

Abstract
Eukaryotic protein kinases have evolved to be highly regulated and dynamic molecular switches that are typically kept in an inactive state and then activated in response to extracellular signals. The hallmark signature of an active kinase is a hydrophobic spine called the regulatory (R) spine, which consists of four residues, two in the N-lobe and two in the C-lobe. RS1 is in the catalytic loop, RS2 is the Phe in the DFG motif, RS3 is at the C-terminus of the αC-Helix, and RS4 is at the beginning of β4. Assembly of the R-spine is typically facilitated by phosphorylation of the Activation Loop. The assembled R-spine brings together all of the functional motifs that are essential for transferring the phosphate from ATP to a tethered protein substrate. This includes the G-Loop, which anchors the ATP, the catalytic loop, the DFG motif fused to the Activation Loop, and the αC-Helix. We focus here on the properties of the αC-Helix showing 1) how residues communicate with different parts of the molecule, 2) how it is recruited to the active site as a consequence of assembling of the R-spine, and 3) how it is regulated by linkers/motifs/proteins that lie outside the conserved kinase core.
Keywords: protein kinases, cAMP-dependent protein kinase (PKA), regulatory spines (R-spines), αC-Helix
1. Introduction
The eukaryotic protein kinases have evolved to be highly dynamic and regulated molecular switches. To achieve this they have developed a set of conserved motifs that span the entire kinase core. Those motifs that are essential for catalysis were hijacked from the eukaryotic like kinases (ELKs), but other motifs were recruited to integrate regulation as a key feature [1, 2]. In general the ELKs have evolved to be efficient enzymes, and this is true for most of the metabolic enzymes. In contrast, the EPKs have evolved to be dynamic molecular switches, like the GTPases. Two new features are built into the EPKs. One is the large and dynamic Activation Loop that is typically regulated by phosphorylation and the other is the helical subdomain that follows the αF Helix and consists of the αG-αH-αI Helices [3]. These two segments are connected by a highly conserved ion pair between a Glu at the end of the Activation Loop and an Arg that follows the αH Helix[4]. This ion pair is an important conserved feature of the EPKs, and many disease susceptibility mutations lie around this region [5].
Although the conserved motifs that define the EPKs were first recognized by Hanks and Hunter when only a handful of kinase sequences were known, their careful designation of 11 subdomains based on these conserved sequence motifs has remained as a good way to assess the elements of secondary structure[6]. When the first protein kinase structure was solved[7], these subdomains could for the first time be mapped onto the EPK fold of cAMP-dependent protein kinase (PKA) and suddenly the functional importance of these motifs became more clear. The fold and the subdomain map provide a template for the entire kinome, which includes over 500 protein kinases as well as many splice variants[8]. All of the kinases have now also been aligned according to structure in the ProKinO data base so that the numbering of the motifs in any kinase can now be easily compared and also correlated with disease phenotypes [9, 10]. For our purposes we use the PKA numbering as a frame of reference.
As we look at these conserved motifs that are displayed throughout the protein kinase core, and as we solve more and more protein kinase structures, we are gaining an appreciation of the functional importance of these motifs not only for structure and function but also for activation. In addition, we are learning how these motifs are integrated in a correlated way to create a dynamic allosteric network [11]. Once again it becomes increasingly clear that the protein kinases, like the GTPases, have evolved to be highly dynamic and regulated switches. In some cases the kinase can contribute to downstream signaling without actually transferring the phosphate while in other cases transfer of the phosphate is essential [12-15]. In either case, however, these conformational switches are highly regulated. One can also no longer just go from structure to function where the end states are defined by a crystal structure. Instead we must consider dynamics as an integral intermediate step that links structure and function. There are two levels of dynamics: one relates to activation and the assembly of the active kinase [12] while the other is associated with the dynamics of the catalytic cycle[16]. To understand these dynamic processes one needs to look more deeply at the conserved motifs that were initially defined. In particular one needs to look at not only the electrostatic and hydrogen bonding residues in these motifs but also at the conservation of hydrophobic residues.
The appreciation of the hydrophobic residues and of a conserved EPK hydrophobic architecture came about several years ago when we compared active and inactive kinases and searched, in particular, for features that defined an active kinase. Through this computational analysis, we discovered an unusual mechanism for the assembly of an active kinase that involved key hydrophobic residues whose functional importance had not been previously appreciated [17]. By comparing a set of active and inactive kinases, we identified a “regulatory spine” or R-spine that is assembled in every active kinase but broken in inactive kinases[18] (Figure 1). The R-spine can be broken in several ways, but in an active kinase it will always be assembled. The R-spine consists of four hydrophobic residues (RS1-RS4) that span the N- and C-lobes of the kinase core. RS1 is in the catalytic loop, RS2 is the Phe in the DFG motif, RS3 is at the C-terminus of the αC-Helix, and RS4 is at the beginning of β4. Our recent Community Map (ComMap) analysis of the PKA catalytic (C) subunit shows how the various motifs in the active kinase are integrated into a highly dynamic unit upon the binding of ATP and magnesium [11]. The initial activation of the kinase is embedded within the assembly of the R-spine while the dynamics of the catalytic cycle are captured in the ComMaps. Our focus here will be on the activation mechanism and in particular on the αC-Helix. For this we need to define more carefully the R-spine and the residues and motifs that contribute to the assembly of the R-spine.
