

Abstract
Study Design Broad narrative review.
Objectives Intramedullary spinal cord tumors (IMSCT) are rare neoplasms that can potentially lead to severe neurologic deterioration, decreased function, poor quality of life, or death. As such, a better understanding of these lesions is needed. The following article, part one of a two-part series, addresses IMSCT with regards to their epidemiology, histology, pathophysiology, imaging characteristics, and clinical manifestations.
Methods The authors performed an extensive review of the peer-reviewed literature addressing the aforementioned objectives.
Results Numerous IMSCT exist with varying epidemiology. Each IMSCT has its own hallmark characteristics and may vary with regards to how aggressively they invade the spinal cord. These lesions are often difficult to detect and are often misdiagnosed. Furthermore, radiographically and clinically, these lesions may be difficult to distinguish from one another.
Conclusions Awareness and understanding of IMSCT is imperative to facilitate an early diagnosis and plan management.
Keywords: intradural, intramedullary, spinal, cord, tumors, epidemiology, ependymoma, astrocytoma
Introduction
Primary spine tumors are rare neoplasms that can lead to significant patient morbidity and mortality.1 2 Intramedullary spinal cord tumors (IMSCTs) are the rarest of these neoplasms and can potentially lead to severe neurologic deterioration, decreased function, poor quality of life, or death.2 Within the IMSCT category, the most common lesions are ependymomas, astrocytomas, and hemangioblastomas, followed by other rare lesions.2 3 Each specific lesion has its own hallmark characteristics, and the tumors vary with regards to the current understanding of their etiology or associations, their imaging and clinical characteristics, and/or how aggressively they invade the spinal cord. Due to the rarity of occurrence, these lesions are often difficult to detect and are often misdiagnosed, leading to delays in the appropriate patient care.4 Additionally, despite the hallmark characteristics and current diagnostic abilities, these lesions remain both radiographically and clinically difficult to distinguish from one another.5 Preoperative neurologic status and tumor histology are two of the most important variables affecting treatment outcome with these lesions, and thus awareness of these lesions is imperative to facilitate an early diagnosis.6 7 8 9 Early diagnosis allows early referral for treatment, which can be the critical difference in the treatment outcome. The following is a broad narrative review of the literature addressing IMSCTs with regards to their epidemiology, histology, pathophysiology, imaging characteristics, and clinical manifestations.
Epidemiology
Incidence
Primary spinal cord tumors are 10 times less common than their cranial counterparts, although they are histopathologically similar. Spine tumors are historically classified as (1) extradural, (2) intradural extramedullary, and (3) intradural intramedullary.1 Extradural tumors are the most common spinal tumors and are usually of metastatic origin.3 4 Intradural intramedullary lesions comprise 20 to 30% of all primary spinal cord tumors.4 The remaining 70 to 80% of primary intradural tumors are intradural extramedullary tumors.3 Intradural tumors are most likely to be found in the thoracic region, followed by the cervical and then lumbar spine.
Gliomas make up 80% of all intramedullary tumors. The gliomas are further subdivided into astrocytomas (60 to 70%) and ependymomas (30 to 40%).3 Astrocytomas have a higher occurrence in children than adults, whereas ependymomas are more common in adults than children.10 Hemangioblastomas are the third most frequent IMSC tumor, comprising 2 to 15% of all intramedullary tumors.11 Metastatic intramedullary tumors are rare but present in 2% of all intramedullary tumors. Autopsy series have reported that 0.9 to 2.1% of all patients with cancer exhibit intramedullary spinal cord metastases.12 13 Thus, the incidence of these tumors may be higher than initially predicted. Of the intramedullary tumors with metastatic origins, 40 to 60% and 14% arise from primary neoplasms of the lung and breast, respectively.13 14 15 The remaining constituents of IMSCTs include lipomas (1%) and other rare tumors that have been documented sparingly in the literature (Table 1).
Table 1. Types of intramedullary spinal cord tumors.
| Tumor class | Associated tumors |
|---|---|
| Neuroepithelial tissue | Astrocytic • Astrocytoma • Glioblastoma Embryonal • Primitive neuroectodermal Ependymal • Ependymoma • Subependymoma Mixed glioma Neuronal and mixed neuronal-glial • Gangliocytoma • Ganglioglioma • Ganglioneuroblastoma Oligodendroglial • Oligodendroglioma Uncertain origin • Polar spongioblastoma |
| Spinal nerves | Neurofibroma Schwannoma |
| Nonmeningothelial, mesenchymal | Hemangioblastoma Lipoma Melanoma Sarcoma |
| Germ cell tumors | Germinoma Teratoma |
| Cysts and tumorlike lesions | Dermoid Epidermoid |
| Hematopoietic neoplasms | Primary central nervous system lymphoma (microglial) |
| Metastatic tumors and other rare neoplasms |
Genetic Factors
Several genetic factors are associated with IMSCTs. Understanding the various genetic mutations assists in illustrating the clinical manifestations, progression, and management of these tumors. Additionally, through a brief understanding of tumor genetics, the association of spinal tumors with lesions elsewhere in the body can be appreciated. The clinical syndromes currently associated with IMSCTs include neurofibromatosis 1, 2 (NF-1, NF-2) and Von Hippel-Lindau disease (VHL).
Neurofibromatosis
Neurofibromatosis is a common autosomal-dominant disorder with 100% penetrance in familial lines.16 Two subtypes have been established: NF-1 and NF-2. Fifty percent of patients with neurofibromatosis possess a family history of the disorder with new mutations developing in the remaining constituents.
The reported prevalence of NF-1 (also known as von Reckinghausen disease or peripheral neurofibromatosis) is 1 in 3,000 to 4,000 individuals.17 Although the incidence of IMSCT in individuals with NF-1 is speculative, it has been suggested that almost 19% of subjects diagnosed with NF-1 develop IMSCT.18 Expressivity varies due to the heterogenetic characteristics of the disorder. The mutation is located on the long arm of chromosome 17 (17q), which encodes for neurofibromin, a negative regulator of the RAS cellular proliferation pathway.19 Neurofibromin is a tumor suppressor gene, whereby mutation disinhibits the RAS pathway leading to increased cellular division and proliferation and eventual tumor development.19 In the presence of NF-1, multiple neurofibromas may appear. Although many tumors are associated with NF-1, in relation to IMSCTs, astrocytomas are the most likely to develop (Table 2).18 20
Table 2. Genetic basis of intramedullary spinal cord tumors.
| Genetic disorder | Associated tumor |
|---|---|
| Neurofibromatosis-1 | Acute nonlymphocytic leukemias Astrocytomas Carcinoid tumors Hematomas Hypothalamic gliomas Malignant nerve sheath tumors Meningiomas Neurofibromas Optic nerve gliomas Pheochromocytomas Primitive neuroectodermal tumors Rhabdomyosarcomas Wilms tumor |
| Neurofibromatosis-2 | Bilateral acoustic schwannomas Ependymomas Gliomas Meningiomas Schwannomas |
| Von Hippel-Landau | Adrenal pheochromocytomas Central nervous system hemangioblastoma (medulla oblongata or spinal cord) Epididymal cystadenomas Lindau tumor (cerebellum hemangioblastoma) Nephritic cysts Pancreatic cysts Renal cell carcinomas Renal cysts Retinal hemangioblastomas |
Less prevalent than NF-1, NF-2 (also known as central neurofibromatosis) occurs in 1 in 40,000 individuals and represents 2.5% of patients with IMSCT (Table 2).18 The location of NF-2 has been identified as a mutation of the “Merlin” gene on chromosome 22q12.19 NF-2 is largely associated with ependymomas and occasionally meningiomas (extramedullary).18 The severe form of the condition exhibits multiple tumors with an earlier onset and a rapid clinical deterioration. The milder form is characterized by fewer tumors with a later onset and a gradual clinical progression.
