

Abstract
Rationale
Cardiac resynchronization therapy (CRT) is the only heart failure (HF) therapy documented to improve left ventricular (LV) function and reduce mortality. The underlying mechanisms are incompletely understood. While β-adrenergic signaling has been studied extensively, the effect of CRT on cholinergic signaling is unexplored.
Objective
We hypothesized that remodeling of cholinergic signaling plays an important role in the aberrant calcium signaling and depressed contractile and β-adrenergic responsiveness in dyssynchronous HF (DHF) that are restored by CRT.
Methods and Results
Canine tachypaced DHF and CRT models were generated to interrogate responses specific to dyssynchronous vs. resynchronized ventricular contraction during hemodynamic decompensation. Echocardiographic, electrocardiographic and invasive hemodynamic data were collected from normal controls, DHF and CRT models. LV tissue was used for biochemical analyses and functional measurements (calcium transient, sarcomere shortening) from isolated myocytes (N=42–104 myocytes/model; 6–9 hearts/model). Human LV myocardium was obtained for biochemical analyses from explanted failing (N=18) and non-failing (N=7) hearts. The M2 subtype of muscarinic acetylcholine receptors (M2-mAChR) was upregulated in human and canine HF compared to non-failing controls. CRT attenuated the increased M2-mAChR expression and Gαi-coupling, and enhanced M3-mAChR expression in association with enhanced calcium cycling, sarcomere shortening and β-adrenergic responsiveness. Despite model-dependent remodeling, cholinergic stimulation completely abolished isoproterenol-induced triggered activity in both DHF and CRT myocytes.
Conclusions
Remodeling of cholinergic signaling is a critical pathological component of human and canine HF. Differential remodeling of cholinergic signaling represents a novel mechanism for enhancing sympathovagal balance with CRT and may identify new targets for treatment of systolic HF.
Keywords: Parasympathetic, muscarinic receptor, acetylcholine, sympathetic, autonomic nervous system, cardiac resynchronization therapy, heart failure, arrhythmia (mechanisms), vagal stimulation
INTRODUCTION
Heart failure (HF) is a leading cause of death worldwide. Despite contemporary medical advances, about half of HF patients die within five years of diagnosis1. Pharmacological approaches improve HF symptoms and delay mortality but do not reverse disease progression. Cardiac resynchronization therapy (CRT) is the only approach documented to improve left ventricular (LV) function and reduce mortality2–4, albeit by mechanisms that are incompletely understood5. Improving this understanding may help us improve upon the response to CRT and identify new therapeutic targets that extend the benefits of CRT to a wider HF population.
CRT has salutary effects beyond restoration of electromechanical synchrony that involve remodeling of β-adrenergic signaling pathways and restoring sympathovagal balance6–8. While mechanisms leading to depressed β-adrenergic signaling have been studied extensively, far less is known about concurrent functional alterations in cholinergic (parasympathetic/muscarinic) signaling or its role in the HF phenotype9. Evidence from animal models10, 11 and ongoing clinical trials12–14 suggests modulating cholinergic activity and restoring sympathovagal balance has salutary effects in HF, but the underlying molecular mechanisms have not been established9. The effect of CRT on cholinergic signaling is unexplored.
Because β-adrenergic and cholinergic signaling pathways are intimately coupled, we hypothesized that remodeling of cholinergic signaling plays an important role in the aberrant calcium signaling and depressed contractile responses to β-adrenergic stimulation in dyssynchronous HF (DHF) that are restored by CRT.
METHODS
We studied three canine models: (1) normal controls (N=8); (2) DHF (N=10), which were first subjected to ablation of the left bundle branch and then, 6 weeks of right atrial (RA) tachypacing at 200 beats per minute; (3) CRT (N=10), which was developed as DHF for the first 3 weeks followed by biventricular tachypacing (LV lateral and RV anteroapical epicardium) for the next 3 weeks. Echocardiography, electrocardiography (ECG) and tissue Doppler (longitudinal strain speckle tracking with four-chamber views) were performed at 3 and 6 weeks to assess dyssynchrony (variance of peak systolic strain timing) as previously described6. The ECGs and accuracy of automated detection of RR and QT intervals were manually reviewed with the aid of a graphical display using applications written in Matlab (MathWorks, Inc, Natick, MA). Heart rate variability (HRV) in the time domain was calculated as the standard deviation of NN intervals (SDNN) as previously described15. At terminal study, dogs were anesthetized with pentobarbital, pacing was suspended, and a micromanometer (Millar Instruments Inc.) was advanced into the LV to record pressure. The chest was opened and hearts were rapidly removed under cold cardioplegia. The mid-myocardial layer from the LV lateral wall, i.e., region between the left anterior descending and circumflex arteries, was frozen for tissue analysis or perfused for myocyte isolation as previously described6, 8.
Freshly isolated myocytes were loaded with the ratiometric calcium indicator, Indo-1 AM (Life Technologies, Grand Island, NY) at room temperature. Myocyte sarcomere shortening (SS) and whole-cell calcium transients (CaT) were assessed with an inverted Olympus microscope equipped with fluorescence imaging (MyoCam, IonOptix) using different solution exposure protocols (Online Fig. I). Functional studies were performed at 37°C with field stimulation at 1 Hz, as previously described6. Calcium recordings and sarcomere shortening were assessed at 0.5 and 2 Hz, and with 2 mM extracellular calcium, and yielded concordant findings. Isoproterenol (Iso), carbamylcholine (CCh), pirenzapine (M1-mAChR antagonist), 4-DAMP (M3-mAChR antagonist), atropine and pertussis toxin (PTX) were purchased from Sigma-Aldrich Inc. (St. Louis, MO). J 104129 fumarate, an antagonist with higher specificity than 4-DAMP for M3-mAChR, was purchased from Tocris Bioscience (Bristol, United Kingdom) to confirm findings in some experiments with 4-DAMP.
Myocardium was homogenized in lysis buffer (Cell Signaling Technology), and 50 to 100 μg of total protein were loaded for gel electrophoresis as previously described8. mRNA expression was assessed by Taqman real-time polymerase chain reaction (PCR) using the Path-ID Multiplex One Step RT-PCR kit (Applied Biosystems) as previously described8. The canine-specific primer and probe sequences for M2-mAChR were as follows: cCHRM2-F (GGACAATTGGTTATTGGCTTTGTTA), cCHRM2-R (GTGGCGTTACAAAGTGCATAGC), cCHRM2-T (ATCAACAGCACCATCAATCCCGCC).
The antibodies used for Western blots were: M2-mAChR (Sigma M9558 1:1500 for canine samples and Millipore Ab5166 1:1500 for human samples), M3-mAChR Santa Cruz sc-9108 1:1200), Gαq/11 (Santa Cruz sc 392 1:250 dilution) and Cx43 (Chemicon mab3068 1:1000). The same antibodies for M2 and Cx43 were used in immunostaining at dilutions of 1:100 and 1:50, respectively. Western blot analysis from LV myocardial samples have been previously reported6, 8. LV tissue was obtained from explanted failing and non-failing hearts deemed unsuitable for transplantation after approval from the Institutional Review Boards at the Johns Hopkins University and University of Munich, Germany. Immunohistochemistry analysis was performed and representative sections were selected by a senior pathologist not involved in the study and blinded to the canine model groups.
