

SUMMARY
The outer membrane protein Ail (attachment invasion locus) is a virulence factor of Yersinia pestis that mediates cell invasion, cell attachment and complement resistance. Here we describe its three-dimensional backbone structure determined in decyl-phosphocholine (DePC) micelles by NMR spectroscopy. The NMR structure was calculated using the membrane function of the implicit solvation potential, eefxPot, which we have developed to facilitate NMR structure calculations in a physically realistic environment. We show that the eefxPot force field guides the protein towards its native fold. The resulting structures provide information about the membrane-embedded global position of Ail, and have higher accuracy, higher precision and improved conformational properties, compared to the structures calculated with the standard repulsive potential.
Keywords: Ail, Yersinia pestis, membrane protein, structure, NMR, implicit solvation
Yersinia pestis, the bacterium that causes plague, is an extremely pathogenic organism with a long history of precipitating massive human pandemics (Perry and Fetherston 1997; Cornelis 2000; Brubaker 2004). Y. pestis is transmitted both via the bite of infected fleas (bubonic plague) and via aerosols (pneumonic plague) (Perry and Fetherston 1997), and can proliferate in diverse environments. This critical aspect of its virulence is governed by a number of virulence attributes that include Ail (attachment invasion locus), an outer membrane protein with established roles in cell invasion, cell attachment, complement resistance and virulence (Miller et al. 1990; Bartra et al. 2008; Felek et al. ; Hinnebusch et al. ; Kolodziejek et al. 2010).
Previous studies have shown that Y. pestis Ail binds the human extracellular matrix proteins fibronectin, laminin and heparan sulfate proteoglycans (Tsang et al. 2010; Yamashita et al. 2011; Tsang et al. 2012), as well as the complement regulatory component C4BP (Ho et al. 2014). The structure of Ail, crystallized from tetraethylene glycol monooctyl ether (C8E4), has been determined at high resolution (Yamashita et al. 2011). The protein folds as an eight-stranded β-barrel with three intracellular loops (IL1-IL3) and four extracellular loops (EL1-EL4) formed by residues that are implicated in ligand binding (Miller et al. 2001; Tsang et al. 2013; Ding et al. 2015b). However, the functionally important extracellular loops EL2 and EL3 were incompletely resolved in the crystal structure.
Using NMR spectroscopy with Ail in decyl-phosphocholine (DePC) micelles, backbone resonances for all extracellular loop sites could be observed and assigned. Heteronuclear 1H/15N NOE experiments showed the presence of backbone flexibility in the extracellular loops, particularly EL2, consistent with their lack of resolution by crystallography (Ding et al. 2015b). Assignments of the backbone N, HN, CA and CB resonances were obtained for all Ail amino acids, with the exception of three residues in IL1 (Ile36, Phe37, Asn38) and two residues in IL2 (Glu83, Tyr84), and deposited in the Biological Magnetic Resonance data Bank (BMRB 25281). The NMR data showed that the Ail β-barrel is stabilized by a strong network of backbone amide hydrogen bonds that resist exchange with water, with stronger hydrogen bonds observed for residues located in a hydrophobic band around the β-barrel, and weaker, readily exchangeable hydrogen bonds, observed for sites that are water-exposed (Ding et al. 2015b). Furthermore, long-range amide hydrogen distances, derived from 1H-1H NOE measurements were measured for both transmembrane and extracellular sites, between all neighboring β-strands, including key contacts between the first and last β-strand (β1-β8) that define the barrel closure.
Here, we have used these NMR restraints to determine the backbone structure of Ail in DePC micelles. Two structures were calculated with XPLOR-NIH (Schwieters et al. 2003; Schwieters et al. 2006), one using the standard, purely repulsive form of the Van der Waals function (REPEL), with the default protein topology and parameter files (protein.par and protein.top), and the other using the implicit membrane function of the potential, eefxPot, with its dedicated protein topology and parameter files (protein_eef.par and protein_eef.top) (Tian et al. 2014, 2015). The structural coordinates and experimental restraints have been deposited in the Protein Data Bank (PDB: 2N2L, 2N2M).