Von Hippel-Lindau Disease
VHL is a rare autosomal-dominant disease with 90% penetrance. Moller first suggested that VHL was a genetic disorder in 1929.90 Briefly summarized, mutation of a tumor suppressor gene located on chromosome 3p25–26 is responsible for this condition.21 22 The VHL tumor suppressor proteins form a protein complex that interacts with elongins B and C and other proteins, marking them for degradation by the cell, which inhibits hypoxia-related cell transcription factors (HIF1a, HIF2a).19 23 The VHL proteins also suppress the hypoxia-induced production of vascular endothelial growth factor, erythropoietin, and platelet-derived growth factor.19 24 Without the VHL protein present, an increased presence of these transcription factors occurs on the cellular level, eventually leading to tumor formation, and given the specifics of the transcription factors involved, these are often highly vascular lesions. VHL is characterized by widespread formation of both benign and malignant tumors throughout the body. According to Melmon and Rosen, VHL is diagnosed if the following manifest: the presence of more than one hemangioblastoma of the central nervous system (CNS), the presence of an isolated lesion associated with a visceral manifestation of the disease, or the presence of one characteristic of the disease and a family history of the disease.25 A wide array of tumors is associated with this disease, and especially common are retinal hemangioblastomas (Table 2). Hemangioblastomas of the medulla oblongata and spinal cord as well as renal cell carcinoma may ensue and lead to significant mortality in 50% of individuals with the disease. Of CNS hemangioblastomas, 80% occur in the posterior fossa and 20% appear in the spinal cord. Furthermore, 10 to 15% of cranial hemangioblastomas are associated with VHL, whereas 25% of spinal cord hemangioblastomas are associated with VHL.1 3 In association with VHL, there is an earlier age of development of hemangioblastomas than in those of sporadic origin.
Syringomyelia
The classification and terminology related to fluid-filled cavities in the spinal cord remains controversial. In this text, we will refer to the tumor-associated fluid-filled cavity within the spinal cord as syringomyelia or syrinx.26 Syrinx occurs with 25 to 58% of patients with IMSCT, and in this association, syringes appear more commonly in the lower cervical and upper thoracic region. They occur most frequently with ependymomas, followed by hemangioblastomas and cavernomas.27 28 29 30 31 Irrespective of tumor type, there is a correlation between the degree of cephalad extension for a tumor and the presence of a syrinx. According to Samii and Klekamp, 49% of syringes occur above the tumor level, 11% occur below tumor level, and 40% are bipolar. A small percentage involves the medulla oblongata.29 The presence of a syrinx is considered a favorable prognostic sign. Noninfiltrative tumors with distinct cleavage planes are more likely to display syringes than more diffuse, infiltrative tumors. After tumor removal is performed, the syrinx will typically dissolve. Moreover, patients presenting with the concomitant finding of syringomyelia with their tumor tend to recover from surgery more rapidly.
As mentioned, the classification and etiology of syringomyelia is a current area of investigation and a continually evolving field, but it can be generally divided into three broad subgroups: (1) resulting from alteration of cerebrospinal fluid (CSF) flow dynamics related to hindbrain disorders, (2) resulting from intramedullary tissue damage secondary to hemorrhage or infarction, and (3) resulting from direct secretory ability of intramedullary tumor.26 The latter two mechanisms of syrinx formation pertain to IMSCT.27 32 33 34 35 36 37 The resultant syringes from hemorrhage and infarction can be seen occasionally in ependymomas, which often hemorrhage, and especially so in highly vascular lesions such as hemangioblastoma and cavernoma, which are the most common lesions to have associated syrinx. The secretory theory proposes that pathologic tumor vessels create transudation and the secretion of fluid from tumor cells, which thus account for syrinx development. Because the chemical composition of the tumor syrinx has an elevated protein concentration compared with CSF and syrinx fluid associated with non-IMSCT, the syringomyelia may be inherent to the tumor. These mechanisms of syrinx formation in relation to IMSCT are wholly different from those related to CSF obstruction and hindbrain disorders, which include such theories as Gardner's “water hammer” theory, Williams' “suck and slosh” theory, Ball and Dayan's theory of infiltration through perivascular (Virchow-Robbin) spaces, and Oldfield's theory, which combines features of all of the above.26 35 37 However, tumor obstruction of the CSF dynamics and subsequent relative blockage in CSF flow at the local level may also have a role in the pathogenesis of syrinx formation by increasing the focal transmedullary pressure in addition to the two main tumor-related etiologies of hemorrhage/infarction and secretory ability.29 38
Pathophysiology
Astrocytomas
Astrocytomas are red, gray, glossy tumors that are characterized by a poorly defined plane and are generally infiltrative in nature. Astrocytomas are the second most common IMSCT in adults at 30 to 35% of tumors and the most common in children at 90% of tumors.1 10 39 Of all astrocytomas arising in the CNS, 3% occur in the spinal cord.40 They primarily occur in the cervical spine, and they often involve multiple spinal segments due to the expansive nature of these tumors (Fig. 1). Nearly 20% of such lesions are associated with syrinx formation.29 Astrocytomas have an association with NF-1, occur predominantly in males, and rarely manifest in patients over the age of 60.41 Adults mainly exhibit high-grade lesions, whereas low-grade lesions are associated with the younger population.42 Malignant degeneration develops in 25% of adult astrocytomas.43
Fig. 1.
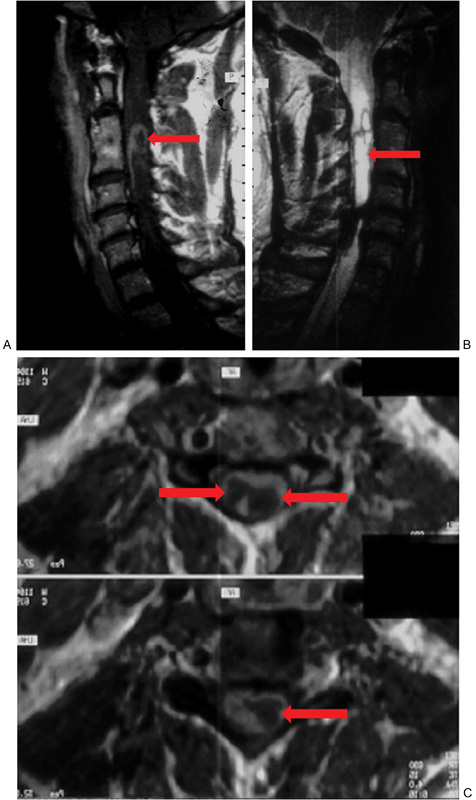
(A) Sagittal T1-weighted post–gadolinium contrast cervical magnetic resonance image (MRI) noting a heterogeneously enhancing intramedullary astrocytoma (arrow). (B) Sagittal T2-weighted cervical MRI demonstrating an intramedullary astrocytoma (arrow). (C) Axial T1-weighted post–gadolinium contrast cervical MRI illustrating a cystic astrocytoma; the cystic portions are highlighted by arrows and are hypointense compared with the enhancing surrounding rim.
