

Abstract
DEPDC1 is a recently identified novel tumor-related gene that is upregulated in several types of cancer and contributes to tumorigenesis. In this study, we have investigated the expression pattern and functional implications of DEPDC1 during cell cycle progression. Expression studies using synchronized cells demonstrated that DEPDC1 is highly expressed in the mitotic phase of the cell cycle. Immunofluorescence assays showed that DEPDC1 is predominantly localized in the nucleus during interphase and is redistributed into the whole cell upon nuclear membrane breakdown in metaphase. Subsequently, siRNA-mediated knockdown of DEPDC1 caused a significant mitotic arrest. Moreover, knockdown of DEPDC1 resulted in remarkable mitotic defects such as abnormal multiple nuclei and multipolar spindle structures accompanied by the upregulation of the A20 gene as well as several cell cycle-related genes such as CCNB1 and CCNB2. Taken together, our current observations strongly suggest that this novel cancerous gene, DEPDC1, plays a pivotal role in the regulation of proper mitotic progression. [BMB Reports 2015; 48(7): 413-418]
Keywords: Cancer, Cell cycle, DEPDC1, Mitosis, Nuclear protein
INTRODUCTION
DEP domain containing 1 (DEPDC1) is a highly conserved protein among many species ranging from Caenorhabditis elegans to mammals. It was first reported to be aberrantly upregulated in bladder cancer and plays an essential role in the growth of bladder cancer cells (1-3). Subsequent reports further demonstrated that DEPDC1 was also overexpressed in other types of cancers including breast cancer, multiple myeloma and hepatocellular carcinomas (4-6) and has prognostic value for predicting outcomes in patients with multiple myeloma, hepatocellular carcinomas and lung cancer (5-7). Consequently, knockdown of DEPDC1 inhibited growth and induced apoptosis in bladder cancer and myeloma cells (1, 5). Notably, a very recent report showed that DEPDC1 participates in the anti-tubulin drug-induced apoptotic cell death pathway by promoting JNK-dependent degradation of the BCL-2 family protein MCL1 (8, 9). These studies strongly suggested that this newly identified cancerous gene, DEPDC1, plays a pivotal role in tumorigenesis, and might serve as a novel potential target in the diagnosis and/or treatment of various cancers.
Cancer is frequently viewed as a cell cycle disease. Accumulating evidence strongly suggests that the vast majority of human cancers arise from serious defects in accurate cell cycle regulation, which consequently leads to uncontrolled cell growth (10). Numerous tumor-related genes such as Plk1 and FOXM1 have been shown to play crucial roles in both cell cycle progression and tumorigenesis (11-13). However, whether DEPDC1 plays an important role in cell cycle progression remained unclear.
In the present study, we have for the first time examined the expression profile of DEPDC1 during the cell cycle and investigated the functional role of DEPDC1 in the regulation of cell cycle progression in HeLa cells.
RESULTS
DEPDC1 is highly expressed in the mitotic phase during the cell cycle
In the first step of our study, we examined the temporal expression profile of the endogenous DEPDC1 during the cell cycle progression. To this end, human cervical carcinoma-derived HeLa cells were synchronized at the G1/S boundary by a double-thymidine block and at the mitotic phase by a thymidine-nocodazole block (Fig. 1A). At the indicated time points after being released from synchronization, the cells were collected and subjected to quantitative RT-PCR analysis. As clearly shown in Fig. 1B and 1C, DEPDC1 was highly expressed in M-phase cells at the mRNA level, whereas its expression de creased significantly when the cells entered the G1 or S phase. In addition, immunoblotting analysis further demonstrated that both of the two DEPDC1 isoforms were highly expressed in the mitotic phase (Fig. 1D and E).