Structure calculations in the implicit membrane environment of eefxPot
NMR-restrained structure calculations are typically performed in a force-field where all nonbonded interactions are represented by a single, purely repulsive term, that does not include van der Waals attractive forces, electrostatic forces or solvation energy. This simplified potential is very successful for facilitating simulated annealing structure calculations from fully extended templates (Nilges et al. 1988; Clore and Gronenborn 1989), but does not provide a physically realistic solvent environment, especially for membrane proteins that reside in the highly heterogeneous and anisotropic environment of the lipid bilayer membrane.
The eefxPot potential, on the other hand, includes terms for Lennard-Jones van der Waals energy (both repulsive and attractive forces), electrostatic energy and solvation free energy. The potential is based on the CHARMM effective energy function for implicit water and membrane solvation (Lazaridis and Karplus 1999; Lazaridis 2003). While the first version of eefxPot was limited to bulk water solvation (Tian et al. 2014), the most recent development includes the membrane and supports structure calculations of membrane proteins in an implicit membrane environment (Tian et al. 2015).
The membrane of eefxPot is modeled by a function that modulates the solvation and electrostatic parameters of all atoms according to their absolute distance along the membrane normal (coincident with the coordinate z axis) from the hydrophobic membrane center (z = 0). This membrane profile function depends on the membrane hydrophobic thickness (T) and a parameter (n) that defines the size of the region over which the environment changes from hydrophobic to hydrophilic. The values of T=25 Å and n=10, used in this study, give a membrane thickness that mimics the relatively thin bacterial outer membrane and a membrane profile similar to that derived from neutron and X-ray scattering (Luzzati and Husson 1962; Marsh 2013; Nagle and Tristram-Nagle 2000; Kucerka et al. 2011; Nagle 2013). The potential is accessible from the Python interface of XPLOR-NIH by calling eefxPot and its accessory module eefxPotTools. It may be downloaded from the web (http://nmr.cit.nih.gov/xplor-nih/) with the latest release of XPLOR-NIH (version 2.39).
Structures of Ail were calculated using two simulated annealing protocols, the first to fold 100 structures from an initially extended polypeptide conformation, and the second to refine 100 structures from the best folded structure. The final ensembles were generated by selecting the 10 best refined structures with lowest restraining energy terms, least number of violations and, in the case of eefxPot, proper membrane embedding.
The fold and refine protocols are based on the internal variable module (IVM) (Schwieters and Clore 2001) and share the same basic architecture comprising four stages: (i) torsion angle dynamics at high-temperature (3,500 K for fold, 3,000 K for refine), for a time of 32 ps or 32,000 timesteps (fold protocol), or a time of 20 ps or 20,000 timesteps (refine protocol); (ii) torsion angle dynamics with simulated annealing from the initial high temperature value to 25 K, in steps of 12.5 K, for a time of 0.4 ps or 200 timesteps per temperature step; (iii) 500 steps of Powell torsion angle minimization; and (iv) 500 steps of Powell Cartesian minimization.
Dihedral angle restraints were derived from analysis of the assigned chemical shifts with the program TALOS-N (Shen and Bax 2013) and imposed with the XPLOR term CDIH. Amide hydrogen bonds and amide hydrogen NOE distances were imposed with the distance restraint term noePot, side chain rotamers were restrained with the torsionDB statistical torsion angle potential (Bermejo et al. 2012), and standard potentials were implemented to define chemical bond lengths, bond angles, and atomic radii. In the high temperature stage, experimental restraints were applied with force constants of kCDIH=100 kcal mol−1 rad−2 (dihedral angles) and kDIST=2 kcal mol−1 Å−2 (distances). During simulated annealing, kCDIH was set to 200 kcal mol−1 rad−2 and kDIST was increased geometrically from 2 to 30 kcal mol−1 Å−2. The torsionDB force constant was set to ktDB=0.02 kcal mol−1 at high temperature stage and ramped geometrically from 0.02 to 2 kcal mol−1 during simulated annealing.