Figure 1. Assembly of the hydrophobic R-spine defines the fundamental architecture of an active kinase.
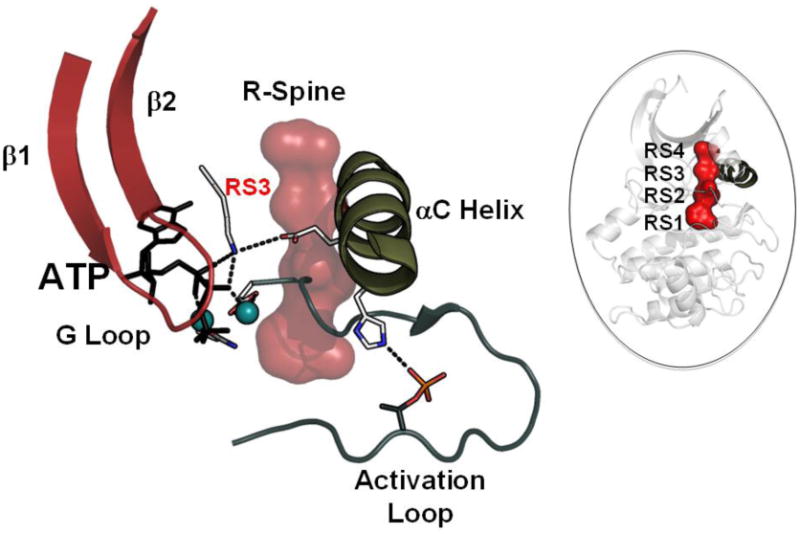
The assembly of the R-spine integrates all of the active site residues and leaves the kinase poised for catalysis. PKA is used as a prototypical kinase. We focus here on the αC Helix and why its correct alignment is essential for activation. Right. The four R-spine residues, shown here in red for PKA, are aligned in every active kinase. Left. Relationship of the αC Helix to the G-Loop and the Activation Loop. Beta strands 1 and 2 anchor the adenine ring of ATP while the G-Loop anchors the γ phosphate of ATP. The DFG motif that precedes the Activation Loop is positioned correctly at the active site when the Phe (RS2) is aligned with the rest of the R-spine. The Activation Loop phosphate is typically anchored by a basic residue at the N-terminus of the assembled αC Helix. RS3 in the αC Helix is also one of R-spine residues; it stacks directly on top of the DFG phenylalanine RS2 in the active kinase. Anchoring of the αC Helix to the R-spine is thus an essential step in the activation process. Both DFG and αC must be aligned.
2. Dynamic assembly of an active kinase is driven by the R-spine
2.1 Loops converge with the αC Helix to create the active site
The way in which the assembly of the hydrophobic R-spine defines the fundamental architecture that organizes the entire active site is summarized in Figure 1. In an active kinase two critical loops converge around the R-spine – the Glycine-rich loop and the DFG loop that is fused to the Activation Loop. The Glycine Loop (G-loop) is a unique feature of the EPKs and is actually quite distinct from the P-Loop, which is a feature of the Walker motif that is found in ATP-ases and other nucleotide binding proteins [19]. The G-Loop assumes different conformations and is sensitive to nucleotides and inhibitors. It is firmly anchored to β strands 1 and 2 and is recruited to the active site by another spine, the catalytic spine (C-spine), which is completed by the adenine ring of ATP[20]. For this review, however, we focus on the R-spine, which is dynamically assembled. In contrast to the G-Loop, the FG motif fused to the Activation Loop is highly dynamic and is typically correctly assembled only in the active kinase. In most cases assembly of the Activation Loop is facilitated by phosphorylation [21-24], and assembly of the Activation Loop leads to the correct positioning of the DFG motif. The DFG motif is actually also a loop that that bridges β8 and β9 and was initially referred to as the magnesium-positioning loop. The DFG Phe is the RS2 residue in the R-spine. The αC Helix, which also contains an R-spine residue (RS3) is also dynamically recruited to the active site by the assembly of the R-spine. Both the DFG motif and the αC Helix must be correctly aligned for the kinase to be active. Only then can a protein substrate be delivered to the bound ATP.
The assembly of an active kinase is extremely dynamic and highly regulated, and the precise mechanism for regulation is unique to each kinase. Although in terms of activation much attention has focused on the “DFG in” and “DFG-out” motifs, there are, in fact, a number of ways that the R-spine can be broken, and in many cases the “DFG in” conformation does not reflect an active conformation. Whether one describes the “DFG in” vs. the “DFG out” or the “αC in” vs. the “αC out” is irrelevant; it is the assembly of the R-spine in its entirety that must be considered. The assembly of the integrated R-spine provides a mechanistic explanation for the assembly of an active kinase. It is the underlying mechanism for every EPK although the details of the assembly of the R-spine and in particular how this process is regulated will be different for each kinase. Our focus here is on the αC helix, which corresponds to subdomain III in the Hanks and Hunter nomenclature. We focus on the highly integrative role that this helix plays where each residue has a defined function. Some residues face towards the conserved core and contribute to the integration of the active site where phosphoryl transfer takes place while others are anchored to the phosphorylated activation loop. The surface that is most highly regulated, however, interacts with the tails and linkers and other domains that flank the kinase core and in some cases like cdk2 with another subunit, cyclin. For our comparative analysis of the αC Helix we use several different kinases as examples: PKA, ERK2, cdk2, Src, IRK and BRaf.