Fig. 1. DEPDC1 is highly expressed in mitosis. (A) Cell synchronization. HeLa cells were unsynchronized or synchronized at the G1/S boundary by using a double-thymidine block or in mitosis with a thymidine-nocodazole block. Cell cycle profile was determined by FACS analysis. (B) DEPDC1 mRNA expression after double-thymidine block release. HeLa cells were synchronized at the G1/S boundary by using a double-thymidine block. At the indicated time points following release from the second thymidine block, total RNA was prepared and subjected to quantitative RT-PCR. (C) DEPDC1 mRNA expression after thymidine-nocodazole block release. HeLa cells were synchronized in mitosis by using a thymidine-nocodazole block. At the indicated time points following release from the nocodazole block, total RNA was prepared and subjected to quantitative RT-PCR. (D) DEPDC1 protein is highly expressed in mitosis. Whole cell lysates were prepared from unsynchronized or synchronized HeLa cells and subjected to immunoblotting. (E) Western blot quantification. Intensity of individual bands was quantified using Quantity One software (Bio-Rad, Version 4.3.1), and expressed relative to GAPDH signal as a measure of protein relative abundance in the different samples.

Subcellular distribution of DEPDC1 during the cell cycle
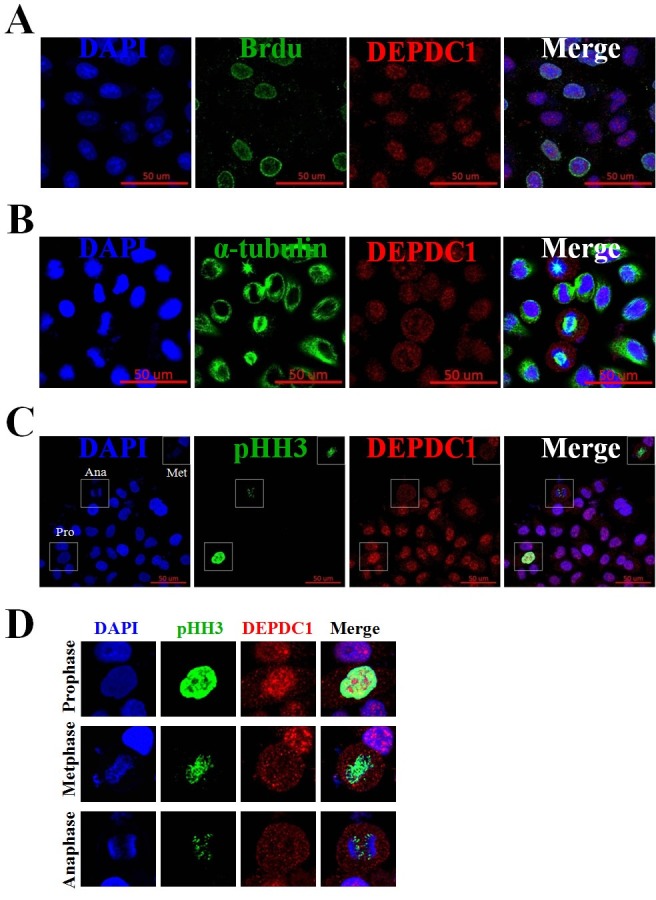
Next, we examined the spatial expression profile of the endogenous DEPDC1 during the cell cycle progression. To determine the spatial expression of DEPDC1 in S phase cells, HeLa cells were incubated in the presence of BrdU, and the cells were subjected to immunofluorescence staining with anti-DEPDC1 and anti-BrdU antibodies. As shown in Fig. 2A, DEPDC1 was expressed in all BrdU labeled S-phase cells and predominantly localized in the nucleus. To determine the spatial expression of DEPDC1 in M-phase cells, asynchronously growing HeLa cells were fixed and simultaneously stained with anti-DEPDC1 and anti-α-tubulin or anti-phospho-histone H3 (Ser10) antibodies. Intriguingly, as seen in Fig. 2B and 2C, DEPDC1 was found to be localized in the cytoplasm during mitosis. High magnification images of DEPDC1 expression further clearly demonstrated that DEPDC1 remained localized in the nucleus in prophase, and was redistributed into the whole cell upon nuclear membrane breakdown in metaphase and anaphase cells (Fig. 2D).