Ependymomas
Ependymomas are soft, encapsulated, reddish gray or yellow tumors with modest vascularity that present in the third or fourth decade of life. Half of all ependymomas are located in the region spine and a slight majority are located in the cord (55%), relative to the cauda equina (45%).1 They are not sex discriminant.44 45 The majority of them are classified as benign tumors, and ependymomas have a propensity to grow slowly. Spinal ependymomas are primarily present in the cervical or cervicothoracic region.46 47 The cysts associated with these tumors are predominantly found in the superior margin of the tumor and have an increased incidence in the cervical region (Fig. 2A).29 43 48 Approximately 65% of these tumors are associated with syrinx formation.29
Fig. 2.
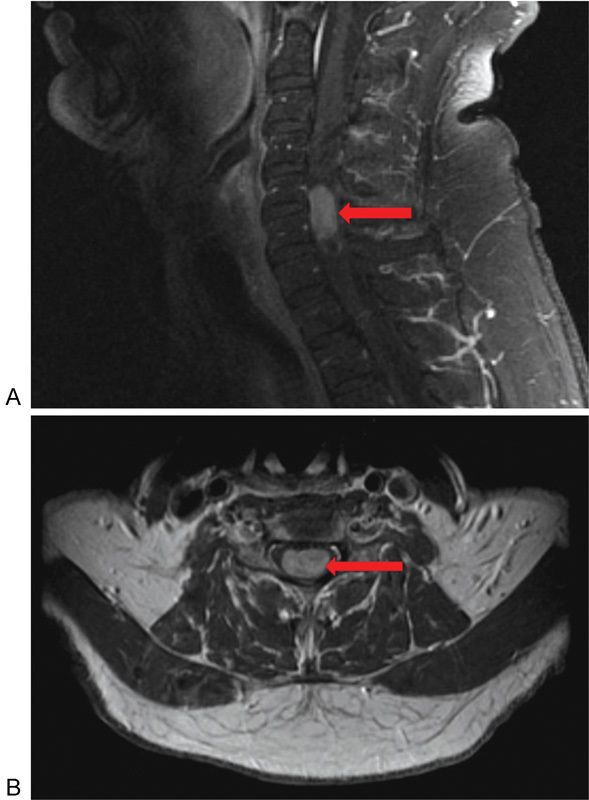
(A) Sagittal T1-weighted post–gadolinium contrast cervical magnetic resonance image illustrating the presence of a homogeneously enhancing ependymoma at the levels of C5 and C6 (arrow). (B) Corresponding axial view noting the centromedullary location of the ependymoma (arrow).
Originating from ependymal cells, ependymomas are more centromedullary located as compared with astrocytomas, and they tend to blend with the cord (Fig. 2B). They appear as a focal enlargement within the spinal cord. Ependymomas have a mean extension of three to four vertebral bodies, whereas astrocytomas average five to six segmental levels.49 Abnormalities of chromosome 22 are associated with ependymomas, and this leads to their association with NF-2.19 Various histologic subtypes of ependymomas exist. The most common is the cellular variant or the “typical” form, which is a World Health Organization (WHO) grade II tumor that is well circumscribed. Pathologic analysis reveals both true ependymal rosettes and more commonly perivascular pseudorosettes.50 Tanycytic ependymomas are also WHO grade II, are less common overall, and are found more commonly in the spinal cord than in the brain. Histologically, tanycytic ependymomas lack ependymal rosettes and their pseudorosettes are vaguely apparent, which can lead to misdiagnosis as pilocytic astrocytoma (WHO grade I).19 Myxopapillary ependymomas are WHO grade I tumors commonly found at the filum terminale, but these are considered extramedullary (Fig. 3). Rarely anaplastic ependymomas, WHO grade III, can be encountered, which develop more rapidly than lower grades, can occasionally develop from malignant degeneration, and have an overall poor prognosis.19 Ependymomas may also be found outside the nervous system in the soft tissue, ovaries, and mediastinum. Subependymomas (WHO grade I), which may be histologically related to ependymomas, have also been reported as rare IMSCT.51
Fig. 3.
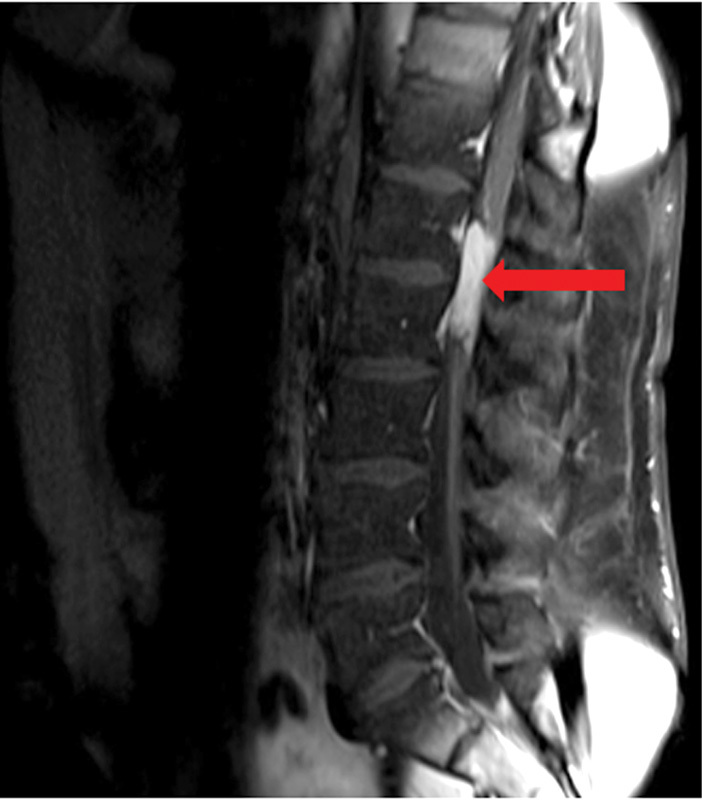
Sagittal T1-weighted post–gadolinium contrast lumbar magnetic resonance image illustrating the presence of a homogenously enhancing ependymoma at the levels of L1 and L2 (arrow).
Hemangioblastoma
Hemangioblastomas are small benign richly vascularized solitary neoplasms that rarely extend beyond one or two segments. Although more often located in the cerebellum, hemangioblastomas are also found in the spinal cord, predominantly located at the posterior or posterolateral region of the spinal canal (Fig. 4). Most hemangioblastomas develop sporadically but, as previously mentioned, they are associated with VHL, especially when presenting in the spinal cord. They constitute between 2 and 8% of all IMSCTs.1 Commonly, they are located in the cervical spine but may be found in any portion of the neuroaxis. Cyst formation is evident within 50 to 70% of tumors, and the tumors have an association with syrinx.52 53 54 Hemangioblastomas are more commonly found in men.52 53 54 55 Although hemangioblastomas are not age discriminant, most occur during the fourth decade of life. It is possible that individuals with a hemangioblastoma can remain asymptomatic throughout life, with tumor only apparent at autopsy.55
Fig. 4.