2.2 Active and inactive kinases
By superimposing several kinases in their active and inactive states, Figure 2 emphasizes not only the conserved features of the active kinases, but also the highly dynamic nature of the Activation Loop and the R-spine in the inactive kinases. In the active kinases on the left the R-spine is assembled and the Activation Loop is in a stable conformation. In this state there is very close communication between the αC Helix and the Activation Loop as highlighted by the arrow. On the right in Figure 2 are the superimposed inactive kinases. In Figure 3 where the active and inactive kinases are displayed separately one can better appreciate how the assembly and disassembly of the R-spine is quite different for each kinase. In the inactive kinases the Activation Loops are highly disordered and very different, and the R-spine is disassembled differently in each kinase. The αC Helix is typically in an “out” conformation and in some cases the αC Helix is even partially disordered. In all cases communication between the αC Helix and the Activation Loop is broken.
Figure 2. Assembly of the active kinase is a highly dynamic process.
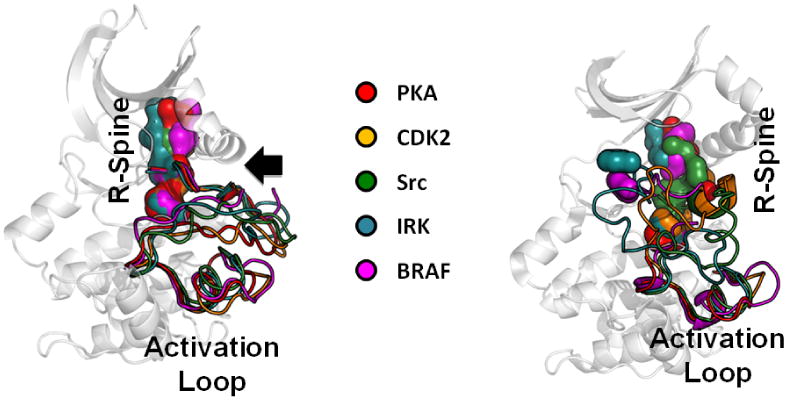
A comparison of active (right) and inactive (left) kinases emphasizes the highly dynamic properties of the EPK family. By superimposing a set of active and inactive kinases one can not only appreciate how dynamic the activation loop is in the absence of phosphorylation but also how different the kinases are from each other. This highly dynamic and regulated Activation Loop is one of the unique features that distinguishes the EPKs from the ELKs. The R-spines are also broken in different ways.
Figure 3. Assembly and disassembly of the R-Spine drives the activation of the kinase.
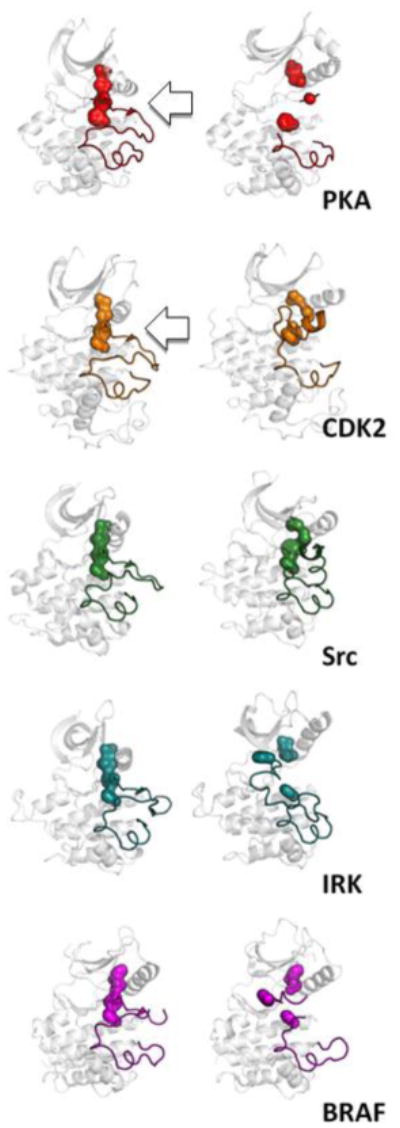
Although in all cases, the R-spine must be correctly aligned for the kinase to be fully active, the R-spine can be broken in different ways. Five different examples demonstrate the diverse ways in which the R-spine can be broken. In each case the active kinase is on the left (PKA PDBID:1ATP, CDK2 PDBID:1E9H, Src PDBID:1Y57, IRK PDBID:1IR3, BRAF PDBID:4E26) and the inactive kinase where the spine is broken is on the right (PKA PDBID:4DFY, CDK2 PDBID:1B39, Src PDBID:2SRC, IRK PDBID:1IRK, BRAF PDBID:1UWH).
Although we have many protein kinase structures, most are bound to inhibitors or are inactive. In only relatively few cases do we have a complete understanding of the interface between the αC Helix and the Activation Loop in an active kinase let alone in an inactive kinase where there is often considerable disorder. Most likely this disorder is enhanced because we usually have structures of only the kinase domain. In many cases it is likely that the inactive state could actually be very stable due to interactions with other domains or proteins that are missing when one sees only the kinase domain. We use PKA and cdk2 as examples of kinases here we have a good understanding of both the active and the inactive conformations and focus in particular how the αC Helix is engaged with the kinase core as well as with regions that lie outside the kinase core. In the case of PKA this includes the two tails (C-tail and N-tail) that are fused to the two ends of the kinase core, while in Cdk2 the same space is filled by cyclin. Although many features, in particular around the catalytic cleft of all the active kinases, are shared and conserved, the comparison of PKA and cdk2 also highlights the unique features of each kinase and how they are regulated by motifs that lie outside the conserved core.