Fig. 2. Immunofluorescence assays of DEPDC1 expression during cell cycle. (A) PRR11 expression in S phase of the cell cycle. Asynchronously growing HeLa cells were incubated in the presence of BrdU for 30 min and then stained simultaneously with anti-BrdU and anti-DEPDC1 antibodies. Cell nuclei were stained with DAPI (blue). Scale bar, 50 μm. (B) and (C) Expression of DEPDC1 in M phase of the cell cycle. Exponentially growing cells were fixed and simultaneously stained with anti-DEPDC1 (red) and anti-α-tubulin (green) or anti-phospho-histone H3 (green). Cell nuclei were stained with DAPI (blue). (D) Higher magnification images of DEPDC1 expression in prophase, metaphase and anaphase as indicated in (C).
Knockdown of DEPDC1 leads to mitotic arrest and defects
Furthermore, we employed siRNA-mediated knockdown strategy to evaluate the potential role of DEPDC1 in cell cycle progression. For this purpose, HeLa cells were transiently transfected with control siRNA or with siRNA against DEPDC1. Forty-eight hours after transfection, total RNA and whole cell lysates were prepared and subjected to quantitative RT-PCR and immunoblotting, respectively. As shown in Fig. 3A, siRNA targeting DEPDC1 efficiently knocked down the endogenous DEPDC1, whereas control siRNA had an undetectable effect on the expression level of the endogenous DEPDC1. Flow cytometry of HeLa cells transfected with siRNA against DEPDC1 showed a marginal increase in G2/M fractions compared with control siRNA-transfected cells (Fig. 3B and 3C). To determine the stage of the cell cycle when DEPDC1-knocked down cells were arrested, we performed indirect immunofluorescence staining with anti-phospho-histone H3 antibody. As shown in Fig. 3D and 3E, knockdown of DEPDC1 caused a remarkable increase in the number of phospho-histone H3-positive mitotic cells compared with the control cells. Thus, it is likely that DEPDC1 is required for proper mitotic progression.
Fig. 3. Knockdown of DEPDC1 induces mitotic arrest and defects. (A) siRNA-mediated silencing of DEPDC1. HeLa cells were transiently transfected with a negative control siRNA (NC siRNA) or with siRNA against DEPDC1. Forty-eight hours after transfection, total RNA and whole cell lysates were prepared and subjected to RT-PCR (left) and immunoblotting (right), respectively. (B) The effects of DEPDC1 depletion on cell cycle profile. HeLa cells were transiently transfected as in (A). Forty-eight hours after transfection, cells were harvested, fixed in ethanol, stained with propidium iodide (PI) and subsequently subjected to FACS analysis. Representative histograms of cell cycle analysis are shown. (C) Statistical analysis of percentage of cells with G1, S or G2/M DNA content that correspond with data in (B) are shown. (D) Knockdown of DEPDC1 causes mitotic arrest and defects. Exponentially growing HeLa cells were transiently transfected with the indicated siRNAs. Forty-eight hours after transfection, cells were fixed and stained with anti-phospho-histone H3 antibody (green). DAPI was used to stain cell nuclei (blue). Red arrow indicates cells with irregular nucleus. Scale bar, 50 μm. (E) and (F) Statistical analysis of percentage of phospho-histone H3 positive cells and cells with irregular nuclei that correspond with data in (D) are shown. (G) Knockdown of DEPDC1 causes mitotic defects. HeLa cells were transiently transfected with the indicated siRNAs. Forty-eight hours after transfection, cells were fixed and stained with anti-α-tubulin antibody (green). DAPI was used to stain cell nuclei (blue). Red arrow indicates cells with abnormal multipolar spindle structure. Note that the different cell sizes and numbers are due to the uneven cell distribution of cell seeding. Scale bar, 50 μm.