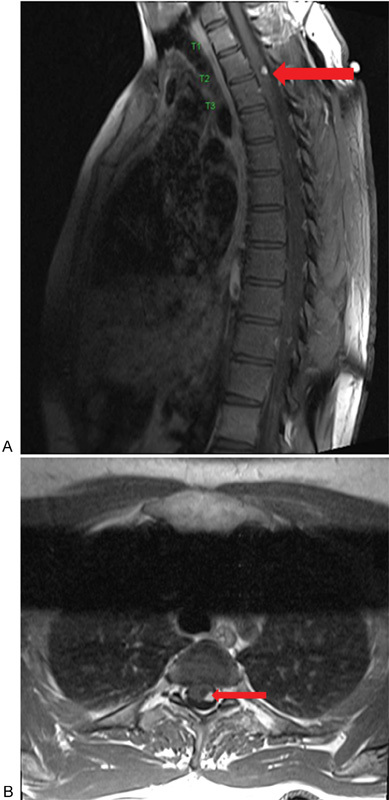
(A) Sagittal T1-weighted post–gadolinium contrast magnetic resonance image noting an avidly homogenous enhancing nodule representing a hemangioblastoma at the level of T2–T3 (arrow). (B) Corresponding axial view noting the involvement of the tumor in relation to the cord (arrow).
Hemangioblastomas are composed of a dense network of vascular capillary channels containing endothelial cells, pericytes, and lipid-laden stromal cells.56 The cells of origin remain unknown, but based on the genetics of VHL and mutations found in sporadic lesions, they are likely vascular endothelial growth factor-secreting undifferentiated mesenchymal cells.1 19 Two histologic forms exist: reticular and cellular. The former is composed of irregular nuclei with conspicuous vessels. Conversely, the cellular form exhibits fewer conspicuous vessels with varying stromal cells; this form bears a similarity to astrocytomas and may be difficult to discriminate from these lesions.
Hemangioblastomas are well-demarcated tumors that are amenable to resection. With complete resection, the prognosis is excellent. Pain and sensory changes are the most frequent presenting complaints. Imaging is a crucial factor in surgical planning and should be thoroughly evaluated. An associated cord widening is commonly found adjacent to the tumor, possibly attributed to edema related to increased blood flow in surrounding draining veins or within the tumor creating vascular congestion. After tumor removal, widening regresses. In addition, spinal angiography is recommended to delineate the characteristics of the spinal draining veins.57
Intramedullary Spinal Cord Metastases
The literature supports the belief that metastatic IMSCTs are rare. More recently, combining routine magnetic resonance imaging (MRI) and autopsy observation, it appears that metastatic IMSCTs may be more common than initially reported. These fast spreading tumors may escape diagnosis, as patients bearing these lesions may be asymptomatic. Although imaging is helpful in demarcating the location and extent of these tumors, the proper diagnosis is accomplished by surgical biopsy and the presence of a primary source of cancer.
The mechanism of cord infiltration of metastatic IMSCT is ambiguous. Because the majority of metastatic IMSCTs metastasize from the lung, the tumor is thought to spread from hematogenous dissemination leading to arterial embolization.15 58 59 Other proposed theories of tumor seeding involve retrograde infiltration via the spinal cord venous system (Batson's plexus),60 spread through perforating veins in the bone, metastatic antidromic cellular migration via nerve root to the spinal cord, CSF dissemination through intraspinal perineural sheaths, and finally, penetration of the spinal cord parenchyma via penetrating vessels within the Virchow-Robin spaces.14
Lipomas
Lipomas are rare congenital tumors that constitute 1% of intraspinal tumors. Commonly found in the cauda equina and conus medullaris, these tumors tend to violate the posterior cord and are usually extramedullary.61 62 Lipomas are commonly associated with spinal dysraphism and are thought to arise from premature disjunction of the cutaneous ectoderm from the neural ectoderm prior to neural tube closure, allowing mesenchymal cells to infiltrate into the neural groove.1 These tumors possess a higher water content than other intramedullary tumors and tend to attach firmly to the dura; their cellular content is indistinguishable from normal adipose tissue.62 63 64 These tumors are slow-growing, and they become symptomatic due to focal mass effect. They present no cleavage plane from the surrounding cord with blending of the neural and fatty tissue.1 65 As a result, complete tumor resection is extremely difficult, and some neurologic damage may be expected when attempting resection. Although little is known about these tumors, patients tend not to improve from their baseline neurologic state but the long-term prognosis is excellent with 90% progression-free survival at 16 years following total or near total excision and 35% progression-free survival at 10 years following a debulking procedure.1 Developmentally, these tumors prevent normal maturation of the surrounding neural tissue.
Other Rare Neoplasms
Numerous additional IMSCTs exist but are extremely rare. Cavernomas are rare vascular lesions that may hemorrhage repeatedly during the lifetime of an individual, and they can have associated syringes. Intramedullary lymphomas may occur in isolation or with other central nervous system lesions. They respond to chemotherapy. Gangliogliomas are benign tumors consisting of glial and neuronal cells that are estimated to comprise 3.8% of CNS tumors found in the upper cervical cord.66 67 Oligodendrogliomas possess ill-defined borders and less extensive cord growth compared with other IMSCTs; fewer than 50 cases have been literature reported. Other IMSCT neoplasms include melanocytomas, melanomas, fibrosarcomas, peripheral neuroectodermal tumors, and amyloid angiopathies (Table 1).1 68 69 70
Imaging
Radiographic evaluation is necessary to determine the location and extent of tumor involvement and may help to differentiate between lesions. A role still exists for plain radiographs in evaluation as they can illustrate bony erosions or evaluate for scoliosis. The presence of scoliosis can be indicative as advanced IMSCT can present with scoliosis even in adults.71 72 In adults, widening of the spinal canal is usually not noted except with large myxopapillary ependymoma in the conus medullaris or filum terminale. In children, widening of the spinal canal and kyphoscoliosis are more frequently encountered.71 73 74 75 Myelography has been utilized in the past to evaluate the cord contours; however, cord widening is sometimes difficult to appreciate and the procedure may exacerbate symptoms. Myelography has been supplanted by MRI except in rare circumstances.
MRI is the preferred modality in identifying and characterizing IMSCT. Each tumor has a defined T1-, T2-, and gadolinium-enhanced T1-weighted imaging pattern. MRI may be helpful in defining the presence or absence of a cord-tumor interface, illustrating associated cysts or syringomyelia, which may support the ability to achieve a favorable resection. With modern imaging capabilities, differentiation between ependymomas, astrocytomas, and even hemangioblastomas remains difficult; however, several radiographic features may help in this distinction.76 A recently published article by Arima et al scrutinized the ability of MRI to accurately differentiate between these three most common IMSCTs and were able to achieve an 89% accuracy.76 Although this appears to be a high rate of diagnostic accuracy, it does not yet achieve a value that allows treatment based on MRI features alone. Looking at gliomas specifically (ependymomas and astrocytomas), both lesions demonstrate fusiform configuration over numerous vertebral segments, and they both appear hypo- or isointense on T1-weighted images and hyperintense on T2-weighted images. Furthermore, these two tumors enhance with contrast. The rare subependymoma shares these MRI T1- and T2-weighted features, can have associated syrinx, and is usually eccentrically located and nonenhancing; contrast enhancement is faint if present.51
Ependymomas tend to be centrally located within the cord and display symmetric expansion with diffuse heterogeneous enhancement (Fig. 2). They tend to occupy the whole width of the cord and generally produce enhanced margins (Fig. 2). Astrocytomas, in comparison, tend to be eccentrically positioned, may display an exophytic component, and can be nonenhancing or heterogeneously enhancing or even have an enhancing nodule (Fig. 1).76 77 Radiographically, they do not present with well-defined borders but large satellite cysts may be visualized.78 79 80 Polar cysts may appear and have a low signal intensity on T1-weighted images and high signal intensity on T2-weighted images. Intradural hemorrhage may be seen in both types of glioma but are more likely in ependymoma, with the imaging characteristics dependent on the age of the hemorrhage and possible secondary development of syrinx.
