

Abstract
Leptin is an adipokine that has been linked with the cardiovascular complications resulting from obesity such as hypertension and heart disease. Obese patients have high levels of circulating leptin due to increased fat mass. Clinical and population studies have correlated high levels of circulating leptin with the development of cardiac hypertrophy in obesity. Leptin has also been demonstrated to increase the growth of cultured cardiomyocytes. However, several animal studies of obese leptin deficient mice have not supported a role for leptin in promoting cardiac hypertrophy so the role of leptin in this pathological process remains unclear. Leptin is also an important hormone in the regulation of cardiac metabolism where it supports oxidation of glucose and fatty acids. In addition, leptin plays a critical role in protecting the heart from excess lipid accumulation and the formation of toxic lipids in obesity a condition known as cardiac lipotoxicity. This paper focuses on the data supporting and refuting leptin’s role in promoting cardiac hypertrophy as well as its important role in the regulation of cardiac metabolism and protection against cardiac lipotoxicity.
Keywords: Leptin, Leptin receptor, Lipotoxicity, Cardiotoxicity, Obesity, Diabetes
Core tip: Leptin is a hormone derived from adipocytes which regulates food intake and body weight. It is present at high levels in obese individuals where it can impact organs such as the heart. Leptin has been shown to both promote and protect the heart against obesity induced heart disease. This review examines the controversial role of leptin in the development of cardiac hypertrophy as well as its important role in regulating cardiac metabolism and protecting the heart against obesity induced lipotoxicity.
INTRODUCTION
Leptin is a hormone most abundantly produced by white adipocytes which then acts in the hypothalamus of the brain to decrease appetite and increase energy expenditure. Leptin was discovered in the early 1990’s after genetic mapping of a mutation in the gene found in a specific strain of obese mice, the ob/ob mouse, which was originally described in the 1950’s[1,2]. These mice are characterized by having no leptin which results in marked hyperphagia, decreased energy expenditure and obesity. Another strain of obese mice called db/db mouse was subsequently found to have a mutation in the ObR gene encoding the leptin receptor[3]. This strain of mice is characterized by having very high levels of circulating leptin due to lack of functional leptin receptors, marked hyperphagia, decreased energy expenditure and obesity. There are also several rat strains with defective leptin receptor such as the Zucker fatty (fa/fa) rat and the Koletsky fatty rat[4,5]. Recently, a zinc-finger approach was utilized to create a rat model of leptin receptor deficiency on a salt-sensitive hypertension background[6]. All of these models as well as development of cell type/tissue-specific knockouts of the ObR gene have greatly increased our knowledge about the physiological role of leptin[7].
While leptin is mainly expressed in adipose tissue, it is also expressed in peripheral organs such as the heart[8]. Leptin receptors are also highly expressed in the heart of several species including humans[9,10]. In the rat heart, the long form of the leptin receptor is expressed in addition to other shorter isoforms[11]. Reverse-transcriptase polymerase chain reaction of the mouse heart readily reveals expression of all isoforms of the leptin receptor similar to the expression pattern found in the brain[12] (Figure 1). The long form of the leptin receptor, ObRb, activates signaling through the Janus kinases (JAK)/ signal transducers and activators of transcription (STAT) pathway and other Src Homology 2 domain containing proteins such as suppressor of cytokine signalling and SHP-2 (Src-like homology 2 domain containing protein tyrosine phosphatase) and STAT[13]. Short leptin receptor isoforms (ObRa, ObRc, ObRd) contain a box 1 motif which is able to bind JAK and activate other signal transduction cascades[13]. The ObRe which is also referred to as the soluble leptin receptor can regulate serum leptin concentration and also serves as a carrier protein delivering the hormone to its membrane receptors[14]. Not only are leptin receptors expressed in the heart but they are also regulated by various stimuli. Cardiac ischemia has been reported to have varying effects on expression of leptin receptors, with studies demonstrating that a 30 min ischemic period was associated with a decrease in leptin receptor expression and another study reporting that a 40 min ischemic period increased leptin receptor expression[11,15]. The specific role of leptin in cardiac ischemia was addressed in an elegant study utilizing cardiac-specific deletion of leptin receptors. Cardiac-specific deletion of leptin receptors resulted in a decrease in contractile function and metabolism of glucose and in an increase in mortality and morbidity following cardiac ischemia[16]. These results highlight the important cardio-protective function of leptin in cardiac ischemia due in part to its role in the regulation of cardiac metabolism which will be discussed in greater detail below. Leptin receptors in the heart are also regulated by pressure and stretch. It has been reported that pressure-overload induced cardiac hypertrophy resulted in a significant increase in the long form of the leptin receptor (ObR-B) but not the short form (ObR-A) in the heart[17]. The controversial role of leptin in cardiac hypertrophy will be addressed in the following section below.
Figure 1.
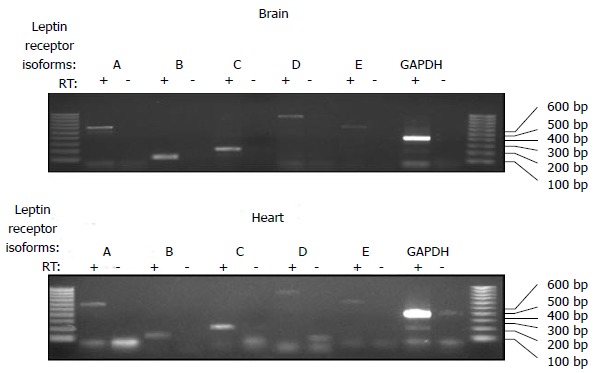
Comparison of leptin receptor isoforms in the mouse brain and heart. RNA was isolated from the brain and heart and reverse transcribed into cDNA. Polymerase chain reaction was then performed using primers specific for each mouse isoform of the leptin receptor as previously described[12]. All 5 of the leptin receptor isoforms were detected in the mouse heart as well as the brain.
THE ROLE OF LEPTIN IN CARDIAC HYPERTROPHY
Leptin has several functions in the heart including stimulation of fatty acid and glucose metabolism, prevention of steatosis, and protection against apoptosis (Figure 2). It also can raise blood pressure and heart rate through central mechanisms and promotes cardiac inflammation (Figure 2). Although obesity is associated with hyperleptinemia, increased cardiac mass and left ventricular (LV) wall thickness, it is unclear if leptin can directly cause cardiac hypertrophy (Figure 2). Epidemiologic studies have demonstrated positive correlations between plasma leptin levels and LV hypertrophy[18]. However, most of these observations are confounded by the fact that increased body mass and plasma leptin levels are highly correlated[19]. Obesity is also usually accompanied by hypertension which is the most common cause of cardiac hypertrophy[20]. In addition to increased blood pressure, obesity may cause cardiac hypertrophy by several other mechanisms including neurohormonal (renin-angiotensin-aldosterone system) and sympathetic nervous system activation, insulin resistance and hyperglycemia, and increased blood volume[21,22]. The exact roles of leptin in regulating cardiac structural changes in obesity such as hypertrophy are not well understood. In fact, differential hypertrophic and antihypertrophic effects of leptin have been reported and may be related to temporal effects or synergistic interactions with other obesity-associated factors.
Figure 2.
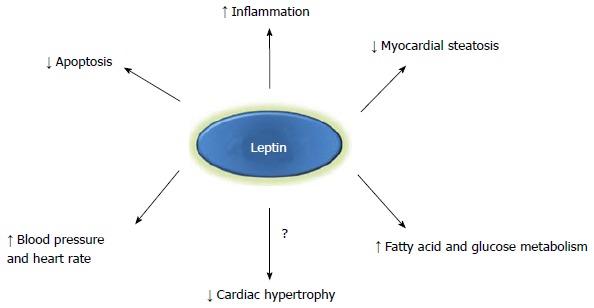
Potential mechanisms by which leptin may mediate cardiac function. Leptin may exert cardioprotective or maladaptive effects through hemodynamic factors such as increased heart rate and blood pressure, metabolic changes including augmented fatty acid or glucose utilization, reduced cardiac apoptosis, or structural cardiac changes such reduced cardiac lipid accumulation and possibly attenuated myocardial hypertrophy. Increased inflammation may be beneficial in some cardiac conditions (i.e., post-myocardial infarction) depending on the timing and extent of the inflammatory response.
Does leptin directly cause cardiac hypertrophy?
One of the earliest studies demonstrating a pro-hypertrophic effect of leptin comes from an experiment by Rajapurohitam et al[23] in which cultured neonatal rat ventricular myocytes were treated with varying concentrations of leptin. The authors observed a 42% increase in cell surface area 24 h after administration of 3.1 nmol/L leptin. Exposure to leptin also significantly increased cell size in cultured human and neonatal rat cardiomyocytes[24,25]. Leptin treatment increased matrix metalloproteinase-2 activity and collagen III and IV mRNA expression but resulted in no change in total collagen synthesis. Tajmir et al[26] demonstrated hyperplasia of both murine and human cardiomyocytes in response to leptin treatment which appeared to be mediated by activation of extracellular signal-regulated kinase (ERK) 1/2 and phosphatidylinositol-3 kinase. However, studies by Piñieiro et al[27] did not observe any effect of Leptn to increase cell size of murine HL-1 cardiomyocytes while these in vitro results suggest leptin contributes to adverse cardiac remodeling and hypertrophy, the results from whole animal and human studies are not that clear regarding the direct role of leptin to cause cardiac hypertrophy.
