dilution (1 h; RT). Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI). After washing with TBST, the sections were mounted with Fluoromount-G (Southern Biotech). Slides were visualized using an Olympus FV1000 imaging system.
HPLC Measurement of BH4.
Hearts were homogenized in ice-cold 0.1 N HCl. BH4 and BH2 were then oxidized to biopterin and pterin using a 2% (wt/vol) KI/3% (wt/vol) iodine solution in acid or basic conditions. Samples were loaded onto an Atlantis T3 reverse phase column (5 μm; 4.6 × 150 mm) (Waters Corporation). Isocratic elution was performed at a flow rate of 1.2 mL/min, using a buffer consisting of 100 mM KH2PO4 (pH 2.5), 6 mM citric acid, 2.5 mM 1-octanesulfonic acid (OSA), and 2% (vol/vol) methanol (9). Analytes were detected via fluorescence spectroscopy (wavelength ex. 348 nm; em. 444 nm).
HPLC Measurement of 2’-P-ADPR Levels and CD38 Activity.
Hearts were homogenized in ice-cold buffer (Hepes 20 mM, pH 7.3, Sucrose 250 mM, EDTA 0.1 mM, β-2 Mercaptoethanol 2.5 mM), and samples were subjected to chloroform treatment (1:1 volume) followed by centrifugation at 16,000 × g (2×; 5 min). The aqueous phase was collected and filtered using a Costar Spin-X 0.45 µm pore size filter. The effluent from Costar Spin-X tubes was diluted 1:3 with mobile phase (0.04 M Sodium Phosphate/1% methanol, pH 7.0). Filtrate was injected onto a TSKgel ODS-80TM column (25 cm × 4.6 mm) (Supelco) and separation achieved as described with UV/Vis detection at 254 nm (36) (SI Appendix, SI Methods). 2’-P-ADPR standard was prepared by incubating NADP+ with ADP ribosyl cyclase of A. californica. CD38 activity was measured by in vitro assays of 2’-P-ADPR formation. After homogenization, samples were centrifuged at 16,000 × g (2×; 5 min) at 4 °C. The supernatants were kept on ice and a HiTrap desalting column used to remove enzyme substrate. We then added 25 µL of homogenate (∼60 µg protein) to the reaction mixture (Tris buffer 50 mM, pH 7.4 with 100 µM NADP+), with a 10-min incubation at 37 °C. Upon reaction completion, samples were placed on ice and centrifuged using Costar Spin-X 0.45 µm pore size filters (3 min). HPLC measurements of 2’-P-ADPR were performed on the filtrate.
Measurements of NOS Activity.
NOS activity was measured by l-[14C]arginine to l-[14C]citrulline conversion assay as previously reported (10, 19) (SI Appendix, SI Methods).
EPR Spin Trapping of NO and O2•–.
EPR spin trapping of NO was performed using Fe-MGD (0.5 mM, 2.5 mM) in perfusate with 10 mg/mL BSA. Spin trapping of O2•– was performed using 50 mM DMPO in perfusate with heart effluent immediately mixed with 100 mM methyl-β-cyclodextrin. Measurements were performed with and without SOD1 (200 U/mL) or l-NAME (1 mM) (13) using a Bruker EMX spectrometer (SI Appendix, SI Methods).
Stable Knockdown of CD38 in RAECs.
Stable knockdown of CD38 was performed using shRNA plasmid provided by Hon-Cheung Lee, University of Hong Kong, Hong Kong (37), as described in SI Appendix, SI Methods.
H/R Model.
RAECs were washed with PBS and kept in serum-free DMEM in a hypoxic environment created by placing the flasks containing cells at confluence into a Billups–Rothenberg incubator chamber flushed with a 95% N2/5% CO2 gas as detailed in SI Appendix, SI Methods.
Statistics.
Results were expressed as mean ± SE. Statistical significance was determined by ANOVA (followed by Newman–Keuls test) for multiple groups. Paired or unpaired t tests were used for comparison between two groups.
Supplementary Material
Acknowledgments
This work was supported by NIH Grants HL63744, HL65608, HL38324, and EB016096 (to J.L.Z.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1505556112/-/DCSupplemental.