Hemangioblastomas are richly vascularized tumors that can have significant surrounding edema.81 They are associated with syringomyelia, can be found at any spinal level, and are most commonly sporadic. In patients with VHL, they are associated with additional visceral lesions or neuraxis hemangioblastomas. On MRI, they have mural nodules, which appear isointense on T1-weighted images and hyperintense on T2-weighted images with homogenous contrast enhancement, in contrast to the usual heterogenous contrast pattern of ependymomas and astrocytomas (Fig. 4).76 Vascular imaging or even spinal angiography may be useful to delineate feeding vessels and illustrate associated dilated pial veins due to vascular shunting and can be helpful for consideration of preoperative embolization in these highly vascular lesions.
Metastatic IMSCTs are well encapsulated and are unlikely to present with cystic change or intralesional hemorrhage.82 They most commonly present as single lesions in the thoracic cord but can be multiple or present in the cervical and lumbar spine.82 Metastatic lesions are most likely eccentrically located in the cord and expand the cord parenchyma, which was historically visible on myelography and illustrated clearly on MRI.14 82 83 MRI is the imaging tool of choice, and recently described “rim” and “flame” signs have been described to help differentiate non-CNS intramedullary metastases from primary spinal cord tumors, which occur significantly more frequently either alone or in combination in metastases than in primary cord tumors with high specificity but low sensitivity (specificity 97% for either alone and 100% when both present).84 The rim sign is defined by Rykken et al as a complete or partial rim of gadolinium enhancement, and the flame sign is defined as an ill-defined flame-shaped gadolinium-enhancing region at the superior or inferior margin of an otherwise well-defined lesion.84 Metastases are most likely to be T1 isointense and T2 hyperintense relative to the cord. The amount of edema may be out of proportion to the size of the tumor, and this is evidenced by extensive T2 hyperintensity, which can be on average 3.6-fold larger than that of the enhancing portion of the lesion.82 The estimated prevalence of spinal cord metastases from a postmortem series ranged from 0.9 to 2.1% of patients with cancer.84 Any patient with a known history of cancer and an intramedullary spinal cord lesion should have metastatic IMSCT as part of the differential diagnosis.
Common IMSCTs may be difficult to differentiate from the rare tumors radiographically. Intramedullary lymphomas are rare tumors often linked to other supratentorial lesions. Oligodendrogliomas are characterized by ill-defined borders, no peritumoral edema, and slight T1-weighted image hyperintensity. Lipomas have imaging characteristics similar to fat (high T1-weighted image intensity).
Clinical Manifestations
IMSCTs present with a wide array of symptoms that vary in intensity and chronicity. The clinical features of each tumor are related to the growth rate, location, and longitudinal extent of the tumor.19 In general, spinal cord lesions should be suspected when patients present with bilateral motor and sensory symptoms not involving the head and face, often with other upper motor neuron symptoms consistent with a myelopathic syndrome.1 Hydrocephalus occurs in 1 to 8% of patients with IMSCT.85 86 The hydrocephalus may result from tumoral obstruction of the subarachnoid CSF flow or impaired CSF absorption induced by nondisseminated subarachnoid space tumors that increase CSF protein levels.85 Focal block of CSF flow by a tumor (or abscess formation) with increased CSF protein and presence of xanthochromia is known as Froin syndrome, which can lead to a “dry” lumbar puncture.87
The most common tumor presentation is back or neck pain. This is hypothesized to result from dural distension and irritation. The pain is of constant intensity and varies between individual patients; it is classically worsened in the recumbent position. Nerve root compression can produce weakness, spasticity, and clumsiness.88 Centrally located lesions can produce myelopathic symptoms. If the tumor extends cranially, cranial nerve involvement is possible. Paresthesias or dysesthesias can present unilaterally, often starting distally and progressing proximally before affecting the opposite side. Also, radiculopathy is implicated with lumbosacral involvement.88
Additional factors can contribute to the symptomology of IMSCT. Among them are age, degenerative changes of the spinal column, spinal canal size, medical comorbidities, and tethering structures, which may alter sensory and motor function. Tethering effects of the dentate ligament and dorsal and ventral roots occur as a response to cord distension by the tumor mass. Compromise of the corticospinal tract produces upper motor neuron deficit. Decrease in temperature sensation and pain results from spinothalamic tract encroachment. Compression of the dorsal columns can manifest with defects in proprioception and gait abnormalities. Tumors affecting the autonomic pathways produce disturbances of the sympathetic and parasympathetic system. Severe cord defects can also complicate respiratory, bowel, bladder, or sexual function.19 88
Damage to cranial nerves is also a possibility with tumors located in the upper cervical spine. The hypoglossal nerve can be subject to compression by tumors located laterally at the foramen magnum leading to ipsilateral tongue paresis and atrophy. Arising from the C1–C5 anterior horn cells, the accessory nerve travels cephalad through the foramen magnum via the subarachnoid space between the ventral and dorsal rootlets and unites with its cranial counterpart. Thus, tumors around the upper cervical spinal cord can compress the accessory nerve, leading to weakness and atrophy of the sternocleidomastoid and trapezius muscles.89
Conclusion
IMSCTs are rare manifestations that can severely impair neurologic function and the quality of life. The proper diagnosis of IMSCT is essential to provide the best treatment and obtain the optimal outcome. The preoperative functional status, tumor histology, and degree of tumor invasion dictate postoperative results. Therefore, prompt diagnosis and intervention are essential. However, the manifestation of symptoms can be misleading and the diagnosis can be missed. Overall, various types of IMSCT exist, and the management of these tumors demand individualized scrutiny and understanding.
Acknowledgment
We would like to thank the Hong Kong Theme-Based Research Scheme (T12-708/12N) for their support of this work.