During the eefxPot fold protocol, the 32 ps high temperature stage was divided into three blocks. This was done to prevent fatal atomic overlap in the early stages of calculations. Two initial 3 ps segments of dynamics were performed using REPEL with eefxPot turned off, the first with only CA-CA atomic interactions active, the van der Waals force constant set to Crep=0.004 kcal mol−1 Å−4 and the van der Waals radius scale factor set to krep=1.2, and the second with all atom-atom interactions turned on, Crep=4 kcal mol−1 Å−4 and krep=0.8. The third 26 ps segment of dynamics at high temperature, as well as the subsequent simulated annealing stage, were performed without REPEL, using exclusively eefxPot with its scale set to 0.004 at high temperature and then ramped from 0.004 to 1 during simulated annealing. The protein center of mass was positioned at the membrane center (z = 0) before each block of the high temperature stage, while the protein position was left unrestrained during simulated annealing.
We have found that the timing and force constants of the fold protocol are particularly important for transmembrane β-barrels such as Ail, especially in cases where relatively few experimental restraints are available. In these proteins, sequential amino acids alternate in character between hydrophobic and hydrophilic and the membrane-spanning structural unit is formed by nonsequential residues far apart in the amino acid sequence. In nature, β-barrel folding and membrane insertion are assisted by dedicated machinery (Kleinschmidt 2015); here, they were assisted by extending the high temperature stage to allow attractive van der Waals forces to work, and imposing dihedral angle restraints more strongly in the early stages of the protocol. Once folded, the structure and membrane position of Ail were stably sustained by eefxPot.
Structure of Ail in DePC micelles
Although the membrane model of eefxPot is intended to be used with restraints measured for membrane proteins in lipid bilayer membranes, it can also be applied to restraints measured in detergent micelles. In this study, the structure of Ail was determined in 170 mM DePC, at 45°C. This detergent gives the best solution NMR spectra (Ding et al. 2015b), but high quality solution NMR spectra can also be obtained for Ail incorporated in small lipid bilayer nanodiscs (Ding et al. 2015b), and larger nanodiscs reconstituted with Ail can be harvested by centrifugation as sedimented solutes to yield high quality solid-state NMR spectra (Ding et al. 2015a). Interestingly, even though the high detergent concentrations required for solution NMR spectroscopy are not compatible with the ligand binding activity of Ail (Ding et al. 2015b), it is notable that the solution and solid-state NMR spectra from micelles and nanodiscs all have very similar chemical shifts, indicating that the protein adopts the same overall fold in all three environments.
The NMR structure of Ail in micelles (Fig. 1) is also similar to the crystal structure (Yamashita et al. 2011). Both REPEL and eefxPot produce good structures, with acceptable conformational properties and statistics (Fig. 2; Table 1), but the eefxPot structures have higher accuracy and precision and also provide a view of the protein embedded in the membrane. The extracellular loops are more disordered than those of the crystal structure where they form an extended βsheet that extends out from the membrane. While the extent of loop dynamics will have to be assessed in detergent-free lipid bilayers, loop flexibility is consistent with the versatile ligand-binding properties of Ail and with the incomplete observation of EL2 and EL3 electron density in the crystals.
Figure 1. Structure of Ail determined by NMR in DePC micelles.
Structures were calculated using eefxPot (cyan) or REPEL (gray), or taken from the crystallographic coordinates (pink; PDB 3QRA). The eefxPot membrane is depicted as horizontal lines separated by the membrane thickness T=25Å. Each ensemble is aligned to its lowest energy structure. (A-C) Ensembles of 10 best structures calculated with REPEL (A) or eefxPot (B, C). The crystal structure is aligned to the lowest energy model of each ensemble. (D) Lowest energy structure from eefxPot, showing the four extracellular loops (EL1-EL4) and the two juxtaposed patches of positivley charged residues near the extracellular membrane surface. The red-cyan color spectrum reflects the 1H/2H exchange profile of amide hydrogens that resist (red) or undergo (cyan) exchange with solvent. Structures were rendered with PyMol (DeLano 2005)
Figure 2.