2.3 Regulated assembly of the R-spine can be hijacked by disease-driving mutations
The assembly of the R-spine is typically initiated by some activation event that unleashes the inhibited kinase and allows it to toggle between active and inactive conformations or between the assembled and disassembled R-spine[15]. Once unleashed the stable spine can be assembled and/or stabilized by a variety of events that include dimerization and activation loop phosphorylation as well as other complex phosphorylation events that lie outside the kinase core. An analysis of the hydrophobic space around the R-spine shows a set of “shell” residues. The spine residues are referred to as RS1, RS2 and RS3. RS2, while the shell residues are referred to as Sh1, Sh2, and Sh3. Sh2 is also known as the gatekeeper (M116 in PKA) while Sh3 Met120 in PKA) lies just beyond the gatekeeper and is part of the linker that joins the N-and C-Lobes. Sh1 (Val104 in PKA) lies just before RS4 in the αC-β4. The role of hydrophobicity in this region of the core was mapped[25], and using BRaf as a model system for exploring the assembly of the R-spine, we discovered a number of single hydrophobic mutations that could drive the assembly of the spine and create a constitutively active kinase[13]. Many of these mutations are found in mutant kinases in cancer[12]. The spine architecture, coupled with the assembly of the R-spine and the community map[11], provide a mechanistic understanding of how mutations far from the active site can drive the constitutive assembly of an active kinase that is no longer regulated. Here we focus specifically on the multi-facetted functions of the αC Helix.
3. Anatomy of the αC Helix
The αC Helix can be thought of as a contiguous set of wheels (or as a screw) where every cog in the wheel reaches out and touches a different part of the molecule. While we can analyze the interactions that each spoke makes, it is the integration that is achieved by the entire helix wheel when it is correctly aligned that needs to be understood. Simply analyzing the primary and secondary and tertiary interactions is not sufficient to provide this dynamic portrait. Neither is crystallography where we can hopefully trap relevant static structures in a lattice. This tells us little about dynamics, and often the biologically important segments are disordered and/or removed to provide higher resolution. NMR provides us with a dynamic residue-specific profile but this needs to be integrated with the other information [16]. Small angle X-ray and neutron scattering can also provide a profile of protein dynamics in solution but this is low resolution. In an effort to understand and quantify these dynamic features we used a community analysis method to study different states of the PKA catalytic subunit (Apo/open, ATPMg/open, ATPMg/closed, and ATPMg2/closed). This analysis shows dramatically how the αC Helix nucleates communication with the entire molecule and provides a plausible explanation for why mutations that lie far from the active site can disrupt the entire functioning kinase [11]. Here we focus just on the properties of the αC Helix that allow it to play such a central role.
3.1 Functional motifs in the αC Helix
One can appreciate the complexity of the αC Helix by simply walking down the helix or by viewing it as a wheel with each spoke of the wheel having a distinct and well-defined function. To appreciate the dynamic coupling of the αC Helix with the rest of the protein, hydrophobic, charged, and hydrogen bonding contributions all need to be considered. To emphasize the diversity of the functions that are integrated by the αC-Helix we initially highlight three residues that are at the beginning, middle and end of the helix (Figure 4). The αC Helix contains one highly conserved charged residue, Glu91, that binds to a conserved residue in β strand 3, Lys72[26]. This ion pair serves as a signature motif for an active kinase. Without either of these residues, Glu91 and Lys72, the kinase cannot support steady state catalysis and until recently this has been sufficient to classify the kinase as a pseudokinase. Glu91 is in the middle of the αC Helix, and its function is conserved in all kinases. At the C-terminus of the αC Helix is the RS3 spine residue (Leu95 in PKA), and this is also functionally conserved in all kinases although the nature of the hydrophobic residue can vary. Having a Leu or Val, for example, at this position, which is typical, provides for more flexibility, while introducing a stronger hydrophobic residue such as Phe, Ile, or Met can drive the constitutive assembly of the R-spine and this is a mutation that can be found in cancer genes [12]. The N-terminus of the αC-Helix interacts typically with the activation loop. Often, as in the case of PKA and cdk2, there is a basic residue here (His87 for PKA) that interacts with a phosphate in the Activation Loop when the kinase is in a closed conformation. It is not clear whether this drives the assembly of the active kinase or stabilizes the already assembled R-spine, but kinase activity is usually enhanced when the Activation Loop is phosphorylated[27]. However, docking of a basic residue at the N-terminus of the αC Helix to the Activation Loop phosphate is a critical anchor point that nucleates communication across the entire protein [11]. There are some kinases that are not phosphorylated on the activation loop, and in these cases the nature of the interaction of the N-terminus of the C-Helix with the Activation loop is different. Figure 4 not only shows how the assembled R-spine interacts with different parts of the protein but also shows how the αC Helix brings the G-Loop in close proximity to the Activation Loop at the active site cleft. Assembly of the R-spine drags both the DFG motif and the αC Helix into their correct active alignment so that the entire molecule is poised to transfer the phosphoryl moiety from ATP to the protein substrate.
Figure 4. Motifs that define the αC-Helix.