Footnotes
Disclosures Dino Samartzis, none Christopher C. Gillis, none Patrick Shih, none John E. O'Toole, none Richard G. Fessler, none
References
- 1.Mechtler L L, Nandigam K. Spinal cord tumors: new views and future directions. Neurol Clin. 2013;31(1):241–268. doi: 10.1016/j.ncl.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Fisher C G, Goldschlager T, Boriani S. et al. A novel scientific model for rare and often neglected neoplastic conditions. Evid Based Spine Care J. 2013;4(2):160–162. doi: 10.1055/s-0033-1357365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grimm S, Chamberlain M C. Adult primary spinal cord tumors. Expert Rev Neurother. 2009;9(10):1487–1495. doi: 10.1586/ern.09.101. [DOI] [PubMed] [Google Scholar]
- 4.Duong L M, McCarthy B J, McLendon R E. et al. Descriptive epidemiology of malignant and nonmalignant primary spinal cord, spinal meninges, and cauda equina tumors, United States, 2004–2007. Cancer. 2012;118(17):4220–4227. doi: 10.1002/cncr.27390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Setzer M, Murtagh R D, Murtagh F R. et al. Diffusion tensor imaging tractography in patients with intramedullary tumors: comparison with intraoperative findings and value for prediction of tumor resectability. J Neurosurg Spine. 2010;13(3):371–380. doi: 10.3171/2010.3.SPINE09399. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed R, Menezes A H, Awe O O, Torner J C. Long-term disease and neurological outcomes in patients with pediatric intramedullary spinal cord tumors. J Neurosurg Pediatr. 2014;13(6):600–612. doi: 10.3171/2014.1.PEDS13316. [DOI] [PubMed] [Google Scholar]
- 7.Lee S H, Chung C K, Kim C H. et al. Long-term outcomes of surgical resection with or without adjuvant radiation therapy for treatment of spinal ependymoma: a retrospective multicenter study by the Korea Spinal Oncology Research Group. Neuro-oncol. 2013;15(7):921–929. doi: 10.1093/neuonc/not038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson D A, Fusco D J, Uschold T D, Spetzler R F, Chang S W. Survival and functional outcome after surgical resection of intramedullary spinal cord metastases. World Neurosurg. 2012;77(2):370–374. doi: 10.1016/j.wneu.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 9.Wong A P, Dahdaleh N S, Fessler R G. et al. Risk factors and long-term survival in adult patients with primary malignant spinal cord astrocytomas. J Neurooncol. 2013;115(3):493–503. doi: 10.1007/s11060-013-1251-y. [DOI] [PubMed] [Google Scholar]
- 10.DeSousa A L, Kalsbeck J E, Mealey J Jr, Campbell R L, Hockey A. Intraspinal tumors in children. A review of 81 cases. J Neurosurg. 1979;51(4):437–445. doi: 10.3171/jns.1979.51.4.0437. [DOI] [PubMed] [Google Scholar]
- 11.Mandigo C E, Ogden A T, Angevine P D, McCormick P C. Operative management of spinal hemangioblastoma. Neurosurgery. 2009;65(6):1166–1177. doi: 10.1227/01.NEU.0000359306.74674.C4. [DOI] [PubMed] [Google Scholar]
- 12.Costigan D A, Winkelman M D. Intramedullary spinal cord metastasis. A clinicopathological study of 13 cases. J Neurosurg. 1985;62(2):227–233. doi: 10.3171/jns.1985.62.2.0227. [DOI] [PubMed] [Google Scholar]
- 13.Chason J L, Walker F B, Landers J W. Metastatic carcinoma in the central nervous system and dorsal root ganglia. A prospective autopsy study. Cancer. 1963;16:781–787. doi: 10.1002/1097-0142(196306)16:6<781::aid-cncr2820160614>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 14.Edelson R N, Deck M DF, Posner J B. Intramedullary spinal cord metastases. Clinical and radiographic findings in nine cases. Neurology. 1972;22(12):1222–1231. doi: 10.1212/wnl.22.12.1222. [DOI] [PubMed] [Google Scholar]
- 15.Jellinger K, Kothbauer P, Sunder-Plassmann E, Weiss R. Intramedullary spinal cord metastases. J Neurol. 1979;220(1):31–41. doi: 10.1007/BF00313146. [DOI] [PubMed] [Google Scholar]
- 16.Mapstone T B. Neurofibromatosis and central nervous system tumors in childhood. Neurosurg Clin N Am. 1992;3(4):771–779. [PubMed] [Google Scholar]
- 17.Riccardi V M, Eichner J E. Baltimore, MD: Johns Hopkins University Press; 1986. Neurofibromatosis: Phenotype, Natural History and Pathogenesis. [Google Scholar]
- 18.Lee M, Rezai A R, Freed D, Epstein F J. Intramedullary spinal cord tumors in neurofibromatosis. Neurosurgery. 1996;38(1):32–37. doi: 10.1097/00006123-199601000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Lyon, France: International Agency for Research on Cancer (IARC); 2007. WHO Classification of Tumors of the Central Nervous System. 4th ed. [Google Scholar]
- 20.Yagi T, Ohata K, Haque M, Hakuba A. Intramedullary spinal cord tumour associated with neurofibromatosis type 1. Acta Neurochir (Wien) 1997;139(11):1055–1060. doi: 10.1007/BF01411560. [DOI] [PubMed] [Google Scholar]
- 21.Decker H J, Neuhaus C, Jauch A. et al. Detection of a germline mutation and somatic homozygous loss of the von Hippel-Lindau tumor-suppressor gene in a family with a de novo mutation. A combined genetic study, including cytogenetics, PCR/SSCP, FISH, and CGH. Hum Genet. 1996;97(6):770–776. doi: 10.1007/BF02346188. [DOI] [PubMed] [Google Scholar]
- 22.Kley N, Whaley J, Seizinger B R. Neurofibromatosis type 2 and von Hippel-Lindau disease: from gene cloning to function. Glia. 1995;15(3):297–307. doi: 10.1002/glia.440150310. [DOI] [PubMed] [Google Scholar]
- 23.Stebbins C E, Kaelin W GJ Jr, Pavletich N P. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284(5413):455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- 24.Gnarra J R, Zhou S, Merrill M J. et al. Post-transcriptional regulation of vascular endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc Natl Acad Sci U S A. 1996;93(20):10589–10594. doi: 10.1073/pnas.93.20.10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melmon K L, Rosen S W. Lindau's disease. Review of literature and study of a large kindred. Am J Med. 1964;36:595–617. doi: 10.1016/0002-9343(64)90107-x. [DOI] [PubMed] [Google Scholar]
- 26.Blegvad C, Grotenhuis J A, Juhler M. Syringomyelia: a practical, clinical concept for classification. Acta Neurochir (Wien) 2014;156(11):2127–2138. doi: 10.1007/s00701-014-2229-z. [DOI] [PubMed] [Google Scholar]
- 27.Barnett H JM, Foster J B, Hudgson P. Philadelphia, PA: W.B. Saunders; 1973. Syringomyelia. [Google Scholar]