Fractional changes are presented as mean±SEM and compared using a paired t-test. Categorical comparisons were performed using a chi-square test. Absolute values are reported in box and whisker plots (mean, median, interquartile range, minimum and maximum). Comparisons between multiple experimental groups were performed by one-way analysis of variance (ANOVA) and a Tukey test. In vivo data at multiple time points were analyzed by repeated-measures ANOVA.
RESULTS
The canine DHF model6–8 was generated by left bundle branch ablation to disrupt synchronous activation combined with rapid RA pacing for six weeks. CRT was produced by switching to biventricular tachypacing from weeks 4–6. Because both models were tachypaced, they developed similar global LV dysfunction (Fig. 1a) and were designed to interrogate responses specific to dyssynchronous vs. resynchronized ventricular contraction during hemodynamic decompensation. Compared to non-HF controls, DHF animals demonstrated significant regional dyssynchrony of LV shortening and depressed ejection fraction, stroke volume, dP/dtmax, and heart rate variability (Fig. 1b–h). These hemodynamic and electrophysiological changes were significantly improved by CRT.
Figure 1. Echocardiographic, hemodynamic and electrophysiological characteristics of normal, DHF and CRT animals before sacrifice at 6 weeks.
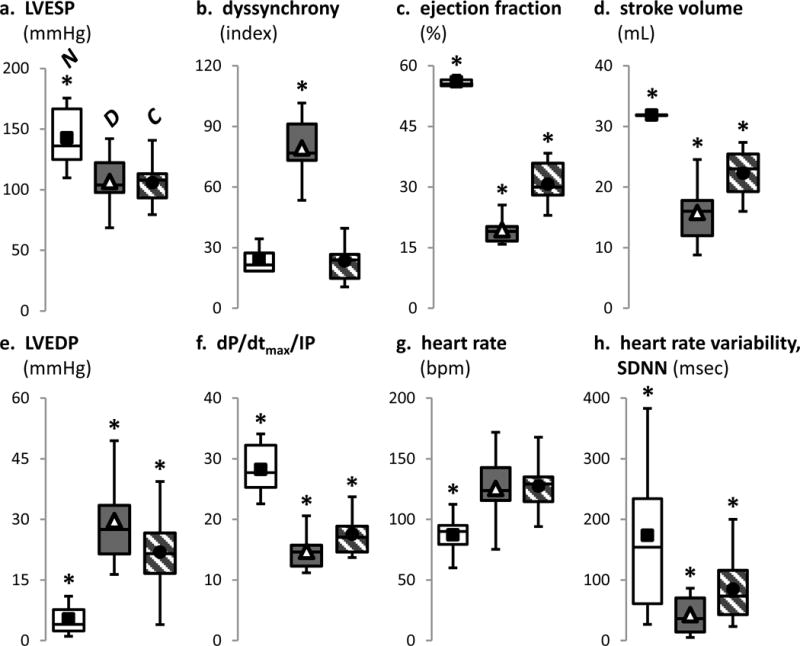
Invasive pressure measurements revealed LV end-systolic pressures (panel a) were similar between DHF (N=10) and CRT (N=10), but both were reduced compared to normal controls (N=8). Regional LV longitudinal strain (b) derived from echocardiographic speckle tracking analysis revealed similar, simultaneous strains in all regions for normal controls, but in DHF, septal shortening preceded the lateral wall with reciprocal septal stretch when the lateral wall contracted; restoration of synchrony was observed with CRT. Echocardiography-derived ejection fractions (c) and stroke volumes (d) were decreased in DHF and improved by CRT, but both remained lower than normal controls. Invasive pressure measurements revealed higher LV end-diastolic pressures (e) and lower dP/dtmax [normalized to instantaneous developed pressure (IP)] (f) in DHF compared to CRT and normal controls. Despite a similar increase in heart rate (g) in DHF and CRT, the heart rate variability (h) was significantly lower in DHF compared to normal and CRT.
In all panels of this figure, *p<0.01 vs. all other groups.
We hypothesized that during tonic β-adrenergic stimulation, acute cholinergic stimulation suppresses CaT and SS in DHF more than in normal or CRT. In the continued presence of isoproterenol (Iso), cholinergic stimulation with carbamylcholine (CCh) markedly depressed peak CaT and SS amplitudes in DHF myocytes by 59% and 74%, respectively. These responses were only modestly diminished in normal controls and even less so in CRT (Fig. 2a–b, Online Fig. IIa). Reversal of depression by atropine indicated an mAChR-specific effect16, 17. Some DHF cells were so sensitive to cholinergic stimulation that contraction was arrested despite continued isoproterenol exposure, and full restoration of the CaT and contraction required atropine (Supplementary Video I). Cholinergic stimulation also prolonged the CaT and SS more in DHF than in normal or CRT myocytes (Fig. 2a, c, Online Fig. IIb).
Figure 2. Response to cholinergic stimulation in the setting of tonic β-adrenergic stimulation.