References
- 1.Yetik-Anacak G, Catravas JD. Nitric oxide and the endothelium: History and impact on cardiovascular disease. Vascul Pharmacol. 2006;45(5):268–276. doi: 10.1016/j.vph.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Mehta JL, Nichols WW, Donnelly WH, Lawson DL, Saldeen TG. Impaired canine coronary vasodilator response to acetylcholine and bradykinin after occlusion-reperfusion. Circ Res. 1989;64(1):43–54. doi: 10.1161/01.res.64.1.43. [DOI] [PubMed] [Google Scholar]
- 3.VanBenthuysen KM, McMurtry IF, Horwitz LD. Reperfusion after acute coronary occlusion in dogs impairs endothelium-dependent relaxation to acetylcholine and augments contractile reactivity in vitro. J Clin Invest. 1987;79(1):265–274. doi: 10.1172/JCI112793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolli R. Mechanism of myocardial “stunning”. Circulation. 1990;82(3):723–738. doi: 10.1161/01.cir.82.3.723. [DOI] [PubMed] [Google Scholar]
- 5.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci USA. 1987;84(5):1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327(6122):524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 7.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA. 1987;84(24):9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci USA. 1996;93(13):6770–6774. doi: 10.1073/pnas.93.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Pascali F, Hemann C, Samons K, Chen CA, Zweier JL. Hypoxia and reoxygenation induce endothelial nitric oxide synthase uncoupling in endothelial cells through tetrahydrobiopterin depletion and S-glutathionylation. Biochemistry. 2014;53(22):3679–3688. doi: 10.1021/bi500076r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dumitrescu C, et al. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci USA. 2007;104(38):15081–15086. doi: 10.1073/pnas.0702986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cunnington C, et al. Systemic and vascular oxidation limits the efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation. 2012;125(11):1356–1366. doi: 10.1161/CIRCULATIONAHA.111.038919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Velayutham M, Li H, Kuppusamy P, Zweier JL. Mapping ischemic risk region and necrosis in the isolated heart using EPR imaging. Magn Reson Med. 2003;49(6):1181–1187. doi: 10.1002/mrm.10473. [DOI] [PubMed] [Google Scholar]
- 13.Giraldez RR, Panda A, Zweier JL. Endothelial dysfunction does not require loss of endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2000;278(6):H2020–H2027. doi: 10.1152/ajpheart.2000.278.6.H2020. [DOI] [PubMed] [Google Scholar]
- 14.Vu CQ, Lu PJ, Chen CS, Jacobson MK. 2′-Phospho-cyclic ADP-ribose, a calcium-mobilizing agent derived from NADP. J Biol Chem. 1996;271(9):4747–4754. [PubMed] [Google Scholar]
- 15.Okayama H, Ueda K, Hayaishi O. NAD glycohydrolases from rat liver nuclei. Methods Enzymol. 1980;66:151–154. doi: 10.1016/0076-6879(80)66453-2. [DOI] [PubMed] [Google Scholar]
- 16.Xu M, et al. Contribution of NADPH oxidase to membrane CD38 internalization and activation in coronary arterial myocytes. PLoS One. 2013;8(8):e71212. doi: 10.1371/journal.pone.0071212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang G, et al. Cyclic ADP ribose-mediated Ca2+ signaling in mediating endothelial nitric oxide production in bovine coronary arteries. Am J Physiol Heart Circ Physiol. 2006;290(3):H1172–H1181. doi: 10.1152/ajpheart.00441.2005. [DOI] [PubMed] [Google Scholar]
- 18.Zhang AY, Yi F, Teggatz EG, Zou AP, Li PL. Enhanced production and action of cyclic ADP-ribose during oxidative stress in small bovine coronary arterial smooth muscle. Microvasc Res. 2004;67(2):159–167. doi: 10.1016/j.mvr.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Giraldez RR, Panda A, Xia Y, Sanders SP, Zweier JL. Decreased nitric-oxide synthase activity causes impaired endothelium-dependent relaxation in the postischemic heart. J Biol Chem. 1997;272(34):21420–21426. doi: 10.1074/jbc.272.34.21420. [DOI] [PubMed] [Google Scholar]
- 20.Masano T, et al. Beneficial effects of exogenous tetrahydrobiopterin on left ventricular remodeling after myocardial infarction in rats: The possible role of oxidative stress caused by uncoupled endothelial nitric oxide synthase. Circ J. 2008;72(9):1512–1519. doi: 10.1253/circj.cj-08-0072. [DOI] [PubMed] [Google Scholar]