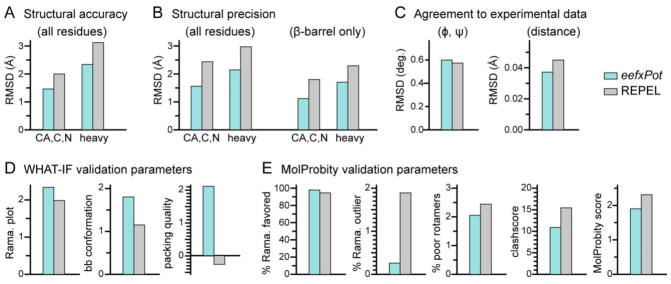
Structure statistics and validation metrics. Bars represent results obtained with eefxPot (cyan) or REPEL (gray). Statistics were evaluated for the ten structures in each refined ensemble. (A) Strucural accuracy evaluated as pairwise RMSD of backbone or heavy atoms between the calculated structures and the crystal structure (PDB: 3QRA). (B) Structural precision evaluated as average pairwise RMSD of backbone or heavy atoms, for all residues or β-barrel residues only. (C) Agreement between calculated structures and experimental dihedral angle (φ, ψ) and distance (NOE and H-bond) restraints. (D) WHAT-IF validation statistics for Ramachandran plot appearance, backbone conformation and protein packing quality. (E) MolProbity validation statistics for percent of residues in favored regions of the Ramachandran plot, percent of residues in unfavored regions of the Ramachandran plot, percent of residues with poor sidechain torsion angles, clashscore, and overall Molprobity score. The MolProbity clashscore and MolProbity score are costs: the lower the better.
Table 1.
Structure and NMR restraints statistics.a
| eefxPot | REPEL | |
|---|---|---|
| Experiment Restraints | ||
| dihedral angles | 294 | 294 |
| NOE distances (i-j>5) | 146 | 146 |
| H-bonds | 77 | 77 |
| RMSD Agreement with Experimental restraints | ||
| dihedral angle (deg) | 0.598±0.039 | 0.570±0.094 |
| NOE (Å) | 0.040±0.003 | 0.044±0.003 |
| H-bond (Å) | 0.028±0.006 | 0.047±0.003 |
| Idealized Covalent Geometry RMSD | ||
| bonds (Å) | 0.995±0.001 | 1.036±0.000 |
| angles (deg) | 0.267±0.002 | 0.537±0.002 |
| impropers (deg) | 0.320±0.008 | 0.645±0.005 |
| Coordinate Precision as average RMSD from mean (Å)b | ||
| backbone CA, C, N atoms | 1.586 | 2.451 |
| all heavy atoms | 2.164 | 2.960 |
| Ramachandran Plot ϕ/ψ Angle Statistics | ||
| most favored regions | 92.9% | 92.2% |
| additional allowed regions | 5.8% | 4.8% |
| generously allowed regions | 0.8% | 1.7% |
| disallowed regions | 0.5% | 1.4% |
Calculated for 10 structures, out of 100 refined structures, selected for lowest energy and lowest experimental restraint violations.
Calculated for all 156 protein residues.
In some of the REPEL structures the hydrophilic extracellular loops fold back towards the membrane and against the barrel body (Fig. 1A). Such conformations are not expected to exist in the native lipid bilayer membrane environment where the loops are known to engage ligands in the water phase. Previous NMR structure calculations of membrane proteins have sought to eliminate such anomalous conformations by establishing an artificial water-membrane boundary through the use of harmonic coordinate restraints (Teriete et al. 2007) or plane distance restraints (Xu et al. 2008; Fox et al. 2014; Gong et al. 2015), or with an empirical potential based on the preferred membrane insertion depth of each amino acid (Shi et al. 2009). However, these approaches do not provide a physical representation of the water-membrane environment. Notably, the structures calculated with eefxPot do not exhibit such unfavorable conformations, showing that the potential effectively guides the water-soluble extracellular loops away from the hydrophobic membrane interior and into the water phase.
In the eefxPot structure, the Ail β-barrel adopts a transmembrane tilt of 7±10° (Fig. 1B) and a membrane insertion depth consistent with the 1H/2H exchange data (Fig. 1D). Although the transmembrane tilt of the β-barrel will have to be confirmed experimentally, we note that eefxPot produces an accurate transmembrane position of the OmpX β-barrel, as assessed by cross validation with experimental 1H/15N solid-state NMR orientation restraints (Tian et al. 2015). The transmembrane core of the Ail β-barrel is well defined by the 1H/2H exchange data and delimited by two bands of aromatic residues, each located at the membrane-water interfaces. The membrane-embedded position of the barrel places a ring of positively charged Lys and Arg sidechains near the extracellular membrane-water interface (Fig. 1D). In the crystal structure, these sites can associate with sucrose octasulfate, a heparin analog small molecule (Yamashita et al. 2011). Their location at the membrane surface suggests that they can also play a role in mediating the interactions of Ail with other membrane components such as phospholipids and lipopolysaccharides.