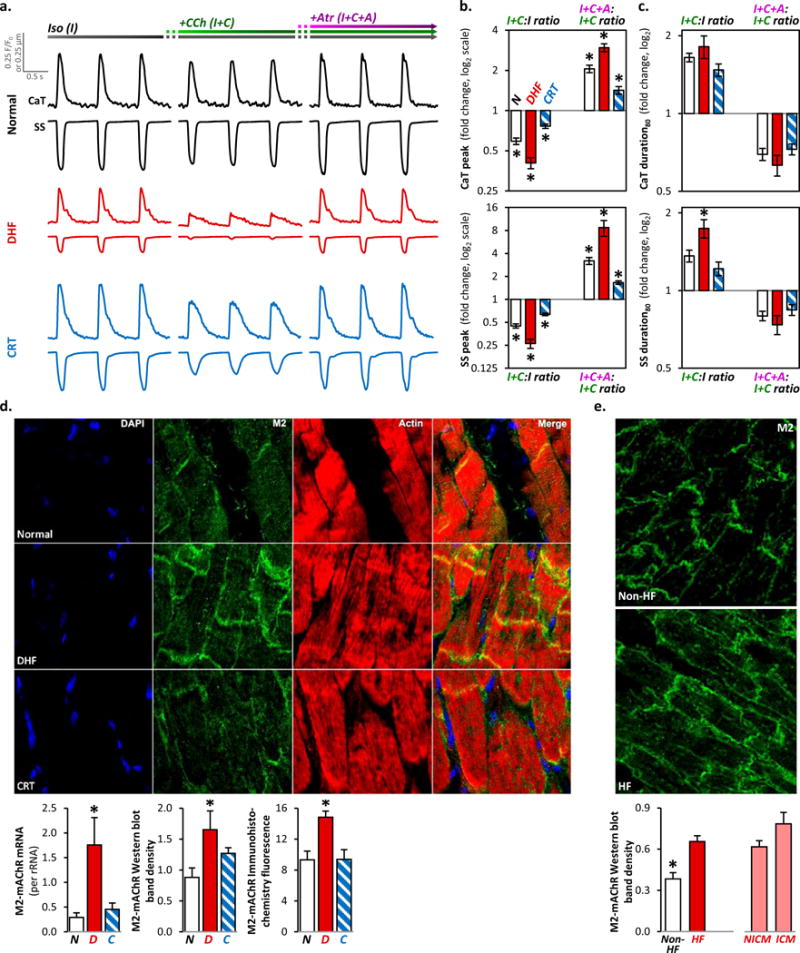
-
β-adrenergic stimulation with isoproterenol [(I), left column];cholinergic stimulation with carbamylcholine (CCh) in the continued presence of isoproterenol [(I+C), middle column];
b. The ratio of the peak responses to CCh added to isoproterenol compared to isoproterenol alone (I+C:I) on CaT (top panel) and SS (bottom) in normal control (empty bar), DHF (red) and CRT (blue) myocytes is plotted on the left column (log2 scale; mean±SEM; n=30–52 myocytes from N=6–9 hearts for each bar). Similarly, the ratio of I+C+A to I+C (I+C+A:I+C) is plotted on the right column. The individual data points are plotted in Online Figure IIa. Cholinergic stimulation reduced the respective peak CaT and SS by 59±4% and 74±4% in DHF; by 41±3% and 55±3% in normal control; and by 23±3% and 36±2% in CRT myocytes. In 15% of DHF myocytes, contraction was arrested despite continued isoproterenol exposure, and subsequent exposure to atropine fully restored CaT and contraction (Supplementary Video 1); these data were not included in this analysis.
c. The ratio of the 80% duration of CaT and SS are plotted in a format similar to panel b. The individual data points are plotted in Online Figure IIb. Cholinergic stimulation markedly prolonged the respective CaT and SS by 81±18% and 74±14% in DHF; by 65±6% and 36±7% in normal control; and by 47±8% and 22±7% in CRT myocytes.
d. Representative immunohistochemical staining sections of canine LV tissue (top) revealed increased M2-mAChR density in DHF myocytes at intercalated discs and sarcolemma. Quantitative analyses of RT-PCR, Western blot and immunohistochemical fluorescence data in canine LV tissue (bottom) revealed increased mRNA and protein expression of M2-mAChR in DHF but not in CRT myocytes (N=5 hearts/group). Similar results were obtained with Western blot analysis of cardiomyocytes isolated on a perchol gradient (data not shown), suggesting these M2-mAChR protein changes occur within cardiomyocytes.
e. Representative immunohistochemistry staining sections of human LV tissue samples from failing and non-failing hearts are shown on top. In failing human LV, M2-mAChR expression was upregulated and redistributed from the intercalated discs to the entire cell border (“lateralization”), similar to that observed in canines. Western blot analysis of tissue lysates from human LV (bottom) revealed 2-fold higher M2-mAChR protein expression in HF (N=18) compared to non-HF (N=7), regardless of ischemic (ICM; N=4) or non-ischemic cardiomyopathy (NICM; N=14).
In all panels of this figure, *p<0.01 vs. all other groups.
To identify the molecular basis of the model-dependent cholinergic responses, we performed immunohistochemistry, western blot and RT-PCR analyses. We observed an increase in M2-muscarinic acetylcholine receptor (M2-mAChR18, 19) mRNA and protein expression with dyssynchrony that was reversed by resynchronization (Fig. 2d). The canine DHF model exhibits similar changes in β-adrenergic signaling as human cardiomyopathy6–8. Thus, we performed similar experiments on LV myocardium from human hearts failing of ischemic and nonischemic etiologies. We observed similar increases in immunoreactive protein expression and subcellular localization of M2-mAChR (Fig. 1e), suggesting M2-mAChR remodeling is a generalizable pathophysiological feature of HF.
Does tonic cholinergic stimulation alter acute β-adrenergic responses? This question is germane to the effect of vagal nerve stimulation (VNS) in HF14. Under normal resting conditions, cholinergic signaling is the predominant autonomic influence on the heart9. In the continued presence of cholinergic stimulation, CaT and SS responses to β-adrenergic stimulation were markedly depressed in DHF (by 42% and 56%) and less inhibited in normal and CRT myocytes (Fig. 3a–c, Online Fig. IIIa–b).
Figure 3. Response to β-adrenergic stimulation in the setting of tonic cholinergic stimulation.
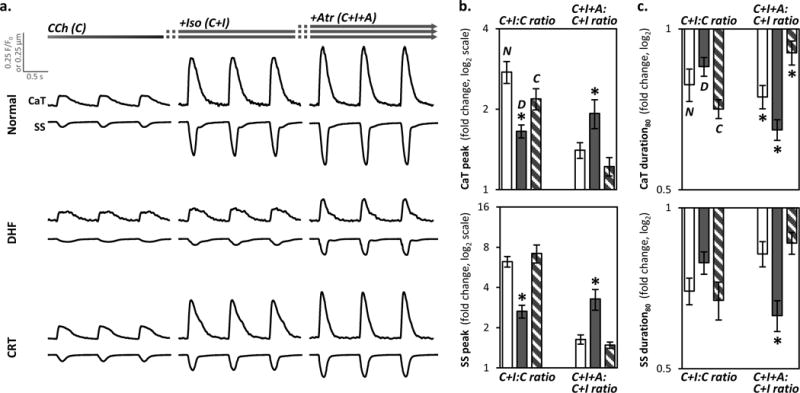
- CCh [(C), left column];
- isoproterenol [(C+I), middle column];
b. The ratio of the peak responses for C+I:C and C+I+A:C+I for CaT (top panel) and SS (bottom) in normal control (empty bar), DHF (filled) and CRT (striped) myocytes is plotted in a format similar to Figure 2b (n=19–32 myocytes/bar; N=6–9 hearts/bar). The individual data points are plotted in Online Figure IIIa. β-adrenergic stimulation markedly increased the respective peak CaT and SS by 176±26% and 525±57% in normal control; and by 119±20% and 620±111% in CRT myocytes; but by only 65±9% and 165±29% in DHF. Addition of atropine caused an additional increase in respective peak CaT and SS by 93±24% and 228±58% in DHF myocytes whereas normal and CRT myocytes showed little response (22–63%).
c. The ratio of the 80% duration of CaT and SS are plotted in a format similar to panel b. The individual data points are plotted in Online Figure IIIb. In the continued presence of CCh, isoproterenol shortened the respective durations of CaT and SS by 15±3% and 21±4% in DHF; by 21±4 and 30±4% in normal controls; and by 29±3% and 33±5% in CRT myocytes. Atropine further shortened the durations by 35±3% and 37±4% in DHF; by 25±5% and 18±4% in normal control; and by 10±3% and 14±5% in CRT myocytes.
