

Graphical abstract
Keywords: Aurora-A; Imidazo[4,5-b]pyridine; Aurora kinase
Abstract
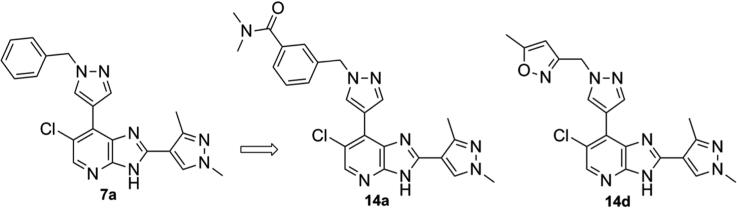
Introduction of a 1-benzyl-1H-pyrazol-4-yl moiety at C7 of the imidazo[4,5-b]pyridine scaffold provided 7a which inhibited a range of kinases including Aurora-A. Modification of the benzyl group in 7a, and subsequent co-crystallisation of the resulting analogues with Aurora-A indicated distinct differences in binding mode dependent upon the pyrazole N-substituent. Compounds 7a and 14d interact with the P-loop whereas 14a and 14b engage with Thr217 in the post-hinge region. These crystallographic insights provide options for the design of compounds interacting with the DFG motif or with Thr217.
Over the last decade, extensive research has been directed towards the discovery of small molecule inhibitors of the Aurora protein kinases as anticancer agents. This effort led to the identification of multiple structurally diverse chemotypes for Aurora kinase modulation.1–5 In addition to small molecules inhibiting all three Aurora isoforms A, B and C, several compounds displaying Aurora isoform selectivity have also been reported including AZD1152 which selectively inhibits Aurora-B,6 and the selective Aurora-A inhibitor MLN8237.7 Several of these small-molecule inhibitors (e.g., PHA-739358,8,9 AZD1152 and MLN8237) have progressed through preclinical development into clinical evaluation for the treatment of a range of human malignancies.1–5
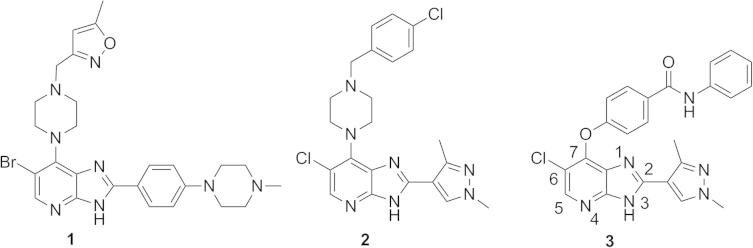
We have previously reported imidazo[4,5-b]pyridine-based inhibitors of Aurora kinases including 1 (CCT137690),10 the dual FLT3/Aurora kinase inhibitor 2 (CCT241736),11 and compound 3 which selectively inhibits Aurora-A over Aurora-B12 (Fig. 1). In our work related to the discovery of 3, modification of the imidazo[4,5-b]pyridine scaffold at C7 included the introduction of a 1-benzyl-1H-pyrazol-4-yl moiety that provided entry into 7-(pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine-based derivatives. Herein, we report our medicinal chemistry effort aimed at improving the pharmacological profile for this class of compounds, and present kinome profiling data that indicate promiscuous kinase inhibition for this subseries. In addition, we report ligand/Aurora-A protein crystallographic data that show different orientations for the substituent on the pyrazole ring suggesting that the 7-(pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine scaffold could be utilised for the design of compounds that additionally interact with the DFG motif or with Thr217, the latter tactic has been demonstrated to enhance Aurora-A over Aurora-B selectivity.12
Figure 1.
Imidazo[4,5-b]pyridine-based inhibitors of Aurora kinases.
Synthesis of 7-substituted imidazo[4,5-b]pyridine derivatives 7a–e (Table 1) is shown in Scheme 1. Key intermediates 5a–e were obtained via Suzuki reaction between 4-chloro-3-nitropyridin-2-amine (4) and the requisite boronic acid or boronic acid pinacol ester coupling partner. 1-(3,4-Difluorobenzyl)-, 1-(4-fluorobenzyl)-, and 1-(4-chlorobenzyl)-1H-pyrazole-4-boronic acid pinacol esters were prepared by heating 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (11) with the appropriate benzyl bromide at 80 °C for 2–3 h in acetonitrile in the presence of Cs2CO3, reaction conditions previously reported for the synthesis of 1-benzyl-3-heterocyclic pyrazoles.13 (1-Benzyl-1H-pyrazol-4-yl)boronic acid and (1-(4-fluorophenyl)-1H-pyrazol-4-yl)boronic acid, required for the synthesis of 5a and 5e respectively, were commercially available. N-chlorosuccinimide-mediated C5-pyridine chlorination was followed by reaction with 1,3-dimethyl-1H-pyrazole-4-carbaldehyde in the presence of Na2S2O4, as previously described,11,12,14 to afford the imidazo[4,5-b]pyridine derivatives 7a–e (Scheme 1, Table 1).