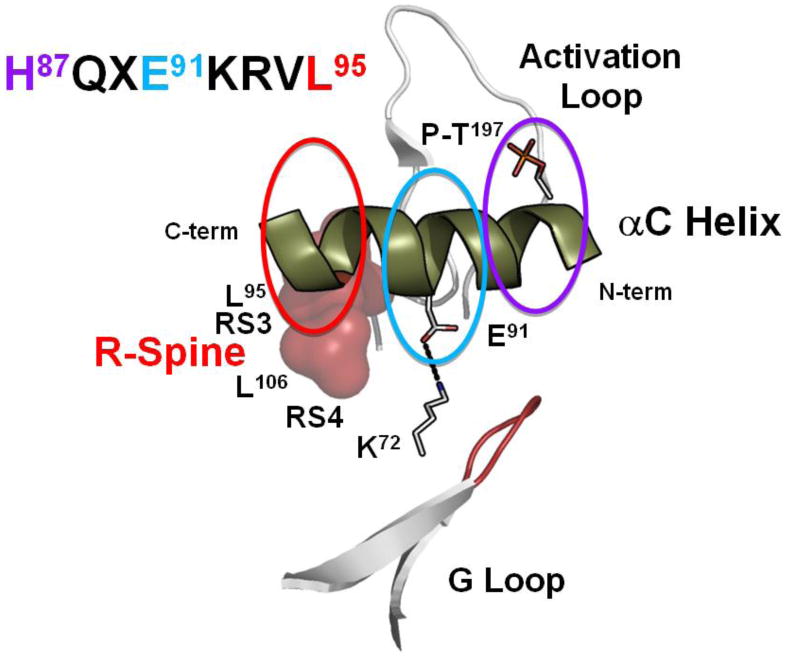
The extended αC Helix allows one to appreciate how essential residues are spread across the entire helix. While every residue is functionally important three specific features are highlighted here. The sequence of the αC Helix is shown on the top left with three key residues highlighted in red, teal and purple. RS3/L95 (red) lies at the C-terminus, the conserved salt bridge to Lys72, Glu91 (teal), is in the middle, and His87 (purple) at the N-terminus is anchored to the activation loop phosphate, (P)-Thr197.
The top panel in Figure 5 shows how the αC Helix is anchored to the R-spine by its RS3 residue in five different active protein kinases. Rotating the αC Helix 90° shows how the RS3 and RS4 spine residues interact in the assembled spine and also how another highly conserved loop, the αC-β4 Loop, is formed. In contrast to the G-Loop and the DFG motif, the αC-β4 loop is extremely stable. This combined segment extending from the beginning of the αC Helix to RS4 at the beginning of β4 can be thought of as the communication “hub” of the any active protein kinase. These residues not only communicate with the active site of the kinase core, they also interact with the tails and linkers that are fused to the kinase core and often they interact with other molecules or domains. These interfaces that are formed and broken as the kinase shuttles between its active and inactive conformations are poorly understood but extremely important.
Figure 5. Integration of the R-spine with the core is conserved in all kinases.
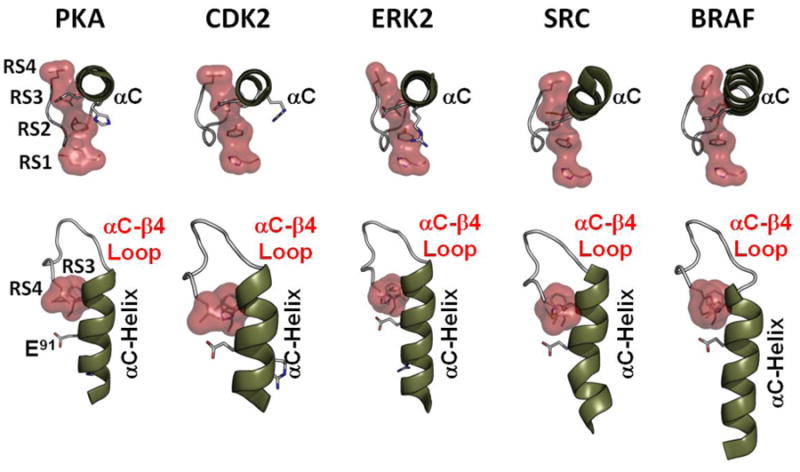
The R-spine residues (Tyr164/RS1, Phe184/RS1. Leu95/RS3, and Leu106/RS4) are indicated as red shells. The top panel shows how all five kinases are anchored to the spine through their RS3 residue that is at the C-terminus of the αC helix. The bottom panel shows how RS3 and RS4 are coupled at the C-terminal end of the αC Helix. The αC-β4 Loop is another conserved loop that is very stable in both the active and inactive kinases.