Human patient studies have reported associations of plasma leptin levels with cardiac hypertrophy. In hypertensive insulin-resistant men, fasting plasma leptin levels were positively correlated with myocardial wall thickness, but not with LV mass. This relationship was significant even after controlling for BMI, waist-to-hip ratio and blood pressure suggesting an independent effect of leptin on cardiac structure[28]. In another study, LV mass was found to be positively correlated with leptin levels after controlling for body mass index (BMI)[29]. After gastric bypass surgery and profound weight loss, there were significant reductions in BMI, insulin resistance and leptin levels, but only leptin levels were significantly correlated with the decrease in LV mass on multivariable analyses. These clinical findings suggest that leptin may contribute to the LV hypertrophic process. In a study of 36 hypertensive men, plasma leptin was significantly predictive of echocardiographic wall thickness independent of 24 h ambulatory blood pressure. However, other significant predictors in this model included insulin sensitivity and night-time diastolic blood pressure[18].
The mechanisms by which leptin may contribute to myocardial hypertrophy are poorly understood. In addition to its powerful effects to regulate appetite and body weight, leptin also has a powerful effect to activate the sympathetic nervous system via central nervous system pathways. Chronic leptin infusion increased arterial blood pressure which increases cardiac afterload and which would lead to increased cardiac hypertrophy over the long-term[30]. Leptin is also associated with increased heart rate which would also tend to increase myocardial workload and promote hypertrophy[31]. In addition to these effects, leptin may also contribute to endothelial dysfunction and vascular stiffness which could also contribute to cardiac hypertrophy[32]. It is important to note, however, that many of the reported effects of leptin are based on either short-term animal studies, in vitro experiments or epidemiologic data which makes it difficult to determine the direct role of leptin in regulating cardiac hypertrophy.
While it is clear that obesity is associated with cardiac hypertrophy, the role of leptin as a mediator or cause is still under investigation. Evidence strongly supporting an antihypertrophic role of leptin comes from an elegant experiment by Barouch et al[33] in which they evaluated LV structure and function, including LV wall thickness and mass, in ob/ob and db/db mice. To differentiate the direct effects of leptin on cardiac hypertrophy from the effects of obesity, the investigators subjected ob/ob mice to intravenous leptin infusion or caloric restriction. Administration of leptin significantly reduced wall thickness and reduced myocyte size by approximately 25%. While both the leptin-treated ob/ob mice and the calorie-restricted mice lost a similar amount of body weight, the pair fed group had no significant reduction in LV mass or wall thickness suggesting a leptin dependent effect in the reversal of myocardial hypertrophy. Additionally, the hypertrophic LV changes in the ob/ob mouse are not related to changes in blood pressure since these mice are normotensive[34]. Another important observation of this study was that the increase in myocardial wall thickness was not related to fatty infiltration of the heart muscle, as cardiac myocyte size was found to be increased in ob/ob mice[33].
Additional evidence for an antihypertrophic effect of leptin comes from experiments performed in our lab[35]. We evaluated the direct effect of leptin on myocardial lipid accumulation and LV hypertrophy in db/db mice and transgenic db/db “rescue” mice in which the normal rat leptin receptor was overexpressed or “rescued” in a cardiomyocyte-specific manner. After 30 wk of study including serial metabolic parameters and echocardiographic assessments, both the db/db and “rescue” mice were morbidly obese, hyperglycemic, and had high plasma triglycerides compared to lean control mice. The db/db mice developed significant cardiac hypertrophy and increased LV wall thickness. The “rescue” mice, in which cardiac leptin signaling was restored, had lower heart weights and LV wall thickness compared to db/db mice suggesting an antihypertrophic effect of leptin. If leptin had a direct hypertrophic effect, our db/db cardiac leptin receptor rescue mice would be primed for an increase in myocardial mass in this setting. db/db mice have very elevated circulating leptin levels, and our transgenic “rescue” mice had evidence of increased leptin signaling in the heart as indicated by elevated levels of phosphorylated STAT3. If increased leptin signaling directly leads to cardiac hypertrophy our transgenic “rescue” model would have developed an increase in myocardial mass and wall thickness due to high circulating leptin and augmented leptin receptor responsiveness. One limitation of this study was that we did not specifically evaluate myocyte sizes but instead measured wall thickness and heart weight[35].
In summary, the available data on the effects of leptin on cardiac growth and hypertrophy are conflicting and are summarized in Table 1. Hyperleptinemia is associated with cardiac hypertrophy but the presence of many confounding factors makes it difficult to establish a causal relationship. Furthermore, acute and chronic effects of leptin differentially regulate myocyte growth. Obesity and subsequent leptin resistance may play an important role in this relationship. Animal studies suggest that hyperleptinemia does not directly cause cardiac hypertrophy but may rather play an integral role in cardiac structural alterations that occur in response to obesity and the associated hemodynamic and metabolic changes. Additional, well controlled studies are warranted to better delineate the mechanisms by which leptin may regulate cardiac structural remodeling.
Table 1.
Effects of leptin on cardiac mass and left ventricular hypertrophy
| Ref. | Findings | Effect of leptin on LVH | |
| In vitro experiments | Rajapurohitam et al[23] | Exposure of cultured neonatal rat ventricular myocytes to leptin (0.31 to 31.4 nmol/L) increased cell area by 42% | Pro-hypertrophic |
| Xu et al[25] | Exposure of cultured neonatal rat cardiomyocytes to leptin (1-1000 ng) for 4 h increased cell surface area | Pro-hypertrophic | |
| Piñieiro et al[27] | Exposure of murine HL-1 cells to leptin did not increase cell size of cardiomyocytes | Neutral | |
| Madani et al[24] | Treatment of human pediatric cardiomyocytes with 6 nmol/L leptin increased cell size by 60% | Pro-hypertrophic | |
| Tajmir et al[26] | Treatment of HL-1 cells with 60 nmol/L leptin increased cell numbers 2.3-fold | Pro-hypertrophic | |
| In vivo experiments | Barouch et al[33] | 6-mo-old leptin deficient ob/ob mice had increased myocyte diameters compared with wild-type mice. Leptin (iv) treatment in ob/ob mice completely reversed LVH and normalized wall thickness as well as reduced cellular hypertrophy by approximately 25%. Pair-feeding did not significantly reduce LV mass despite similar weight loss | Anti-hypertrophic |
| Hall et al[35] | db/db mice developed LVH (increased wall thickness and heart weights). Transgenic db/db mice with cardiomyocyte-specific leptin receptor rescue did not cause LVH; in fact the heart weights were reduced | Anti-hypertrophic | |
| Epidemiologic studies | Paolisso et al[28] | Plasma leptin level was correlated (n = 55 males) with interventricular wall (r = 0.34) and posterior wall (r = 0.38) thicknesses after adjusting for BMI and waist/hip ratio | Pro-hypertrophic |
| Paolisso et al[18] | Study of 36 hypertensive patients demonstrating increased LV wall thickness (but not LV mass) measured by echo was associated with plasma leptin independent of BMI or waist/hip ratio (P = 0.001) | Pro-hypertrophic | |
| Perego et al[29] | Study of 31 obese subjects undergoing gastric bypass surgery demonstrated leptin was independently associated with LV mass (β = 10.66, P = 0.001). One year after surgery, decrease in LV mass only correlated with the decrease in leptin levels (P = 0.01) | Pro-hypertrophic | |
| Lieb et al[40] | Cross-sectional analysis of 432 aged (> 70 yr) participants in the Framingham Heart Study demonstrated leptin concentrations were inversely correlated with LV mass (β = -0.134, P = 0.02), left atrial size (β = -0.131, P = 0.04) and LV wall thickness (β = -0.134, P = 0.02) measured by echo | Anti-hypertrophic | |
| Martin et al[41] | In 1464 MESA Study participants who underwent cardiac magnetic resonance imaging, a 1-SD increment in leptin was associated with smaller LV mass (β = -4.66%, P < 0.01), LV volume (β = -5.87, P < 0.01), and reduced odds ratio for presence of LVH (OR = 0.65, P < 0.01) after adjustment for age, gender, race, height, and weight | Anti-hypertrophic |
LVH: Left ventricular hypertrophy; BMI: Body mass index.
Leptin and cardiac function
In addition to its role in regulating cardiac structural changes, leptin may also be an important factor in regulating cardiac function. Leptin has been associated with pathophysiologic cardiovascular conditions including coronary artery disease and congestive heart failure[36,37]. Leptin has important effects on systemic hemodynamics and myocardial metabolism (as discussed in detail below) which may also have profound effects to regulate cardiac function. Similar to its potential implication in cardiac hypertrophy, the effects of hyperleptinemia on cardiac function have been difficult to assess given the number of confounding factors associated with obesity that all have detrimental effects on the heart. As obesity and its co-morbid conditions such as hypertension and diabetes are increasingly prevalent, understanding the relationships and mechanisms by which each of these conditions impacts the development and progression of congestive heart failure has important clinical implications for its prevention and treatment.