Table 1.
| Compd | R | Aurora-A IC50 (μM) | Aurora-B IC50 (μM) | MLM/HLM% met/30 min |
|---|---|---|---|---|
| 7a |  |
0.212 ± 0.107 | 0.461 ± 0.147 | 40/43 |
| 7b | 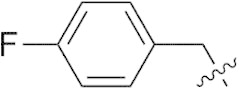 |
0.182 | 0.347 | 52/39 |
| 7c | 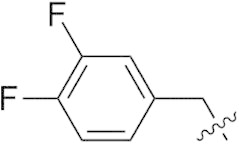 |
0.322 | 0.458 | 31/28 |
| 7d | 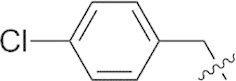 |
0.190 | 0.271 | 37/16 |
| 7e | 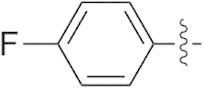 |
>10 | >10 | 31/20 |
Scheme 1.
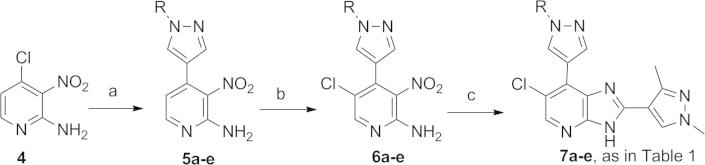
Reagents and conditions: (a) boronic acid or boronic acid pinacol ester, THF/H2O, Pd(dppf)Cl2, Na2CO3, 80 °C, 3.5–26 h or DME, Pd(dppf)Cl2, 1 M aqueous Na2CO3, 150 °C, microwave irradiation, 15 min (for the introduction of 1-(4-fluorophenyl)-1H-pyrazol-4-yl); (b) N-chlorosuccinimide, CH3CN, reflux, 3.5–7 h; (c) EtOH, 1,3-dimethyl-1H-pyrazole-4-carbaldehyde, 1 M aqueous Na2S2O4, 80 °C, 16–20 h.
The 2-amino-3-nitro-pyridine derivative 9 (Scheme 2), key intermediate for the synthesis of 14a and 14b (Table 2), was obtained from 4-chloro-3-nitropyridin-2-amine (4) by a Suzuki cross-coupling reaction to (1-(3-(methoxycarbonyl)benzyl)-1H-pyrazol-4-yl)boronic acid pinacol ester, prepared by reacting 11 with methyl 3-(bromomethyl)benzoate13, followed by C5-pyridine chlorination with N-chlorosuccinimide (Scheme 2). An attempt to prepare 10a directly from the methyl ester intermediate 9 upon treatment with dimethylamine under microwave irradiation resulted in formation of the corresponding carboxylic acid. Coupling of this acid with dimethylamine via HATU carboxyl activation provided 10a (Scheme 2). Starting from the methyl ester intermediate 9, access to 10b was achieved by alkaline ester hydrolysis followed by HATU-mediated coupling with 1-methylpiperazine (Scheme 2). Imidazo[4,5-b]pyridine ring formation to afford 14a and 14b (Table 2) was effected by reacting 10a and 10b with 1,3-dimethyl-1H-pyrazole-4-carbaldehyde in the presence of Na2S2O4 as previously described.11,12,14 The ortho-substituted derivative 14c (Table 2) was prepared by a procedure analogous to that described for its meta-isomer 14b (Scheme 2, Table 2).
Scheme 2.
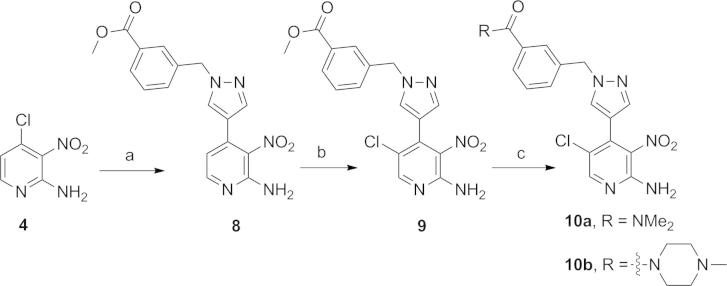
Reagents and conditions: (a) (1-(3-(methoxycarbonyl)benzyl)-1H-pyrazol-4-yl)boronic acid pinacol ester, DMF/H2O, Pd(dppf)Cl2, Na2CO3, 120 °C, microwave irradiation, 0.5 h; (b) N-chlorosuccinimide, CH3CN, reflux, 3.5 h; (c) for 10a: (i) 2 M Me2NH in THF, 105 °C, microwave irradiation, 37 h then remove volatiles; (ii) DMF, HATU, iPr2NEt, room temp, 1.7 h then 2 M Me2NH in THF, room temp, 2.3 h; for 10b: (i) 1 M aqueous KOH, MeOH, 50 °C, 4.5 h; (ii) DMF, HATU, iPr2NEt, room temp, 1 h then 1-methylpiperazine, room temp, 6 h.