Intriguingly, the above indirect immunofluorescence analysis also demonstrated that DEPDC1-depleted cells exhibit obvious mitotic defects with multiple nuclei structure (Fig. 3D and 3F). To confirm the observations, we performed indirect immunofluorescence staining with anti-α-tubulin antibody. As shown in Fig. 3G, some DEPDC1-knocked down cells did display abnormal multipolar spindle structure, which might subsequently lead to the cells pulling chromosomes in several directions, resulting in multiple nuclei in one cell.
DEPDC1 does not co-localize with centrosome
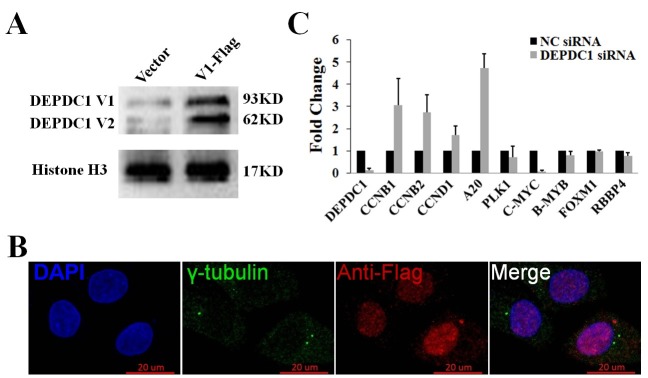
Since the centrosome is an organelle that serves as the main microtubule organizing center (MTOC) and the anchor for the mitotic spindle, we wondered whether DEPDC1 could be a centrosome-associated protein. To this end, HeLa cells were transiently transfected with the mammalian expression construct of the DEPDC1 isoform 1 fused with a C-terminal Flag peptide (DEPDC1-V1-Flag, Fig. 4A). As shown in Fig. 4B, however, we did not detect the co-localization of DEPDC1 with the specific centrosomal marker γ-tubulin in cells, indicating that DEPDC1 might be involved in mitotic spindle formation through an as yet unknown indirect mechanism.
Fig. 4. DEPDC1 is not associated with centrosome. (A) Overexpression of DEPDC1 in HeLa cells. HeLa cells were transiently transfected with an empty vector or the expression plasmid for Flag-DEPDC1. Twenty four hours after transfection, whole cell lysates were prepared from unsynchronized or synchronized HeLa cells and subjected to immunoblotting. (B) HeLa cells were transiently transfected with the expression plasmid for Flag-DEPDC1. Twenty four hours after transfection, cells were fixed and stained with anti-γ-tubulin antibody (green) and anti-Flag antibody (red). DAPI was used to stain cell nuclei (blue). Scale bar, 20 μm. (C) Knockdown of DEPDC1 results in the upregulation of A20. HeLa cells were transiently transfected with the negative control siRNA (NC siRNA) or with siRNA against DEPDC1. Forty-eight hours after transfection, total RNAs were prepared and subjected to quantitative RT-PCR.
Knockdown of DEPDC1 results in the upregulation of A20
DEPDC1 has been shown to act as a transcription repressor inhibiting the transcription of A20, a negative regulator of the NF-κB signaling pathway (2). Notably, accumulating evidence suggests that NF-κB plays an important role in mitotic progression through the regulation of cell cycle-related genes such as CCNB1 and CCNB2 (16-18). Thus, we determined that the transcription of A20 and several NF-κB regulated cell cycle genes upon DEPDC1 silencing. As shown in Fig. 4C, knockdown of DEPDC1 caused remarkable upregulation of A20 as well as CCNB1 and CCNB2, suggesting that DEPDC1 might regulate mitotic progression via regulating A20 transcription.