Elevated leptin levels have been observed in patients with dilated cardiomyopathy and have been suggested to be a marker of heart failure progression[38]. In a prospective study of 4080 older men followed for 9 years, increased BMI and circulating leptin levels were independent predictors of incident heart failure. After adjustment for BMI and other potential mediators, increased leptin levels remained significantly associated with an increased risk for heart failure in men without pre-existing coronary artery disease[39]. Leptin levels were also associated with incident congestive heart failure and cardiovascular disease in an elderly cohort from the Framingham Heart Study. However, after adjustment for BMI the association with congestive heart failure was negated[40]. More recently, investigators from the Multi-Ethnic Study of Atherosclerosis demonstrated that leptin levels were not associated with incident cardiovascular events after adjustment for cardiovascular risk factors and BMI[41]. Based on these epidemiologic data, it remains unclear whether leptin is associated with development of heart failure, and if so, whether it plays a causal or compensatory role?
Leptin exerts physiologic effects that may be detrimental in states of cardiac dysfunction or heart failure. Leptin’s hemodynamic effects generally increase myocardial workload via activation of the sympathetic nervous system. These effects include increasing resting heart rate and blood pressure[30]. Leptin may therefore act synergistically with other factors associated with obesity such as hyperglycemia, inflammation, and oxidative stress to accelerate the development of cardiovascular disease. However, it is possible that the chronic effects of leptin may have adverse consequences on myocardial function and the acute effects may provide a compensatory response to cardiac insults such as ischemia or heart failure.
Evidence for an acute beneficial effect of leptin comes from studies in experimentally-induced myocardial infarction and heart failure. McGaffin et al[16] induced anterior myocardial infarctions in control mice and in mice with cardiac-specific deletion of the leptin receptor. Mice lacking the leptin receptor specifically in the heart developed more LV dysfunction and had higher mortality after induction of myocardial infarction. The disruption of leptin signaling was associated with more LV dilation, hypertrophy, inflammation and adverse cardiac remodeling post-myocardial infarction. These investigators also demonstrated that many of the beneficial effects of leptin in this setting may be mediated via the AMP-activated protein kinase (AMPK) pathway.
We have studied the acute protective effects of leptin in a model of heart failure induced by Cre-recombinase activation[42]. Activation of Cre-recombinase is a widespread molecular tool used to conditionally delete or express genes in a tissue-specific and temporal manner. This technique has been somewhat limited due to observations by our lab and others that induction of Cre-recombinase activity in the heart can lead to transient LV dysfunction and a dramatic drop in ejection fraction[43,44]. Specifically, we reported that conditional deletion of the cardiac leptin receptor resulted in severe cardiogenic shock and death of the animals which was most likely related to impaired myocardial energy metabolism[42]. These results emphasize the important role that leptin plays in cardiac metabolism which will be discussed in further detail in the next section.
Further evidence for a beneficial effect of leptin on cardiac function comes from experiments showing aged ob/ob and db/db mice have increased cardiac myocyte apoptosis and decreased survival compared with wild-type controls[45]. Leptin treatment significantly reduced apoptosis in ob/ob mice as well as in isolated myocytes. Although ob/ob and db/db mice generally have normal LV systolic function, they appear to have LV diastolic functional abnormalities[35,46]. These studies suggest that intact cardiac leptin signaling is important for normal cardiac function and may be protective against cardiac insults such as ischemia.
ROLE OF LEPTIN IN THE REGULATION OF CARDIAC METABOLISM
The role of leptin in the control of whole body energy homeostasis in humans is well established[47]. The effects of leptin on myocardial metabolism, and the consequences for cardiac adaptation in disease states, are far less well understood. Our knowledge in that area comes from studies performed on live rodents and isolated heart preparations. These studies have focused on the metabolism of glucose and free fatty acids, the main substrates for energy provision of the heart. Altogether, the investigations have revealed a dichotomy between central and peripheral actions of leptin (Figure 3). However, variations in the age or strain of animals, and in the experimental conditions employed, have made it difficult to identify with certainty the molecular regulatory pathways involved. It should also be noted that most studies used male animals only. Consequently, it is unknown whether the metabolic actions of leptin on the heart are characterized by a sexual dimorphism. This part of the review will focus first on the cardiometabolic effects mediated by leptin signaling in the brain. The metabolic consequence of leptin signaling activation in cardiac muscle will then be reviewed. Lastly, the effects of leptin on cardiac metabolism in disease states, and their impact on contractile function, will be discussed.
Figure 3.
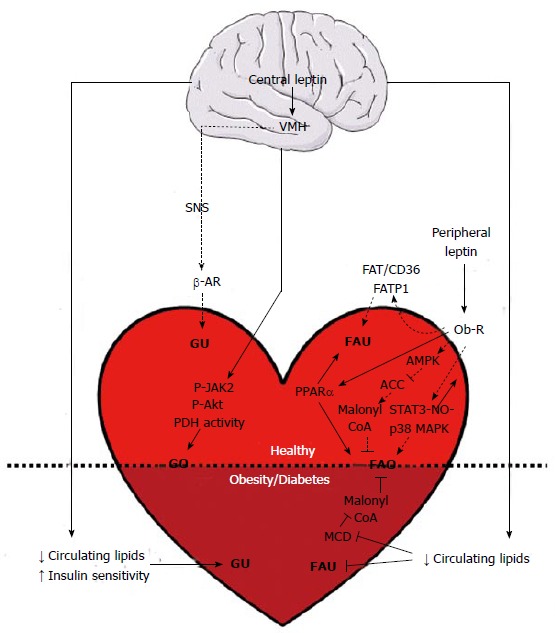
Cardiometabolic effects of leptin in health and obesity. Triangular and flat arrowheads represent stimulatory and inhibitory effects on the designed targets, respectively. Dotted lines indicate acute leptin effects (appearing between less than an hour and several hours of treatment), while plain lines represent chronic leptin effects (reported after several days or weeks of treatment). ACC: Acetyl-CoA carboxylase; AMPK: AMP-activated protein kinase; β-AR: Beta-adrenergic receptor; FAO: Fatty acid oxidation; FAT/CD36: Fatty acid translocase/cluster of differentiation 36; FATP1: Fatty acid transport protein 1; FAU: Fatty acid uptake; GO: Glucose oxidation; GU: Glucose uptake; MAPK: Mitogen-activated protein kinase; MCD: Malonyl-CoA decarboxylase; NO: Nitric oxide; Ob-R: Leptin receptor; P-Akt: Phosphorylated Akt kinase; PDH: Pyruvate dehydrogenase; P-JAK2: Phosphorylated Janus kinase 2; PPARα: Peroxisome proliferator-activated receptor alpha; SNS: Sympathetic nervous system; STAT3: Signal transducer and activator of transcription 3; VMH: Ventromedial hypothalamus. Horizontal black line demarcates differences between the healthy heart and the heart in obesity/diabetes.
Centrally mediated effects of leptin on cardiac metabolism
Pioneering work performed with the leptin-deficient ob/ob mice demonstrated that chronic intraperitoneal leptin injection at a sub-active dose for the reduction of body weight gain or hyperinsulinemia was sufficient to normalize blood glucose levels[48]. It was later demonstrated in the same animal model that acute intravenous infusion of leptin increased glucose turnover and stimulated the uptake of glucose in peripheral organs, including the heart. The 6-fold increase in myocardial glucose uptake did not significantly impact heart rate[49]. Both intravenous and intracerebroventricular (icv) infusions of leptin were found to similarly increase glucose turnover in C57BL/6J wild-type mice[50]. A single bolus injection of leptin in the ventromedial hypothalamus of young Sprague Dawley rats also resulted in a 4-fold increase in myocardial glucose uptake[51]. This increase in glucose uptake was shown to be additive to the one induced by an intravenous administration of insulin[42]. Based on these observations it was concluded that this rapid increase in peripheral glucose utilization is governed by a central mechanism, independent from insulin, and involving the activation of sympathetic nerves and the local activation of β-adrenergic receptors on target tissues[52]. A week of daily icv leptin administration in C57BL mice on a low-fat diet also resulted in an increase in myocardial glucose oxidation that was associated with increased phosphorylation of JAK2 and Akt kinases, and with increased pyruvate dehydrogenase activity (Figure 3). Rates of palmitate oxidation were not significantly altered, and here again, the switch toward higher glucose utilization did not modify mechanical function of the heart[53]. Thus, in rodents with normal leptin sensitivity, both acute and chronic activation of central leptin signaling favor myocardial glucose utilization through mechanisms stimulating the uptake and the oxidation of this substrate. Although the involvement of other hormonal signals cannot be ruled out, the effect seems to be independent from insulin secretion or from a change in insulin sensitivity.