3.2 Surfaces of the αC-Helix
To highlight the different surfaces of the αC Helix we compare two well-characterized protein kinases, PKA and cdk2. Figure 6 shows the classical helix wheel for PKA and cdk2 where every residue is shown. Also highlighted are the interactions that are mediated by different surfaces of the wheel. The N-terminus, for example, facilitates interactions with the Activation Loop in the active kinase, and this communication is usually completely missing in the inactive kinase when the Activation Loop is disordered or ordered differently. The surface that includes the conserved glutamic acid faces the active site cleft and is always anchored in every active kinase to the conserved Lys in β3. The surface at the C-terminus that follows RS3 typically interacts with regions that lie outside the conserved core and these interactions are unique for every kinase. In Figure 7 we show just the key residues that nucleate interactions with different parts of the kinase using PKA as a prototypical kinase. This includes interaction of His87 with the activation Loop, the K72-E91 ion pair, and in particular the residues that follow RS3 and mediate interactions with non-core parts of the protein. In addition to being well characterized in their different structural states, PKA and cdk2 are complete structures, not just a kinase core. In the case of PKA the full length kinase includes a C-terminal and an N-terminal tail and deletion of either tail is inactivating. The C-Tail in PKA wraps around both the C-Lobe and the N-Lobe of PKA and is conserved in all AGC kinases[28]. The N-Tail in PKA consists of a long amphipathic helix that is also anchored to both lobes of the kinase core. It is not conserved in other AGC kinases but is essential for the mammalian PKA C-subunit[29]. Mutation of specific residues in either tail is also inactivating. In the case of cdk2, the active kinase is assembled by association with a cyclin[30] and it is the cyclin that stabilizes the active conformation. These two kinases demonstrate why the surfaces of the αC Helix that interact with segments that lie outside the core are essential for regulation of the kinase. Our understanding of these surfaces in most other kinases is incomplete.
Figure 6. Functional motifs are embedded along the entire length of the αC Helix.
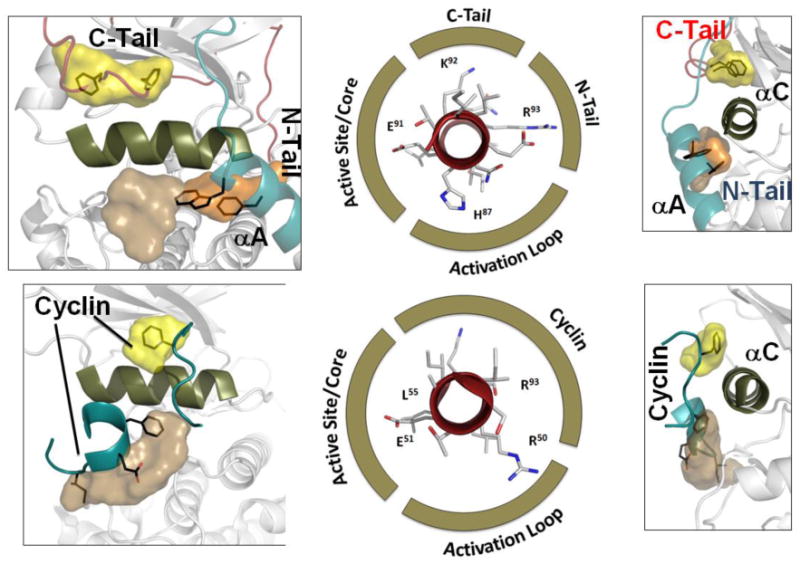
Each residue along the helix wheel has a specific binding partner/surface. While Glu91 goes to Lys72 in β3 and brings together the ATP binding motif with the catalytic machinery enabled by Asp184, His87 points toward the activation loop phosphate. In contrast to these surfaces which bridge to the core, the outer surface of the αC Helix is positioned by residues that lie outside the conserved core or by other proteins or domains that bind to this region. Two such pockets on the outer surface of the αC Helix were identified in a computational screen. These pockets are indicated as yellow and tan surfaces. While these pockets are conserved in all EPKs, the chemical nature of each pocket differs. In the case of PKA and other AGC kinases, one of the pockets (yellow) is filled by the hydrophobic FXXF motif that likes at the end of the C-Tail. A Trp in the N-terminal αA Helix partially fills the other pocket. In contrast, the activating cyclin fills both pockets in cdk2 emphasizing the diversity of each kinase. The critical residues that interact with the activation loop phosphate, the K72, and the outer surface are highlighted in the helix wheel in the center.
Figure 7. Complimentary surfaces in the αC Helix interact with both the core and with linkers, tails, and other segments that lie outside the conserved core.
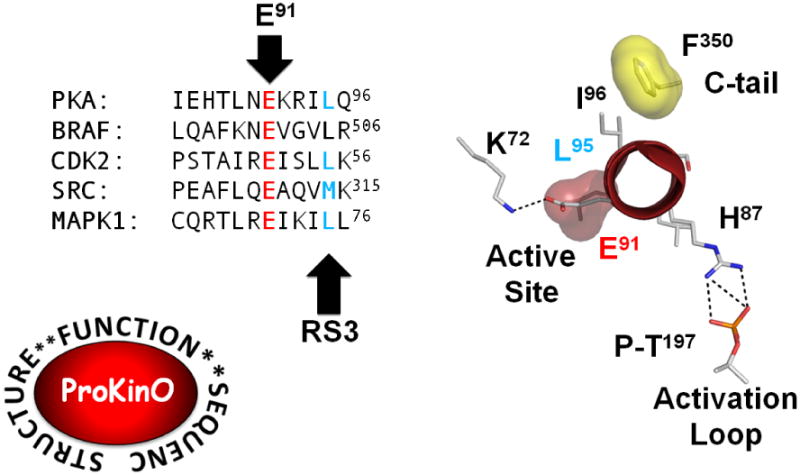
The sequences of the αC Helix for the kinases shown in Figure 2 are aligned here. The conserved Glu is in red and the conserved RS3 R-spine residue is shown in teal. Bot tresidues are also highlighted by an arrow. The three key residues/region hi-lighted in Figure 4 are also displayed here in a helical wheel presentation. The conserved interaction of Glu91 with Lys72 shows how one surface faces towards the active site and serves to integrate conserved motifs that are important for catalysis. In contrast His87 interacts with the Activation Loop phosphate. On the top is shown the docking site for the hydrophobic HF motif at the end of the αC Helix. The αA Helix, in particular Trp30 (not shown in the figure), is wedged tightly between the αC Helix and the activation loop. These two interactions involved regions that lie outside the conserved kinase core but are nevertheless essential for activity.