DISCUSSION
Several studies have demonstrated that DEPDC1 is aberrantly overexpressed and has prognostic value in several different types of cancers, indicating its involvement in carcinogenesis and its potential as a diagnostic and/or therapeutic target (1-7). Given the close correlation between carcinogenesis and cell cycle dysregulation, it is possible that DEPDC1 participates in cell cycle progression. To test this hypothesis, we employed human cervical carcinoma-derived HeLa cells as a model cell system to investigate the expression pattern and functional implications of DEPDC1 in the cell cycle. According to our observations, DEPDC1 plays a pivotal role in mitotic progression as the expression profile showed a remarkably elevated expression in the mitotic phase and functional analysis revealed that depletion of DEPDC1 resulted in mitotic arrest and obvious mitotic defects such as abnormal multiple nuclei and multipolar spindle structure. These DEPDC1 depletion-induced phenotypes were also observed in other types of cancer cells (data not shown). Therefore, given that cancer is closely linked to cell cycle dysregulation (10-13), we deduced that overexpression of DEPDC1 might facilitate and/or even promote carcinogenesis at least partially through dysregulation of the cell cycle progression.
Previous reports showed that DEPDC1 interacts and colocalizes with zinc finger transcription repressor ZNF224 to repress the transcription of A20, resulting in the activation of the NF-κB anti-apoptotic pathway in bladder cancer (2). Depletion of IKKα, an upstream regulatory kinase of NF-κB, has been reported to induce an increase in cells in G2/M, and this effect is due to up-regulation of CCNB1 (16, 18). Consistent with these reports, we found that DEPDC1 knockdown resulted in significant upregulation of A20 as well as several NF-κB regulated cell cycle genes such as CCNB1 and CCNB2 in HeLa cells undergoing mitotic arrest. In addition, a very recent report suggested that DEPDC1 could regulate the JNK signaling pathway (8, 9). Notably, in addition to its predominant role in cancer and apoptosis, JNK was also shown to be activated during mitosis and to regulate mitotic events (19, 20). Thus, further studies are needed to investigate whether and/or how DEPDC1 regulates NF-κB and JNK signaling in cell cycle progression as well as in tumorigenesis.
MATERIALS AND METHODS
Cell line and culture
Human cervical carcinoma-derived HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Invitrogen), penicillin (50 U/ml), and streptomycin (50 lg/ml). Cells were maintained at 37℃ in a humidified atmosphere of 5% CO2.
Cell synchronization
Hela cells were synchronized at the G1/S boundary by using double-thymidine block or in mitosis with thymidine-nocodazole block as described previously (13, 14). Synchronization and the cell cycle state were examined and monitored by fluorescence-activated cell sorter analysis.
FACS analysis
Cell cycle distribution was determined by flow cytometry after staining cells with propidium iodide. In brief, floating and adherent cells were collected by trypsin digestion and low speed centrifugation, washed with ice-cold phosphate-buffered saline (PBS), and fixed with 70% ethanol. The cells were then treated with 50 μg/ml of RNase A and 50 μg/ml of propidium iodide for 30 min at room temperature. The stained cells were analyzed using a FACScan flow cytometer (BD Biosciences).
siRNA-mediated knockdown
siRNAs used in the present study were chemically synthesized and provided by Shanghai GenePharma. The sequences of the siRNA duplexes for the negative control were 5'-UUCUCCGAACGUGUCACGUTT-3' (sense) and 5'-ACGUGACACGUUCGGAGAATT-3' (antisense), and those for DEPDC1 were 5'-GGAAGAUGUUGAAGAAGUUTT-3' (sense) and 5'-AACUUCUUCAACAUCUUCCTT-3' (antisense). The indicated siRNAs were transiently transfected into cells using Lipofectamine RNAiMAX transfection reagent (Invitrogen) following the manufacturer’s instructions. Cells were then collected and subjected to subsequent analysis 24 to 72 h after transfection.
RNA isolation and quantitative real-time PCR analysis
Total RNA was prepared using Total RNA Kit I (Omega Bio-Tek) according to the manufacturer's instructions, and reverse transcription of 1 μg of total RNA was carried out using random primers and PrimeScript (Takara) following the manufacturer's instructions. The resultant cDNA was amplified by quantitative real-time PCR using SYBR Premix Ex TaqTM (Takara) according to the manufacturer's recommendations. The relative expression level of the target gene compared with that of the housekeeping gene, GAPDH, was calculated using the 2−ΔΔCt method (14,15). The sequences of primers used in the present study are listed in Supplementary Table 1.