Peripheral effects of leptin on cardiac metabolism
Unlike the reports on its central actions, there is little evidence to support a stimulatory effect of peripheral leptin on myocardial glucose metabolism: While a small but significant increase in glucose uptake was reported when hearts of Wistar rats were perfused in the Langendorff mode with a low dose of leptin (1 ng/mL), the effect may have been caused by the absence of fatty acids in the perfusate[54]. Indeed, in hearts of male Sprague Dawley rats perfused in the working mode with both glucose and palmitate, rates of glucose oxidation were unaffected by the presence of a pharmacological dose of leptin (60 ng/mL). Conversely, the total oxidation of fatty acids, from both exogenous (palmitate) and endogenous (triacylglycerol stores) origin, increased by 82%[55]. Using similar experimental conditions, Sharma and colleagues confirmed the existence of a leptin-mediated increase in exogenous palmitate oxidation that occurred in absence of changes for the rates of glucose oxidation[56]. Experiments performed with HL-1 cardiomyocytes partly corroborate these results: while a 1 h incubation with leptin failed to modify basal or insulin-stimulated glucose uptake and oxidation, it resulted in increased palmitate uptake and oxidation[57]. The increase in palmitate uptake was linked to the upregulation of the fatty acid transporters FATP1 and CD36. It is noteworthy that the incubation of neonatal rat ventricular myocytes with leptin for 72 h induced the expression of peroxisome proliferator-activated receptor alpha (PPARα), a key activator of fatty acid metabolism in the heart[58]. However, while increased fatty acid oxidation in HL-1 cells was traced to an increase in AMP-activated protein kinase activity and to the subsequent inhibition of malonyl-CoA production (a potent endogenous inhibitor of mitochondrial fatty acid uptake), this mechanism was not induced in the isolated rat heart[55]. Instead, in the intact heart, leptin was found to stimulate fatty acid oxidation by a STAT-3-nitric oxide-p38 MAPK-dependent mechanism (Figure 3)[56]. In conclusion, based on experimental settings where both glucose and fatty acids were present, the activation of myocardial leptin signaling rapidly stimulates fatty acid uptake and oxidation without affecting glucose oxidation. In the long term, leptin may also promote fatty acid oxidation through the upregulation of PPARα.
Metabolic effects of leptin in disease states
Obesity and diabetes are characterized by an increased reliance of the heart on fatty acid oxidation for energy provision. Sustained high rates of fatty acid uptake and oxidation inhibit both basal rates of glucose oxidation and insulin-stimulated glucose utilization, leading to a dramatic reduction in cardiac mechanical efficiency (work performed per unit of oxygen consumed)[59,60]. This metabolic remodeling has been observed in ob/ob mice, and persists even when the isolated hearts are perfused under low fatty acid condition[61]. However, this metabolic remodeling is reversible, and glucose intolerant patients undergoing a modest weight loss present with reduced myocardial fatty acid uptake and with improved cardiac mechanical function[62]. Sloan and colleagues elegantly demonstrated the importance of hypothalamic leptin signaling in the regulation of the balance of myocardial substrate selection during weight loss. By combining calorie restriction with leptin treatments in ob/ob mice, they showed that the hormone is necessary to normalize basal myocardial palmitate oxidation and to restore the insulin-mediated switch to glucose utilization[63]. The authors attributed their results to the leptin-mediated inhibition of the rise in circulating free fatty acids caused by calorie restriction, thereby leading to the normalization of myocardial fatty acid oxidation gene expression and to the improvement of myocardial insulin sensitivity. In accordance with these results, Keung et al[53] observed that chronic central leptin treatment (via intracerebroventricular infusion) of C57BL mice inhibited the increase in myocardial fatty acid oxidation caused by high-fat feeding. The effect was also linked to an improvement in insulin sensitivity and to a decrease in circulating lipid levels, with a subsequent reduction in the expression of cardiac malonyl-CoA decarboxylase, the enzyme that degrades malonyl-CoA (Figure 3). Although the absolute rates of myocardial glucose oxidation were unaffected, this resulted in an increased contribution of glucose metabolism to Krebs cycle activity[53]. Lastly, in a rat model of insulin-dependent diabetes, increased glucose uptake in cardiac muscle as well as in several other organs, together with the suppression of hepatic glucose output, is part of the mechanism by which the activation of central leptin signaling normalizes glycemia[64].
Heart failure also elicits disturbances in the balance between fatty acid and glucose oxidation. Severe heart failure has generally been associated with increased glucose oxidation and decreased fatty acid oxidation, a switch in substrate meant to improve mechanical efficiency of the stressed heart[65]. The expression of leptin and of its receptor increases more than 4-fold in the failing human heart, suggesting increased activity of this signaling pathway as the condition progresses[66]. In a murine model with heart failure from ischemic origin, cardiomyocyte-specific deletion of the leptin receptor exacerbated the deterioration of myocardial structure and function. While myocardial metabolism was normal in the unstressed heart, cardiac specific loss of leptin receptors completely inhibited the switch toward increased glycolysis and glucose oxidation post myocardial infarction and enhanced the development of heart failure[16,67]. These results indicate that both central and cardiac leptin signaling play an important role in metabolic adaptation of the heart in heart failure. The beneficial effects of leptin are achieved either by favoring the return to a normal energy balance in dysregulated metabolic states, or by facilitating the transition toward a state of improved mechanical efficiency.
What is lipotoxicity?
Excess fatty acids as occurs in individuals who consume too many calories or expend too few calories are normally stored as triglycerides in white adipose tissue. However, when there is a defect in the amount of adipose tissue as seen in lipodystrophy or an excessive amount of fatty acids are consumed which exceed the ability of white adipose tissue to expand as seen in obesity, fatty acids can start to accumulate in organs such as the heart. With obesity, the substrate preference and utilization of the heart becomes altered such that substrate utilization is shifted towards fatty acids. This switch towards fatty acid metabolism is promoted by the increased expression of proteins involved in fatty acid oxidation such as carnitine palmitoyltransferase-1 (CPT1). These alterations in normal fatty acid oxidation can promote the formation of toxic lipids like ceramide and contribute to cardiac dysfunction observed in obesity[68-71]. Several studies have demonstrated that increases in ceramide production which arises by condensation of unoxidized palmitoyl-CoA and serine causes cells including cardiac myocytes to undergo apoptosis and die[72,73]. Diacylglycerol (DAG) is another lipid that can mediate fatty acid-induced toxicity. DAG acyl transferase (DGAT) is the enzyme responsible for the addition of the final fatty acid onto DAG to convert it to triglyceride. Transgenic mice which overexpress DGAT in the heart have increased lipid accumulation in the form of increased triglyceride levels but they are protected from lipotoxic induced cardiac dysfunction[74]. Thus, the specific role of increased triglyceride accumulation in the development of cardiac lipotoxicity remains controversial.
Role of lipotoxicity in heart disease
Descriptions of fat storage in the heart date all the way back to the 1800’s and were thoroughly described by Smith et al[75] in the 1930’s. Over 10 years ago Sharma and colleagues described intramyocardial lipid accumulation in human heart failure that was identical to that found in the zucker diabetic fatty (ZDF) rat[76]. These studies clearly demonstrated that increased myocardial lipid accumulation was associated with an upregulation of PPARα responsive genes and an increase in the inflammatory marker tumor necrosis factor-α (TNF-α) both of which are thought to contribute to the cardiac contractile dysfunction observed in both ZDF rats and human patients[76]. Recent advances in cardiac imaging techniques has resulted in the measurement of cardiac triglyceride levels in various patient populations with mixed results regarding the significance of increased cardiac triglyceride accumulation on cardiac function. Several studies in overweight and insulin resistant patients have positively correlated increased myocardial triglyceride levels with alterations in cardiac structure and function[77,78]. Myocardial triglyceride accumulation has also been found to contribute to the pathology of severe aortic stenosis[79]. While these studies have implicated cardiac triglyceride accumulation to alterations in cardiac function several studies have not reported such a correlation. McGavock et al[80] reported increased myocardial triglyceride accumulation in the absence of any changes in cardiac function in patients with type II diabetes. Likewise studies by Nyman correlated increased epicardial and pericardial fats but not intramyocardial triglyceride accumulation with alterations of cardiac function in male patients with metabolic syndrome[81]. Lastly, studies by Liu et al[82] reported that although myocardial triglyceride levels correlated with increases in BMI, they failed to correlate with alterations in cardiac function in healthy African-American males. Although increases in cardiac triglyceride levels have been documented in several pathological conditions as well as in the metabolic syndrome and type II diabetes, their specific role in altering cardiac function in these conditions is still unresolved.
There are several experimental models of cardiac lipotoxicity in rats and mice. The most studied are the models of leptin signaling deficiency such as the ZDF rat and the ob/ob and db/db mouse models[83]. Several other models of cardiac lipotoxicity have been developed in which myocardial fatty acid uptake is increased above normal by overexpression of fatty acid transport protein 1 (FATP1) or cardiac specific expression of long chain acyl CoA synthase 1 (ACS1)[84,85]. Models of cardiac lipotoxicity have also been created by overexpression of enzymes CPT1 and the transcription factor PPAR-α stimulating the β-oxidation of fatty acids. Interestingly, both cardiac specific deletion and overexpression of PPAR-α result in cardiac lipid accumulation with the deletion of PPAR-α decreasing expression of critical enzymes involved in β-oxidation of fatty acids and overexpression of PPAR-α resulting in increased transport of fatty acids into the heart[86,87].
While the mechanism of cardiac lipid accumulation in these various models may differ, the end result by which lipids cause cardiac dysfunction is similar in these various models. The accumulation of lipids such as ceramide and DAG to toxic levels promotes cardiomyocyte apoptosis via increases in reactive oxygen species production which induces non-coding RNAs such as growth arrested DNA-damage inducible gene (GADD)[88,89]. Accumulation of these toxic lipids also promotes endoplasmic reticulum stress which induces eukaryotic elongation factor and leads to cell death[90]. Excess lipid accumulation in the heart also interferes with insulin signaling resulting in cardiac insulin resistance and down-regulation of the IRS1/PI3K/Akt pathway which is a protective pathway in the heart[91].