In all panels of this figure, *p<0.05 vs. all other groups.
With cholinergic stimulation alone, peak CaT and SS amplitudes were decreased in DHF (by 18% and 27%), but remained unchanged in normal and CRT myocytes (Fig. 4a–b, Online Fig. IVa). Cholinergic stimulation prolonged CaT and SS in DHF but shortened them in normal and CRT myocytes (Fig. 4c, Online Fig. IVb). Reversal of cholinergic responses by atropine was not recapitulated by M1- or M3-mAChR inhibition (data not shown), suggesting these effects are mediated via M2-mAChR, consistent with changes in subtype functional expression (Fig. 1d).
Figure 4. Response to cholinergic stimulation alone.
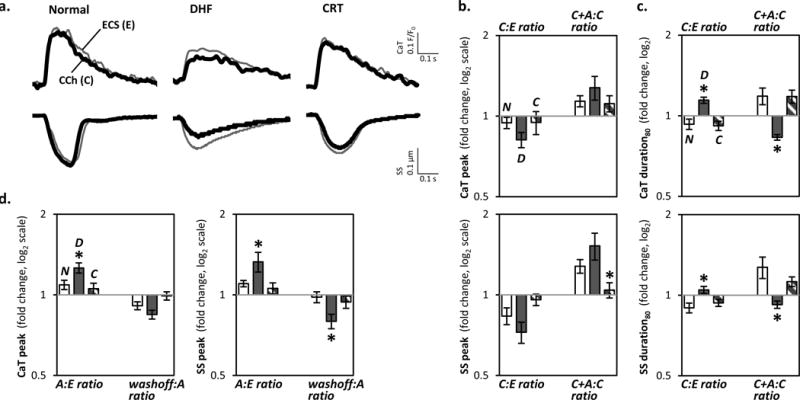
a. Representative CaT and SS are shown from normal controls, DHF, and CRT myocytes sequentially exposed to standard Tyrode’s extracellular solution [(ECS; E), thin gray line] followed by CCh [(C), thick black line].
b. The ratio of the peak responses for C:E and after the addition of atropine to CCh compared to CCh alone (C+A:C) for CaT (top panel) and SS (bottom) in normal control (empty bar), DHF (filled) and CRT (striped) myocytes is plotted in a format similar to Figure 3b (n=20–30 myocytes/bar; N=6–9 hearts/bar). The individual data points are plotted in Online Figure IVa. Cholinergic stimulation decreased the respective peak CaT and SS by 18±5% and 27±7% in DHF but had little or no effect in normal and CRT myocytes. These effects were reversed by subsequent addition of atropine.
c. The ratio of the 80% duration of CaT and SS are plotted in a format similar to panel b. The individual data points are plotted in Online Figure IVb. Cholinergic stimulation prolonged the CaT and SS durations by 14±3% and 5±3% in DHF whereas in normal and CRT myocytes, the CaT and SS durations was either shortened or unchanged.
d. The ratio of the peak responses to atropine (A:E) and washout for CaT (top panel) and SS (bottom) in normal control (empty bar), DHF (filled) and CRT (striped) myocytes is plotted in a format similar to panel b (n=8 myocytes/bar; N=3 hearts/bar). Atropine increased the peak CaT (left panel) and SS (right) by 26±5% and 33±11% in DHF, and by 9±4% and 10±3% in normal controls, but had no effect in CRT myocytes. These effects were reversed by washing off atropine.
In all panels of this figure, *p<0.05 vs. all other groups.
Does M2-mAChR remodeling have functional effects independent of receptor activation? Exposure of DHF myocytes to atropine alone reversibly increased peak CaT and SS amplitudes (by 26% and 33%) (Fig. 4d). These atropine-induced effects were infrequently observed in CRT and normal cells, suggesting DHF hearts are biased towards M2-mAChR-Gαi coupled signaling both in the absence and presence of cholinergic stimulation.
How does M2-mAChR-Gαi remodeling alter arrhythmic risk? In 30–50% of myocytes from all models, isoproterenol alone induced after-transients and after-contractions (Fig. 5a), consistent with findings from a recent study on normal canine myocytes20. Cholinergic stimulation completely abolished these disturbances in DHF and CRT, with little effect on peak CaT and SS in CRT myocytes. In the presence of tonic cholinergic stimulation, isoproterenol did not induce after-transients and after-contractions until exposure to atropine (Fig. 5b). Compared to DHF, early after-transients and after-contractions were more frequently seen in normal and CRT myocytes. The atropine effect was not recapitulated by M1- or M3-mAChR inhibition (data not shown). In myocytes from all models pretreated with pertussis toxin (PTX), isoproterenol promptly induced after-transients and after-contractions that were not affected by cholinergic stimulation (Fig. 5c).
Figure 5. Cholinergic stimulation suppresses after-transients and after-contractions trigged by β-adrenergic stimulation.
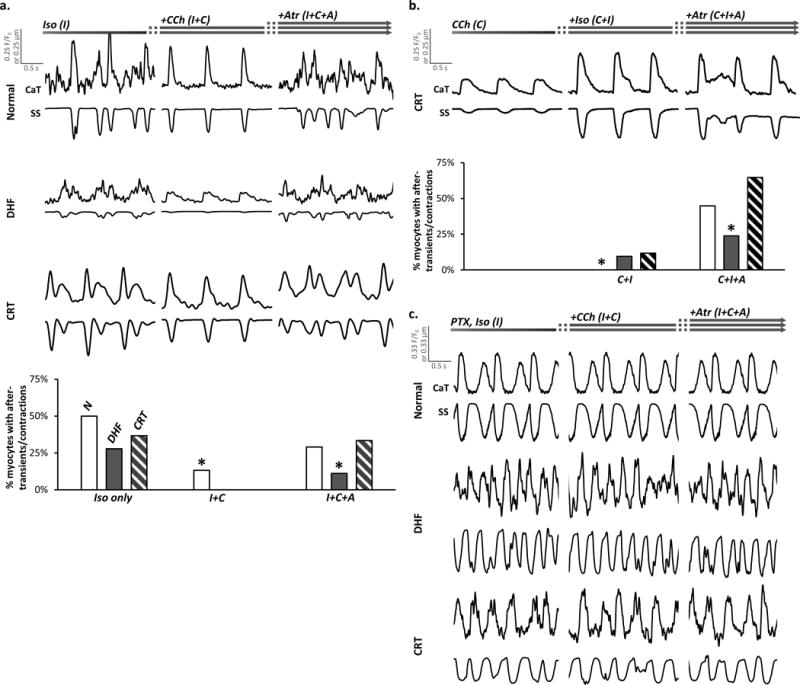
a. Representative CaT and SS from normal control, DHF and CRT myocytes are plotted (top) in a format similar to Figure 2a. Exposure to isoproterenol (I) induced after-transients and after-contractions that subsequently, were suppressed by CCh (I+C) and recurred with atropine (I+C+A). The bar graph (bottom) shows the percent of normal, DHF and CRT myocytes that demonstrated triggered activity (after-transients and after-contractions) in response to the corresponding solution exposures depicted at the top of the panel (n=42–104 myocytes/group; N=6–9 hearts/group). Myocytes that demonstrated isoproterenol-induced triggered activity were used only for analysis in this section and excluded from the analyses shown in Figures 2–4 and 6.