Effect of eefxPot on structural accuracy, precision and quality
The NMR structures of Ail calculated with eefxPot have significantly closer agreement to the crystal structure (Fig. 2A, Table 1). As transmembrane β-barrel conformations are relatively refractory, the crystal structure of Ail, which was determined with rather high resolution, is likely a good representative of the native conformation, thus, we infer that closer agreement to the crystal structure reflects higher structural accuracy. The precision of the eefxPot structural ensemble is also significantly higher than the REPEL ensemble (Fig. 2B, Table 1). Taken together, these results reflect the ability of eefxPot to effectively guide the conformation of Ail towards native fold, as observed for other soluble and membrane proteins (Tian et al. 2014, 2015).
Furthermore, although introducing an additional term in the target energy function can worsen the agreement between calculated structures and the other experimental and conformational energy terms, we find that the improvements in accuracy and precision afforded by eefxPot are obtained without sacrificing agreement with either conformational or experimental restraints. Both eefxPot and REPEL structures have comparable RMSDs to the experimental restraints (Fig. 2C, Table 1), well within the acceptable range, with the eefxPot structures showing agreements that are better for distance restraints and only slightly lower for dihedral angles.
Assessment of the backbone conformations, side chain conformations and nonbonded atomic interactions by means of WHAT-IF (Vriend 1990; Doreleijers et al. 2012) and MolProbity (Chen et al. 2010) metrics shows that eefxPot yields structures with overall higher conformational quality (Fig. 2D, Fig. 2E), reflected in increased population of the favored regions of Ramachandran space and lower number of Ramachandran outliers. Furthermore, although both eefxPot and REPEL produce acceptable χ1/χ2 rotamer normality scores, an effect due primarily to the torsionDB potential of XPLOR-NIH (Bermejo et al. 2012), the eefxPot structures appear to have fewer poor rotamers.
The validation metrics further show that eefxPot improves the quality of nonbonded atomic interactions. The structures of Ail calculated with eefxPot show improvements in both the WHAT-IF packing quality, which reflects the atomic distributions around different molecular fragments (Vriend and Sander 1993), and the MolProbity clashscore, which reflects the number of serious atomic overlaps per thousand atoms (Word et al. 1999). These key metrics provide database-independent estimates of the quality of nonbonded atomic interactions or atomic packing. The overall improvement is reflected in the lower MolProbity score - the lower the better – that is obtained with eefxPot.
Conclusions
We conclude that the implicit membrane environment of eefxPot supports the native structure of the outer membrane protein Ail. The resulting structures have higher accuracy, higher precision and better overall quality. Although eefxPot is designed to be used with experimental restraints measured in detergent-free lipid bilayer samples, the results obtained with Ail show that it is compatible with measurements made in detergent micelles, where it is useful for imposing boundaries between the water and membrane environments and to establish proper protein membrane-embedded topology. The structure of Ail calculated with this implicit membrane model informs our understanding of the protein in the bacterial outer membrane.
It is also important to note that since the lipid bilayer membrane of eefxPot is modeled implicitly, its ability to reflect the structural and physical properties of natural membranes is limited (Tian et al. 2015). The principal limitations of the model are related to its inadequate representation of the hydrophobic matching phenomenon. In nature, match of a protein’s amino acid sequence to the hydrophobic environment of the surrounding membrane is achieved by adjusting the protein’s global position (tilt and rotation) in the membrane and/or adjusting the lipid bilayer’s structure and order (Mouritsen and Bloom 1984; de Planque and Killian 2003). While the first mechanism is reproduced very well by eefxPot, the second is not, because membrane deformation is difficult to model implicitly.
Implicit membrane models cannot adequately reproduce the effects of lipid composition, aqueous phase composition, temperature, etc., on the structural and physical properties of lipid bilayers. Membrane proteins that form water-containing cavities also pose a challenge, although this can be addressed by mathematically shaping the implicit membrane to form a water-filled pore (Lazaridis 2005). In the case of detergent micelles, the structure of the hydrophobic environment departs even further from the continuous membrane model of eefxPot, as it is far from planar, much more fluid, and contains much more water than a lipid bilayer. A solution to this problem may be to perform calculations where the water phase is represented implicitly with eefxPot, and both the protein and the detergent molecules are treated in atomistic detail, as described for molecular dynamics (MD) simulations (Versace and Lazaridis 2015).