Immunoblotting analysis
Cells were lysed in RIPA lysis buffer (Santa Cruz Biotechnology) supplemented with protease inhibitor mixture (Roche Applied Science). Protein concentrations of the lysates were determined by BCA reagent (Applygen Technologies). Equal amounts of the lysates (30 μg of protein) were denatured at 100℃ for 5 min, separated by 10% standard SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and electro-transferred onto polyvinylidene difluoride membranes (Millipore). The membranes were blocked with 5% non-fat dry milk in Tris-buffered saline (TBS) containing 0.1% Tween 20 at 4℃ overnight. After blocking, the membranes were probed with the indicated primary antibodies at room temperature for 1 h, followed by incubation with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 h. The proteins were finally visualized by enhanced chemiluminescence (ECL, Amersham). Band densities were quantified using Quantity One software (v. 4.3.1, Bio-Rad, Hercules, CA). The antibodies used in this study are listed in Supplementary Table 2.
Indirect immunofluorescent staining
Cells were seeded on glass coverslips, fixed in freshly prepared ice-cold 4% paraformaldehyde in PBS at room temperature for 10 min, permeabilized in 0.5% Triton X-100 in PBS at room temperature for 10 min, and then blocked with 2% bovine serum albumin (BSA) plus 5% FBS in PBS at room temperature for 1 h. After washing in PBS, the cells were incubated with primary antibodies at room temperature for 1 h, followed by incubation with the corresponding secondary antibodies at room temperature for 1 h. The cells were then mounted with medium containing 4’6-diamidino-2-phenylindole (DAPI, Vector Laboratories), and the preparations were visualized with an Olympus BX51 fluorescence microscope and a Zeiss confocal LSM 768 microscope. The antibodies used for immunoflurescence assay are listed in Supplementary Table 2.
DEPDC1 expression plasmids and transient transfection
The expression construct of DEPDC1-V1-Flag, the DEPDC1 isoform 1 fused with a C-terminal Flag peptide, was kindly provided by Dr. Toyomasa Katagiri (1-3). For transient transfection, cells were seeded at a density of 0.8 × 105 cells/24-well tissue culture plate or 2.5 × 105 cells/6-well tissue culture plate and incubated overnight. The cells were then transiently transfected with the indicated plasmids using LipofectamineⓇ 2000 transfection reagent (Invitrogen) following the manufacturer’s protocols.
Acknowledgments
This work was supported in part by a grant-in-aid from the National Natural Science Foundation of China (30801356 and 81001097 to YB, http://www.nsfc.gov.cn/), the National clinical key discipline construction project (No. 2012-649), and the Chongqing medical research key project (No. 2013-1-004 to JZ). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kanehira M, Harada Y, Takata R, et al. Involvement of upregulation of DEPDC1 (DEP domain containing 1) in bladder carcinogenesis. Oncogene. (2007);26:6448–6455. doi: 10.1038/sj.onc.1210466. [DOI] [PubMed] [Google Scholar]
- 2.Harada Y, Kanehira M, Fujisawa Y, et al. Cell-permeable peptide DEPDC1-ZNF224 interferes with transcriptional repression and oncogenicity in bladder cancer cells. Cancer Res. (2010);70:5829–5839. doi: 10.1158/0008-5472.CAN-10-0255. [DOI] [PubMed] [Google Scholar]
- 3.Obara W, Ohsawa R, Kanehira M, et al. Cancer peptide vaccine therapy developed from oncoantigens identified through genome-wide expression profile analysis for bladder cancer. Jpn J Clin Oncol. (2012);42:591–600. doi: 10.1093/jjco/hys069. [DOI] [PubMed] [Google Scholar]