Table 2.
| Compd | R | Aurora-A IC50 (μM) | Aurora-B IC50 (μM) | MLM/HLM% met/30 min |
|---|---|---|---|---|
| 14a | 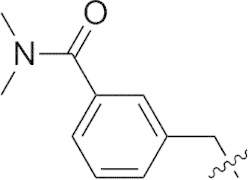 |
0.036 | 0.296 | 72/72 |
| 14b | 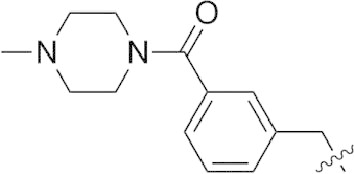 |
0.046 | 0.235 | 97/87 |
| 14c | 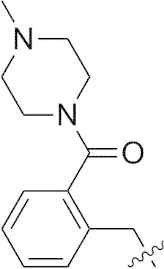 |
0.126 | 0.354 | 98/82 |
| 14d | 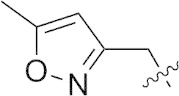 |
0.035 | 0.075 | 35/37 |
| 14e | 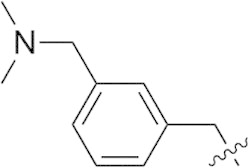 |
0.014 | 0.017 | 97/95 |
| 14f | 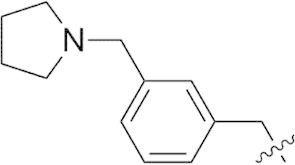 |
0.017 | 0.031 | 100/92 |
| 14g | 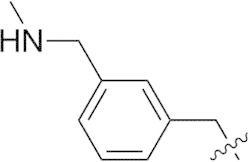 |
0.018 | 0.032 | 53/38 |
Access to 2-amino-3-nitro-pyridine derivative 13 (Scheme 3), key intermediate for the preparation of 14e–g, was achieved by alkylation of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (11) with 3-(bromomethyl)benzaldehyde13 followed by a Suzuki cross-coupling reaction with 4-chloro-3-nitropyridin-2-amine (4) (Scheme 3). Reductive amination of the benzaldehyde moiety in 13 was accomplished under standard conditions (NaBH(OAc)3/AcOH with methylamine or pyrrolidine, and NaBH3CN with dimethylamine). The synthesis of 14e–g (Table 2) was finalised by the C5-pyridine chlorination of the requisite intermediate followed by imidazo[4,5-b]pyridine ring formation as described for the preparation of 7a–e (Scheme 1). Finally, the isoxazole derivative 14d (Table 2) was prepared from 4 by a route analogous to that described for the synthesis of 7a–e (Scheme 1). For this synthetic sequence, (1-((5-methylisoxazol-3-yl)methyl)-1H-pyrazol-4-yl)boronic acid pinacol ester, required for the Suzuki cross-coupling reaction, was prepared by alkylation of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (11) with 3-(bromomethyl)-5-methylisoxazole.13
Scheme 3.
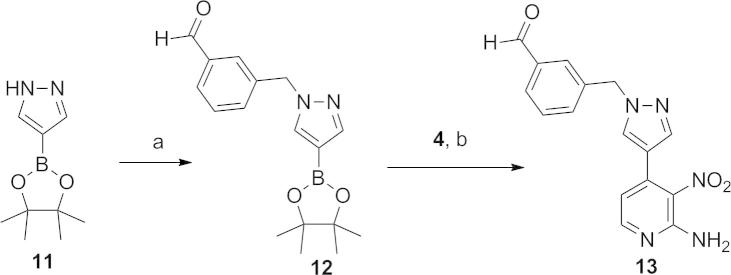
Reagents and conditions: (a) 3-(bromomethyl)benzaldehyde, CH3CN, Cs2CO3, 80 °C, 2 h; (b) THF/H2O, Pd(dppf)Cl2, Na2CO3, 80 °C, 2 h.
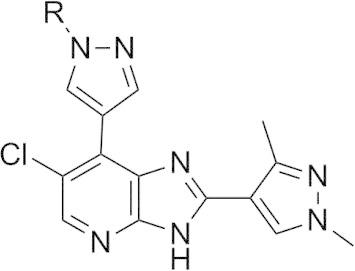
As part of our medicinal chemistry programme to discover selective inhibitors of Aurora-A,12 we introduced the 1-benzyl-1H-pyrazol-4-yl moiety at C7 of the imidazo[4,5-b]pyridine scaffold to provide 7a (Table 1). We previously reported 1,3-dimethyl-1H-pyrazol-4-yl as a preferred substituent at the C2 position of the imidazo[4,5-b]pyridine scaffold11 and this moiety was therefore incorporated into the design of 7a and analogues presented in this study. In biochemical assays, 7a inhibited both Aurora-A and Aurora-B with IC50 values of 0.212 and 0.461 μM, respectively. A slightly improved level of target inhibition was observed in HeLa cervical cancer cells where 7a inhibited the autophosphorylation of Aurora-A at T288 (a cell-based biomarker for Aurora-A inhibition)12,15,16 with an IC50 value of 0.087 μM and histone H3 phosphorylation at S10 (a cell-based biomarker for Aurora-B inhibition)12,15,16 with an IC50 value of 0.223 μM. Regarding cell growth inhibition, 7a inhibited the growth of SW620 and HCT116 human colon carcinoma cells (GI50 = 0.18 and 0.15 μM, respectively).19 Compound 7a displayed moderate mouse and human liver microsomal stability (40% and 43% metabolised, respectively, after a 30 min incubation). This in vitro profile prompted us to investigate this C7-pyrazole substituted imidazo[4,5-b]pyridine subseries in more detail. Firstly, we attempted to increase biochemical potency against Aurora-A, and also to improve microsomal stability.