- 28.Pullicino P, Kendall B E. Computed tomography of “cystic” intramedullary lesions. Neuroradiology. 1982;23(3):117–121. doi: 10.1007/BF00347553. [DOI] [PubMed] [Google Scholar]
- 29.Samii M Klekamp J Surgical results of 100 intramedullary tumors in relation to accompanying syringomyelia Neurosurgery 1994355865–873., discussion 873 [DOI] [PubMed] [Google Scholar]
- 30.Goy A M, Pinto R S, Raghavendra B N, Epstein F J, Kricheff I I. Intramedullary spinal cord tumors: MR imaging, with emphasis on associated cysts. Radiology. 1986;161(2):381–386. doi: 10.1148/radiology.161.2.3763905. [DOI] [PubMed] [Google Scholar]
- 31.Scotti G, Scialfa G, Colombo N, Landoni L. Magnetic resonance diagnosis of intramedullary tumors of the spinal cord. Neuroradiology. 1987;29(2):130–135. doi: 10.1007/BF00327537. [DOI] [PubMed] [Google Scholar]
- 32.Mehdorn H M, Stolke D. Cervical intramedullary cavernous angioma with MRI-proven haemorrhages. J Neurol. 1991;238(8):420–426. doi: 10.1007/BF00314647. [DOI] [PubMed] [Google Scholar]
- 33.Mork S J, Loken A C. Ependymoma: a follow-up study of 101 cases. Cancer. 1977;40(2):907–915. doi: 10.1002/1097-0142(197708)40:2<907::aid-cncr2820400247>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 34.Russell D S, Rubinstein L J. Baltimore, MD: Williams & Wilkins; 1989. Pathology of Tumors of the Nervous System. 5th ed. [Google Scholar]
- 35.Gardner W J. Hydrodynamic mechanism of syringomyelia: its relationship to myelocele. J Neurol Neurosurg Psychiatry. 1965;28:247–259. doi: 10.1136/jnnp.28.3.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kiwitt J CW, Lanksch W R, Fritsch H. et al. Magnetic resonance tomography of solid spinal cord tumors with extensive secondary syringomyelia. Adv Neurosurg. 1988;16:211–215. [Google Scholar]
- 37.Williams B. On the pathogenesis of syringomyelia: a review. J R Soc Med. 1980;73(11):798–806. doi: 10.1177/014107688007301109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winkelman M D, Adelstein D J, Karlins N L. Intramedullary spinal cord metastasis. Diagnostic and therapeutic considerations. Arch Neurol. 1987;44(5):526–531. doi: 10.1001/archneur.1987.00520170054022. [DOI] [PubMed] [Google Scholar]
- 39.Epstein F J, Farmer J P. Pediatric spinal cord tumor surgery. Neurosurg Clin N Am. 1990;1(3):569–590. [PubMed] [Google Scholar]
- 40.Sloof J L, Kernohan J W, MacCarthy C S. Philadelphia, PA: Saunders; 1964. Primary Intramedullary Tumors of the Spinal Cord and Filum Terminale. [Google Scholar]
- 41.Minehan K J, Shaw E G, Scheithauer B W, Davis D L, Onofrio B M. Spinal cord astrocytoma: pathological and treatment considerations. J Neurosurg. 1995;83(4):590–595. doi: 10.3171/jns.1995.83.4.0590. [DOI] [PubMed] [Google Scholar]
- 42.Houten J K, Cooper P R. Spinal cord astrocytomas: presentation, management and outcome. J Neurooncol. 2000;47(3):219–224. doi: 10.1023/a:1006466422143. [DOI] [PubMed] [Google Scholar]
- 43.Cooper P R. Outcome after operative treatment of intramedullary spinal cord tumors in adults: intermediate and long-term results in 51 patients. Neurosurgery. 1989;25(6):855–859. doi: 10.1097/00006123-198912000-00001. [DOI] [PubMed] [Google Scholar]
- 44.Citron N, Edgar M A, Sheehy J, Thomas D GT. Intramedullary spinal cord tumours presenting as scoliosis. J Bone Joint Surg Br. 1984;66(4):513–517. doi: 10.1302/0301-620X.66B4.6746684. [DOI] [PubMed] [Google Scholar]
- 45.Garrido E, Stein B M. Microsurgical removal of intramedullary spinal cord tumors. Surg Neurol. 1977;7(4):215–219. [PubMed] [Google Scholar]
- 46.Brotchi J, Fischer G. Spinal cord ependymomas. Neurosurg Focus. 1998;4(5):e2. doi: 10.3171/foc.1998.4.5.5. [DOI] [PubMed] [Google Scholar]
- 47.Hanbali F Fourney D R Marmor E et al. Spinal cord ependymoma: radical surgical resection and outcome Neurosurgery 20025151162–1172., discussion 1172–1174 [DOI] [PubMed] [Google Scholar]
- 48.McCormick P C, Torres R, Post K D, Stein B M. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72(4):523–532. doi: 10.3171/jns.1990.72.4.0523. [DOI] [PubMed] [Google Scholar]
- 49.Balériaux D LF. Spinal cord tumors. Eur Radiol. 1999;9(7):1252–1258. doi: 10.1007/s003300050831. [DOI] [PubMed] [Google Scholar]
- 50.Sekula R F, Sandhu F A, Oliverio P J, Henderson F C. Intramedullary tumors of the cord. Semin Spine Surg. 2000;12:21–29. [Google Scholar]
- 51.Wu Z, Iwanami A, Yasuda A, Mikami S, Toyama Y, Nakamura M. Intramedullary cervicothoracic subependymoma: report of three cases and review of the literature. J Orthop Sci. 2014 doi: 10.1007/s00776-014-0585-4. [DOI] [PubMed] [Google Scholar]
- 52.Murota T Symon L Surgical management of hemangioblastoma of the spinal cord: a report of 18 cases Neurosurgery 1989255699–707., discussion 708 [DOI] [PubMed] [Google Scholar]
- 53.Browne T R, Adams R D, Roberson G H. Hemangioblastoma of the spinal cord. Review and report of five cases. Arch Neurol. 1976;33(6):435–441. doi: 10.1001/archneur.1976.00500060041009. [DOI] [PubMed] [Google Scholar]
- 54.Yasargil M G, Antic J, Laciga R, de Preux J, Fideler R W, Boone S C. The microsurgical removal of intramedullary spinal hemangioblastomas. Report of twelve cases and a review of the literature. Surg Neurol. 1976;6(3):141–148. [PubMed] [Google Scholar]
- 55.Wyburn-Mason R. London, UK: Henry Kimpton; 1943. The Vascular Abnormalities and Tumors of the Spinal Cord and Its Membranes. [Google Scholar]
- 56.Richard S Campello C Taillandier L Parker F Resche F; French VHL Study Group. Haemangioblastoma of the central nervous system in von Hippel-Lindau disease J Intern Med 19982436547–553. [DOI] [PubMed] [Google Scholar]
- 57.Nemoto Y, Inoue Y, Tashiro T. et al. Intramedullary spinal cord tumors: significance of associated hemorrhage at MR imaging. Radiology. 1992;182(3):793–796. doi: 10.1148/radiology.182.3.1535896. [DOI] [PubMed] [Google Scholar]
- 58.Olson M E, Chernik N L, Posner J B. Infiltration of the leptomeninges by systemic cancer. A clinical and pathologic study. Arch Neurol. 1974;30(2):122–137. doi: 10.1001/archneur.1974.00490320010002. [DOI] [PubMed] [Google Scholar]
- 59.Price R A, Johnson W W. The central nervous system in childhood leukemia. I. The arachnoid. Cancer. 1973;31(3):520–533. doi: 10.1002/1097-0142(197303)31:3<520::aid-cncr2820310306>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 60.Batson V O. The role of the vertebral veins in metastatic processes. Ann Intern Med. 1942;16:38–45. [Google Scholar]