- 4.Kretschmer C, Sterner-Kock A, Siedentopf F, et al. Identification of early molecular markers for breast cancer. Mol Cancer. (2011);10:15. doi: 10.1186/1476-4598-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kassambara A, Schoenhals M, Moreaux J, et al. Inhibition of DEPDC1A, a bad prognostic marker in multiple myeloma, delays growth and induces mature plasma cell markers in malignant plasma cells. PLoS One. (2013);8:e62752. doi: 10.1371/journal.pone.0062752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan SG, Liao WJ, Yang JJ, et al. DEP Domain Containing 1 is a Novel Diagnostic Marker and Prognostic Predictor for Hepatocellular Carcinoma. Asian Pac J Cancer Prev. (2014);15:10917–10922. doi: 10.7314/APJCP.2014.15.24.10917. [DOI] [PubMed] [Google Scholar]
- 7.Okayama H, Kohno T, Ishii Y, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALKnegative lung adenocarcinomas. Cancer Res. (2012);72:100–111. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- 8.Sendoel A, Maida S, Zheng X, et al. DEPDC1/LET-99 participates in an evolutionarily conserved pathway for anti-tubulin drug-induced apoptosis. Nat Cell Biol. (2014);16:812–820. doi: 10.1038/ncb3010. [DOI] [PubMed] [Google Scholar]
- 9.Denning DP, Hirose T. Anti-tubulins DEPendably induce apoptosis. Nat Cell Biol. (2014);16:741–743. doi: 10.1038/ncb3012. [DOI] [PubMed] [Google Scholar]
- 10.Williams GH, Stoeber K. The cell cycle and cancer. J Pathol. (2012);226:352–364. doi: 10.1002/path.3022. [DOI] [PubMed] [Google Scholar]
- 11.Malumbres M, Carnero A. Cell cycle deregulation: a common motif in cancer. Prog Cell Cycle Res. (2003);5:5–18. [PubMed] [Google Scholar]
- 12.Perez de Castro I, dede Carcer G, Malumbres M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis. (2007);28:899–912. doi: 10.1093/carcin/bgm019. [DOI] [PubMed] [Google Scholar]
- 13.Ji Y, Xie M, Lan H, et al. PRR11 is a novel gene implicated in cell cycle progression and lung cancer. Int J Biochem Cell Biol. (2013);45:645–656. doi: 10.1016/j.biocel.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Bu Y, Suenaga Y, Okoshi R, et al. NFBD1/MDC1 participates in the regulation of G2/M transition in mammalian cells. Biochem Biophys Res Commun. (2010);397:157–162. doi: 10.1016/j.bbrc.2010.05.063. [DOI] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. (2001);25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Prajapati S, Tu Z, Yamamoto Y, et al. IKK α regulates the mitotic phase of the cell cycle by modulating Aurora A phosphorylation. Cell Cycle. (2006);5:2371–2380. doi: 10.4161/cc.5.20.3359. [DOI] [PubMed] [Google Scholar]
- 17.Cude K, Wang Y, Choi HJ, et al. Regulation of the G2-M cell cycle progression by the ERK5-NFκB signaling pathway. J Cell Biol. (2007);177:253–264. doi: 10.1083/jcb.200609166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ledoux AC, Perkins ND. NF-κB and the cell cycle. Biochem Soc Trans. (2014);42:76–81. doi: 10.1042/BST20130156. [DOI] [PubMed] [Google Scholar]
- 19.Ribas VT, Gonçalves BS, Linden R, et al. Activation of c-Jun N-terminal kinase (JNK) during mitosis in retinal progenitor cells. PLoS One. (2012);7:e34483. doi: 10.1371/journal.pone.0034483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Almuedo-Castillo M, Crespo X, Seebeck F, et al. JNK controls the onset of mitosis in planarian stem cells and triggers apoptotic cell death required for regeneration and remodeling. PLoS Genet. (2014);10:e1004400. doi: 10.1371/journal.pgen.1004400. [DOI] [PMC free article] [PubMed] [Google Scholar]