Introduction of a p-F substituent in the phenyl ring of 7a provided compound 7b which displayed a similar Aurora-A/B inhibitory profile, and human liver microsomal stability to that of 7a. No improvement in Aurora-A inhibitory potency was observed with the introduction of an additional fluorine (compound 7c, Table 1), however the p-chlorobenzyl counterpart (compound 7d, Table 1) displayed similar Aurora inhibitory activities to that of 7b with improved human liver microsomal stability (16% metabolised after 30 min incubation with human liver microsomes). For a better understanding of the microsomal stability data, the protein binding for 7a, 7d and 7e in microsomal incubations was measured. The percentage of 7a unbound in MLM and HLM was determined as 6.71 and 4.89, respectively. Compounds 7d and 7e were more highly bound: for 7d the percentage unbound was 0.50 in both MLM and HLM and for 7e was 0.16 and 0.10, respectively. Based on these data, it is possible that the observed improvement in HLM metabolic stability for 7d compared to 7a is due to lower fraction unbound for the former; notably 7a and 7d display similar MLM stabilities despite the lower fraction unbound for 7d. In HeLa cells, 7d inhibited both Aurora-A (p-T288 IC50 = 0.040 μM) and Aurora-B (p-HH3 IC50 = 0.330 μM); potencies similar to those observed with compound 7a. The direct attachment of a p-fluorophenyl group to the N1-pyrazole was detrimental to Aurora inhibition, with compound 7e inhibiting Aurora-A and Aurora-B with IC50 values greater than 10 μM (Table 1). Cell growth inhibition studies revealed that compounds 7b–d maintained the inhibitory potencies seen with 7a; for example, 7d inhibited the growth of SW620 and HCT116 cells (GI50 = 0.26 and 0.24 μM, respectively). Likewise, 7b and 7c showed potent HCT116 cell growth inhibition (GI50 = 0.20 and 0.13 μM, respectively). We postulated that potent cell growth inhibitory activity relative to biochemical Aurora-A and Aurora-B modulation may be attributable to gain of off-target kinase inhibition in this sub-series, although we acknowledge that translation to more potent cellular inhibition of Aurora-A versus Aurora-A biochemical potency may also be a contributing factor. The potent cell-based activity prompted us to investigate kinome selectivity profiles for this class of compound by screening 7a and 7d in a 102-kinase panel at a concentration of 1 μM.20 Indeed, both compounds inhibited a range of kinases including ERK8, GSK3β, MLK1, JAK2, TrkA and VEGFR greater than 80% (Table S1, Supplementary data) with Gini coefficients21 of 0.273 for 7a and 0.364 for 7d. These Gini coefficient values are significantly lower to those of previously reported 7-(piperazin-1-yl)-3H-imidazo[4,5-b]pyridine- and 7-phenoxy-3H-imidazo[4,5-b]pyridine-based inhibitors of Aurora kinases.10,12 The Gini coefficient for compound 3 was reported as 0.719,12 and the corresponding value for compound 1 is 0.560. To understand the potential for further improvement, we obtained the crystal structure of 7a bound to Aurora-A22 (Fig. 3, Table S3, Supplementary data). This structure shows the ligand in the ATP binding site with the pyridine nitrogen atom hydrogen bonded to the backbone NH of Ala213 and the imidazole NH interacting with the carbonyl of Ala213, consistent with previous reports of the hinge binding mode for the imidazo[4,5-b]pyridine scaffold.10–12 Similar to crystal structures of previous compounds 1, 15 and 16 (Figs. 1 and 2; compounds 51 and 40c in Ref. 10, and compound 21a in Ref. 11, respectively) the N-benzyl substituent on the C7-pyrazole is oriented towards the P-loop, making Van der Waals contacts with Val147 and Gly142 (Fig. 3).
Figure 3.
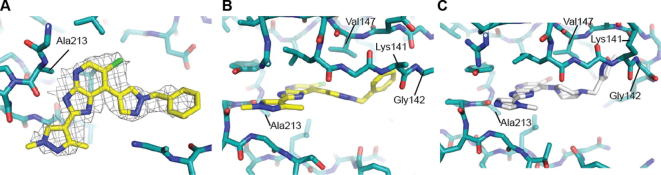
Crystal structure of 7a bound to Aurora-A at 3.1 Å resolution. (A) The ATP binding site viewed from above. The P-loop has been removed to provide a clear view of the ligand, shown with carbon atoms coloured yellow, and the surrounding protein, shown with carbon atoms coloured teal. The final 2mFo-DFc electron density map is shown as a wire-mesh, contoured at 1 σ. (B) Side view with the P-loop shown and key interacting residues labelled. (C) Crystal structure of 16 (compound 21a in Ref. 11; carbon atoms coloured white) bound to Aurora-A with the P-loop shown and key interacting residues labelled. Structure taken from the Protein Data Bank (PDB) ID 4B0G.
Figure 2.