3.3 Conserved pockets on the kinase core
A comparative analysis of the surface of the conserved kinase core revealed a set of very well defined pockets that are distributed all around the surface [31]. These can be thought of as “allosteric” pockets. The chemical nature of these allosteric pockets is kinase-specific but the location of each pocket is conserved. Many of these pockets are filled by well-defined motifs in different kinases; however, a few pockets were defined as “orphans” as there function was not known. Two pockets in particular are associated with the αC Helix. One lies on the top of the αC Helix (shown in yellow), and the other lies near the bottom (shown in tan) in the orientation of the helix that is shown in Figure 6. In this orientation the αC Helix is shown in an N- to C- orientation in contrast to Figure 4 where the orientation is C- to N. Using PKA and cdk2 as examples, we can see that these surfaces are occupied by segments that lie outside the conserved core.
In PKA the yellow pocket is filled by the hydrophobic motif (FXXF) that is at the end of the C-Tail and is a conserved feature of all AGC kinases[28]. This same pocket is filled by cyclin in the cdk2:cyclinD complex. The tan pocket is filled in part by W30 and F26 in PKA. These residues are at the end of the αA helix and are wedged between the αC Helix and the Activation Loop. Mutation of W30 to Ala is inactivating[29]. In cdk2 this pocket is again filled by the cyclin which functions as a activating regulatory subunit. The two panels on the right show clearly the surface of the αC Helix that is complimented by segments that lie outside the conserved kinase core, and this surface is still very poorly understood for most protein kinases.
3.4. Core and Non-core segments interact with and regulate the αC Helix
The sequences of the αC Helix is compared in five different kinases in Figure 7. The ProKinO data base provides a vert rapid way to align any kinase based of the structure alignment of functionally conserved residues. This allows one not only to rapidly compare any kinase but also to search for disease related mutations in specific regions. By analogy with PKA one can see how a few specific residues brig together conserved residues at the active site cleft. The surface including E91 and H87 interact in a conserved way with the active site and with the activation loop and thee interations are more-or-less conserved in every kinase when the R-spien is assembled. The other surfaces, however, that lie beyond RS3 (L95) interact with residues that oie outside the conserved core. In some cases like PKA these surfaces are bound to the C- and N-terminal tails, while in other kinase like cdk2 these surfaces are filled by cyclin. In both cases, however, the αC Helix is regulated by segments that lie outside the core. In most cases we do not know how these surfaces are filled In either the inactive or in the active kinase.
4. Assembly of the R-spine
Assembly of the spine is typically initiated by some activation event that unleashes the inhibited kinase and allows it to toggle between active and inactive conformations or between the assembled and disassembled R-spine[15]. Once unleashed the stable spine is assembled by a variety of events that include dimerization and activation loop phosphorylation as well as other complex phosphorylation events that lie outside the kinase core. An analysis of the hydrophobic space around the R-spine shows a set of “shell” residues. The spine residues are referred to as RS1, RS2 and RS3. RS2, while the shell residues are referred to as Sh1, Sh2, and Sh3. Sh2 is also known as the gatekeeper while Sh3 lies just beyond the gatekeeper and is part of the linker that joins the N-and C-Lobes. The role of hydrophobicity in this region of the core was mapped[25], and using BRaf as a model system for exploring the assembly of the R-spine, we discovered a number of single hydrophobic mutations that could drive the assembly of the spine and create a constitutively active kinase[13]. Many of these mutations are found in mutant kinases in cancer[12]. The spine architecture, coupled with the assembly of the R-spine and the Community Map[11], provide a mechanistic understanding of how mutations far from the active site can drive the constitutive assembly of an active kinase that is no longer regulated. Here we have focused specifically on the multi-facetted functions of the αC Helix.
5. Conclusions/Challenges
Because the protein kinases are associated with so many diseases, especially cancers, they have become major therapeutic targets. While correlation of diseases with particular kinase genes is well known, a major challenge is to achieve specificity. Most inhibitors target the active site and in particular the ATP binding pocket, but as seen here, the ATP pocket is well conserved in the family making it difficult to achieve strict specificity. Another problem is the acquisition of resistance mutations. Many of these mutations are related to the R-spine and help to stabilize the active kinase. Cancer mutations. Allosteric sites.