Role of leptin in protection against cardiac lipotoxicity
Plasma leptin levels are elevated in individuals with obesity due to expansion of white adipose tissue mass. In obesity, it is hypothesized that there is central resistance to the effects of leptin on appetite and energy expenditure which results in increased adiposity and an increase in plasma leptin levels[7]. The increase in plasma leptin levels is also believed by some investigators to be an underlying cause for cardiovascular pathology in obesity[92,93]. However, an alternative hypothesis put forth by Unger proposes that increased plasma leptin levels are a protective mechanism preventing steatosis of organs such as the liver, pancreas, and heart in obesity[94].
Much of what is known about the role of leptin in protecting against lipotoxicity is derived from strains of leptin and leptin receptor deficient rodents. For example, leptin receptor deficient rats and mice are characterized by marked steatosis of peripherial organs such as the heart. Previous studies by Sharma et al[76] have reported that intracardial lipid accumulation and subsequent alterations in cardiac function and gene expression seen in the ZDF rat are remarkably similar to that observed in human patients with heart failure. Likewise studies in leptin receptor deficient db/db mice have also reported increased cardiac triglyceride accumulation that is associated with the development of cardiomyopathy and alteration of cardiac metabolism[95-97]. The leptin deficient ob/ob mouse also displays severely increased cardiac triglyceride levels associated with diastolic dysfunction[46]. Interestingly, the alterations in cardiac metabolism and cardiac lipid accumulation characterizing the ob/ob mouse appear to be totally dependent on leptin insufficiency as calorie restriction used to normalize body weight does not improve the increase in plasma fatty acid levels, the enhanced uptake of fatty acids by the heart or the increase in cardiac lipid accumulation[63]. Both central and peripheral administration of leptin restored myocardial insulin sensitivity and decreased myocardial fatty acid transport and lipid accumulation independently of calorie restriction[63].
The important role of leptin to protect against lipotoxicity was first demonstrated in the liver and pancreas where adenoviral restoration of leptin receptor in these tissues in ZDF rats decreased triglyceride accumulation and protected against lipotoxic injury[98,99]. In the heart, increases in plasma leptin levels achieved by adenoviral overexpression of leptin in the liver were able to normalize cardiac triglyceride levels and restore normal cardiac function and histology in cardiac specific acyl CoA synthase transgenic mice which are a model of severe cardiac steatosis[100]. We recently reported that cardiac specific overexpression of leptin receptors normalized cardiac triglyceride levels and diastolic function in db/db “rescue” mice despite these mice being severely obese, hyperglycemic, and hyperlipidemic[35]. This effect was associated with enhanced STAT3 phosphorylation in the hearts suggesting that activation of this pathway is involved in the protection against lipid accumulation in the heart[35].
At the molecular level, leptin can protect against cardiac lipotoxicity through several pathways. One mechanism by which leptin protects against cardiac lipotoxicity is through induction of fatty acid oxidation. Fatty acid oxidation is highly regulated by the AMPK pathway. AMPK can phosphorylate acetyl-CoA carboxylase (ACC) and malonyl CoA decarboxylase (MCD). Phosphorylation of these proteins has opposite effects on their activity which results in decreased levels of malonyl CoA which is the first committed step in lipogenesis and a powerful inhibitor of carnitine palmityl transferase-1 (CPT-1)-mediated fatty acid oxidation (Figure 4). Previous studies in skeletal muscle have demonstrated that leptin can increase AMPK phosphorylation to promote fatty acid oxidation[101]. However, acute leptin treatment (60 min) in the isolated working rat heart stimulated fatty acid oxidation without any changes in AMPK phosphorylation state, ACC activity, or malonyl-CoA levels[55]. These results suggest that leptin may stimulate cardiac fatty acid oxidation through a mechanism that does not include AMPK activation; however, the effects of chronic leptin exposure in vivo have not been determined. Leptin may also promote fatty acid oxidation in the heart by decreasing the sensitivity of CPT-1 to malonyl CoA via an Akt related signaling pathway[102]. Uncoupling proteins are another potential pathway by which leptin may increase fatty acid oxidation. Leptin can act centrally to increase uncoupling protein levels via β-adrenergic mediated mechanism,and the hormone may also directly regulate uncoupling protein levels in skeletal muscle and the heart[103]. Leptin can also increase cardiac fatty acid oxidation through a STAT-3–nitric oxide–p38 MAPK-dependent mechanism (Figure 4)[56]. Leptin may also protect against cardiac lipotoxicity by its actions on fatty acid transport into cardiac myocytes. Leptin has been demonstrated to decrease the levels of both the fatty acid transport protein, fatty acid translocase (FAT/CD36), and plasma membrane-associated fatty acid-binding protein (FABPpm) in skeletal muscle; whereas, it has been reported to increase both FAT/CD36 as well as FATP1 in cultured mouse cardiomyocytes[57]. These results from cultured cells are in opposition to studies in leptin deficient ob/ob mice in which the levels of expression of genes that stimulate fatty acid uptake were increased in the heart[46]. Although the exact mechanism(s) need to be worked out, it is more than likely that the stimulation of fatty acid oxidation by leptin in the heart reduces the amount of lipid intermediates such as ceramide and DAG below toxic levels[104]. This in turn decreases lipid mediated apoptosis and protects cardiac function in obesity (Figure 4).
Figure 4.
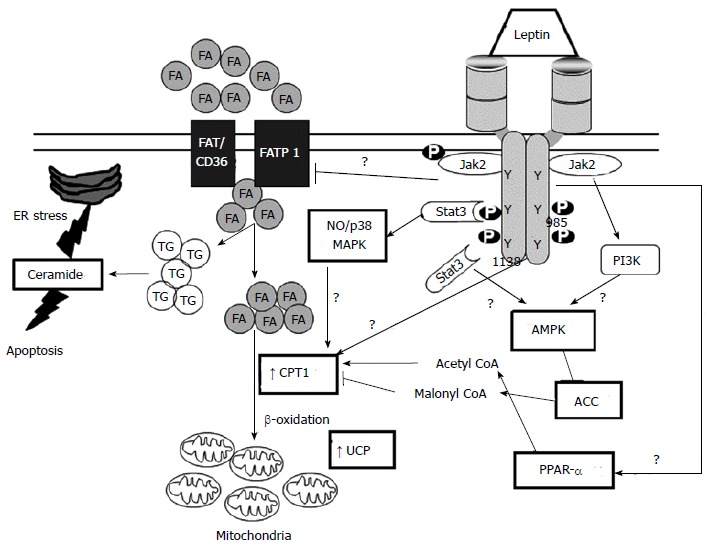
Leptin and cardiac lipotoxicity. Triangular and flat arrowheads represent stimulatory and inhibitory effects on the designed targets, respectively. Heavier lines represent proposed actions of leptin. ACC: Acetyl-CoA carboxylase; AMPK: AMP-activated protein kinase; CPT-1: Carnitine palmityl transferase-1; ER: Endoplasmatic reticulum; FA: Fatty acids; FAT/CD36: Fatty acid translocase/cluster of differentiation 36; FATP1: Fatty acid transport protein 1; NO: Nitric oxide; P-JAK2: Phosphorylated Janus kinase 2; PPARα: Peroxisome proliferator-activated receptor alpha; STAT3: Signal transducer and activator of transcription 3; TG: Triglycerides; UCP: Uncoupling protein.
CONCLUSION
Leptin is a hormone derived from adipose tissue which undoubtedly plays an essential role in the regulation of body weight and appetite. However, emerging studies have demonstrated that leptin is also a critical hormone for the cardiovascular system which can regulate metabolism and function of the heart. It is clear that leptin exerts diverse functions in the heart (Figure 2). While some of leptin’s effects can be deleterious to cardiac function primarily through its central actions on blood pressure and heart rate, its potential growth effects on the heart to promote cardiac hypertrophy are not clear. Leptin does have beneficial actions on myocardial fatty acid and glucose metabolism and the loss of cardiac leptin signaling can adversely affect the heart’s response to stresses such as transient ischemia. Leptin may also directly protect the heart against excessive lipid accumulation in obesity. The development of leptin receptor antagonists has resulted in some investigators suggesting that blockade of leptin signaling in the heart may be beneficial in obesity to attenuate cardiac hypertrophy and improve cardiac function[105-107]. However, given leptin’s beneficial actions on cardiac metabolism and lipid accumulation, any intervention on leptin’s action in the heart must be considered very carefully. Clearly, more animal studies are needed to unravel the biological roles and mechanistic actions of leptin before any interventional studies specifically targeting leptin in the heart are undertaken.
Footnotes
Supported by the National Heart, Lung and Blood Institute, Nos. PO1HL-051971 and R00HL112952; and the National Institute of General Medical Sciences, No. P20GM-104357; and the American Heart Association, No. 14SDG20490339.