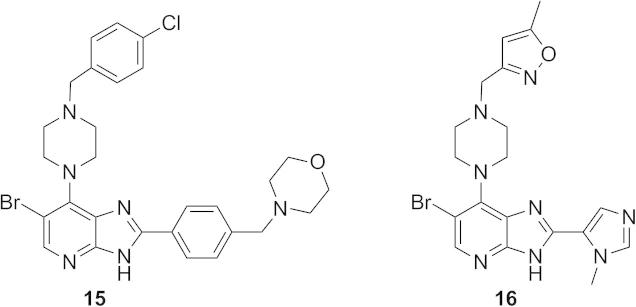
Imidazo[4,5-b]pyridine-based inhibitors of Aurora kinases co-crystallised with Aurora-A: compound 15: PDB ID 2X6D;10 compound 16: PDB ID 4B0G.11
In vivo mouse pharmacokinetic profiling of 7d revealed low oral bioavailability (16%) with moderate clearance (0.016 L/h, 13.3 mL/min/kg) and volume of distribution (0.02 L, 1.0 L/kg). Similar pharmacokinetic parameters were observed for 7a with low oral bioavailability (13%), moderate clearance (0.012 L/h, 10.0 mL/min/kg) and volume of distribution (0.01 L, 0.5 L/kg). The mouse plasma protein binding for 7a and 7d was determined as 99.48% and >99.9%, respectively. We determined low kinetic solubility for both 7a and 7d (<0.0001 mg/mL in phosphate buffer, pH = 6.8)23 which may be a contributing factor to the observed low oral bioavailability. In an attempt to improve the aqueous solubility, the Aurora inhibitory potency and kinase selectivity of this sub-series, we explored the introduction of basic substituents such as 1-methylpiperazine and pyrrolidine as well as replacement of the phenyl ring with a more polar heterocycle. This approach was guided by the ligand/protein interactions observed in the protein crystal structure of 7a, which showed that the N-benzyl substituent on the C7-pyrazole is oriented towards the P-loop (Fig. 4). It was anticipated that the introduction of small substituents on the phenyl ring in 7a would be well tolerated without altering its orientation in the kinase active site. Alternatively, we envisaged that Thr217 in Aurora-A may be accessed via an appropriate pyrazole N1-benzyl derivatisation such as a bulky amido substituent, which would form a favourable hydrogen bond interaction with the side chain hydroxyl of Thr217. Similar approaches were previously applied by us in the design of selective inhibitors of Aurora-A.12,16
Figure 4.

Comparison of crystal structure of Aurora-A bound to 7a and published crystal structures from this series. (A) Overlay of structures showing superposed protein backbones (all coloured teal) with 7a (carbon atoms coloured yellow), and published compounds 15, 1 and 16 taken from PDB IDs 2X6D, 2X6E and 4B0G, respectively. (B–E) Magnified view of the interactions between compounds 7a, 15, 1 and 16 respectively and the P-loop with the protein surface shown.
Replacement of the phenyl ring in 7a with 5-methylisoxazole (compound 14d) led to a significant improvement in Aurora inhibition in our biochemical assays (Aurora-A IC50 = 0.035 μM, Aurora-B IC50 = 0.075 μM; Table 2) but promiscuous kinase inhibition remained. In a panel of 105 kinases at a compound concentration of 1 μM, 22 proteins including ERK8, MLK1, JAK2, TrkA and VEGFR were inhibited by ⩾80% (Gini coefficient = 0.320, Table S2, Supplementary data). Compound 14d inhibited the growth of SW620 and HCT116 cells with GI50 values of 0.35 and 0.34 μM respectively, similar to the cell growth inhibition observed with 7d. The crystal structure of 14d bound to Aurora-A22,24,25 (Fig. 5, Table S3, Supplementary data) shows a similar binding mode to that adopted by 7a; however, the P-loop of Aurora-A adopts a different conformation, stabilised by interactions with the 5-methylisoxazole; this P-loop conformation is very similar to that previously observed for compound 1 (compound 51 in Ref. 10).
Figure 5.
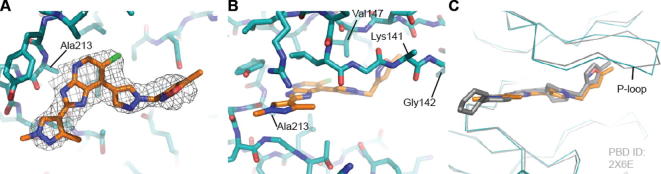
Crystal structure of 14d bound to Aurora-A at 3.1 Å resolution. (A) The ATP binding site viewed from above. The P-loop has been removed to provide a clear view of the ligand, shown with carbon atoms coloured orange and the surrounding protein, shown with carbon atoms coloured teal. The final 2mFo-DFc electron density map is shown as a wire-mesh, contoured at 1 σ. (B) Side view with the P-loop shown and key interacting residues labelled. (C) Overlay of the structures of Aurora-A bound to compound 14d (coloured as before) and compound 1 (compound and protein coloured grey).