b. Representative CaT and SS from CRT myocytes are plotted (top) in a format similar to Figure 3a. In the presence of CCh (C), addition of isoproterenol (C+I) did not induce after-transients and after-contractions until addition of atropine (C+I+A). The percent of normal, DHF and CRT myocytes demonstrating triggered activity are plotted corresponding to the protocol panel c (n=25–63 myocytes/group; N=6–9 hearts/group). Again, the effects of atropine were not recapitulated by pirenzapine or 4-DAMP (data not shown). Myocytes that demonstrated isoproterenol-induced triggered activity were used only for analysis in this section and excluded from the analyses shown in Figures 2–4 and 6.
c. Representative CaT and SS from normal control, DHF and CRT myocytes pretreated with pertussis toxin (PTX) to inhibit Gαi are plotted in a format similar to panel a. In all myocytes from all models, sustained after-transients and after-contractions were noted with isoproterenol regardless of exposure to CCh.
In all panels of this figure, *p<0.05 vs. all other groups.
To characterize the role of M3-mAChR-Gαi signaling, we pretreated myocytes with PTX to inhibit Gαi signaling. With tonic β-adrenergic stimulation, PTX completely abolished the negative inotropic effects from cholinergic stimulation in all models (Fig. 6a, Online Fig. Va). In the presence of PTX and pirenzapine, an M1-mAChR-specific inhibitor, cholinergic stimulation increased peak CaT and SS in CRT but not in DHF cells (Fig. 6b); this effect was suppressed by atropine (Online Fig. Vb) or M3-mAChR inhibition (Fig. 6b, Online Fig. Vc). Consistent with these findings, immunohistochemical analyses demonstrated increased M3-mAChR expression, prominently at the intercalated discs in CRT (Fig. 6c). Western blot analyses revealed increased M3-mAChR protein without any apparent effect on Gαq/11 expression (Fig. 6d).
Figure 6. Cholinergic stimulation mediates positive and negative inotropic effects via distinct muscarinic receptor subtypes.
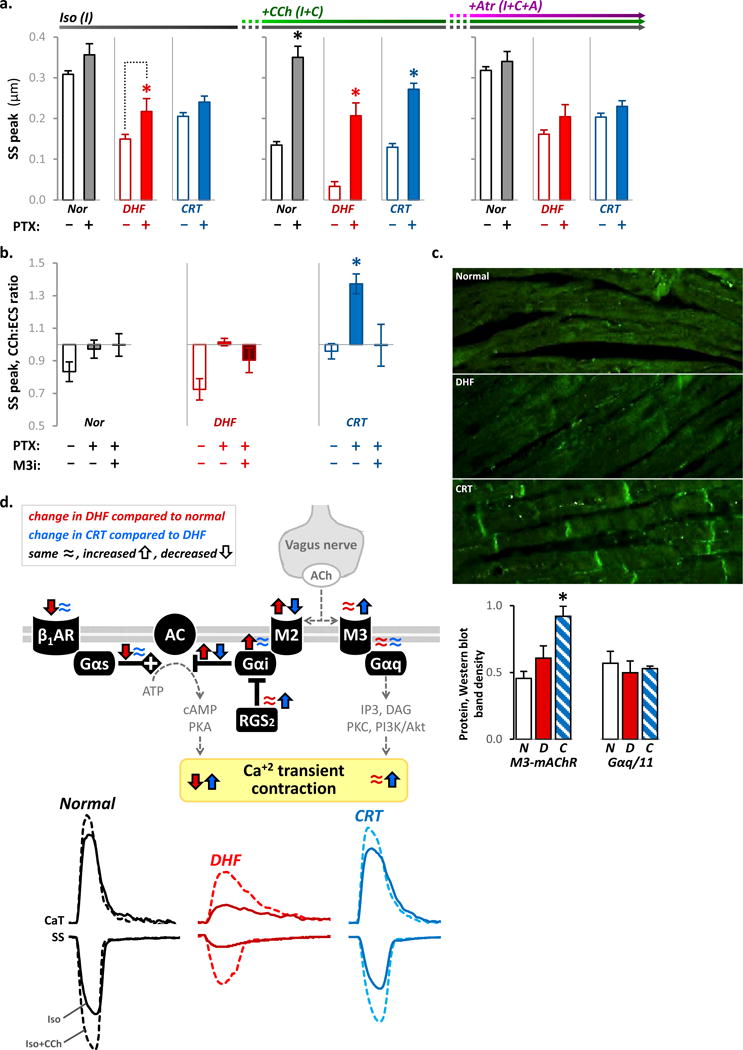
a. The peak SS responses (mean±SEM) corresponding to the indicated solution exchange protocol are compared in the absence (empty bars) or presence (filled) of PTX for normal control (black), DHF (red bars) and CRT (blue) myocytes (n=30–52 myocytes from N=6–9 hearts for each bar). The individual data points are plotted in Online Figure Va. PTX increased the peak SS response to isoproterenol (left column) in DHF myocytes, but had no effect in normal and CRT myocytes. This is consistent with enhanced baseline Gαi activity in DHF. In the continued presence of isoproterenol, pre-treatment with PTX abolished the negative inotropic effects of cholinergic stimulation in all groups (middle column). The peak SS after addition of atropine was not significantly different with and without PTX for normal (p=0.43), DHF (p=0.13) and CRT (p=0.32) myocytes (right column). These data suggest that in the presence of saturating β-adrenegic stimulation, the negative inotropic effect from cholinergic stimulation is mediated via M2-mAChR-Gαi signaling.
b. The ratio of the peak SS responses to CCh alone compared to ECS (C:E) using the same protocol as in Figure 4b are plotted in the absence and presence of PTX and a M3-mAChR-specific inhibitor (M3i) (n=8–30 myocytes/bar; N=3–9 hearts/bar). All myocytes were continuously perfused with pirenzapine to block M1-mAChR-specific effects. The individual data points are plotted in Online Figure Vb–c. Compared to the absence of PTX, cholinergic stimulation in the presence of PTX increased the peak SS by 45±8% in CRT myocytes, but this effect was abolished with M3i. In DHF myocytes, PTX abolished the negative inotropic effect from cholinergic stimulation, but M3i had no significant effect. These data suggest that CRT myocytes are biased towards M3-mAChR-mediated positive inotropic effect whereas normal and DHF myocytes are not.
c. Representative immunohistochemical staining sections of canine mid-myocardial tissue from the LV lateral wall (top) revealed increased M3-mAChR density in CRT myocytes at the intercalated discs. Western blots of tissue lysates (5 hearts per group) revealed CRT increased M3-mAChR protein expression without any change in Gαq/11 protein expression.