Nevertheless, while these limitations are significant in MD simulations performed free of the influence of experimental restraints, they become much less important when eefxPot is used for its intended purpose of assisting NMR-restrained structure calculations and the force field is not alone in determining protein structure. NMR structures calculated with eefxPot, in turn, can provide much improved starting points for MD simulations with all-atom force fields, designed to obtain atomic-level structure refinement and information about protein-lipid or protein-water interactions.
In all cases of soluble and membrane proteins tested so far (Tian et al. 2014, 2015), eefxPot outperforms the standard REPEL force field: it yields structures with higher accuracy, precision and conformational quality and is very effective at guiding proteins towards their native fold, even in the absence of large numbers of experimental restraints. Furthermore, as eefxPot is developed for XPLOR-NIH, it is fully compatible with all the experimental restraining terms available in the program, as well as with the full range of molecular parameters and topologies relevant for NMR, such as paramagnetic spin labels, for example.
These initial results are quite compelling in demonstrating that eefxPot is useful for NMR calculations. Indeed, there seems to be no reason not to use it, especially since it is as computationally efficient as REPEL and requires no or minimal additional setup. Further developments of the eefxPot membrane profile function to model water occlusion, surface charge, and micelle-like spherical or oblong structures will further extend its applications to NMR studies of membrane proteins.
Acknowledgments
This research was supported by grants from the National Institutes of Health (NIH: R01 GM110658, R01 GM100265, P41 EB002031, P30 CA030199). CDS was supported by funds from the NIH Intramural Research Program of The Center for Information Technology.
References
- Bartra SS, Styer KL, O’Bryant DM, Nilles ML, Hinnebusch BJ, Aballay A, Plano GV. Resistance of Yersinia pestis to complement-dependent killing is mediated by the Ail outer membrane protein. Infect Immun. 2008;76 (2):612–622. doi: 10.1128/IAI.01125-07. IAI.01125-07 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermejo GA, Clore GM, Schwieters CD. Smooth statistical torsion angle potential derived from a large conformational database via adaptive kernel density estimation improves the quality of NMR protein structures. Protein Sci. 2012;21 (12):1824–1836. doi: 10.1002/pro.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker RR. The recent emergence of plague: a process of felonious evolution. Microb Ecol. 2004;47 (3):293–299. doi: 10.1007/s00248-003-1022-y. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66 (Pt 1):12–21. doi: 10.1107/S0907444909042073. S0907444909042073 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Gronenborn AM. Determination of three-dimensional structures of proteins and nucleic acids in solution by nuclear magnetic resonance spectroscopy. Crit Rev Biochem Mol Biol. 1989;24 (5):479–564. doi: 10.3109/10409238909086962. [DOI] [PubMed] [Google Scholar]
- Cornelis GR. Molecular and cell biology aspects of plague. Proc Natl Acad Sci U S A. 2000;97(16):8778–8783. doi: 10.1073/pnas.97.16.8778. 97/16/8778 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Planque MR, Killian JA. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol Membr Biol. 2003;20 (4):271–284. doi: 10.1080/09687680310001605352. [DOI] [PubMed] [Google Scholar]
- DeLano WL. PyMol. 2005 www.pymol.org.