The m-dimethylbenzamide derivative 14a (Table 2) was a more potent inhibitor of Aurora-A compared to 7a but its human liver microsomal stability was lower (72% metabolised after a 30 min incubation). A similar trend was observed with the m-1-methylpiperazine derivative 14b which was also a more potent inhibitor of Aurora-A compared to 7a but with poor metabolic stability in both mouse and human liver microsomes (Table 2). Both 14a and 14b were more potent (by 8- and 5-fold, respectively) inhibitors of Aurora-A relative to Aurora-B (Table 2). The crystal structures of 14a and 14b bound to Aurora-A22,24,25 were determined (Table S3, Supplementary data; Figs. 6 and 7) and revealed a consistent difference in the binding mode of these ligands when compared to 7a and 14d. Bulkier substituents on the pyrazole group no longer interact with the pocket formed by the two β-strands of the P-loop. Instead, these bulkier groups are positioned between the β1 strand of the P-loop and the post-hinge region. This orientation places them in close proximity to Thr 217, with a minimum interaction distance of 3.8 Å, which may explain the selectivity of these compounds towards Aurora-A. Notably, Aurora-B and Aurora-C have a glutamic acid at the equivalent position to Thr217, and computational modelling of this Thr to Glu replacement suggests that the bulkier R-groups of 14a and 14b may invoke a steric clash with a glutamic acid at position 217 (Fig. 6C, Fig. 7C).12
Figure 6.
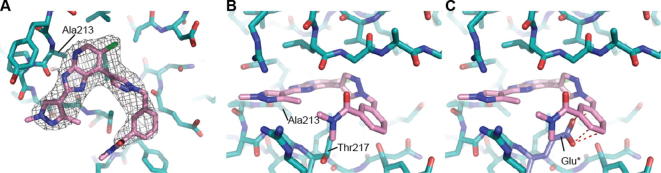
Crystal structure of 14a bound to Aurora-A at 2.8 Å resolution. (A) The ATP binding site viewed from above. The P-loop has been removed to provide a clear view of the ligand, shown with carbon atoms coloured pink and the surrounding protein, shown with carbon atoms coloured teal. The final 2mFo-DFc electron density map is shown as a wire-mesh, contoured at 1 σ. (B) Side view with the P-loop shown and Ala213, Thr217 labelled. (C) Modelling a glutamic acid in position 217 (Glu∗), as found in Aurora-B, leads to steric clashes with the ligand, modelled clash shown as red dashes.
Figure 7.
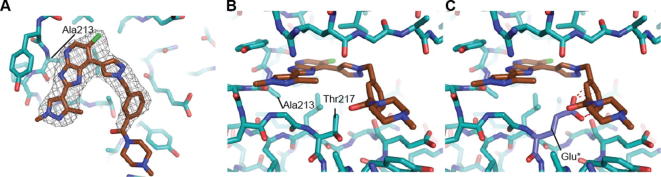
Crystal structure of 14b bound to Aurora-A at 2.9 Å resolution. (A) The ATP binding site viewed from above. The P-loop has been removed to provide a clear view of the ligand, shown with carbon atoms coloured brown and the surrounding protein, shown with carbon atoms coloured teal. The final 2mFo-DFc electron density map is shown as a wire-mesh, contoured at 1 σ. (B) Side view with the P-loop shown and Ala213, Thr217 residues labelled. (C) Modelling a glutamic acid in position 217 (Glu∗), as found in Aurora-B, leads to steric clashes with the ligand, modelled clash shown as red dashes.
The o-regioisomer of 14b (compound 14c) displayed a similar profile to that of 14b although its potency against Aurora-A was marginally lower and selectivity over Aurora-B was eroded (Table 2). The introduction of the smaller m-dimethylamino basic substituent (compound 14e) led to potent inhibition of both Aurora-A and Aurora-B although mouse and human liver microsomal stability was poor (Table 2). Likewise, the m-pyrrolidine derivative 14f potently inhibited both Aurora-A (IC50 = 0.017 μM) and Aurora-B (IC50 = 0.031 μM) but was also highly metabolised (Table 2). The potent Aurora-A and Aurora-B activity of these compounds suggests that they do not adopt a binding mode involving interaction with Thr217 in Aurora-A similar to 14a and 14b, but might bind similarly to 7a and 14d. Unfortunately, we have been unable to resolve the binding mode of compounds 14e and 14f by co-crystal structure determination to verify this hypothesis. The m-methylamino counterpart of 14e (compound 14g) also displayed potent inhibition of both Aurora-A (IC50 = 0.018 μM) and Aurora-B (IC50 = 0.032 μM) and demonstrated improved metabolic stability compared to 14e and 14f (Table 2). Compound 14g inhibited the growth of SW620 and HCT116 cells (GI50 = 0.34 and 0.23 μM, respectively); however, profiling in a 105-kinase panel at a concentration of 1 μM revealed 30 kinases including ERK8, GSK3β, MLK1, JAK2, TrkA and VEGFR that were inhibited by ⩾80% (Gini coefficient = 0.267, Table S2, Supplementary data). Based on these findings, our interest in this 7-(pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine-based sub-series was discontinued and our efforts focussed on compound 3 (Fig. 1) as a highly selective inhibitor of Aurora-A kinase.12 However, the kinase inhibitory profiles of compounds 7a, 7d, 14d, and 14g suggest that the 7-(pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine scaffold could serve as the basis for a multitargeted kinase inhibitor design. In addition, the ligand/Aurora-A protein crystallographic data indicate distinct differences in the orientation of the pyrazole N-substituent. The phenyl group in 7a and the 5-methylisoxazole in 14d point towards the P-loop, whereas the bulkier benzamido substituted phenyl rings in 14a and 14b position between the β1 strand of the P-loop in close proximity to Thr217 and also to the DFG motif. These ligand/protein crystallographic insights could therefore be further exploited in the design of compounds interacting with the DFG motif or in enhancing the selectivity for Aurora-A inhibition over Aurora-B.