d. Proposed mechanism for autonomic remodeling in DHF and with CRT. Cholinergic stimulation can produce both inhibitory and stimulatory calcium and contractile responses in the heart via well-characterized M2-mAChR-Gαi and M3-mAChR-Gαq coupled signaling, respectively. DHF (red arrows and tracings) is associated with down-regulation of β1-adrenergic receptors (β1AR) and inhibition of adenylate cyclase (AC) from direct interactions with the α subunit of the PTX‐sensitive inhibitory G protein (Gαi) selectively coupled to M2-mAChRs. Coordinated increases in M2-mAChR-Gαi-coupled expression and signaling chronically inhibits basal AC-mediated downstream signaling and markedly impairs the efficiency of β-adrenergic responsiveness, resulting in smaller amplitudes and prolonged relaxation of CaT and SS. CRT (blue arrows and tracings) reverses this phenotype by differentially remodeling cholinergic signaling. By concurrently decreasing M2-mAChR and increasing RGS2 expression, CRT decreases the negative inotropic effects of Gαi signaling. Further, CRT increases M3-mAChR-Gαq-mediated signaling associated with positive inotropic responses and putative cardioprotective effects.
In all panels of this figure, *p<0.05 vs. all other groups.
DISCUSSION
We have identified up regulated M2-mAChR18, 19 expression and function in human and canine HF compared to non-failing controls. CRT attenuated M2-mAChR expression and Gαi17, 21, 22-coupling and enhanced M3-mAChR23, 24 expression in association with enhanced calcium cycling and sarcomere shortening. These changes in cholinergic signaling represent a novel mechanism for enhancing sympathovagal balance in CRT and may identify new targets for treatment of systolic HF.
Tonic β-adrenergic stimulation and increased Gαi expression are a hallmark of DHF. CRT reverses this phenotype by up regulating RGS2 and inhibiting Gαi signaling without decreasing Gαi expression, thereby improving calcium handling and sarcomere contraction6, 7. The primary M2-mAChR subtype18, 19 in the heart is selectively coupled to Gαi17 and acts via well-characterized second messenger pathways. Coordinated increases in M2-mAChR and Gαi expression and coupling have been noted in the LV from synchronized failing hearts21, 22 but the functional significance was not known. Whether this coordinated remodeling is also a feature of dyssynchronized and resynchronized HF had not been explored.
Our results indicate DHF hearts are biased towards M2-mAChR-Gαi coupled signaling, even in the absence of cholinergic stimulation. Extensive in vitro and ex vivo evidence indicates Gαi-coupled cardiac M2-mAChRs are activated in proportion to Gαi expression25 and in this setting, atropine may act as an inverse agonist16. These chronically-activated M2-mAChRs may be susceptible to agonist-induced desensitization26, 27, a well-characterized phenomenon that may be a mechanism for the improved hemodynamics14 noted with tonic cholinergic stimulation in ongoing clinical HF trials12, 13, 28.
It is plausible M2-mAChR remodeling occurs early on in HF as a compensatory mechanism to heightened sympathetic tone that, over the long-term, contributes to the pathology of HF, perhaps by depressing myocyte function, calcium handling and β-adrenergic responsiveness. Increased cholinergic tone has been noted early in HF development29 and cholinergic transdifferentiation of cardiac sympathetic neurons has been observed in some HF models30. By decreasing M2-mAChR-Gαi-mediated signaling, CRT improves β-adrenergic responsiveness. This, along with functional inhibition of Gαi by RGS26, 7 results in positive inotropic effects due to improved calcium handling and sarcomere response to β-adrenergic stimulation.
How does M2-mAChR-Gαi remodeling alter arrhythmic risk? Ventricular arrhythmias are a major cause of death in HF patients31, 32. Since the first report in 185933, extensive evidence from animal and clinical studies indicates β-adrenergic signaling increases arrhythmic risk31, 32 and cholinergic stimulation protects the heart from lethal arrhythmias9, 34. Despite model-dependent remodeling, there appears to be a large margin of safety at the cellular level for M2-AChR-Gαi-coupled signaling to protect and rescue normal, DHF and CRT hearts from arrhythmias. These results provide a mechanistic basis for prior observations, i.e., increased arrhythmias with β-adrenergic stimulation and PTX35, antiarrhythmic effects of cholinergic stimulation9, 33, and may have important implications for VNS9–14, 28 and development of new antiarrhythmic therapies34.
Whereas the highly prevalent M2-mAChR18, 19 subtype is selectively coupled to Gαi17, the relatively scarce M3-mAChR23, 24 is highly specific for stimulatory Gαq with putative cardioprotective effects36–38. Recent new insights into the molecular structure, function, pharmacology and fundamental physiological role of M3-mAChRs have identified them as a major target for drug development36, 39. Our results indicate CRT may exert beneficial effects via M3-mAChR-Gαq signaling, including enhanced calcium handling, sarcomere responsiveness and positive inotropy. Notably, CRT increased M3-mAChR expression at the intercalated discs. In cardiomyocytes, M3-mAChR activation during ischemia preserves the phosphorylated levels of sarcolemmal connexin 43 to provide delayed cardioprotection40. Further, M3-mAChR-Gαq signaling augments IP3/DAG-mediated calcium release, PKC-mediated phosphorylation, PI3-kinase/Akt-mediated reduced apoptosis, and RGS2 expression36–38, 41. CRT has similar effects, including increasing RGS2 expression particularly in clinical responders7 that may be exerted via M3-mAChR-Gαq signaling. We could not specifically address this here because an in vitro model of CRT does not currently exist.
The notion that cholinergic signaling has a relatively limited effect on LV function belies much evidence9. Our results indicate remodeling of cholinergic signaling is a critical pathological component of human and canine HF, and differential remodeling of cholinergic signaling is paramount for restoration of autonomic balance by CRT (Fig. 4e). The novel mechanisms identified herein offers an opportunity to apply targeted down-regulation of M2-mAChR and/or up-regulation of M3-mAChR in HF patients who are not CRT responders5 or candidates. Moreover, the beneficial effects of CRT might be enhanced by VNS9–13, 28 and remodeling of the key signaling components reported herein may represent mechanistic pathways engaged by VNS and open new avenues for pharmacological or pacing treatments for HF.
Supplementary Material
Novelty and Significance.
What Is Known?
In healthy heart cells, parasympathetic activation “tunes” β1-adrenergic receptor (β-AR) signaling, via acetylcholine acting at the muscarinic receptor (mAChRs) to suppress Ca+2 transients and contraction.
In the failing heart, compensatory increases in β-AR signaling ultimately is maladaptive.
While β-AR signaling has been extensively studied, the role of mAChR signaling in the failing heart is unknown, and the effect of cardiac resynchronization therapy (CRT) on mAChR signaling has been unexplored.
What New Information Does This Article Contribute?
Compared to non-failing controls, M2-mAChR expression is markedly upregulated in left ventricular (LV) myocytes isolated from failing canine and human hearts.