- 21.Panda K, Adak S, Konas D, Sharma M, Stuehr DJ. A conserved aspartate (Asp-1393) regulates NADPH reduction of neuronal nitric-oxide synthase: Implications for catalysis. J Biol Chem. 2004;279(18):18323–18333. doi: 10.1074/jbc.M310391200. [DOI] [PubMed] [Google Scholar]
- 22.Armarego WL, Randles D, Taguchi H. Peroxidase catalysed aerobic degradation of 5,6,7,8-tetrahydrobiopterin at physiological pH. Eur J Biochem. 1983;135(3):393–403. doi: 10.1111/j.1432-1033.1983.tb07666.x. [DOI] [PubMed] [Google Scholar]
- 23.Thai TL, Arendshorst WJ. Mice lacking the ADP ribosyl cyclase CD38 exhibit attenuated renal vasoconstriction to angiotensin II, endothelin-1, and norepinephrine. Am J Physiol Renal Physiol. 2009;297(1):F169–F176. doi: 10.1152/ajprenal.00079.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chidambaram N, Wong ET, Chang CF. Differential oligomerization of membrane-bound CD38/ADP-ribosyl cyclase in porcine heart microsomes. Biochem Mol Biol Int. 1998;44(6):1225–1233. doi: 10.1080/15216549800202322. [DOI] [PubMed] [Google Scholar]
- 25.Cockayne DA, et al. Mice deficient for the ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood. 1998;92(4):1324–1333. [PubMed] [Google Scholar]
- 26.Reinherz EL, Kung PC, Goldstein G, Levey RH, Schlossman SF. Discrete stages of human intrathymic differentiation: Analysis of normal thymocytes and leukemic lymphoblasts of T-cell lineage. Proc Natl Acad Sci USA. 1980;77(3):1588–1592. doi: 10.1073/pnas.77.3.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao YJ, Zhang HM, Lam CM, Hao Q, Lee HC. Cytosolic CD38 protein forms intact disulfides and is active in elevating intracellular cyclic ADP-ribose. J Biol Chem. 2011;286(25):22170–22177. doi: 10.1074/jbc.M111.228379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Churchill GC, et al. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell. 2002;111(5):703–708. doi: 10.1016/s0092-8674(02)01082-6. [DOI] [PubMed] [Google Scholar]
- 29.Yamasaki M, et al. Organelle selection determines agonist-specific Ca2+ signals in pancreatic acinar and beta cells. J Biol Chem. 2004;279(8):7234–7240. doi: 10.1074/jbc.M311088200. [DOI] [PubMed] [Google Scholar]
- 30.Josephson RA, Silverman HS, Lakatta EG, Stern MD, Zweier JL. Study of the mechanisms of hydrogen peroxide and hydroxyl free radical-induced cellular injury and calcium overload in cardiac myocytes. J Biol Chem. 1991;266(4):2354–2361. [PubMed] [Google Scholar]
- 31.Corretti MC, et al. Glycolytic inhibition and calcium overload as consequences of exogenously generated free radicals in rabbit hearts. J Clin Invest. 1991;88(3):1014–1025. doi: 10.1172/JCI115361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talukder MA, Zweier JL, Periasamy M. Targeting calcium transport in ischaemic heart disease. Cardiovasc Res. 2009;84(3):345–352. doi: 10.1093/cvr/cvp264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burgoyne JR, Mongue-Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111(8):1091–1106. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- 34.Talukder MAH, et al. Is reduced SERCA2a expression detrimental or beneficial to postischemic cardiac function and injury? Evidence from heterozygous SERCA2a knockout mice. (Translated from English) Am J Physiol-Heart C. 2008;294(3):H1426–H1434. doi: 10.1152/ajpheart.01016.2007. [DOI] [PubMed] [Google Scholar]
- 35.Klaidman LK, Leung AC, Adams JD., Jr High-performance liquid chromatography analysis of oxidized and reduced pyridine dinucleotides in specific brain regions. Anal Biochem. 1995;228(2):312–317. doi: 10.1006/abio.1995.1356. [DOI] [PubMed] [Google Scholar]
- 36.Broetto-Biazon AC, et al. Transformation and actions of extracellular NADP(+) in the rat liver. Mol Cell Biochem. 2008;317(1-2):85–95. doi: 10.1007/s11010-008-9834-1. [DOI] [PubMed] [Google Scholar]
- 37.Yue J, et al. CD38/cADPR/Ca2+ pathway promotes cell proliferation and delays nerve growth factor-induced differentiation in PC12 cells. J Biol Chem. 2009;284(43):29335–29342. doi: 10.1074/jbc.M109.049767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.