- Ding Y, Fujimoto LM, Yao Y, Marassi FM. Solid-state NMR of the Yersinia pestis outer membrane protein Ail in lipid bilayer nanodiscs sedimented by ultracentrifugation. J Biomol NMR. 2015a;61 (3–4):275–286. doi: 10.1007/s10858-014-9893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Fujimoto LM, Yao Y, Plano GV, Marassi FM. Influence of the lipid membrane environment on structure and activity of the outer membrane protein Ail from Yersinia pestis. Biochim Biophys Acta. 2015b;1848 (2):712–720. doi: 10.1016/j.bbamem.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doreleijers JF, Sousa da Silva AW, Krieger E, Nabuurs SB, Spronk CA, Stevens TJ, Vranken WF, Vriend G, Vuister GW. CING: an integrated residue-based structure validation program suite. J Biomol NMR. 2012;54 (3):267–283. doi: 10.1007/s10858-012-9669-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felek S, Tsang TM, Krukonis ES. Three Yersinia pestis adhesins facilitate Yop delivery to eukaryotic cells and contribute to plague virulence. Infect Immun. 2010;78 (10):4134–4150. doi: 10.1128/IAI.00167-10. IAI.00167-10 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DA, Larsson P, Lo RH, Kroncke BM, Kasson PM, Columbus L. Structure of the Neisserial outer membrane protein Opa(6)(0): loop flexibility essential to receptor recognition and bacterial engulfment. J Am Chem Soc. 2014;136 (28):9938–9946. doi: 10.1021/ja503093y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong XM, Ding Y, Yu J, Yao Y, Marassi FM. Structure of the Na,K-ATPase regulatory protein FXYD2b in micelles: implications for membrane-water interfacial arginines. Biochim Biophys Acta. 2015;1848 (1 Pt B):299–306. doi: 10.1016/j.bbamem.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch BJ, Jarrett CO, Callison JA, Gardner D, Buchanan SK, Plano GV. Role of the Yersinia pestis Ail Protein in Preventing a Protective Polymorphonuclear Leukocyte Response During Bubonic Plague. Infect Immun. 2011 doi: 10.1128/IAI.05307-11. IAI.05307-11 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DK, Skurnik M, Blom AM, Meri S. Yersinia pestis Ail recruitment of C4b-binding protein leads to factor I-mediated inactivation of covalently and noncovalently bound C4b. Eur J Immunol. 2014;44 (3):742–751. doi: 10.1002/eji.201343552. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt JH. Folding of beta-barrel membrane proteins in lipid bilayers - Unassisted and assisted folding and insertion. Biochim Biophys Acta. 2015 doi: 10.1016/j.bbamem.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Kolodziejek AM, Schnider DR, Rohde HN, Wojtowicz AJ, Bohach GA, Minnich SA, Hovde CJ. Outer membrane protein X (Ail) contributes to Yersinia pestis virulence in pneumonic plague and its activity is dependent on the lipopolysaccharide core length. Infect Immun. 2010;78 (12):5233–5243. doi: 10.1128/IAI.00783-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucerka N, Nieh MP, Katsaras J. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochim Biophys Acta. 2011;1808 (11):2761–2771. doi: 10.1016/j.bbamem.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Lazaridis T. Effective energy function for proteins in lipid membranes. Proteins. 2003;52 (2):176–192. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- Lazaridis T. Structural Determinants of Transmembrane β-Barrels. Journal of chemical theory and computation. 2005;1 (4):716–722. doi: 10.1021/ct050055x. [DOI] [PubMed] [Google Scholar]
- Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35(2):133–152. doi: 10.1002/(SICI)1097-0134(19990501)35:2<133::AID-PROT1>3.0.CO;2-N. [pii] [DOI] [PubMed] [Google Scholar]
- Luzzati V, Husson F. The structure of the liquid-crystalline phasis of lipid-water systems. J Cell Biol. 1962;12:207–219. doi: 10.1083/jcb.12.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D. Handbook of lipid bilayers 2013 [Google Scholar]
- Miller VL, Beer KB, Heusipp G, Young BM, Wachtel MR. Identification of regions of Ail required for the invasion and serum resistance phenotypes. Mol Microbiol. 2001;41 (5):1053–1062. doi: 10.1046/j.1365-2958.2001.02575.x. [DOI] [PubMed] [Google Scholar]