Acknowledgments
This work was supported by CRUK Grant numbers C309/A8274 and C309/A11566. S.L. is also supported by Breakthrough Breast Cancer. R.B. acknowledges the support of a Royal Society Research Fellowship, Breakthrough Breast Cancer Project Grant AURA 05/06 and MRC CASE studentship MR/K016903/1. We acknowledge NHS funding to the NIHR Biomedical Research Centre at The Institute of Cancer Research and the Royal Marsden NHS Foundation Trust. We also thank Dr Amin Mirza, Mr Meirion Richards, Dr Maggie Liu for their assistance with NMR, mass spectrometry and HPLC, and Dr Charlotte Dodson for Aurora-A protein production. We thank Diamond Light Source for beamtime and the staff of beamlines I03 and I04 for assistance with data collection.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2015.08.003.
Contributor Information
Vassilios Bavetsias, Email: vassilios.bavetsias@icr.ac.uk.
Richard Bayliss, Email: richard.bayliss@leicester.ac.uk.
Julian Blagg, Email: julian.blagg@icr.ac.uk.
Supplementary data
Supplementary Tables S1–S3.
References and notes
- 1.Pollard J.R., Mortimore M. J. Med. Chem. 2009;52:2629. doi: 10.1021/jm8012129. [DOI] [PubMed] [Google Scholar]
- 2.Green M.R., Woolery J.E., Mahadevan D. Expert Opin. Drug Discov. 2011;6:291. doi: 10.1517/17460441.2011.555395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheung C.H.A., Coumar M.S., Chang J.-Y., Hsieh H.-P. Expert Opin. Ther. Patents. 2011;21:857. doi: 10.1517/13543776.2011.574614. [DOI] [PubMed] [Google Scholar]
- 4.Kollareddy M., Zheleva D., Dzubak P., Brahmkshatriya P.S., Lepsik M., Hajduch M. Invest New Drugs. 2012;30:2411. doi: 10.1007/s10637-012-9798-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gautschi O., Heighway J., Mack P.C., Purnell P.R., Lara P.N., Jr., Gandara D.R. Clin. Cancer Res. 2008;14:1639. doi: 10.1158/1078-0432.CCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 6.Mortlock A.A., Foote K.M., Heron N.M., Jung F.H., Pasquet G., Lohmann J.-J.M., Warin N., Renaud F., De Savi C., Roberts N.J., Johnson T., Dousson C.B., Hill G.B., Perkins D., Hatter G., Wilkinson R.W., Wedge S.R., Heaton S.P., Odedra R., Keen N.J., Crafter C., Brown E., Thompson K., Brightwell S., Khatri L., Brady M.C., Kearney S., McKillop D., Rhead S., Parry T., Green S. J. Med. Chem. 2007;50:2213. doi: 10.1021/jm061335f. [DOI] [PubMed] [Google Scholar]
- 7.Manfredi M.G., Ecsedy J.A., Chakravarty A., Silverman L., Zhang M., Hoar K.M., Stroud S.G., Chen W., Shinde V., Huck J.J., Wysong D.R., Janowick D.A., Hyer M.L., LeRoy P.J., Gershman R.E., Silva M.D., Germanos M.S., Bolen J.B., Claiborne C.F., Sells T.B. Clin. Cancer Res. 2011;17:7614. doi: 10.1158/1078-0432.CCR-11-1536. [DOI] [PubMed] [Google Scholar]
- 8.Fancelli D., Moll J., Varasi M., Bravo R., Artico R., Berta D., Bindi S., Cameron A., Candiani I., Cappella P., Carpinelli P., Croci W., Forte B., Giorgini M.L., Klapwijk J., Marsiglio A., Pesenti E., Rocchetti M., Roletto F., Severino D., Soncini C., Storici P., Tonani R., Zugnoni P., Vianello P. J. Med. Chem. 2006;49:7247. doi: 10.1021/jm060897w. [DOI] [PubMed] [Google Scholar]
- 9.Caprinelli P., Ceruti R., Giorgini M.L., Cappella P., Gianellini L., Croci V., Degrassi A., Texido G., Rocchetti M., Vianello P., Rusconi L., Storici P., Zugnoni P., Arrigoni C., Soncini C., Alli C., Patton V., Marsiglio A., Ballinari D., Pesenti E., Fancelli D., Moll J. Mol. Cancer Ther. 2007;6:3158. doi: 10.1158/1535-7163.MCT-07-0444. [DOI] [PubMed] [Google Scholar]
- 10.Bavetsias V., Large J.M., Sun C., Bouloc N., Kosmopoulou M., Matteucci M., Wilsher N.E., Martins V., Reynisson J., Atrash B., Faisal A., Urban F., Valenti M., de Haven Brandon A., Box G., Raynaud F.I., Workman P., Eccles S.A., Bayliss R., Blagg J., Linardopoulos S., McDonald E. J. Med. Chem. 2010;53:5213. doi: 10.1021/jm100262j. [DOI] [PubMed] [Google Scholar]