Conflict-of-interest statement: The authors declare no financial conflict of interest including: fees for serving as a speaker or as consultant and/or an adcisory bord member. Ownership of any stocks and/or shares or patents releted to research had ribed in this article.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 3, 2015
First decision: June 3, 2015
Article in press: July 23, 2015
P- Reviewer: De Ponti R, Lin GM S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Lane PW, Dickie MM. The effect of restricted food intake on the life span of genetically obese mice. J Nutr. 1958;64:549–554. doi: 10.1093/jn/64.4.549. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 3.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 4.Phillips MS, Liu Q, Hammond HA, Dugan V, Hey PJ, Caskey CJ, Hess JF. Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet. 1996;13:18–19. doi: 10.1038/ng0596-18. [DOI] [PubMed] [Google Scholar]
- 5.Wu-Peng XS, Chua SC, Okada N, Liu SM, Nicolson M, Leibel RL. Phenotype of the obese Koletsky (f) rat due to Tyr763Stop mutation in the extracellular domain of the leptin receptor (Lepr): evidence for deficient plasma-to-CSF transport of leptin in both the Zucker and Koletsky obese rat. Diabetes. 1997;46:513–518. doi: 10.2337/diab.46.3.513. [DOI] [PubMed] [Google Scholar]
- 6.McPherson K, White TN, Johnson A, Geurts A, Jacob H, Garrett M, Williams J. Initial characterization of leptin receptor knockout Dahl salt-sensitive (SS) rats. FASEB J. 2014;28:1121. Available from: http://www.fasebj.org/content/28/1_Supplement/1121.2. [Google Scholar]
- 7.Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest. 2001;108:1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green ED, Maffei M, Braden VV, Proenca R, DeSilva U, Zhang Y, Chua SC, Leibel RL, Weissenbach J, Friedman JM. The human obese (OB) gene: RNA expression pattern and mapping on the physical, cytogenetic, and genetic maps of chromosome 7. Genome Res. 1995;5:5–12. doi: 10.1101/gr.5.1.5. [DOI] [PubMed] [Google Scholar]
- 9.Lin J, Barb CR, Matteri RL, Kraeling RR, Chen X, Meinersmann RJ, Rampacek GB. Long form leptin receptor mRNA expression in the brain, pituitary, and other tissues in the pig. Domest Anim Endocrinol. 2000;19:53–61. doi: 10.1016/s0739-7240(00)00064-3. [DOI] [PubMed] [Google Scholar]
- 10.Kielar D, Clark JS, Ciechanowicz A, Kurzawski G, Sulikowski T, Naruszewicz M. Leptin receptor isoforms expressed in human adipose tissue. Metabolism. 1998;47:844–847. doi: 10.1016/s0026-0495(98)90124-x. [DOI] [PubMed] [Google Scholar]
- 11.Purdham DM, Zou MX, Rajapurohitam V, Karmazyn M. Rat heart is a site of leptin production and action. Am J Physiol Heart Circ Physiol. 2004;287:H2877–H2884. doi: 10.1152/ajpheart.00499.2004. [DOI] [PubMed] [Google Scholar]
- 12.Fei H, Okano HJ, Li C, Lee GH, Zhao C, Darnell R, Friedman JM. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci USA. 1997;94:7001–7005. doi: 10.1073/pnas.94.13.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sweeney G. Leptin signalling. Cell Signal. 2002;14:655–663. doi: 10.1016/s0898-6568(02)00006-2. [DOI] [PubMed] [Google Scholar]
- 14.Cohen P, Yang G, Yu X, Soukas AA, Wolfish CS, Friedman JM, Li C. Induction of leptin receptor expression in the liver by leptin and food deprivation. J Biol Chem. 2005;280:10034–10039. doi: 10.1074/jbc.M413684200. [DOI] [PubMed] [Google Scholar]
- 15.Matsui H, Motooka M, Koike H, Inoue M, Iwasaki T, Suzuki T, Kurabayashi M, Yokoyama T. Ischemia/reperfusion in rat heart induces leptin and leptin receptor gene expression. Life Sci. 2007;80:672–680. doi: 10.1016/j.lfs.2006.10.027. [DOI] [PubMed] [Google Scholar]
- 16.McGaffin KR, Witham WG, Yester KA, Romano LC, O’Doherty RM, McTiernan CF, O’Donnell CP. Cardiac-specific leptin receptor deletion exacerbates ischaemic heart failure in mice. Cardiovasc Res. 2011;89:60–71. doi: 10.1093/cvr/cvq288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsui H, Yokoyama T, Tanaka C, Sunaga H, Koitabashi N, Takizawa T, Arai M, Kurabayashi M. Pressure mediated hypertrophy and mechanical stretch up-regulate expression of the long form of leptin receptor (ob-Rb) in rat cardiac myocytes. BMC Cell Biol. 2012;13:37. doi: 10.1186/1471-2121-13-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paolisso G, Tagliamonte MR, Galderisi M, Zito GA, D’Errico A, Marfella R, Carella C, de Divitiis O, Varricchio M. Plasma leptin concentration, insulin sensitivity, and 24-hour ambulatory blood pressure and left ventricular geometry. Am J Hypertens. 2001;14:114–120. doi: 10.1016/s0895-7061(00)01241-3. [DOI] [PubMed] [Google Scholar]
- 19.Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 20.Vasan RS, Levy D. The role of hypertension in the pathogenesis of heart failure. A clinical mechanistic overview. Arch Intern Med. 1996;156:1789–1796. [PubMed] [Google Scholar]
- 21.Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, Smith G, Stec DE. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem. 2010;285:17271–17276. doi: 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vasan RS. Cardiac function and obesity. Heart. 2003;89:1127–1129. doi: 10.1136/heart.89.10.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajapurohitam V, Gan XT, Kirshenbaum LA, Karmazyn M. The obesity-associated peptide leptin induces hypertrophy in neonatal rat ventricular myocytes. Circ Res. 2003;93:277–279. doi: 10.1161/01.RES.0000089255.37804.72. [DOI] [PubMed] [Google Scholar]
- 24.Madani S, De Girolamo S, Muñoz DM, Li RK, Sweeney G. Direct effects of leptin on size and extracellular matrix components of human pediatric ventricular myocytes. Cardiovasc Res. 2006;69:716–725. doi: 10.1016/j.cardiores.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 25.Xu FP, Chen MS, Wang YZ, Yi Q, Lin SB, Chen AF, Luo JD. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation. 2004;110:1269–1275. doi: 10.1161/01.CIR.0000140766.52771.6D. [DOI] [PubMed] [Google Scholar]
- 26.Tajmir P, Ceddia RB, Li RK, Coe IR, Sweeney G. Leptin increases cardiomyocyte hyperplasia via extracellular signal-regulated kinase- and phosphatidylinositol 3-kinase-dependent signaling pathways. Endocrinology. 2004;145:1550–1555. doi: 10.1210/en.2003-1128. [DOI] [PubMed] [Google Scholar]
- 27.Piñieiro R, Iglesias MJ, Eiras S, Viñuela J, Lago F, González-Juanatey JR. Leptin does not induce hypertrophy, cell cycle alterations, or production of MCP-1 in cultured rat and mouse cardiomyocytes. Endocr Res. 2005;31:375–386. doi: 10.1080/07435800500456937. [DOI] [PubMed] [Google Scholar]
- 28.Paolisso G, Tagliamonte MR, Galderisi M, Zito GA, Petrocelli A, Carella C, de Divitiis O, Varricchio M. Plasma leptin level is associated with myocardial wall thickness in hypertensive insulin-resistant men. Hypertension. 1999;34:1047–1052. doi: 10.1161/01.hyp.34.5.1047. [DOI] [PubMed] [Google Scholar]
- 29.Perego L, Pizzocri P, Corradi D, Maisano F, Paganelli M, Fiorina P, Barbieri M, Morabito A, Paolisso G, Folli F, et al. Circulating leptin correlates with left ventricular mass in morbid (grade III) obesity before and after weight loss induced by bariatric surgery: a potential role for leptin in mediating human left ventricular hypertrophy. J Clin Endocrinol Metab. 2005;90:4087–4093. doi: 10.1210/jc.2004-1963. [DOI] [PubMed] [Google Scholar]
- 30.Shek EW, Brands MW, Hall JE. Chronic leptin infusion increases arterial pressure. Hypertension. 1998;31:409–414. doi: 10.1161/01.hyp.31.1.409. [DOI] [PubMed] [Google Scholar]
- 31.Carlyle M, Jones OB, Kuo JJ, Hall JE. Chronic cardiovascular and renal actions of leptin: role of adrenergic activity. Hypertension. 2002;39:496–501. doi: 10.1161/hy0202.104398. [DOI] [PubMed] [Google Scholar]
- 32.Singhal A, Farooqi IS, Cole TJ, O’Rahilly S, Fewtrell M, Kattenhorn M, Lucas A, Deanfield J. Influence of leptin on arterial distensibility: a novel link between obesity and cardiovascular disease? Circulation. 2002;106:1919–1924. doi: 10.1161/01.cir.0000033219.24717.52. [DOI] [PubMed] [Google Scholar]
- 33.Barouch LA, Berkowitz DE, Harrison RW, O’Donnell CP, Hare JM. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation. 2003;108:754–759. doi: 10.1161/01.CIR.0000083716.82622.FD. [DOI] [PubMed] [Google Scholar]
- 34.Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG. Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens. 1999;17:1949–1953. doi: 10.1097/00004872-199917121-00026. [DOI] [PubMed] [Google Scholar]