- Miller VL, Bliska JB, Falkow S. Nucleotide sequence of the Yersinia enterocolitica ail gene and characterization of the Ail protein product. J Bacteriol. 1990;172 (2):1062–1069. doi: 10.1128/jb.172.2.1062-1069.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouritsen OG, Bloom M. Mattress model of lipid-protein interactions in membranes. Biophys J. 1984;46 (2):141–153. doi: 10.1016/S0006-3495(84)84007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle JF. Introductory lecture: basic quantities in model biomembranes. Faraday Discuss. 2013;161:11–29. doi: 10.1039/c2fd20121f. discussion 113–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle JF, Tristram-Nagle S. Structure of lipid bilayers. Biochim Biophys Acta. 2000;1469(3):159–195. doi: 10.1016/s0304-4157(00)00016-2. S0304-4157(00)00016-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilges M, Gronenborn AM, Brunger AT, Clore GM. Determination of three-dimensional structures of proteins by simulated annealing with interproton distance restraints. Application to crambin, potato carboxypeptidase inhibitor and barley serine proteinase inhibitor 2. Protein Eng. 1988;2 (1):27–38. doi: 10.1093/protein/2.1.27. [DOI] [PubMed] [Google Scholar]
- Perry RD, Fetherston JD. Yersinia pestis--etiologic agent of plague. Clin Microbiol Rev. 1997;10 (1):35–66. doi: 10.1128/cmr.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters CD, Clore GM. Internal coordinates for molecular dynamics and minimization in structure determination and refinement. J Magn Reson. 2001;152 (2):288–302. doi: 10.1006/jmre.2001.2413. [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Marius Clore G. Using Xplor-NIH for NMR molecular structure determination. Prog Nucl Magn Reson Spectrosc. 2006;48 (1):47–62. http://dx.doi.org/10.1016/j.pnmrs.2005.10.001. [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160(1):65–73. doi: 10.1016/s1090-7807(02)00014-9. S1090780702000149 [pii] [DOI] [PubMed] [Google Scholar]
- Shen Y, Bax A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR. 2013;56 (3):227–241. doi: 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Traaseth NJ, Verardi R, Cembran A, Gao J, Veglia G. A refinement protocol to determine structure, topology, and depth of insertion of membrane proteins using hybrid solution and solid-state NMR restraints. J Biomol NMR. 2009;44 (4):195–205. doi: 10.1007/s10858-009-9328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teriete P, Franzin CM, Choi J, Marassi FM. Structure of the Na,K-ATPase regulatory protein FXYD1 in micelles. Biochemistry. 2007;46 (23):6774–6783. doi: 10.1021/bi700391b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Schwieters CD, Opella SJ, Marassi FM. A practical implicit solvent potential for NMR structure calculation. J Magn Reson. 2014;243:54–64. doi: 10.1016/j.jmr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Schwieters CD, Opella SJ, Marassi FM. A Practical Implicit Membrane Potential for NMR Structure Calculations of Membrane Proteins. Biophys J. 2015 doi: 10.1016/j.bpj.2015.06.047. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang TM, Annis DS, Kronshage M, Fenno JT, Usselman LD, Mosher DF, Krukonis ES. Ail protein binds ninth type III fibronectin repeat (9FNIII) within central 120-kDa region of fibronectin to facilitate cell binding by Yersinia pestis. J Biol Chem. 2012;287 (20):16759–16767. doi: 10.1074/jbc.M112.358978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang TM, Felek S, Krukonis ES. Ail binding to fibronectin facilitates Yersinia pestis binding to host cells and Yop delivery. Infect Immun. 2010;78 (8):3358–3368. doi: 10.1128/IAI.00238-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang TM, Wiese JS, Felek S, Kronshage M, Krukonis ES. Ail proteins of Yersinia pestis and Y. pseudotuberculosis have different cell binding and invasion activities. PLoS One. 2013;8 (12):e83621. doi: 10.1371/journal.pone.0083621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versace RE, Lazaridis T. Modeling Protein-Micelle Systems in Implicit Water. J Phys Chem B. 2015 doi: 10.1021/acs.jpcb.5b00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8 (1):52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- Vriend G, Sander C. Quality control of protein models: directional atomic contact analysis. J Appl Crystallogr. 1993;26 (1):47–60. doi: 10.1107/S0021889892008240. [DOI] [Google Scholar]
- Word JM, Lovell SC, LaBean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. J Mol Biol. 1999;285 (4):1711–1733. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, Chou JJ, Wucherpfennig KW. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell. 2008;135 (4):702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, Lukacik P, Barnard TJ, Noinaj N, Felek S, Tsang TM, Krukonis ES, Hinnebusch BJ, Buchanan SK. Structural insights into Ail-mediated adhesion in Yersinia pestis. Structure. 2011;19 (11):1672–1682. doi: 10.1016/j.str.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]