- 11.Bavetsias V., Crumpler S., Sun C., Avery S., Atrash B., Faisal A., Moore A.S., Kosmopoulou M., Brown N., Sheldrake P.W., Bush K., Henley A., Box G., Valenti M., de Haven Brandon A., Raynaud F.I., Workman P., Eccles S.A., Bayliss R., Linardopoulos S., Blagg J. J. Med. Chem. 2012;55:8721. doi: 10.1021/jm300952s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bavetsias V., Faisal A., Crumpler S., Brown N., Kosmopoulou M., Joshi A., Atrash B., Pérez-Fuertes Y., Schmitt J.A., Boxall K.J., Burke R., Sun C., Avery S., Bush K., Henley A., Raynaud F.I., Workman P., Bayliss R., Linardopoulos S., Blagg J. J. Med Chem. 2013;56:9122. doi: 10.1021/jm401115g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curtis M.P., Sammons M.F., Piotrowski D.W. Tetrahedron Lett. 2009;50:5479. [Google Scholar]
- 14.Yang D., Fokas D., Li J., Yu L., Baldino C.M. Synthesis. 2005:47. [Google Scholar]
- 15.Manfredi M.G., Ecsedy J.A., Meetze K.A., Balani S.K., Burenkova O., Chen W., Galvin K.M., Hoar K.M., Huck J.J., LeRoy P.J., Ray E.T., Sells T.B., Stringer B., Stroud S.G., Vos T.J., Weatherhead G.S., Wysong D.R., Zhang M., Bolen J.B., Claiborne C.F. Proc. Natl. Acad. Sci. U.S.A. 2007;104:4106. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouloc N., Large J.M., Kosmopoulou M., Sun C., Faisal A., Matteucci M., Reynisson J., Brown N., Atrash B., Blagg J., McDonald E., Linardopoulos S., Bayliss R., Bavetsias V. Bioorg. Med. Chem. Lett. 2010;20:5988. doi: 10.1016/j.bmcl.2010.08.091. [DOI] [PubMed] [Google Scholar]
- 17.Aurora-A and Aurora-B IC50 values were determined by using a microfluidic assay which monitors the separation of a phosphorylated product from its substrate, as described in Ref. 12.
- 18.MLM and HLM microsomal stabilities were determined as described in Ref. 12.
- 19.Chan F., Sun C., Perumal M., Nguyen Q.-D., Bavetsias V., McDonald E., Martins V., Wilsher N., Raynaud F.I., Valenti M., Eccles S., te Poele R., Workman P., Aboagye E.O., Linardopoulos S. Mol. Cancer Ther. 2007;6:3147. doi: 10.1158/1535-7163.MCT-07-2156. GI50 values were determined after 72 h incubation as described in the following reference: [DOI] [PubMed] [Google Scholar]
- 20.Kinase selectivity profiling for compounds 7a, 7d, 14d and 14g was carried out at the International Centre for Protein Kinase Profiling, Division of Signal Transduction Therapy, University of Dundee.
- 21.Graczyk P.P. J. Med. Chem. 2007;50:5773. doi: 10.1021/jm070562u. [DOI] [PubMed] [Google Scholar]
- 22.The crystal structure of Aurora-A in complex with compound 7a was determined as in Ref. 12. Other co-crystal structures used a mutant form of Aurora-A kinase domain (C290A, C393A), which has enhanced expression and generates superior crystals (Refs. 24,25). Hexagonal bipyramidal shaped crystals of Aurora A C290A,C393A were grown at 18 °C by hanging drop vapour diffusion in a 1 μL + 1 μL mixture of mother liquor (0.1 M Tris, pH 8.9; 0.2 M lithium sulfate; 30% w/v PEG 400) and Aurora A (12 mg/mL, 1 mM inhibitor compound, 5 mM MgCl2). Crystals were cryoprotected using 25% (v/v) ethylene glycol in mother liquor before cryo-cooling in liquid nitrogen. The structure was solved using molecular replacement with PDB:4CEG as the model and refinement was carried out as described in Ref. 12. Coordinates and structure factors have been deposited in the protein data bank with accession codes 5AAD, 5AAE, 5AAF, 5AAG (corresponding to the Aurora-A co-crystal structures of 7a, 14d, 14a and 14b, respectively).
- 23.Solubility measurements were performed by Pharmorphix® Solid State Services, Member of the Sigma–Aldrich Group, Cambridge, UK.
- 24.Rowan F.C., Richards M., Bibby R.A., Thompson A., Bayliss R., Blagg J. ACS Chem. Biol. 2013;8:2184. doi: 10.1021/cb400425t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burgess S.G., Bayliss R. Acta Crystallogr. F Struct. Biol. Commun. 2015;71:315. doi: 10.1107/S2053230X15002290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables S1–S3.