- 35.Hall ME, Maready MW, Hall JE, Stec DE. Rescue of cardiac leptin receptors in db/db mice prevents myocardial triglyceride accumulation. Am J Physiol Endocrinol Metab. 2014;307:E316–E325. doi: 10.1152/ajpendo.00005.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolk R, Berger P, Lennon RJ, Brilakis ES, Johnson BD, Somers VK. Plasma leptin and prognosis in patients with established coronary atherosclerosis. J Am Coll Cardiol. 2004;44:1819–1824. doi: 10.1016/j.jacc.2004.07.050. [DOI] [PubMed] [Google Scholar]
- 37.Schulze PC, Kratzsch J, Linke A, Schoene N, Adams V, Gielen S, Erbs S, Moebius-Winkler S, Schuler G. Elevated serum levels of leptin and soluble leptin receptor in patients with advanced chronic heart failure. Eur J Heart Fail. 2003;5:33–40. doi: 10.1016/s1388-9842(02)00177-0. [DOI] [PubMed] [Google Scholar]
- 38.Bobbert P, Jenke A, Bobbert T, Kühl U, Rauch U, Lassner D, Scheibenbogen C, Poller W, Schultheiss HP, Skurk C. High leptin and resistin expression in chronic heart failure: adverse outcome in patients with dilated and inflammatory cardiomyopathy. Eur J Heart Fail. 2012;14:1265–1275. doi: 10.1093/eurjhf/hfs111. [DOI] [PubMed] [Google Scholar]
- 39.Wannamethee SG, Shaper AG, Whincup PH, Lennon L, Sattar N. Obesity and risk of incident heart failure in older men with and without pre-existing coronary heart disease: does leptin have a role? J Am Coll Cardiol. 2011;58:1870–1877. doi: 10.1016/j.jacc.2011.06.057. [DOI] [PubMed] [Google Scholar]
- 40.Lieb W, Xanthakis V, Sullivan LM, Aragam J, Pencina MJ, Larson MG, Benjamin EJ, Vasan RS. Longitudinal tracking of left ventricular mass over the adult life course:clinical correlates of short-and long-term change in the framingham offspring study. Circulation. 2009;119:3085–3092. doi: 10.1161/CIRCULATIONAHA.108.824243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin SS, Blaha MJ, Muse ED, Qasim AN, Reilly MP, Blumenthal RS, Nasir K, Criqui MH, McClelland RL, Hughes-Austin JM, et al. Leptin and incident cardiovascular disease: the Multi-ethnic Study of Atherosclerosis (MESA) Atherosclerosis. 2015;239:67–72. doi: 10.1016/j.atherosclerosis.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hall ME, Smith G, Hall JE, Stec DE. Cardiomyocyte-specific deletion of leptin receptors causes lethal heart failure in Cre-recombinase-mediated cardiotoxicity. Am J Physiol Regul Integr Comp Physiol. 2012;303:R1241–R1250. doi: 10.1152/ajpregu.00292.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hall ME, Smith G, Hall JE, Stec DE. Systolic dysfunction in cardiac-specific ligand-inducible MerCreMer transgenic mice. Am J Physiol Heart Circ Physiol. 2011;301:H253–H260. doi: 10.1152/ajpheart.00786.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koitabashi N, Bedja D, Zaiman AL, Pinto YM, Zhang M, Gabrielson KL, Takimoto E, Kass DA. Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circ Res. 2009;105:12–15. doi: 10.1161/CIRCRESAHA.109.198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barouch LA, Gao D, Chen L, Miller KL, Xu W, Phan AC, Kittleson MM, Minhas KM, Berkowitz DE, Wei C, et al. Cardiac myocyte apoptosis is associated with increased DNA damage and decreased survival in murine models of obesity. Circ Res. 2006;98:119–124. doi: 10.1161/01.RES.0000199348.10580.1d. [DOI] [PubMed] [Google Scholar]
- 46.Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003;144:3483–3490. doi: 10.1210/en.2003-0242. [DOI] [PubMed] [Google Scholar]
- 47.Farooqi IS, O’Rahilly S. 20 years of leptin: human disorders of leptin action. J Endocrinol. 2014;223:T63–T70. doi: 10.1530/JOE-14-0480. [DOI] [PubMed] [Google Scholar]
- 48.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 49.Burcelin R, Kamohara S, Li J, Tannenbaum GS, Charron MJ, Friedman JM. Acute intravenous leptin infusion increases glucose turnover but not skeletal muscle glucose uptake in ob/ob mice. Diabetes. 1999;48:1264–1269. doi: 10.2337/diabetes.48.6.1264. [DOI] [PubMed] [Google Scholar]
- 50.Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature. 1997;389:374–377. doi: 10.1038/38717. [DOI] [PubMed] [Google Scholar]
- 51.Minokoshi Y, Haque MS, Shimazu T. Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes. 1999;48:287–291. doi: 10.2337/diabetes.48.2.287. [DOI] [PubMed] [Google Scholar]
- 52.Haque MS, Minokoshi Y, Hamai M, Iwai M, Horiuchi M, Shimazu T. Role of the sympathetic nervous system and insulin in enhancing glucose uptake in peripheral tissues after intrahypothalamic injection of leptin in rats. Diabetes. 1999;48:1706–1712. doi: 10.2337/diabetes.48.9.1706. [DOI] [PubMed] [Google Scholar]
- 53.Keung W, Cadete VJ, Palaniyappan A, Jablonski A, Fischer M, Lopaschuk GD. Intracerebroventricular leptin administration differentially alters cardiac energy metabolism in mice fed a low-fat and high-fat diet. J Cardiovasc Pharmacol. 2011;57:103–113. doi: 10.1097/FJC.0b013e31820014f9. [DOI] [PubMed] [Google Scholar]
- 54.Haap M, Houdali B, Maerker E, Renn W, Machicao F, Hoffmeister HM, Häring HU, Rett K. Insulin-like effect of low-dose leptin on glucose transport in Langendorff rat hearts. Exp Clin Endocrinol Diabetes. 2003;111:139–145. doi: 10.1055/s-2003-39786. [DOI] [PubMed] [Google Scholar]
- 55.Atkinson LL, Fischer MA, Lopaschuk GD. Leptin activates cardiac fatty acid oxidation independent of changes in the AMP-activated protein kinase-acetyl-CoA carboxylase-malonyl-CoA axis. J Biol Chem. 2002;277:29424–29430. doi: 10.1074/jbc.M203813200. [DOI] [PubMed] [Google Scholar]
- 56.Sharma V, Mustafa S, Patel N, Wambolt R, Allard MF, McNeill JH. Stimulation of cardiac fatty acid oxidation by leptin is mediated by a nitric oxide-p38 MAPK-dependent mechanism. Eur J Pharmacol. 2009;617:113–117. doi: 10.1016/j.ejphar.2009.06.037. [DOI] [PubMed] [Google Scholar]
- 57.Palanivel R, Eguchi M, Shuralyova I, Coe I, Sweeney G. Distinct effects of short- and long-term leptin treatment on glucose and fatty acid uptake and metabolism in HL-1 cardiomyocytes. Metabolism. 2006;55:1067–1075. doi: 10.1016/j.metabol.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 58.Hou N, Luo MS, Liu SM, Zhang HN, Xiao Q, Sun P, Zhang GS, Luo JD, Chen MS. Leptin induces hypertrophy through activating the peroxisome proliferator-activated receptor α pathway in cultured neonatal rat cardiomyocytes. Clin Exp Pharmacol Physiol. 2010;37:1087–1095. doi: 10.1111/j.1440-1681.2010.05442.x. [DOI] [PubMed] [Google Scholar]
- 59.Lopaschuk GD, Folmes CD, Stanley WC. Cardiac energy metabolism in obesity. Circ Res. 2007;101:335–347. doi: 10.1161/CIRCRESAHA.107.150417. [DOI] [PubMed] [Google Scholar]
- 60.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 61.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 62.Labbé SM, Noll C, Grenier-Larouche T, Kunach M, Bouffard L, Phoenix S, Guérin B, Baillargeon JP, Langlois MF, Turcotte EE, et al. Improved cardiac function and dietary fatty acid metabolism after modest weight loss in subjects with impaired glucose tolerance. Am J Physiol Endocrinol Metab. 2014;306:E1388–E1396. doi: 10.1152/ajpendo.00638.2013. [DOI] [PubMed] [Google Scholar]
- 63.Sloan C, Tuinei J, Nemetz K, Frandsen J, Soto J, Wride N, Sempokuya T, Alegria L, Bugger H, Abel ED. Central leptin signaling is required to normalize myocardial fatty acid oxidation rates in caloric-restricted ob/ob mice. Diabetes. 2011;60:1424–1434. doi: 10.2337/db10-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.German JP, Thaler JP, Wisse BE, Oh-I S, Sarruf DA, Matsen ME, Fischer JD, Taborsky GJ, Schwartz MW, Morton GJ. Leptin activates a novel CNS mechanism for insulin-independent normalization of severe diabetic hyperglycemia. Endocrinology. 2011;152:394–404. doi: 10.1210/en.2010-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 66.McGaffin KR, Zou B, McTiernan CF, O’Donnell CP. Leptin attenuates cardiac apoptosis after chronic ischaemic injury. Cardiovasc Res. 2009;83:313–324. doi: 10.1093/cvr/cvp071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Witham W, Yester K, O’Donnell CP, McGaffin KR. Restoration of glucose metabolism in leptin-resistant mouse hearts after acute myocardial infarction through the activation of survival kinase pathways. J Mol Cell Cardiol. 2012;53:91–100. doi: 10.1016/j.yjmcc.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 68.Slawik M, Vidal-Puig AJ. Lipotoxicity, overnutrition and energy metabolism in aging. Ageing Res Rev. 2006;5:144–164. doi: 10.1016/j.arr.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 69.McGavock JM, Victor RG, Unger RH, Szczepaniak LS. Adiposity of the heart, revisited. Ann Intern Med. 2006;144:517–524. doi: 10.7326/0003-4819-144-7-200604040-00011. [DOI] [PubMed] [Google Scholar]