Acknowledgments
We would like to acknowledge support from NIH to SST (GM19301) and from the Howard Hughes Medical Institute to SST and ASS. We are especially indebted to the students and fellows in our laboratories who have helped to provide experimental validation for many of the concepts that we have tried to summarize here. In particular we acknowledge Jiancheng Hu in the Shaw lab and Hiruy Meharena, Chris McClendon, and Lalima Ahula in the Taylor lab.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kannan N, Neuwald AF. Did protein kinase regulatory mechanisms evolve through elaboration of a simple structural component? J Mol Biol. 2005;351:956–972. doi: 10.1016/j.jmb.2005.06.057. [DOI] [PubMed] [Google Scholar]
- 2.Taylor SS, Keshwani MM, Steichen JM, Kornev AP. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos T R Soc B. 2012;367:2517–2528. doi: 10.1098/rstb.2012.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor SS, Kornev AP. PKA: Prototype for Dynamic Signaling in Time and Space. In: Wall ME, editor. Quantitative Biology: From Molecular to Cellular Systems. CRC Press; Boca Raton, London, New York: 2012. pp. 267–298. [Google Scholar]
- 4.Yang J, Wu J, Steichen JM, Kornev AP, Deal MS, Li S, Sankaran B, Woods VL, Jr, Taylor SS. A conserved glu-arg salt bridge connects coevolved motifs that define the eukaryotic protein kinase fold. J Mol Biol. 2012;415:666–679. doi: 10.1016/j.jmb.2011.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torkamani A, Kannan N, Taylor SS, Schork NJ. Congenital disease SNPs target lineage specific structural elements in protein kinases. Proc Natl Acad Sci U S A. 2008;105:9011–9016. doi: 10.1073/pnas.0802403105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 7.Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 8.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 9.Gosal G, Kochut KJ, Kannan N. ProKinO: an ontology for integrative analysis of protein kinases in cancer. PLoS One. 2011;6:e28782. doi: 10.1371/journal.pone.0028782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McSkimming DI, Dastgheib S, Talevich E, Narayanan A, Katiyar S, Taylor SS, Kochut K, Kannan N. ProKinO: A Unified Resource for Mining the Cancer Kinome. Hum Mutat. 2015;36:175–186. doi: 10.1002/humu.22726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McClendon CL, Kornev AP, Gilson MK, Taylor SS. Dynamic architecture of a protein kinase. Proc Natl Acad Sci U S A. 2014;111:E4623–4631. doi: 10.1073/pnas.1418402111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu J, Ahuja LG, Meharena HS, Kannan N, Kornev AP, Taylor SS, Shaw AS. Kinase regulation by hydrophobic spine assembly in cancer. Mol Cell Biol. 2015;35:264–276. doi: 10.1128/MCB.00943-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJ, Kornev AP, Taylor SS, Shaw AS. Allosteric Activation of Functionally Asymmetric RAF Kinase Dimers. Cell. 2013;154:1036–1046. doi: 10.1016/j.cell.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J, Yu H, Kornev AP, Zhao J, Filbert EL, Taylor SS, Shaw AS. Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc Natl Acad Sci U S A. 2011;108:6067–6072. doi: 10.1073/pnas.1102554108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaw AS, Kornev AP, Hu J, Ahuja LG, Taylor SS. Kinases and pseudokinases: lessons from RAF. Mol Cell Biol. 2014;34:1538–1546. doi: 10.1128/MCB.00057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masterson LR, Cheng C, Yu T, Tonelli M, Kornev A, Taylor SS, Veglia G. Dynamics connect substrate recognition to catalysis in protein kinase A. Nat Chem Biol. 2010;6:821–828. doi: 10.1038/nchembio.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kornev AP, Haste NM, Taylor SS, Ten Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. P Natl Acad Sci USA. 2006;103:17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proc Natl Acad Sci U S A. 2008;105:14377–14382. doi: 10.1073/pnas.0807988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson LN, Noble ME, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 22.Johnson LN, Lewis RJ. Structural basis for control by phosphorylation. Chemical Reviews. 2001;101:2209–2242. doi: 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- 23.Johnson DA, Akamine P, Radzio-Andzelm E, Madhusudan M, Taylor SS. Dynamics of cAMP-dependent protein kinase. Chem Rev. 2001;101:2243–2270. doi: 10.1021/cr000226k. [DOI] [PubMed] [Google Scholar]
- 24.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 25.Meharena HS, Chang P, Keshwani MM, Oruganty K, Nene AK, Kannan N, Taylor SS, Kornev AP. Deciphering the structural basis of eukaryotic protein kinase regulation. PLoS Biol. 2013;11:e1001680. doi: 10.1371/journal.pbio.1001680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zoller MJ, Nelson NC, Taylor SS. Affinity labeling of cAMP-dependent protein kinase with p-fluorosulfonylbenzoyl adenosine. Covalent modification of lysine 71. J Biol Chem. 1981;256:10837–10842. [PubMed] [Google Scholar]
- 27.Steichen JM, Iyer GH, Li S, Saldanha SA, Deal MS, Woods VL, Jr, Taylor SS. Global consequences of activation loop phosphorylation on protein kinase A. J Biol Chem. 2010;285:3825–3832. doi: 10.1074/jbc.M109.061820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kannan N, Haste N, Taylor SS, Neuwald AF. The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proc Natl Acad Sci U S A. 2007;104:1272–1277. doi: 10.1073/pnas.0610251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herberg FW, Zimmermann B, McGlone M, Taylor SS. Importance of the A-helix of the catalytic subunit of cAMP-dependent protein kinase for stability and for orienting subdomains at the cleft interface. Protein Sci. 1997;6:569–579. doi: 10.1002/pro.5560060306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 31.Thompson EE, Kornev AP, Kannan N, Kim C, Ten Eyck LF, Taylor SS. Comparative surface geometry of the protein kinase family. Protein Sci. 2009;18:2016–2026. doi: 10.1002/pro.209. [DOI] [PMC free article] [PubMed] [Google Scholar]