In LV myocytes of failing hearts, hyperactive M2-mAChR-Gαi coupled signaling protects against electrical instability (a substrate for lethal arrhythmias) caused by heightened β-AR signaling, but this also reduces mechanical function.
CRT decreases M2- and increases M3- mAChR expression, resulting in improved β-AR responsiveness and mechanical function while maintaining electrical stability.
The development of new and improved HF therapies remains a clinical, research and public health priority. CRT is the only HF therapy to decrease long-term mortality, restore autonomic balance and improve both acute and chronic LV function. The underlying mechanisms are largely unknown. Autonomic imbalance is associated with worsening HF and increased mortality risk, independent of LV function and ventricular arrhythmias. The present study demonstrates a critical role of parasympathetic mAChRs in HF, arrhythmic risk and CRT. It suggests that the beneficial effects of CRT involve differential remodeling of mAChRs… Further understanding these mechanisms can lead to the design and development of new, more effective HF therapies.
Acknowledgments
We gratefully acknowledge Dr. Charles Steenbergen for blinded analysis of immunohistochemistry slides and selection of representative samples, Ms. Deborah DiSilvestre and Dr. Swati Dey for technical assistance with some of the experimental protocols, Drs. Federica Farinelli and Khalid Chakir for myocyte isolation and procurement, and Mr. Rick Tunin for maintaining the animal facility.
SOURCES OF FUNDING
This work was supported by P01 HL 77180 (DAK, BOR, GFT).
Nonstandard Abbreviations and Acronyms
- 4-DAMP
1,1-dimethyl-4-diphenylacetoxypiperidinium iodide
- ANOVA
one-way analysis of variance
- β-AR
β1-adrenergic receptor
- CaT
calcium transient
- CCh
carbamylcholine
- CRT
cardiac resynchronization therapy
- DHF
dyssynchronous heart failure
- ECG
electrocardiography
- ECS
Tyrode’s extracellular solution
- HF
heart failure
- HRV
heart rate variability
- ICM
ischemic cardiomyopathy
- Iso
isoproterenol
- LV
left ventricular
- mAChR
muscarinic acetylcholine receptor
- NICM
non-ischemic cardiomyopathy
- PTX
pertussis toxin
- RA
right atrial
- RT-PCR
real-time polymerase chain reaction
- SDNN
standard deviation of NN intervals
- SS
sarcomere shortening
- VNS
vagus nerve stimulation
Footnotes
DISCLOSURES
None.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics—2013 update: A report from the american heart association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cleland JG, Daubert JC, Erdmann E, Freemantle N, Gras D, Kappenberger L, Tavazzi L. Longer-term effects of cardiac resynchronization therapy on mortality in heart failure [the cardiac resynchronization-heart failure (care-hf) trial extension phase] European heart journal. 2006;27:1928–1932. doi: 10.1093/eurheartj/ehl099. [DOI] [PubMed] [Google Scholar]
- 3.Bristow MR, Saxon LA, Boehmer J, Krueger S, Kass DA, De Marco T, Carson P, DiCarlo L, DeMets D, White BG, DeVries DW, Feldman AM. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. The New England journal of medicine. 2004;350:2140–2150. doi: 10.1056/NEJMoa032423. [DOI] [PubMed] [Google Scholar]
- 4.Cleland JG, Daubert JC, Erdmann E, Freemantle N, Gras D, Kappenberger L, Tavazzi L. The effect of cardiac resynchronization on morbidity and mortality in heart failure. The New England journal of medicine. 2005;352:1539–1549. doi: 10.1056/NEJMoa050496. [DOI] [PubMed] [Google Scholar]
- 5.Kirk JA, Kass DA. Electromechanical dyssynchrony and resynchronization of the failing heart. Circulation research. 2013;113:765–776. doi: 10.1161/CIRCRESAHA.113.300270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakir K, Daya SK, Aiba T, Tunin RS, Dimaano VL, Abraham TP, Jaques-Robinson KM, Lai EW, Pacak K, Zhu WZ, Xiao RP, Tomaselli GF, Kass DA. Mechanisms of enhanced beta-adrenergic reserve from cardiac resynchronization therapy. Circulation. 2009;119:1231–1240. doi: 10.1161/CIRCULATIONAHA.108.774752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakir K, Depry C, Dimaano VL, Zhu WZ, Vanderheyden M, Bartunek J, Abraham TP, Tomaselli GF, Liu SB, Xiang YK, Zhang M, Takimoto E, Dulin N, Xiao RP, Zhang J, Kass DA. Galphas-biased beta2-adrenergic receptor signaling from restoring synchronous contraction in the failing heart. Science translational medicine. 2011;3:100ra188. doi: 10.1126/scitranslmed.3001909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aiba T, Hesketh GG, Barth AS, Liu T, Daya S, Chakir K, Dimaano VL, Abraham TP, O’Rourke B, Akar FG, Kass DA, Tomaselli GF. Electrophysiological consequences of dyssynchronous heart failure and its restoration by resynchronization therapy. Circulation. 2009;119:1220–1230. doi: 10.1161/CIRCULATIONAHA.108.794834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coote JH. Myths and realities of the cardiac vagus. The Journal of physiology. 2013 doi: 10.1113/jphysiol.2013.257758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Popovic ZB, Bibevski S, Fakhry I, Sica DA, Van Wagoner DR, Mazgalev TN. Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model. Circulation Heart failure. 2009;2:692–699. doi: 10.1161/CIRCHEARTFAILURE.109.873968. [DOI] [PubMed] [Google Scholar]
- 11.Hamann JJ, Ruble SB, Stolen C, Wang M, Gupta RC, Rastogi S, Sabbah HN. Vagus nerve stimulation improves left ventricular function in a canine model of chronic heart failure. European journal of heart failure. 2013;15:1319–1326. doi: 10.1093/eurjhf/hft118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hauptman PJ, Schwartz PJ, Gold MR, Borggrefe M, Van Veldhuisen DJ, Starling RC, Mann DL. Rationale and study design of the increase of vagal tone in heart failure study: Inovate-hf. American heart journal. 2012;163:954–962 e951. doi: 10.1016/j.ahj.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 13.De Ferrari GM, Tuinenburg AE, Ruble S, Brugada J, Klein H, Butter C, Wright DJ, Schubert B, Solomon S, Meyer S, Stein K, Ramuzat A, Zannad F. Rationale and study design of the neurocardiac therapy for heart failure study: Nectar-hf. European journal of heart failure. 2014;16:692–699. doi: 10.1002/ejhf.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Premchand RK, Sharma K, Mittal S, Monteiro R, Dixit S, Libbus I, DiCarlo LA, Ardell JL, Rector TS, Amurthur B, KenKnight BH, Anand IS. Autonomic regulation therapy via left or right cervical vagus nerve stimulation in patients with chronic heart failure: Results of the anthem-hf trial. Journal of cardiac failure. 2014 doi: 10.1016/j.cardfail.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 15.Heart rate variability: Standards of measurement, physiological interpretation and clinical use. Task force of the european society of cardiology and the north american society of pacing and electrophysiology. Circulation. 1996;93:1043–1065. [PubMed] [Google Scholar]