- 61.Fromm H, von Wild K. Clinical aspects, operative treatment and rehabilitation of paraplegia caused by lipomas of the spinal cord-with particular emphasis of the “intramedullary” lipomas. Paraplegia. 1974;12(1):15–20. doi: 10.1038/sc.1974.4. [DOI] [PubMed] [Google Scholar]
- 62.Giuffrè R. Intradural spinal lipomas. Review of the literature (99 cases) and report of an additional case. Acta Neurochir (Wien) 1966;14(1):69–95. doi: 10.1007/BF01401892. [DOI] [PubMed] [Google Scholar]
- 63.Swanson H S, Barnett J CJ Jr. Intradural lipomas in children. Pediatrics. 1962;29:911–926. [PubMed] [Google Scholar]
- 64.Eichler M E, Dacey R G. Philadelphia, PA: Lippincott-Raven; 1997. Intramedullary spinal cord tumors; pp. 2089–2102. [Google Scholar]
- 65.Timmer F A, van Rooij W J, Beute G N, Teepen J L. Intramedullary lipoma. Neuroradiology. 1996;38(2):159–160. doi: 10.1007/BF00604807. [DOI] [PubMed] [Google Scholar]
- 66.Hamburger C Büttner A Weis S Ganglioglioma of the spinal cord: report of two rare cases and review of the literature Neurosurgery 19974161410–1415., discussion 1415–1416 [DOI] [PubMed] [Google Scholar]
- 67.Patel U, Pinto R S, Miller D C. et al. MR of spinal cord ganglioglioma. AJNR Am J Neuroradiol. 1998;19(5):879–887. [PMC free article] [PubMed] [Google Scholar]
- 68.McCormick P C, Stein B M. Miscellaneous intradural pathology. Neurosurg Clin N Am. 1990;1(3):687–699. [PubMed] [Google Scholar]
- 69.Schwartz T H, Chang Y, Stein B M. Unusual intramedullary vascular lesion: report of two cases. Neurosurgery. 1997;40(6):1295–1301. doi: 10.1097/00006123-199706000-00036. [DOI] [PubMed] [Google Scholar]
- 70.Deme S, Ang L-C, Skaf G, Rowed D W. Primary intramedullary primitive neuroectodermal tumor of the spinal cord: case report and review of the literature. Neurosurgery. 1997;41(6):1417–1420. doi: 10.1097/00006123-199712000-00040. [DOI] [PubMed] [Google Scholar]
- 71.Fraser R D, Paterson D C, Simpson D A. Orthopaedic aspects of spinal tumors in children. J Bone Joint Surg Br. 1977;59(2):143–151. doi: 10.1302/0301-620X.59B2.873974. [DOI] [PubMed] [Google Scholar]
- 72.Papagelopoulos P J, Peterson H A, Ebersold M J, Emmanuel P R, Choudhury S N, Quast L M. Spinal column deformity and instability after lumbar or thoracolumbar laminectomy for intraspinal tumors in children and young adults. Spine (Phila Pa 1976) 1997;22(4):442–451. doi: 10.1097/00007632-199702150-00019. [DOI] [PubMed] [Google Scholar]
- 73.Haft H, Ransohoff J, Carter S. Spinal cord tumors in children. Pediatrics. 1959;23(6):1152–1159. [PubMed] [Google Scholar]
- 74.Lonstein J E. Post-laminectomy kyphosis. Clin Orthop Relat Res. 1977;(128):93–100. [PubMed] [Google Scholar]
- 75.Tachdjian M O, Matson D D. Orthopaedic aspects of intraspinal tumors in infants and children. J Bone Joint Surg Am. 1965;47:223–248. [PubMed] [Google Scholar]
- 76.Arima H, Hasegawa T, Togawa D. et al. Feasibility of a novel diagnostic chart of intramedullary spinal cord tumors in magnetic resonance imaging. Spinal Cord. 2014;52(10):769–773. doi: 10.1038/sc.2014.127. [DOI] [PubMed] [Google Scholar]
- 77.Parizel P M, Balériaux D, Rodesch G. et al. Gd-DTPA-enhanced MR imaging of spinal tumors. AJR Am J Roentgenol. 1989;152(5):1087–1096. doi: 10.2214/ajr.152.5.1087. [DOI] [PubMed] [Google Scholar]
- 78.Brotchi J Dewitte O Levivier M et al. A survey of 65 tumors within the spinal cord: surgical results and the importance of preoperative magnetic resonance imaging Neurosurgery 1991295651–656., discussion 656–657 [PubMed] [Google Scholar]
- 79.Fine M J, Kricheff I I, Freed D, Epstein F J. Spinal cord ependymomas: MR imaging features. Radiology. 1995;197(3):655–658. doi: 10.1148/radiology.197.3.7480734. [DOI] [PubMed] [Google Scholar]
- 80.Kahan H, Sklar E ML, Post M J, Bruce J H. MR characteristics of histopathologic subtypes of spinal ependymoma. AJNR Am J Neuroradiol. 1996;17(1):143–150. [PMC free article] [PubMed] [Google Scholar]
- 81.Colombo N, Kucharczyk W, Brant-Zawadzki M. et al. Magnetic resonance imaging of spinal cord hemangioblastomas. Acta Radiol Diagn (Stockh) 1986;769:S734. [PubMed] [Google Scholar]
- 82.Rykken J B, Diehn F E, Hunt C H. et al. Intramedullary spinal cord metastases: MRI and relevant clinical features from a 13-year institutional case series. AJNR Am J Neuroradiol. 2013;34(10):2043–2049. doi: 10.3174/ajnr.A3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Choucair A K Myelopathies in the cancer patient: incidence, presentation, diagnosis and management Oncology (Williston Park) 19915725–31., discussion 35–37 [PubMed] [Google Scholar]
- 84.Rykken J B, Diehn F E, Hunt C H. et al. Rim and flame signs: postgadolinium MRI findings specific for non-CNS intramedullary spinal cord metastases. AJNR Am J Neuroradiol. 2013;34(4):908–915. doi: 10.3174/ajnr.A3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mirone G, Cinalli G, Spennato P, Ruggiero C, Aliberti F. Hydrocephalus and spinal cord tumors: a review. Childs Nerv Syst. 2011;27(10):1741–1749. doi: 10.1007/s00381-011-1543-5. [DOI] [PubMed] [Google Scholar]
- 86.Cinalli G, Sainte-Rose C, Lellouch-Tubiana A, Sebag G, Renier D, Pierre-Kahn A. Hydrocephalus associated with intramedullary low-grade glioma. Illustrative cases and review of the literature. J Neurosurg. 1995;83(3):480–485. doi: 10.3171/jns.1995.83.3.0480. [DOI] [PubMed] [Google Scholar]
- 87.Mirza S, Adams W M, Corkhill R A. Froin's syndrome revisited, 100 years on. Pseudo-Froin's syndrome on MRI. Clin Radiol. 2008;63(5):600–604. doi: 10.1016/j.crad.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 88.Abul-Kasim K, Thurnher M M, McKeever P, Sundgren P C. Intradural spinal tumors: current classification and MRI features. Neuroradiology. 2008;50(4):301–314. doi: 10.1007/s00234-007-0345-7. [DOI] [PubMed] [Google Scholar]
- 89.Elhammady M S, Farhat H, Aziz-Sultan M A, Morcos J J. Isolated unilateral hypoglossal nerve palsy secondary to an atlantooccipital joint juxtafacet synovial cyst. J Neurosurg Spine. 2009;10(3):234–239. doi: 10.3171/2008.12.SPINE08158. [DOI] [PubMed] [Google Scholar]
- 90.Moller H U. Familial angiomatosis retinae et cerebelli. Lindau's disease. Acta Opthalmol (Copenh) 1929;7:244–260. [Google Scholar]