- 70.Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 71.Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res. 2008;49:2101–2112. doi: 10.1194/jlr.M800147-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Unger RH, Orci L. Lipoapoptosis: its mechanism and its diseases. Biochim Biophys Acta. 2002;1585:202–212. doi: 10.1016/s1388-1981(02)00342-6. [DOI] [PubMed] [Google Scholar]
- 73.Shimabukuro M, Higa M, Zhou YT, Wang MY, Newgard CB, Unger RH. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem. 1998;273:32487–32490. doi: 10.1074/jbc.273.49.32487. [DOI] [PubMed] [Google Scholar]
- 74.Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH, Goldberg IJ. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. 2009;284:36312–36323. doi: 10.1074/jbc.M109.049817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smith HL, Willius FA. Adiposity of the heart. Arch Intern Med. 1933;52811:931. [Google Scholar]
- 76.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18:1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 77.Szczepaniak LS, Dobbins RL, Metzger GJ, Sartoni-D’Ambrosia G, Arbique D, Vongpatanasin W, Unger R, Victor RG. Myocardial triglycerides and systolic function in humans: in vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn Reson Med. 2003;49:417–423. doi: 10.1002/mrm.10372. [DOI] [PubMed] [Google Scholar]
- 78.Utz W, Engeli S, Haufe S, Kast P, Hermsdorf M, Wiesner S, Pofahl M, Traber J, Luft FC, Boschmann M, et al. Myocardial steatosis, cardiac remodelling and fitness in insulin-sensitive and insulin-resistant obese women. Heart. 2011;97:1585–1589. doi: 10.1136/hrt.2011.224451. [DOI] [PubMed] [Google Scholar]
- 79.Mahmod M, Bull S, Suttie JJ, Pal N, Holloway C, Dass S, Myerson SG, Schneider JE, De Silva R, Petrou M, et al. Myocardial steatosis and left ventricular contractile dysfunction in patients with severe aortic stenosis. Circ Cardiovasc Imaging. 2013;6:808–816. doi: 10.1161/CIRCIMAGING.113.000559. [DOI] [PubMed] [Google Scholar]
- 80.McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation. 2007;116:1170–1175. doi: 10.1161/CIRCULATIONAHA.106.645614. [DOI] [PubMed] [Google Scholar]
- 81.Nyman K, Granér M, Pentikäinen MO, Lundbom J, Hakkarainen A, Sirén R, Nieminen MS, Taskinen MR, Lundbom N, Lauerma K. Cardiac steatosis and left ventricular function in men with metabolic syndrome. J Cardiovasc Magn Reson. 2013;15:103. doi: 10.1186/1532-429X-15-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu CY, Bluemke DA, Gerstenblith G, Zimmerman SL, Li J, Zhu H, Lai S, Lai H. Myocardial steatosis and its association with obesity and regional ventricular dysfunction: evaluated by magnetic resonance tagging and 1H spectroscopy in healthy African Americans. Int J Cardiol. 2014;172:381–387. doi: 10.1016/j.ijcard.2014.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 84.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005;96:225–233. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- 85.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci USA. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Watanabe K, Fujii H, Takahashi T, Kodama M, Aizawa Y, Ohta Y, Ono T, Hasegawa G, Naito M, Nakajima T, et al. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. J Biol Chem. 2000;275:22293–22299. doi: 10.1074/jbc.M000248200. [DOI] [PubMed] [Google Scholar]
- 88.Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. 2001;276:14890–14895. doi: 10.1074/jbc.M010286200. [DOI] [PubMed] [Google Scholar]
- 89.Brookheart RT, Michel CI, Listenberger LL, Ory DS, Schaffer JE. The non-coding RNA gadd7 is a regulator of lipid-induced oxidative and endoplasmic reticulum stress. J Biol Chem. 2009;284:7446–7454. doi: 10.1074/jbc.M806209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Borradaile NM, Buhman KK, Listenberger LL, Magee CJ, Morimoto ET, Ory DS, Schaffer JE. A critical role for eukaryotic elongation factor 1A-1 in lipotoxic cell death. Mol Biol Cell. 2006;17:770–778. doi: 10.1091/mbc.E05-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Miyamoto S, Murphy AN, Brown JH. Akt mediated mitochondrial protection in the heart: metabolic and survival pathways to the rescue. J Bioenerg Biomembr. 2009;41:169–180. doi: 10.1007/s10863-009-9205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang R, Barouch LA. Leptin signaling and obesity: cardiovascular consequences. Circ Res. 2007;101:545–559. doi: 10.1161/CIRCRESAHA.107.156596. [DOI] [PubMed] [Google Scholar]
- 93.Singh M, Bedi US, Singh PP, Arora R, Khosla S. Leptin and the clinical cardiovascular risk. Int J Cardiol. 2010;140:266–271. doi: 10.1016/j.ijcard.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 94.Unger RH. Hyperleptinemia: protecting the heart from lipid overload. Hypertension. 2005;45:1031–1034. doi: 10.1161/01.HYP.0000165683.09053.02. [DOI] [PubMed] [Google Scholar]
- 95.Yue P, Arai T, Terashima M, Sheikh AY, Cao F, Charo D, Hoyt G, Robbins RC, Ashley EA, Wu J, et al. Magnetic resonance imaging of progressive cardiomyopathic changes in the db/db mouse. Am J Physiol Heart Circ Physiol. 2007;292:H2106–H2118. doi: 10.1152/ajpheart.00856.2006. [DOI] [PubMed] [Google Scholar]
- 96.Huynh K, Kiriazis H, Du XJ, Love JE, Jandeleit-Dahm KA, Forbes JM, McMullen JR, Ritchie RH. Coenzyme Q10 attenuates diastolic dysfunction, cardiomyocyte hypertrophy and cardiac fibrosis in the db/db mouse model of type 2 diabetes. Diabetologia. 2012;55:1544–1553. doi: 10.1007/s00125-012-2495-3. [DOI] [PubMed] [Google Scholar]
- 97.Li YJ, Wang PH, Chen C, Zou MH, Wang DW. Improvement of mechanical heart function by trimetazidine in db/db mice. Acta Pharmacol Sin. 2010;31:560–569. doi: 10.1038/aps.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee Y, Wang MY, Kakuma T, Wang ZW, Babcock E, McCorkle K, Higa M, Zhou YT, Unger RH. Liporegulation in diet-induced obesity. The antisteatotic role of hyperleptinemia. J Biol Chem. 2001;276:5629–5635. doi: 10.1074/jbc.M008553200. [DOI] [PubMed] [Google Scholar]
- 99.Wang MY, Koyama K, Shimabukuro M, Mangelsdorf D, Newgard CB, Unger RH. Overexpression of leptin receptors in pancreatic islets of Zucker diabetic fatty rats restores GLUT-2, glucokinase, and glucose-stimulated insulin secretion. Proc Natl Acad Sci USA. 1998;95:11921–11926. doi: 10.1073/pnas.95.20.11921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee Y, Naseem RH, Duplomb L, Park BH, Garry DJ, Richardson JA, Schaffer JE, Unger RH. Hyperleptinemia prevents lipotoxic cardiomyopathy in acyl CoA synthase transgenic mice. Proc Natl Acad Sci USA. 2004;101:13624–13629. doi: 10.1073/pnas.0405499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Minokoshi Y, Kahn BB. Role of AMP-activated protein kinase in leptin-induced fatty acid oxidation in muscle. Biochem Soc Trans. 2003;31:196–201. doi: 10.1042/bst0310196. [DOI] [PubMed] [Google Scholar]
- 102.Guzmán-Ruiz R, Somoza B, Gil-Ortega M, Merino B, Cano V, Attané C, Castan-Laurell I, Valet P, Fernández-Alfonso MS, Ruiz-Gayo M. Sensitivity of cardiac carnitine palmitoyltransferase to malonyl-CoA is regulated by leptin: similarities with a model of endogenous hyperleptinemia. Endocrinology. 2010;151:1010–1018. doi: 10.1210/en.2009-1170. [DOI] [PubMed] [Google Scholar]
- 103.Steinberg GR, Bonen A, Dyck DJ. Fatty acid oxidation and triacylglycerol hydrolysis are enhanced after chronic leptin treatment in rats. Am J Physiol Endocrinol Metab. 2002;282:E593–E600. doi: 10.1152/ajpendo.00303.2001. [DOI] [PubMed] [Google Scholar]
- 104.Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144:5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- 105.Brydon L. Adiposity, leptin and stress reactivity in humans. Biol Psychol. 2011;86:114–120. doi: 10.1016/j.biopsycho.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Martínez-Martínez E, Jurado-López R, Cervantes-Escalera P, Cachofeiro V, Miana M. Leptin, a mediator of cardiac damage associated with obesity. Horm Mol Biol Clin Investig. 2014;18:3–14. doi: 10.1515/hmbci-2013-0060. [DOI] [PubMed] [Google Scholar]
- 107.Karmazyn M, Rajapurohitam V. Leptin as a cardiac pro-hypertrophic factor and its potential role in the development of heart failure. Curr Pharm Des. 2014;20:646–651. doi: 10.2174/13816128113199990023. [DOI] [PubMed] [Google Scholar]