- 16.Eglen RM. Overview of muscarinic receptor subtypes. Handbook of experimental pharmacology. 2012:3–28. doi: 10.1007/978-3-642-23274-9_1. [DOI] [PubMed] [Google Scholar]
- 17.Haga K, Haga T, Ichiyama A, Katada T, Kurose H, Ui M. Functional reconstitution of purified muscarinic receptors and inhibitory guanine nucleotide regulatory protein. Nature. 1985;316:731–733. doi: 10.1038/316731a0. [DOI] [PubMed] [Google Scholar]
- 18.Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T. Structure of the human m2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson DM, Heijman J, Bode EF, Greensmith DJ, van der Linde H, Abi-Gerges N, Eisner DA, Trafford AW, Volders PG. Diastolic spontaneous calcium release from the sarcoplasmic reticulum increases beat-to-beat variability of repolarization in canine ventricular myocytes after beta-adrenergic stimulation. Circulation research. 2013;112:246–256. doi: 10.1161/CIRCRESAHA.112.275735. [DOI] [PubMed] [Google Scholar]
- 21.Vatner DE, Sato N, Galper JB, Vatner SF. Physiological and biochemical evidence for coordinate increases in muscarinic receptors and gi during pacing-induced heart failure. Circulation. 1996;94:102–107. doi: 10.1161/01.cir.94.1.102. [DOI] [PubMed] [Google Scholar]
- 22.Wilkinson M, Giles A, Armour JA, Cardinal R. Ventricular, but not atrial, m2-muscarinic receptors increase in the canine pacing-overdrive model of heart failure. The Canadian journal of cardiology. 1996;12:71–76. [PubMed] [Google Scholar]
- 23.Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, Shaw DE, Weis WI, Wess J, Kobilka BK. Structure and dynamics of the m3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nash MS, Young KW, Challiss RA, Nahorski SR. Intracellular signalling. Receptor-specific messenger oscillations. Nature. 2001;413:381–382. doi: 10.1038/35096643. [DOI] [PubMed] [Google Scholar]
- 25.Spalding TA, Burstein ES. Constitutive activity of muscarinic acetylcholine receptors. Journal of receptor and signal transduction research. 2006;26:61–85. doi: 10.1080/10799890600567349. [DOI] [PubMed] [Google Scholar]
- 26.Murakami S, Inanobe A, Kurachi Y. Short-term desensitization of muscarinic k+ current in the heart. Biophysical journal. 2013;105:1515–1525. doi: 10.1016/j.bpj.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haga T. Molecular properties of muscarinic acetylcholine receptors. Proceedings of the Japan Academy. Series B, Physical and biological sciences. 2013;89:226–256. doi: 10.2183/pjab.89.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dicarlo L, Libbus I, Amurthur B, Kenknight BH, Anand IS. Autonomic regulation therapy for the improvement of left ventricular function and heart failure symptoms: The anthem-hf study. Journal of cardiac failure. 2013;19:655–660. doi: 10.1016/j.cardfail.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Kinugawa T, Dibner-Dunlap ME. Altered vagal and sympathetic control of heart rate in left ventricular dysfunction and heart failure. The American journal of physiology. 1995;268:R310–316. doi: 10.1152/ajpregu.1995.268.2.R310. [DOI] [PubMed] [Google Scholar]
- 30.Kanazawa H, Ieda M, Kimura K, Arai T, Kawaguchi-Manabe H, Matsuhashi T, Endo J, Sano M, Kawakami T, Kimura T, Monkawa T, Hayashi M, Iwanami A, Okano H, Okada Y, Ishibashi-Ueda H, Ogawa S, Fukuda K. Heart failure causes cholinergic transdifferentiation of cardiac sympathetic nerves via gp130-signaling cytokines in rodents. The Journal of clinical investigation. 2010;120:408–421. doi: 10.1172/JCI39778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeMazumder D, Tomaselli GF. Molecular and cellular mechanisms of cardiac arrhythmias. In: Hill JA, Olson EN, editors. Muscle 2-volume set: Fundamental biology and mechanisms of disease. Philadelphia, PA: Elsevier Inc; 2012. [Google Scholar]
- 32.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circulation research. 2004;95:754–763. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 33.Einbrodt PP. Ueber herzreizung und ihr verhaeltnis zum blutdruck. Akademie der Wissenschaften (Vienna) Sitzungsberichte. 1859;38:345–359. [Google Scholar]
- 34.Shen MJ, Zipes DP. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circulation research. 2014;114:1004–1021. doi: 10.1161/CIRCRESAHA.113.302549. [DOI] [PubMed] [Google Scholar]
- 35.Grimm M, Gsell S, Mittmann C, Nose M, Scholz H, Weil J, Eschenhagen T. Inactivation of (gialpha) proteins increases arrhythmogenic effects of beta-adrenergic stimulation in the heart. Journal of molecular and cellular cardiology. 1998;30:1917–1928. doi: 10.1006/jmcc.1998.0769. [DOI] [PubMed] [Google Scholar]
- 36.Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, Baell JB, Sexton PM, Christopoulos A, Shaw DE. Structural basis for modulation of a g-protein-coupled receptor by allosteric drugs. Nature. 2013;503:295–299. doi: 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- 37.Hang P, Zhao J, Qi J, Wang Y, Wu J, Du Z. Novel insights into the pervasive role of m(3) muscarinic receptor in cardiac diseases. Current drug targets. 2013;14:372–377. [PubMed] [Google Scholar]
- 38.Pan Z, Guo Y, Qi H, Fan K, Wang S, Zhao H, Fan Y, Xie J, Guo F, Hou Y, Wang N, Huo R, Zhang Y, Liu Y, Du Z. M3 subtype of muscarinic acetylcholine receptor promotes cardioprotection via the suppression of mir-376b-5p. PloS one. 2012;7:e32571. doi: 10.1371/journal.pone.0032571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kruse AC, Kobilka BK, Gautam D, Sexton PM, Christopoulos A, Wess J. Muscarinic acetylcholine receptors: Novel opportunities for drug development. Nature reviews Drug discovery. 2014;13:549–560. doi: 10.1038/nrd4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao J, Su Y, Zhang Y, Pan Z, Yang L, Chen X, Liu Y, Lu Y, Du Z, Yang B. Activation of cardiac muscarinic m3 receptors induces delayed cardioprotection by preserving phosphorylated connexin43 and up-regulating cyclooxygenase-2 expression. British journal of pharmacology. 2010;159:1217–1225. doi: 10.1111/j.1476-5381.2009.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hao J, Michalek C, Zhang W, Zhu M, Xu X, Mende U. Regulation of cardiomyocyte signaling by rgs proteins: Differential selectivity towards g proteins and susceptibility to regulation. Journal of molecular and cellular cardiology. 2006;41:51–61. doi: 10.1016/j.yjmcc.2006.04.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.